The Plasma Membrane: A Platform for Intra- and Intercellular Redox Signaling


1
Institute of Microbiology and Genetics, Department of Genetics of Eukaryotic Microorganisms, Georg August University Göttingen, Grisebachstr. 8, D-37077 Göttingen, Germany
2
Protein Transport and Secretion Unit, Division of Genetics and Cell Biology, Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS) Ospedale San Raffaele, Università Vita-Salute San Raffaele, 20132 Milan, Italy
*
Authors to whom correspondence should be addressed.
Antioxidants 2018, 7(11), 168; https://doi.org/10.3390/antiox7110168
Submission received: 31 October 2018
/
Revised: 15 November 2018
/
Accepted: 17 November 2018
/
Published: 20 November 2018
(This article belongs to the Special Issue Thiol Redox Systems in Health and Disease)
Abstract
:Membranes are of outmost importance to allow for specific signal transduction due to their ability to localize, amplify, and direct signals. However, due to the double-edged nature of reactive oxygen species (ROS)—toxic at high concentrations but essential signal molecules—subcellular localization of ROS-producing systems to the plasma membrane has been traditionally regarded as a protective strategy to defend cells from unwanted side-effects. Nevertheless, specialized regions, such as lipid rafts and caveolae, house and regulate the activated/inhibited states of important ROS-producing systems and concentrate redox targets, demonstrating that plasma membrane functions may go beyond acting as a securing lipid barrier. This is nicely evinced by nicotinamide adenine dinucleotide phosphate (NADPH)-oxidases (NOX), enzymes whose primary function is to generate ROS and which have been shown to reside in specific lipid compartments. In addition, membrane-inserted bidirectional H2O2-transporters modulate their conductance precisely during the passage of the molecules through the lipid bilayer, ensuring time-scaled delivery of the signal. This review aims to summarize current evidence supporting the role of the plasma membrane as an organizing center that serves as a platform for redox signal transmission, particularly NOX-driven, providing specificity at the same time that limits undesirable oxidative damage in case of malfunction. As an example of malfunction, we explore several pathological situations in which an inflammatory component is present, such as inflammatory bowel disease and neurodegenerative disorders, to illustrate how dysregulation of plasma-membrane-localized redox signaling impacts normal cell physiology.
1. Introduction
When looking to the world surrounding us, it becomes pretty clear that animals and plants are adapted to the particular conditions of the habitats in which they live. Thus, Emperor penguins have four layers of scale-like feathers that isolate them from the cold Antarctic wind, and the Saguaro cactus’s spines point down to conduct into its folds and its base the rare rain that falls in the Sonoran Desert. These adaptations, developed over billions of years and collectively defined as evolution, are preceded by parallel slow changes at the cellular level. However, the extracellular environment is not static through time. Composed by multiple elements and varying even from minute to minute, cells are continuously exposed to an almost infinite combination of environmental states. To adapt to these changes, cells are forced to communicate, implying transmission of messages in the appropriate time-frame to mount an adequate cellular response.
Many of the molecules that are involved in cellular communication never enter the target cell. Instead, these first messengers bind to specific receptors on cell surfaces, triggering a rapid increase in the intracellular levels of a so-called second messenger [1]. This second messenger is normally a small non-protein molecule that readily diffuses inside the cell. Within the cell, it further regulates the activity of multiple signaling proteins, thereby expanding transmission of information downstream through receptor activation in a non-linear way. Amplification, diversification, and distribution of the original signal to all relevant subcellular compartments are thus achieved, ensuring a proper on-time response of the cell as a whole to each stimulus. In addition, the levels of a second messenger can be influenced by multiple independent upstream inputs at the same time, allowing for a more precise modulation of the outcome of a signal. Therefore, cellular response will not only depend on the specific cell stage in which a signal arrives to the plasma membrane, but also on the amplitude, kinetics, and spatial distribution of the second messenger involved.
To grant control of the velocity, duration, and fidelity of transmission, second messengers are characterized by four basic aspects: (i) they are either enzymatically generated or released in a regulated manner into the cytosol through channels; (ii) they are either enzymatically degraded or have their basal levels restored by the action of pumps or through diffusion and reaction with their targets; (iii) their intracellular levels rise and fall within a short time, creating gradients from their origin that determine their effectivity; and (iv) they are specific in action. These features have been defined after decades of intense research on molecules with a widely recognized function as second messengers, such as cyclic adenosine monophosphate (cAMP) [2], diacylglycerol (DAG) [3], nitric oxide (NO) [4], and Ca2+ [5], which represents the prototypic second messenger in living cells. Remarkably, some other molecules, despite the substantial body of evidence that, through the years, has linked them with signal transduction [6], have become accepted as second messengers only recently [7,8]. As one of them, reactive oxygen species (ROS) have been traditionally considered as unavoidable toxic wastes that result from metabolic activity, or as the noxious payback for a life developed under aerobic conditions. Indeed, the collective term ROS has too often been used with laxity to group together all of the molecular intermediates derived from the sequential reduction of molecular oxygen (O2), even though they present notable differences in terms of stability and reactivity.
2. ROS: Signals, Second Messengers, or Simply Foes?
It is generally accepted that the generation of ROS by non-enzymatic mechanisms is a collateral result of ATP synthesis in mitochondria, with production ratios that depend on the cellular metabolic rate and the availability of the initial substrate, O2 [9,10]. However, ROS are also generated on purpose in living cells: to date, 31 ROS-generating protein systems have been described in different locations [11], potentially allowing for enzymatic control of ROS production in response to a stimulus. Importantly, in most cases, a cellular membrane (either the plasma membrane or an organelle membrane) separates these enzymes from their putative targets [12].
O2 in its ground-state is a bi-radical, meaning that it contains two unpaired electrons with parallel spins in its outer valence shell. The unusual electron configuration precludes its direct reaction with many molecules. This includes divalent reductants, implying that the most common mechanism of O2 reduction involves the transfer of a single electron (monovalent reduction). The resulting molecules can be either free radicals (containing an unpaired electron in its outer orbit), including a superoxide anion (O2•−) and a hydroxyl radical (OH•), or non-radical oxidants, such as hydrogen peroxide (H2O2) (Figure 1). These molecules differ in their reactivity, target specificity, half-life, and lipid solubility, and thus are more or less suitable for signaling proposes.
The highly instable and reactive superoxide anion (O2•−) is the most proximal product of the one-electron reduction of O2. It is produced mainly by flavoproteins, such as NADH dehydrogenase in mitochondria [9], NADPH oxidases (NOX) [13], or electron carriers, such as CoQH2 [10,14]. Typical substrates of O2•− are iron–sulfur [Fe-S] clusters, whose activation state is frequently regulated by O2•−-mediated oxidation [15,16]. Independently of [Fe-S]-containing proteins, O2•− oxidative activity has been implicated in cellular processes such as autophagy [17] and epigenetic control of gene expression [18]. The question of whether O2•− is a relevant signal depends mainly on the scavenging enzyme superoxide dismutase (SOD) [19], as the rate constant for the reaction with SODs is very rapid (2 × 109 M−1 s−1) and the cytosolic concentration of these enzymes greatly exceeds the steady-state concentration of their substrate [20]. In other words, O2•− would only be able to operate as a signaling molecule within a very short distance from its site of generation to avoid dismutation by SODs. Notably, the persistence of a particular redox molecule is intimately linked to the redox environment in which it is produced [21], and it would be feasible that O2•− becomes a much more relevant signal when the cellular steady state shifts to a more oxidizing profile [22] in which SODs could be product-inactivated [23].
The product of a spontaneous or SOD-catalyzed O2•− conversion is hydrogen peroxide (H2O2) [24]. The role of this oxidant in signal transduction is nowadays widely accepted, since its chemistry fully adapts to the characteristics that qualify a molecule as a second messenger. First of all, all electrons in H2O2 are paired (Figure 1), entailing that it is uncharged and relatively unreactive at physiological pH, a fact that redounds on a higher lifetime and stability. Second, H2O2 can be produced by several enzymatic systems, mostly through univalent reduction of O2•− but also directly [11], and is degraded by devoted protein scavengers (catalase, peroxiredoxins, glutathione peroxidases) [25]. Finally, additional strategies, such as compartmentalization of H2O2 production and regulated distribution using dedicated membrane channels, are employed to preserve homeostatic control of its levels [26]. In a similar way to intracellular Ca2+ storage and regulated release, this organizational scheme may contribute to achieve spatiotemporal specificity, allowing for the formation of steep gradients that differentially activate redox-sensitive pathways.
Notwithstanding, H2O2 can be further reduced to the hydroxyl radical (OH•) in the presence of reduced transition metals, such as iron and copper (Fenton Reaction). This radical is highly unstable and quite unselective in oxidation of target molecules and cannot, like O2•− and H2O2, be eliminated by an enzymatic reaction [27]. Therefore, its disposal is mainly the result of its reaction with other macromolecules that are situated in the immediate environment. Analogously to O2•−, the reactivity of OH• is not a total impediment to its function as a signal in cells: it is conceivable that, under the extreme oxidative conditions in which OH• generation is favored, its reactive nature is exploited to promote a particular cell response, even to activate cell death mechanisms. In that case, OH• may be considered both a signal and an executioner. If this turns out to be true, the lack of specificity brought about by the fast reaction of OH• might be by-passed by strategical positioning of particular targets in close proximity to its sites of production. Along these lines, several studies have related OH• action with specific functions in plants [28,29] and with differentiation of some human cell lines in vitro [30,31]. Likewise, it has been hypothesized that OH•-mediated crosslinking is the basis of the supramolecular organization of cell structures, such as the plasma membrane [32].
3. Signal Thiol Oxidations Mediated by Hydrogen Peroxide
Over the last decade, the number of reported biological events in which ligand–receptor interaction induces H2O2-dependent responses has grown exponentially. Accountable for this are at least two of its chemical features: on the one hand, H2O2 is a strong two-electron oxidant, but on the other it requires high activation energy to start the oxidation of targets [25]. Therefore, this ROS is considered a poor random reactant in vivo, displaying high selectivity on its reactions [33]. Indeed, H2O2-derived signaling impacts mainly metalloproteins bearing transition metal centers or thiols in specific cysteine or selenocysteine residues [34,35,36], thereby altering their activity and the outcome of the corresponding cellular pathways. Whether a cysteine suits this modification strongly depends on the localization of the residue in the protein, its exposition to the surrounding environment, and its ionization state, but also on other factors, such as solvation, steric hindrance, hydrogen bonding, and formation of cyclic transition states [37,38,39]. Thus, although the largest portion of cysteines within cytoplasmic proteins is unreactive to H2O2, selected protein environments provide specificity for H2O2 signaling. The general chemical reaction with H2O2 is a nucleophilic attack, in which the deprotonated form of the cysteine side chain (-S−), a thiolate, attacks the peroxide bond (O-O) in H2O2 [40]. Stabilization of the negatively charged form of the cysteine is mediated by the presence of positively charged neighboring residues, frequently arginines, decreasing the local pKa [41,42]. The two-electron oxidation of a thiolate by H2O2 yields sulfenic acid, a naturally unstable modification [43] that can be the subject of several fates: (i) spontaneous reversal back to the thiolate, (ii) stabilization due to a favorable structural topology of the protein [44], (iii) enzymatic reduction by thioredoxins [45], or (iv) progression to further chemical oxoforms if the oxidant signal persists [46]. This onward oxidation can result in the formation of sulfinic (-SO2H) or irreversible sulfonic (-SO3H) acids. Besides, the formation of reversible disulfides, sulfenamides, S-glutathionylation, and other modifications can follow sulfenic acid formation, making secondary-derived thiol oxidation products from H2O2 further suitable to serve for signaling purposes [47]. The wide range of possibilities for modification of cysteines driven by the oxidation–reduction of thiolates is an important factor to diversify signaling, adding an incredible level of versatility to H2O2 as a second messenger. Furthermore, emerging evidence indicates that methionine, the second sulfur-containing amino acid, might provide an analogous redox-dependent system [48].
Although the idea that H2O2-mediated signaling mostly relies on oxidation of specific cysteine switches is now firmly established, there are still unanswered questions about how the transmission of the signal proceeds. In vivo, the reactions of H2O2 with glutathione peroxidases (GPX) and peroxiredoxins (PRX) are most likely to occur due to the high rate constants of 6 × 107 and 108 M−1 s−1, respectively [49,50,51]. In comparison, the bulk of the currently identified redox-sensitive cysteinome presents very slow reaction rates, around 20 M−1 s−1 [42,51]. This means that H2O2 must either reach a high localized concentration, be produced for an extended time, or even both, to outcompete signal quenching. Thus, transmission of a redox signal from H2O2 to protein thiolates can theoretically occur mainly if: (i) the target cysteine has a rate constant equal to or higher than that of GPX or PRX (Figure 2A); (ii) the H2O2 source is close enough to the target protein to allow for site-localized oxidation (Figure 2B); (iii) the scavenging proteins are inactivated by over-oxidation, the so-called floodgate model (Figure 2C) [52]; or (iv) a highly reactive thiol protein acts as an intermediary, it is a signaling relay (Figure 2D) [33,53]. Apart from a few instances where PRX have been shown to be the relay transmitting the signal, evidence for these mechanisms is limited. This probably means that, as in those malicious questions in test exams, more than one answer can be true at the same time.
4. The Plasma Membrane as a Platform for Redox Signal Transmission
Redox chemistry cannot be conceived separately to the notion of redox compartmentalization. Current evidence well-supports the existence of distinct redox environments within the cell, such as in mitochondria and endoplasmic reticulum (ER) with respect to the cytoplasm [33,54]. However, while much attention has been directed to the ROS-producing enzymes installed in those organelles, substantially less has been dedicated to the plasma membrane, as an entity preserving the low background of ROS that allows for specific signaling events and as a dock whose architecture facilitates the concentration of many ROS-related proteins (Figure 3). Indeed, not only integral membrane proteins, such NADPH oxidases (NOX), exert their activity at the plasma membrane [55]. Also, other redox enzymatic systems, such as xanthine oxidase (XOR) [56] and nitric oxide synthases (NOS) [57], generate ROS at the outer or inner part of the membrane, respectively. Moreover, independently of the leaflet where ROS are produced, bidirectional H2O2-transporting proteins assure the correct distribution and delivery of the signal [54,58], and further control of redox processes is guaranteed by the localization of scavengers in the vicinity of the plasma membrane [59,60,61,62]. Furthermore, the plasma membrane is the starting point of processes enabling redox signaling inside the cell. For example, specific endosomes, termed redoxosomes, are formed from invaginations on the plasma membrane, allowing for highly spatially localized intracellular ROS production [63].
Therefore, from the perspective of redox biology, the plasma membrane can be defined not merely as an instrumental physical barrier protecting cells from oxidative insults, but also as an organizing center that both directs and maintains redox signal specificity. To explain the manifold implications that this concept has, we give examples for several relevant processes outgoing from a general description of the membrane’s structure.
4.1. The Plasma Membrane: More Than Lipids
Referring to the classical membrane fluid mosaic model of Singer and Nicholson, biological membranes are bilayers of phospholipids that are organized in a hydrophobic center and hydrophilic outer leaflets and therefore serve as diffusion barriers. To allow for a selective interchange of molecules or information, amphipathic proteins are solved in the lipid matrix [64]. Since the late 1990s, this simple view of lipids as solvents for proteins has been overcome and a more complex picture has been accepted. Thus, as result of an asymmetrical distribution of specific lipids, membranes are further organized in lipid rafts (LRs), defined by being detergent-insoluble sphingolipid- and cholesterol-rich domains [65]. These regions have been demonstrated to be active structural signaling organizers rather than simply building blocks, being either enriched with specific components or enabled with the capacity to recruit them upon stimulation. Many transmembrane proteins have been shown to have affinity for LRs, including receptors, ion channels, and transporters, while cytoplasmic proteins associate to LRs typically by post-translational modifications, such as glycosylphosphatidylinositol (GPI)-anchoring, palmitoylation, and myristoylation [66,67]. Indeed, their capacity either to bring together different proteins that cooperate to transfer a signal, or to physically sequestrate others to block unspecific signaling, is crucial to allow for signaling processes [68]. Importantly, a large group of redox-related proteins have been found in LRs, foremost NOXes, whose downstream signaling is interrupted by drugs that disrupt LRs, and phosphatases, the earliest identified redox targets [69,70]. Moreover, some evidence suggests that the formation of LR domains may be itself altered by ROS, either directly by enhancing the activity of enzymes that promote LR clustering [71] or indirectly through their effects on the synthesis of lipids, such as ceramide or cholesterol [72,73].
A special type of LR are caveolar rafts, membrane invaginations generated by caveolin proteins [74]. At least three caveolin isoforms have been identified: caveolin-1 and caveolin-2 are expressed in most cell types, while caveolin-3 is specific of muscle cells [75]. Caveolins not only structurally define caveolae, but act as protein scaffolds to facilitate protein interactions in a restricted area of the plasma membrane. Notably, caveolin-1 has been shown to be phosphorylated by redox-sensitive kinases, such as Fyn, Abl, and Src, in response to ROS [76,77,78], and this modification is able to change its binding partner profile [79,80]. Furthermore, increasing evidence relates intracellular ROS levels to caveolin-1 expression [81], repression of its degradation [82], and membrane trafficking [83], suggesting feedback regulatory processes. Remarkably, caveolae structures have been also recently linked to the formation of redox-active endosomes, so-called redoxosomes. These single-membraned organelles generate ROS in an enclosed environment, thus facilitating co-localization of ROS generators and targets and preventing non-specific ROS-dependent damage reactions [63,84,85]. In mammalian systems, several stimuli have been identified to result in the formation of such redoxosomes, among them interleukin-1-β (IL-1β), tumor necrosis factor α (TNFα), and hypoxia=reoxygenation (H=R) [86,87]. In all those processes, members of the NOX family were identified as the source of O2•− generation within the redoxosome, suggesting a mechanistic conservation of signaling [85]. Intriguingly, localization of some receptors either to the plasma membrane or to endosomes modulates their potential to be activated, thereby regulating which downstream cascades are turned on. As an example, EGF receptor (EGFR)-triggered pathways can be either modulated depending on the presence or absence of endocytosis of the activated EGFR, or independently of localization and activation at the plasma membrane, since the active signaling of EGFR is taking place in the redoxosomes [88,89,90,91]. The discussed underlying mechanisms are divergent ligand-binding capacities due to different lipid compositions in endosomes or fusion of redoxosomes with vesicles harboring second effectors [92]. Besides the described caveolin-dependent formation of redoxosomes, there are indications for a probable clathrin-dependent process. In a recent study dealing with Clostridium difficile toxin B (TcdB)-induced necrosis in diarrhea, the authors speculate about internalization of the toxin together with p22phox, a critical component of some NOXes, to clathrin-coated vesicles, resulting in the formation of redoxosomes, ROS overproduction, and tissue damage [93]. In parallel, the internalization of NOX homologs has been shown to be clathrin-dependent in plants [94].
Apart from LR and caveolae, polyphosphoinositides (PPIn) form anchor points specifically associating proteins to the cytoplasmic leaflet of eukaryotic membranes, and therefore providing platforms for cellular signaling. Various isoforms of PPIn exist, resulting from differential phosphorylation of the inositol ring of phosphatidylinositol (PtdIns) [95]. These PPIn can be recognized by several highly conserved lipid-binding domains in proteins, such as the PH, FERM, FYVE, and PX domains, and thus regulate protein localization impacting its activity [96,97]. Regarding redox signaling, PX–PPIn interactions are crucial to allow the activation of several NOX isoforms [98,99,100,101]. Further, several studies have reported an impact of H2O2 on PPIn formation and hydrolysis [102,103,104,105], probably as result of its known effects on kinases and phosphatases, such as PTEN [106].
Despite that all of the above-described constituents of the plasma membrane have been shown to house important ROS-related systems, their way of facilitating redox signaling events might be quite diverse due to their different dynamics: while both non-caveolar LR and PPIn-anchors are continuously facing changes due to clustering or declustering of components or phosphorylation and dephosphorylation events, the composition of caveolar LRs is stable and hardly rearranged and might only change due to endocytosis events or fusion with vesicles. Thus, it is not surprising that differential targeting of ROS-producing enzymes and redox targets to these lipid-interaction platforms mediates distinct signaling pathways to orchestrate different cell responses.
4.2. NADPH Oxidases and Peroxiporins as a Generator–Facilitator System on the Plasma Membrane
The seven members of the human NADPH oxidase (NOX) family are widely recognized as the most important sources of signaling-competent H2O2. All of them have been identified at the plasma membrane of different cellular types in several tissues (Table 1), allowing for both general and cell-type-specific redox-dependent pathways to occur [13].
Broadly speaking, NOXes catalyze the oxidation of NADPH and the reduction of molecular oxygen via a highly conserved flavocytochrome core: six transmembrane domains hold a heme cluster that transfers electrons from NADPH through to a membrane [107]. A second membrane-spanning subunit, p22phox, provides stability to the complex in the majority of the isoforms (NOX1 to 4) [108]. Due to structural differences, the NOX family is further divided in ‘true’ NOX enzymes and dual oxidases (DUOX). In the DUOX case, an additional seventh transmembrane domain is linked to an N-terminal peroxidase-like domain via a short cytosolic bridge to allow for direct generation of H2O2. In contrast, the final product of NOX1, NOX2, NOX3, and NOX5 is O2•−. To ensure H2O2 production by these NOX family members anyhow, they cooperate in a finely balanced way with SOD enzymes [109,110]. As an exception to the general theme, NOX4 can be cited. This enzyme is—in contrast to all other family members—constitutively active without the need for stimulation [111]. Furthermore, it directly generates H2O2 despite lacking the DUOX-typical domain and it has been described to be mainly an ER-resident enzyme [112]. However, some controversy exists and several studies also report NOX4 localization to many other sites in the cell, including the nucleus and the plasma membrane [113,114].
The other ‘true’ NOXes are plasma membrane proteins that localize both to caveolar and non-caveolar rafts [74]. Some NOX enzymes require a subset of regulatory proteins for full activation of their catalytic subunit [132]. The recruitment of these regulators can be induced via diverse stimuli, allowing for a precisely timed release of H2O2 and resulting in NOX-isoform-specific outcomes [133]. Thus, the NOX1, NOX2, and NOX3 complexes are considered to be inactive or to display low levels of ROS production in membrane LR until their cytoplasmic activators are recruited [134,135]. In the case of NOX2, the best-studied member of the family, ligand-induced stimulation causes translocation to the plasma membrane of the pre-associated regulatory subunits p40phox, p47phox, and p67phox, where they are stabilized by PX domain–PPIn interactions [99]. Another necessary activator, the small GTPase Rac, directly interacts with the membrane through a geranyl lipid modification [136]. As cholesterol depletion impacts more on membrane subunit retention and complex activation rather than on activator translocation [137,138], it has been proposed that localization of all the NOX2 constituents to LRs is a sequential step to membrane binding of the regulatory proteins directed to solve the intricacies of bringing together all of the components in the correct spatial orientation to achieve proper assembly and full activation [134]. Similarly, activation by the NOX1 and NOX3 regulator, NOXO1, is also preceded by a PX–PPIn interaction of the subunit [98]. Instead, for NOX5, which depends on a EF-hand Ca2+-sensitive region for activation rather than on interaction with regulatory proteins, it has been shown recently that its N-terminal region binds to phosphatidylinositol 4,5- bisphosphate (PtdIns(4,5)P2), promoting localization to cholesterol- and caveolin-rich subregions at the plasma membrane [100,101]. Furthermore, it has been speculated that PtdIns(4,5)P2 has an additional function in co-localization of NOX5 with its target F-actin [139]. Finally, it has been shown that DUOX proteins require the presence of two maturation factors, DUOX activator 1 and 2 (DUOXA1 and DUOXA2), to be properly processed and exit the ER to the plasma membrane [140,141]. Once there, DUOXes are activated by Ca2+ mobilization and binding to EF-hand motifs, but whether they are targeted to any kind of LR is currently not known. However, phospholipase C β (PLCβ) and protein kinase Cα (PKCα), two factors inducing Ca2+-mediated activation of DUOXes, are enriched in lipid rafts [142,143], a fact that would predict DUOXes localization to the same site. In support of this hypothesis, a recent publication has linked LR-dependent Ca2+ signaling to DUOX-mediated ROS generation in Drosophila during pathogenic infection [144].
Importantly, the presence of NOX proteins at a particular region of the plasma membrane is not enough to explain the efficiency of their ROS-derived signaling. Thus, a finding of great significance to understand transmission of redox messages may lie in the evidence that H2O2 can be transported in a regulated manner across biological barriers, and that variations in permeability modulate its downstream intracellular effects [26]. For instance, although the redox-driving activity of plasma-membrane-bound NOX downstream of receptor activation is beyond doubt [145,146,147], known cytosolic targets of the NOX-generated H2O2 do not display reaction rates defeating that of PRX [148,149,150]. Furthermore, non-directional diffusion of H2O2 would compromise the possibility of any type of site-localized oxidation, including the floodgate model. PRX-based relays and co-localization with target proteins in intracellular structures, such as redoxosomes, are still possible in this context, but insufficient to explain the whole bunch of NOX-regulated processes.
Peroxiporins are now recognized as a further subclass of the aquaporin (AQP) protein family of membrane channels that are able to permeate H2O2 in addition to water or glycerol [151], whose functional characterization has opened new interesting perspectives on the possible mechanisms of NOX-mediated redox signaling. The net flow of solute through AQPs is dictated only by concentration gradients, implying that they are not active pumps or exchangers like Ca2+ transporters, but bi-directional passage facilitators. Movement of substrates through bilayers has been calculated for water to be accelerated by 5–50-fold in comparison to passive diffusion [152]. Assuming that the size and electrochemical properties of H2O2, including its capacity to form hydrogen bonds (fundamental for traversing the AQP pore), are quite similar to water [153], it is expected that H2O2 transport velocity ranges amongst similar parameters. The peroxiporin functional category currently comprises AQP3 [154], AQP8 [58,155], and AQP9 [154,156], whereas AQP11, an ER-resident AQP, has been also suggested to have a peroxiporin role [157]. In this last case, the idea of the existence of two H2O2-transporting channels, one situated in the plasma membrane and one in the ER, is somewhat appealing, as it resembles very much the scheme followed by Ca2+, in which the second messenger is stored in membrane-protected compartments in order to exclude the molecule from the cytoplasm, allowing for spatiotemporal specificity on signaling in restricted areas of the cell.
Following these lines, and considering the potential harmful consequences of uncontrolled release of H2O2 into the cytosol, it was expected that some type of redox-dependent regulation was operating on peroxiporins. Indeed, early studies indicated that AQP-mediated water transport could be inhibited by oxidative gating [158,159,160]. Two alternative mechanisms were proposed: (i) H2O2 directly oxidizes and thereby inhibits aquaporins; or (ii) H2O2 acts as a signaling molecule activating a pathway that ultimately leads to channel closure. However, no inhibitory effect of H2O2 on the conducting activity of individual AQPs heterologously expressed in yeast or Xenopus oocytes could be recorded [58,161,162,163]. Remarkably, when investigating membrane permeability to H2O2 in conditions of cell stress, Medraño-Fernandez et al. uncovered that a reversible redox-based mechanism modulates the transporting capacity of the peroxiporin AQP8 [26]. Gating involved the combined and coordinated action of H2O2 and the gasotransmitter H2S in a two-step process: First, a reactive cysteine inside the extracellular vestibule of the pore (C53) is primed by sulfenylation through the passage of the transported H2O2 molecules. Then, the moiety is modified by persulfidation induced by the activation of the H2S-producing redox-sensitive enzyme cystathionine-β-synthase (CBS), which is situated in the vicinity of the cytosolic mouth of the channel [164]. The latter modification elongates the side chain of C53 in a way that a histidine (H72), situated in the narrowest part of the pore, is displaced from its original position, leading to channel blockade. The system has been hypothesized to function as a sensor of the levels of H2O2 molecules crossing the plasma membrane [164]. Interestingly, C53 of AQP8 is highly conserved in all known peroxiporins [26,164], suggesting that a similar mechanism of gating could be operating in all of them.
The fact that peroxiporins facilitate H2O2 transport across the plasma membranes gives a new perspective to redefine the hypotheses on ROS-derived signal transduction previously enunciated (Figure 2). Regulated H2O2 transport through channels provides directionality to signals, potentiating the formation of cytosolic gradients with the capacity to restrain redox reactions as a function of site-localized concentration. In this scenario, all four possibilities of redox signal transmission would be feasible, either as independent, contemporary, or hierarchical events. Together, these may result in the activation of different sets of signaling layers to allow for an appropriate response to the stimulus arriving from the plasma membrane (Figure 4): discrete fluxes of H2O2 could oxidize strategically positioned proteins in the immediate proximity of the cytosolic mouth of the transporter (as CBS) [164], even if their velocity of reaction is slower than that of PRX. Higher rates of H2O2 transport would, on the other side, over-oxidize vicinal proteins, including PRX, permitting floodgate model-like signal transmission. Finally, relays would be possible depending on the localization of the putative intermediaries: low H2O2 concentrations would allow for the modification of proteins near the channel via local PRX, while more distant PRX would be oxidized and act as relays whenever fluxes exceed proximal scavengers and diffuse to further positions within the cell.
Notably, current research further points to a functional interaction of NOX enzymes with H2O2-transporting channels: in mouse primary keratinocytes, co-immunoprecipitation of NOX2 with AQP3 has been shown, demonstrating that these two proteins act in concert as H2O2 producer and facilitator to allow for successful redox signaling [165]; analogously, a similar NOX2–AQP8 cooperation axis has been described on B-cells [166]. Moreover, it is important to underline that all putative components of the above-described redox circuitry, H2O2-stimulating receptors [167], NOXes [13], extracellular SODs [168], peroxiporins [169], peroxiredoxins [59], and cysteine targets [170,171,172,173], have been identified in LRs. Even accumulation of homocysteine, the principal CBS substrate, in mice deficient for this enzyme has been linked with oxidative-stress-mediated tissue injury due to exacerbated ROS production by NOXes in LRs [174]. This last observation was attributed in the past to the promotion of LR stability by homocysteine; however, in light of the uncovered functions on controlling H2O2 permeability of CBS, it is also conceivable that the absence of the enzyme negatively impacts termination of ROS-driven signaling in LRs, and that both mechanisms cooperate to cause damage.
Altogether, these data would imply that special domains in plasma membranes are truly operating as a regulatory hub for NOX-derived signaling, linking external stimulus to redox responses, and serving as a platform to orchestrate differential activation of H2O2-mediated pathways. Moreover, the regional association of the ROS-related machinery to particular areas of the plasma membrane may favor cell-to-cell signaling or functional coordination with similar signaling platforms situated in vicinal cells, with or without direct protein–protein interaction (Figure 4, see upper part).
4.3. Other Sources of ROS at the Plasma Membrane
As anticipated before, not only NOXes exert their activity at the plasma membrane. For space constraints, we will dedicate this section only to two further predominant systems in terms of ROS production, xanthine oxidase and nitric oxide synthase, though other minor ROS producers, such as certain lipooxygenases [175] and cyclooxygenases [176], have been reported to be localized in the vicinity of the plasma membrane, at least in part of their life cycle.
Xanthine oxidase. In contrast to aquaporins and NOXes, which are present in several isoforms, the xanthine oxidoreductase (XOR) is a single enzyme mainly expressed in liver, intestine, kidney, and lactating mammary gland epithelial cells, but generally present in low levels in all human tissues [141,142,143,144,145]. Furthermore, its activity is organ-specific. It consists of two identical subunits forming a 300-kDa homodimer containing two iron–sulfur redox centers, a molybdopterin cofactor (Moco), and a flavin adenine dinucleotide (FAD) cofactor [146]. The Moco site is responsible for the main catalytic process of the enzyme, the oxidation of hypoxanthine to xanthine and xanthine to uric acid, but can also metabolite various endogenous substances and xenobiotics [147]. XOR is localized on two main sites in a cell: either in intracellular vesicles, which might be storage points of XOR [148], or the surface of plasma membranes. In the latter case, most of XOR is particularly localized in membrane regions facing closely neighboring cells, suggesting a role in inter-cellular signaling [148,149,150]. Nevertheless, no localization to lipid rafts or caveolae has been shown so far [79]. Interestingly, the mammalian XOR undergoes various changes, thereby switching from a NAD+-dependent xanthine dehydrogenase form (XDH) to the oxidase form (XO) [151]. This formation process is mediated by reversible thiol oxidation (XDH) or limited proteolysis (XO) and results in the generation of two completely different enzymes: whereas the FAD site of XDH converts NAD+ to NADH, XO catalyzes the generation of O2•− and H2O2 from molecular oxygen [152,153]. Furthermore, hypoxic conditions trigger even more drastic changes: the affinity for nitrites increases under these conditions, resulting in the generation of NO and reactive nitrogen species (RNS) [148]. Notwithstanding, the relations between XOR forms and ROS generation are not absolute: XDH might be less efficient than XO in reducing molecular oxygen, but in the absence of its preferred substrate NAD+, the Km value for oxygen for XDH is 600% higher than for XO [154]. Therefore, even a general increase of XOR activity might give rise to higher ROS production [148]. Due to these different XOR variations and the resulting enzymatic products, this enzyme is described to have both cytotoxic and pro-inflammatory activities alike as to trigger pro- and antitumorigenic effects [152]. In general, it is suggested that the main ROS generated by XOR is the O2•−, and thus its activity has mainly been linked to tissue damage and bacterial killing within the immune response. A typical example is the release of XOR from capillary endothelial cells into the blood in response to diseases, such as meningitis and malaria, followed by the establishment of a microvascular inflammatory response [148,155,156,157]. Intriguingly, under inflammatory conditions, O2 and pH values are significantly decreased, resulting in the almost exclusive production of H2O2 by XOR, which give rise to possible roles of XOR signaling regulation during inflammation [158]. Indeed, a crosstalk between inflammatory pathways and XOR signaling has been shown: cytokines, such as tumor necrosis factor α (TNFα), interleukin-1 (IL-1), and interferon-γ (IFN-γ), regulate XOR activity by expression regulation, since the XOR 5’ region exhibits binding sites for all three cytokines [159,160]. At the same time, there is evidence that INF-γ modifies XOR at the post-translational level, increasing thereby its activity up to 8-fold [160,161]. The other way around, ROS released by XOR increases the expression of NF-κB, implying an intensive crosstalk of both proteins and the corresponding pathways [162,163]. Nevertheless, the impact of XOR in signaling shown so far is mainly based on its product NO and on the generated ROS species O2•− and H2O2. NO was, for example, shown to regulate the ryanodine receptor and the Ca2+ ATPase in the heart, two factors of Ca2+ signaling [164,165]. Since these changes occur on reactive thiols in the corresponding proteins, it would be interesting to elucidate possible changes in signaling outcome dependent on the current form of XOR and the corresponding products in detail [166].
Nitric oxide synthase (NOS). NOSes are a family of enzymes responsible for the production of NO and L-citrulline from L-arginine and O2 through the binding of the critical co-factor tetrahydrobiopterin (BH4). Even if NOS-mediated production of nitrogen species is undoubtedly of great importance, space constraints limit us to comment only on the role of these enzymes as generators of ROS, and we refer the interested reader to other excellent reviews on the matter [177,178,179]. In this sense, aside from NO, NOSes also catalyze an “uncoupled” reaction that leads to O2•− production [180,181,182]. The word “uncoupling” indeed refers to the principal situations in which this phenomenon occurs, that is, when NOSes are not coupled either with its cofactor or with its substrate. The ultimate causes of NOS uncoupling are not completely understood, but O2•−-induced oxidation of BH4, changes in NOS phosphorylation, S-glutathionylation of the enzyme, and disruption of its functional structure have been suggested to play a role in the activity [183]. Three isoforms of NOS have been identified: neuronal NOS (nNOS, type I), inducible NOS (iNOS, type II), and endothelial NOS (eNOS, type III) [182,184]. Activity of NOSes is dynamically modulated by differential targeting of the enzyme to diverse subcellular localizations. For example, in resting endothelial cells, most eNOS protein is localized to caveolae by lipid modifications [185]. Similar to NOX subunits, myristoylation at the eNOS N-terminus appears to be an early event resulting in translocation to the plasma membrane, whereas palmitoylation of the enzyme leads to caveolar targeting [186,187]. This process can be reversed by agonist stimulation, causing eNOS intracellular trafficking by depalmitoylation [188], a process that has been suggested as a negative feedback mechanism for downregulating eNOS activity after signaling [179]. Importantly, NOSes are also regulated by caveolin binding (caveolin-1 for eNOS and iNOS and caveolin-3 for nNOS in skeletal muscle), but this interaction is not required to locate NOS to caveolae [177,189,190]. Instead, caveolin inhibits eNOS and nNOS activity by blocking the access of L-arginine to these proteins. Further, caveolin acts as an allosteric competitor for calmodulin, whose Ca2+-dependent binding is a crucial event for activation of these enzymes. For iNOS, the inhibition mechanism is quite diverse: in this case, caveolin promotes iNOS degradation by a proteasome-dependent pathway to terminate signaling [191]. Although it is clear that this NOS inhibitory/stimulatory mechanism and consequent NO production are facilitated in caveolae, it is unknown so far whether the same cycle and the same interactions affect O2•− generation. However, there is evidence for a controlled uncoupled process for eNOS and nNOS, governed as well by protein–protein interactions in the context of caveolae. This would suggest that, although classically related to pathologic stages, uncoupling may well also have a signaling role in physiological conditions [192]. Since both NOXes and NOSes are localized to caveolae, it will be of paramount interest to investigate how these two facts crosstalk and which impact on signaling derives from them. Interestingly, solely the chemical interplay between NO and O2•− will be not necessarily be the only answer: analysis in vitro suggests that S-nitrosothiol formation can be promoted by conjoint NO/ O2•− reactions on targets when both species are generated simultaneously and at low levels [193,194]. As caveolae are preferential sites for S-nitrosylation [195], it can further be speculated that the shut-down of uncoupled NOS activity is a necessary step for NO-driven signaling stimulated by NOXes, aimed to avoid the destructive effects of excessive O2•− production in the microdomain.
5. Dysregulation of Redox Signal Transmission through the Plasma Membrane: Impact on Disease
When ROS are produced in large amounts and bypass the antioxidant capacity of the cell, maladaptive oxidative stress is triggered [196]. As signaling molecules, it is not counterintuitive that dysfunction in ROS production can be a major source of malfunctioning during transduction of information both towards the inside and the outside of the cell. The particular disposition of NOXes at the plasma membrane, facing the extracellular space, and the bi-directional nature of peroxiporins, only reinforce this notion. Therefore, oxidative stress must not be seen as an isolated event, damaging macromolecules in a single cell or causing inadequate activation of intracellular pathways. Most likely, all nearby structures will be damaged, and spurious activation of redox-dependent cellular pathways should be also expected in neighboring cells. A clear example is provided by uncontrolled inflammation, which is not even a cell-type-restrained effect, but a systemic problem disturbing the normal functioning of several cell types residing in the same area.
Indeed, diseases that course with an inflammatory component provide the best model to appreciate to what extent the correct structuration of plasma membrane and incorporated redox signaling systems, from lipids to enzymes, is fundamental to assure timed and reliable signal transduction and to restrain oxidative damage. To illustrate this aspect, we have selected examples of redox imbalance in various tissues involving the NOX/AQP system, as the major source of potentially detrimental ROS in the area.
5.1. Immune System Diseases, NADPH Oxidases, and Peroxiporins
The enormous amount of data about how ROS influence the immune system makes it pretty difficult to offer an integrative and non-confusing view on ROS involvement in normal and pathological immune responses. Broadly speaking, it seems that heterogeneity can be solved by assuming a dual role of ROS in immune-related disease: beneficial at initial stages, but perpetuating the progression of the disease by fostering chronic inflammation [197]. Additionally, as discussed before, when it comes to ROS signaling, an adequate balance of their levels is the key to sustain homeostasis.
One of the most trademark characteristics associated with phagocytes is the activation of a powerful oxidative burst during their antimicrobial functions. This striking feature captured the attention of numerous researchers, leading to the identification of the first ROS-generating complex, NOX2 [198]. Since then, it has not only been confirmed that NOX2 produces ROS on purpose, but also that localization and timing of ROS production seems to be highly organized to respond to different challenges: while upon particle phagocytosis short limited NOX activity occurs at the phagosomal membrane, priming leads to prolonged NOX2 activity and ROS release mostly on the plasma membrane, directing ROS into the extracellular space [199,200,201]. Thus, killing of foreign invaders can run in parallel with other ROS-coordinated pathways, such as inflammation, modulating the strength of the response to be consistent with the scale of the stimulus.
The strongest evidence of the broad importance of NOX2 was gained in studies analyzing its loss of function. Chronic granulomatous disease (CGD) is a rare inherited immunodeficiency caused by mutations in one of the genes encoding for NOX2 complex components [202,203,204,205]. In this syndrome, a defective activation of NOX2 leads to a strongly diminished ROS production even if pathogens can be efficiently internalized [206,207], possibly resulting in life-threatening bacterial and fungal infections in CGD patients [208,209]. Though the link between absence of ROS production and defective killing mechanisms is rather obvious, the fact that hyperinflammation and oxidative damage has been frequently observed in CGD patients has not been so easy to conceal. The prevailing hypothesis is that non-degraded phagocytosed material accumulates in phagosomes, thereby increasing the pro-inflammatory phenotype. Nevertheless, this does not explain why CGD macrophages are showing reduced efferocytosis [210]. Furthermore, CGD macrophages are defective on producing anti-inflammatory mediators, preparing a fertile ground to tissue damage [211]. The discovery that ROS can be efficient second messengers on signaling pathways shed light on these, on the first glance, counterintuitive findings, and explained how alterations in ROS levels are able to favor inflammatory disorders [212,213,214]. Moreover, expression of certain innate immune receptors, such as TLR5, is reduced on the surface of CGD neutrophils [215], indicating that redox-sensitive signaling elements control either the transcription of specific immune-related genes or their trafficking to the plasma membrane. In support of this view, myeloperoxidase (MPO) deficiency is rarely associated with serious infections [216], even if MPO products are faster and more potent as antimicrobial agents than ROS [201], suggesting that the impaired immune response is not only the result of the lack of a defensive mechanism but a consequence of ROS-derived signaling.
In the last decade, this vision has gone beyond innate immunity, extending NOX2-generated ROS functions to adaptive immune-related processes [217,218,219]. For example, NOX2 activity has been related to functional communication between the innate and adaptive arms of the immune system [220], making CGD carriers prone to develop autoimmune diseases, such as polyarthritis, lupus erythematosus, and Crohn-like inflammatory bowel disease [218,221]. Several NOX2-dependent processes have been described to provide successful antigen presentation by dendritic cells: NOX2-dependent O2•− generation allows for antigen presentation of MHC class I to CD8− T-cells [222,223,224,225,226,227] and further inhibits the degradation of antigen in the phagosome [227,228]. A striking similar secretory deficiency as in CGD has been described in mice B-lymphoma cells with a downregulated expression of the peroxiporin AQP8 [166]: unlike their normal counterparts, AQP8-silenced cells continue to express IgM on their surface after LPS stimulation and secrete less IgM polymers. From these findings, it is deduced that AQP8 might channel NOX2-generated ROS to promote B-cell differentiation into secreting plasma cells [166]. This goes in line with the fact that CGD patients have a reduced memory B-cell count, which is reflected in a decreased capability to maintain a long-term memory defense against pathogens [229,230].
Even if little is known about the expression of the different peroxiporin isoforms in immune cells, some evidence indicates that all peroxiporins may have representation in one or another cell type. Thus, human macrophages express AQP3 [231] and possibly AQP9 [232]; B-cells upregulate AQP3 and AQP8 during B-cell differentiation [166,233]; T-cells and immature dendritic cells bear at least AQP3 [233,234], while only AQP9 seems to be present in neutrophils [235]. Most available reports in the literature have constantly implicated AQP3 and AQP9 with migration processes of different immune cells. AQP3, for example, is essential for chemokine-dependent T-cell migration and to establish a sufficient immune response [234], whereas AQP9 knock-out mice show reduced neutrophil migration to the bacterial product fMLP [236].
Altogether, these data are consistent with the concept of a plasma membrane redox signalosome as a platform for signaling, as ROS producers, transporters, effectors, and inhibitors are jointly gathered in a restrained space, facilitating a coordinated and timed response.
5.2. Inflammatory Bowel Disease, NADPH Oxidases, and Peroxiporins
Inflammatory bowel disease (IBD) is the common term for a group of recurring inflammatory conditions in the gastrointestinal tract [237,238]. The main IBD disorders are Crohn’s disease (CD), characterized by inflammatory patches and deep ulcers all along the digestive tract, and ulcerative colitis (UC), which is limited to the mucosal lining of the colon and appears in a continuous pattern. The symptoms can be very similar, typically including abdominal pain, severe diarrhea, fatigue, weight loss, and malnutrition [239]. Although the mechanisms underlying IBD still remain elusive, it is widely accepted that an over-reaction of the immune response in a genetically susceptible background leads to oxidative stress and damage of the intestinal epithelium. The course of the disease further produces maladaptation to environmental factors and intolerance to the residing microbiota, while facilitating the entry of external invaders [240,241], inducing an auto-amplifier state that re-activates the immune system and creates a perfect storm [242]. Thus, the primordial cause of IBD seems to be a malfunction of host defense responses in an organ particularly well-armed to face external threats.
An imbalance in redox homeostasis starting at the plasma membrane may also be pivotal in IBD pathogenesis, as it has been reported that a critical window of optimal ROS production is necessary to permit the constant renewal dynamics of the intestinal lining [243], and that increases in ROS levels cause upregulation of genes involved in innate and adaptive immune responses specifically in the gastrointestinal tract [244,245]. Thus, it is not surprising that dysfunction of NOX enzymes has been now investigated as a potential risk factor for IBD. Both NOX1 and DUOX2 are expressed in the gut epithelium, but with differential distribution. Whereas NOX1 is expressed at high levels only in the colon [246], the DUOX2 protein is located at the apical membrane of enterocytes, with maximal expression in the tip epithelium of ileum, cecum, and colon [247]. This asymmetrical confinement of NOX isoforms seems to reflect their differential functions: while NOX1 fulfills eminently a regulatory role on both Wnt/ β-catenin and Notch pathways controlling proliferation of stem and progenitor epithelial cells [248,249], DUOX2 coordinates the innate defense in the gut of mammals by controlling the circuitry driven by interleukin receptor activation [250,251].
On the peroxiporin side, only the presence of AQP8 and AQP3 has been profusely documented along the gastrointestinal tract, while AQP9 expression has been restrained to a small population of mucus-secreting globet cells in the ilium and duodenum [252]. AQP8 transcript is expressed both in the small and the large intestine, principally in the duodenum, jejunum, and colon [252,253]. Its subcellular distribution at subapical regions of epithelial cells has been mostly related to its putative transephitelial water-transporting capacity. Notably, when analyzing the AQP8 knock-out mice, it was determined that deficiency in this AQP had little effect on the colonic fluid absorption, fecal dehydration, or in the small intestine agonist-stimulated fluid secretion [254]. Instead, in a drug-induced colitis mouse model, mimicking human Crohn’s disease, AQP8 expression is downregulated as inflammation and injury increases [255], a fact that may be better interpreted in terms of its peroxiporin activity. AQP8 was also markedly lower in inflamed colonic biopsies when compared with normal counterparts [256]. Nevertheless, other studies report an upregulation of its expression in samples from Crohn’s disease and ulcerative colitis patients. This contraposition may be explained by an adaptively triggered protein modulation in early stages of disease and later compensatory mechanisms [257].
AQP3 is highly expressed in both the proximal and distal colon, but great orders of magnitude more abundant in the latter [258]. Owing to its localization in the mucosal epithelial cells [259], a role in water transport for the formation of intestinal contents and feces was deduced. However, analogously to AQP8, downregulation of AQP3 levels was observed in the colon of Crohn’s disease and ulcerative colitis patients [257] and in drug-induced colitis as the signs of intestinal inflammation and injury progress [260]. After small bowel resection and improvement of intestinal functions in IBD rats, AQP3 was upregulated during the adaptation [259], indicating a functional significance of its levels in the pathogenesis of IBD [255,258]. In addition, AQP3 has been shown to be crucial for enterocyte proliferation [261], a process induced by the Wnt/β-catenin pathway and mediated by NOX1, as described above: AQP3 knock-out mice showed impaired capacity of proliferation in experimental models of colitis, even leading to significantly reduced mice survival. As intestinal barrier integrity was impaired on these models and improved by oral glycerol administration, glycerol-transporting ability of AQP3 was judged to be responsible for the effects observed [261]. Notwithstanding, AQP3 deletion induced a dramatic increase in Escherichia coli C25 translocation in AQP3 null mice [262], suggesting that production of ROS that represses bacteria replication is diminished, also leaving space to re-interpret data in light of a role of AQP3 as a peroxiporin.
Paradoxically, the same studies that firmly demonstrate a connection between NOX enzymes and IBD, supported by abundant patient data and cell-based assays, suggest that the functional link does not involve exacerbation of inflammation, but rather a decline in ROS production [263]. This would perfectly fit in a scenario in which the principal peroxiporins are downregulated, as presented above. In these circumstances, it will be really interesting to assess whether inhibition of the expression of these AQPs is induced by susceptibility genes to IBD or NOX-related cascades.
5.3. Neurodegenerative Diseases, NADPH Oxidases, and Peroxiporins
The lack of connective tissue in the brain incites cells to be much closer to each other than cells in peripheral tissues. Therefore, extracellular generation of ROS by NOX in the central nervous system (CNS) must be tightly controlled to avoid unwanted effects on adjacent cells in such a sensitive area. Remarkably, together with aberrant protein aggregation, major cellular symptoms of neurodegenerative diseases are oxidative stress and membrane lipid peroxidation, suggesting the cooperation of unregulated ROS fluxes near the plasma membrane barrier on establishing or aggravating these processes.
Transcripts for NOX1, NOX2, and NOX4 are present in total brains [264], but there is no detailed characterization of the expression of NOX isoforms in each neuronal subtype. Exceptions would be the recorded presence of NOX1 in dopaminergic neurons [265], NOX2 expression in CA1 hippocampal neurons [266,267] and in pyramidal neurons of socially isolated rats [268], and NOX4 on dorsal root ganglion neurons [269], in basal ganglion, and cortical neurons after stroke [270]. With respect to other cellular systems of the CNS, microglial cells are constitutively equipped with low levels of NOX2 that substantially increase upon activation [271] in agreement with their phagocytic functions; microglia also express NOX1 [272] and NOX4 [273]. Instead, astrocytes express mostly NOX4, while oligodendrocytes are the only CNS cells in which there is no data on NOX expression [274]. The precise subcellular distribution of NOX enzymes in neuron membranes is still relatively obscure. However, there is evidence for localization of NOX2 at postsynaptic terminals [275,276] in accordance with its proposed role as mediator of long-term potentiation of memory upon activation of NMDA-receptors [277,278]. Regarding AQPs, expression of all peroxiporins [279,280,281,282], including the suspected H2O2 ER transporter AQP11 [283,284], has been detected at distinct brain sites. Interestingly, all of them are upregulated under stress conditions, which goes in hand with high ROS levels, such as hypoxia, ischemia, or tumorigenesis [279,280,281,285,286]. However, the primary substrate specificity of each AQP in the brain is a matter that has not yet been investigated.
Remarkably, early research on the knock-out mice for NOX2 showed that, besides the known CGD phenotype, mice suffered from impaired memory and a synaptic deficit [287]. Though in human patients only a mild decrease in cognitive function has been described [288], a number of studies concentrated on revealing possible connections of NOX2 dysfunction with human diseases manifesting degenerative cognitive defects. A formal link between microglial NOX2 and several inflammatory brain pathologies, such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS), was achieved [264,289]. For example, strong evidence indicates that in AD, neuron-secreted β-amyloid peptide fragments promote the assembly of NOX2 complexes in the plasma membrane of glia cells [290,291] and on neurons themselves [292,293]. The resulting increase of NOX2 activity causes alterations in the synaptic plasticity capacity of the latter, eventually leading to cell death [294]. Accordingly, in a mouse model with increased β-amyloid peptide accumulation, NOX2 deficiency improves the outcome of AD: neuronal oxidative stress and behavioral deficits strongly decrease even if β-amyloid deposits still persist, indicating that microglial NOX2 is not the cause of AD but a key amplifier of its deleterious effects [295,296]. Importantly, an unidentified source of ROS has been also shown to induce protein tau hyperphosphorylation in neurons, the second main hallmark of AD, a fact that surely warrants further investigation [297]. In PD, mitochondria are considered the primary source of oxidative stress [298]. Nevertheless, NOX proteins seem to contribute as well to the overall increase in ROS levels, as indicated by several reports using toxin-induced PD animal models and in vitro cell cultures [299,300,301,302]. As in the case of AD, microglia-mediated loss of dopaminergic neurons depends on NOX2 translocation to the cell surface, but glia cells are not the primary cause of the disease but part of an amplification loop that increases neurotoxicity [303,304]. In the end, expression of NOX2 is increased in microglia cells in the spinal cord of sporadic ALS patients, at both RNA and protein levels, leading to ROS production and oxidative damage [305]. NOX2 involvement has also been confirmed in different animal models, showing that enhanced expression of this protein accelerates disease progression [306], while its deficiency increases lifespan and ameliorates symptoms [305,307].
It is also possible that non-autonomous production of ROS by glia cells is accompanied by a parallel enhanced autocrine production of ROS in neurons. In that sense, a few recent reports have highlighted a possible role for NOX1 in the development of PD in a cell-autonomous way in dopaminergic neurons. Thus, silencing of NOX1 expression significantly reduced typical molecular features of PD, such as α-synuclein protein aggregation and high α-synuclein ubiquitin expression levels, in a drug-induced model of PD [308,309]. Analogously, silencing of the Rac1 subunit prevented neuronal death [265]. On the other hand, a large group of the inherited forms of ALS (about 15–20%) is caused by point mutations in the gene coding for the cytosolic copper/zinc superoxide dismutase (SOD1) [310]. Since NOX1 deficiency significantly increases lifespan in SOD1 mutant mice [307], NOX1 might fulfill a similar role in ALS pathogenesis as in PD.
Relevant intracellular pathways potentially affected by autocrine neuronal ROS signaling will include those depending on the known redox-sensitive transcription factors HIF1α, Nrf2, or NF-κB [274], while an additional role of redox regulation in CNS-specific transcriptional processes is not excluded. Besides, numerous receptors of the CNS and their downstream signaling (mostly kinases and phosphatases) are regulated by redox modulation of cysteine residues, including the NMDA [311], opioid [312], and gamma-aminobutyric acid receptors [313].
6. Conclusions
Although it is now becoming clear that the basis for redox signaling specificity starts with compartmentalization of producers and targets, the study of the localization of redox systems at particular plasma membrane regions has not gained much attention until recently. Notwithstanding, membrane architecture and organization of ROS-generating enzymes in distinct lipid domains has the potential to explain how efficient redox signal transmission could be achieved, while risk of damage intrinsically associated to ROS is minimized. Moreover, the notion that the plasma membrane actively acts as a platform for redox signaling by distributing, concentrating, and/or excluding redox-related proteins may provide insights to better understand the spatio-temporal regulation of ROS-dependent signaling pathways.
Being the main source of signaling-competent ROS in the area, of especial interest is the concomitant analysis of NOX/AQP expression in membrane rafts that has been outlined in the present review for the first time to our knowledge. We propose here that the correct expression and compartmentalization of this system at the plasma membrane is fundamental to adequately orchestrate responses in which ROS molecules operate and thus deserves further investigation. This would be of paramount importance to advance in the comprehension of complex oxidative-stress-related diseases in which intercellular and intracellular pathways seem to cooperate to cause or aggravate damage, such as IBD and neurodegenerative disorders.
Author Contributions
D.E.N. developed the project idea; D.E.N. and I.M.-F. wrote the first draft of the manuscript, conducted the literature review, and revised the manuscript.
Funding
D.E.N. was supported by a postdoctoral fellowship of the German Research Foundation (return fellowship, NO 1230/2-1); I.M.-F. is supported through a postdoctoral fellowship from Associazione Italiana Ricerca sul Cancro (IG 2016-18824 granted to Roberto Sitia).
Acknowledgments
We thank all members of our laboratories, especially our supervisors (Stefanie Pöggeler, Georg August University, Göttingen, Germany, and Roberto Sitia, San Raffaele Scientific Institute, Milan, Italy) for giving us the confidence and time to write this review, and for useful suggestions. Because of space limitations, we apologize to all those colleagues and researchers in the field whose work is not directly cited here.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rall, T.W.; Sutherland, E.W.; Berthet, J. The relationship of epinephrine and glucagon to liver phosphorylase. J. Biol. Chem. 1957, 224, 459–468. [Google Scholar]
- Bassler, J.; Schultz, J.E.; Lupas, A.N. Adenylate cyclases: Receivers, transducers, and generators of signals. Cell Signal. 2018, 46, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Antal, C.E.; Newton, A.C. Spatiotemporal dynamics of phosphorylation in lipid second messenger signaling. Mol. Cell. Proteom. MCP 2013, 12, 3498–3508. [Google Scholar] [CrossRef] [PubMed]
- Cary, S.P.; Winger, J.A.; Derbyshire, E.R.; Marletta, M.A. Nitric oxide signaling: No longer simply on or off. Trends Biochem. Sci. 2006, 31, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Bagur, R.; Hajnóczky, G. Intracellular Ca2+ Sensing: Its Role in Calcium Homeostasis and Signaling. Mol. Cell 2017, 66, 780–788. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G. Redox signaling: Hydrogen peroxide as intracellular messenger. Exp. Mol. Med. 1999, 31, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Landry, W.D.; Cotter, T.G. ROS signalling, NADPH oxidases and cancer. Biochem. Soc. Trans. 2014, 42, 934–938. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Role of metabolic H2O2 generation: Redox signaling and oxidative stress. J. Biol. Chem. 2014, 289, 8735–8741. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Jastroch, M.; Divakaruni, A.S.; Mookerjee, S.; Treberg, J.R.; Brand, M.D. Mitochondrial proton and electron leaks. Essays Biochem. 2010, 47, 53–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Go, Y.M.; Chandler, J.D.; Jones, D.P. The cysteine proteome. Free Radic. Biol. Med. 2015, 84, 227–245. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, J.M.C. (Eds.) Free Radicals in Biology and Medicine, 5th ed.; Oxford University Press: Oxford, UK, 2015. [Google Scholar]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Guarás, A.; Perales-Clemente, E.; Calvo, E.; Acín-Pérez, R.; Loureiro-Lopez, M.; Pujol, C.; Martínez-Carrascoso, I.; Nuñez, E.; García-Marqués, F.; Rodríguez-Hernández, M.A.; et al. The CoQH2/CoQ Ratio Serves as a Sensor of Respiratory Chain Efficiency. Cell Rep. 2016, 15, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Liochev, S.I.; Fridovich, I. Superoxide and iron: Partners in crime. IUBMB Life 1999, 48, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Zou, C.G.; Banerjee, R. Tumor necrosis factor-alpha-induced targeted proteolysis of cystathionine beta-synthase modulates redox homeostasis. J. Biol. Chem. 2003, 278, 16802–16808. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Azad, M.B.; Gibson, S.B. Superoxide is the major reactive oxygen species regulating autophagy. Cell Death Differ. 2009, 16, 1040–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afanas’ev, I. Mechanisms of superoxide signaling in epigenetic processes: Relation to aging and cancer. Aging Dis. 2015, 6, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Case, A.J. On the Origin of Superoxide Dismutase: An Evolutionary Perspective of Superoxide-Mediated Redox Signaling. Antioxidants 2017, 6, 82. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Fridovich, I. Superoxide dismutase: A comparison of rate constants. Arch. Biochem. Biophys. 1973, 158, 396–400. [Google Scholar] [CrossRef]
- Hancock, J.T.; Whiteman, M. Cellular Redox Environment and Its Influence on Redox Signaling Molecules. React. Oxyg Spec. 2018, 5, 78–85. [Google Scholar] [CrossRef]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- Yim, M.B.; Chock, P.B.; Stadtman, E.R. Copper, zinc superoxide dismutase catalyzes hydroxyl radical production from hydrogen peroxide. Proc. Natl. Acad. Sci. USA 1990, 87, 5006–5010. [Google Scholar] [CrossRef] [PubMed]
- Veal, E.A.; Day, A.M.; Morgan, B.A. Hydrogen peroxide sensing and signaling. Mol. Cell 2007, 26, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. The biological chemistry of hydrogen peroxide. Methods Enzymol. 2013, 528, 3–25. [Google Scholar] [PubMed]
- Medraño-Fernandez, I.; Bestetti, S.; Bertolotti, M.; Bienert, G.P.; Bottino, C.; Laforenza, U.; Rubartelli, A.; Sitia, R. Stress Regulates Aquaporin-8 Permeability to Impact Cell Growth and Survival. Antioxid. Redox Signal. 2016, 24, 1031–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imlay, J.A. Pathways of oxidative damage. Ann. Rev. Microbiol. 2003, 57, 395–418. [Google Scholar] [CrossRef] [PubMed]
- Mittler, R.; Berkowitz, G. Hydrogen peroxide, a messenger with too many roles? Redox Rep. Commun. Free Radic. Res. 2001, 6, 69–72. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, A.V.; Matveyeva, N.P.; Yermakov, I.P. Reactive oxygen species are involved in regulation of pollen wall cytomechanics. Plant Biol. 2014, 16, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Nagy, K.; Pasti, G.; Bene, L.; Zs-Nagy, I. Induction of granulocytic maturation in HL-60 human leukemia cells by free radicals: A hypothesis of cell differentiation involving hydroxyl radicals. Free Radic. Res. Commun. 1993, 19, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Chénais, B.; Andriollo, M.; Guiraud, P.; Belhoussine, R.; Jeannesson, P. Oxidative stress involvement in chemically induced differentiation of K562 cells. Free Radic. Biol. Med. 2000, 28, 18–27. [Google Scholar] [CrossRef]
- Nagy, I.Z. On the true role of oxygen free radicals in the living state, aging, and degenerative disorders. Ann. N. Y. Acad. Sci. 2001, 928, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. Biological Production, Detection, and Fate of Hydrogen Peroxide. Antioxid. Redox Signal. 2018, 29, 541–551. [Google Scholar] [CrossRef] [PubMed]
- Putker, M.; Vos, H.R.; van Dorenmalen, K.; de Ruiter, H.; Duran, A.G.; Snel, B.; Burgering, B.M.T.; Vermeulen, M.; Dansen, T.B. Evolutionary Acquisition of Cysteines Determines FOXO Paralog-Specific Redox Signaling. Antioxid. Redox Signal. 2015, 22, 15–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmström, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Marino, S.M.; Gladyshev, V.N. Proteomics: Mapping reactive cysteines. Nat. Chem. Biol. 2011, 7, 72–73. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Sueta, G.; Manta, B.; Botti, H.; Radi, R.; Trujillo, M.; Denicola, A. Factors affecting protein thiol reactivity and specificity in peroxide reduction. Chem. Res. Toxicol. 2011, 24, 434–450. [Google Scholar] [CrossRef] [PubMed]
- Poole, L.B.; Karplus, P.A.; Claiborne, A. Protein sulfenic acids in redox signaling. Ann. Rev. Pharmacol. Toxicol. 2004, 44, 325–347. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, B.; Hecht, H.J.; Flohe, L. Peroxiredoxins. Biol. Chem. 2002, 383, 347–364. [Google Scholar] [CrossRef] [PubMed]
- Zeida, A.; Babbush, R.; Lebrero, M.C.; Trujillo, M.; Radi, R.; Estrin, D.A. Molecular basis of the mechanism of thiol oxidation by hydrogen peroxide in aqueous solution: Challenging the SN2 paradigm. Chem. Res. Toxicol. 2012, 25, 741–746. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Carroll, K.S. Sulfenic acid chemistry, detection and cellular lifetime. Biochim. Biophys. Acta 2014, 1840, 847–875. [Google Scholar] [CrossRef] [PubMed]
- Poole, L.B. The basics of thiols and cysteines in redox biology and chemistry. Free Radic. Biol. Med. 2015, 80, 148–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winterbourn, C.C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008, 4, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Allison, W.S. Formation and reactions of sulfenic acids in proteins. Acc. Chem. Res. 1976, 9, 293–299. [Google Scholar] [CrossRef]
- Lo Conte, M.; Carroll, K.S. The Redox Biochemistry of Protein Sulfenylation and Sulfinylation. J. Biol. Chem. 2013, 288, 26480–26488. [Google Scholar] [CrossRef] [PubMed]
- Hogg, D.R. Chemistry of sulphenic acids and esters. In Sulfenic Acids and Derivatives; Patai, S., Ed.; John Wiley & Sons Ltd.: Hoboken, NJ, USA, 1990; pp. 361–402. [Google Scholar]
- D’Autréaux, B.; Toledano, M.B. ROS as signalling molecules: Mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007, 8, 813–824. [Google Scholar] [CrossRef] [PubMed]
- Drazic, A.; Winter, J. The physiological role of reversible methionine oxidation. Biochim. Biophys. Acta 2014, 1844, 1367–1382. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C.; Metodiewa, D. Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic. Biol. Med. 1999, 27, 322–328. [Google Scholar] [CrossRef]
- Peskin, A.V.; Low, F.M.; Paton, L.N.; Maghzal, G.J.; Hampton, M.B.; Winterbourn, C.C. The high reactivity of peroxiredoxin 2 with H2O2 is not reflected in its reaction with other oxidants and thiol reagents. J. Biol. Chem. 2007, 282, 11885–11892. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C.; Hampton, M.B. Thiol chemistry and specificity in redox signaling. Free Radic. Biol. Med. 2008, 45, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Wood, Z.A.; Poole, L.B.; Karplus, P.A. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science 2003, 300, 650–653. [Google Scholar] [CrossRef] [PubMed]
- Stöcker, S.; Van Laer, K.; Mijuskovic, A.; Dick, T.P. The Conundrum of Hydrogen Peroxide Signaling and the Emerging Role of Peroxiredoxins as Redox Relay Hubs. Antioxid. Redox Signal. 2018, 28, 558–573. [Google Scholar] [CrossRef] [PubMed]
- Ottaviano, F.G.; Handy, D.E.; Loscalzo, J. Redox regulation in the extracellular environment. Circ. J. Off. J. Jpn. Circ. Soc. 2008, 72, 1–16. [Google Scholar] [CrossRef]
- DeYulia, G.J., Jr.; Carcamo, J.M.; Borquez-Ojeda, O.; Shelton, C.C.; Golde, D.W. Hydrogen peroxide generated extracellularly by receptor-ligand interaction facilitates cell signaling. Proc. Natl. Acad. Sci. USA 2005, 102, 5044–5049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radi, R.; Rubbo, H.; Bush, K.; Freeman, B.A. Xanthine oxidase binding to glycosaminoglycans: Kinetics and superoxide dismutase interactions of immobilized xanthine oxidase-heparin complexes. Arch. Biochem. Biophys. 1997, 339, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Sessa, W.C.; Barber, C.M.; Lynch, K.R. Mutation of N-myristoylation site converts endothelial cell nitric oxide synthase from a membrane to a cytosolic protein. Circ. Res. 1993, 72, 921–924. [Google Scholar] [CrossRef] [PubMed]
- Bienert, G.P.; Møller, A.L.; Kristiansen, K.A.; Schulz, A.; Møller, I.M.; Schjoerring, J.K.; Jahn, T.P. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J. Biol. Chem. 2007, 282, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Woo, H.A.; Yim, S.H.; Shin, D.H.; Kang, D.; Yu, D.Y.; Rhee, S.G. Inactivation of peroxiredoxin I by phosphorylation allows localized H2O2 accumulation for cell signaling. Cell 2010, 140, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Januel, C.; El Hentati, F.Z.; Carreras, M.; Arthur, J.R.; Calzada, C.; Lagarde, M.; Vericel, E. Phospholipid-hydroperoxide glutathione peroxidase (GPx-4) localization in resting platelets, and compartmental change during platelet activation. Biochim. Biophys. Acta 2006, 1761, 1228–1234. [Google Scholar] [CrossRef] [PubMed]
- Nozik-Grayck, E.; Suliman, H.B.; Piantadosi, C.A. Extracellular superoxide dismutase. Int.J. Biochem. Cell Biol. 2005, 37, 2466–2471. [Google Scholar] [CrossRef] [PubMed]
- Percy, M.E. Catalase: An old enzyme with a new role? Can. J. Biochem. Cell Biol. 1984, 62, 1006–1014. [Google Scholar] [CrossRef] [PubMed]
- Spencer, N.Y.; Engelhardt, J.F. The basic biology of redoxosomes in cytokine-mediated signal transduction and implications for disease-specific therapies. Biochemistry 2014, 53, 1551–1564. [Google Scholar] [CrossRef] [PubMed]
- Singer, S.J.; Nicholson, G.L. The Fluid Mosaic Model of the Structure of Cell Membranes. Science 1972, 175, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Tillman, T.S.; Cascio, M. Effects of membrane lipids on ion channel structure and function. Cell Biochem. Biophys. 2003, 38, 161–190. [Google Scholar] [CrossRef]
- Caliceti, C.; Zambonin, L.; Rizzo, B.; Fiorentini, D.; Vieceli Dalla Sega, F.; Hrelia, S.; Prata, C. Role of plasma membrane caveolae/lipid rafts in VEGF-induced redox signaling in human leukemia cells. BioMed Res. Int. 2014, 2014, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.Y.; Yi, F.; Zhang, G.; Gulbins, E.; Li, P.L. Lipid raft clustering and redox signaling platform formation in coronary arterial endothelial cells. Hypertension 2006, 47, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Li, P.L.; Zhang, Y.; Yi, F. Lipid raft redox signaling platforms in endothelial dysfunction. Antioxid. Redox Signal. 2007, 9, 1457–1470. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef] [PubMed]
- Dumitru, C.A.; Zhang, Y.; Li, X.; Gulbins, E. Ceramide: A novel player in reactive oxygen species-induced signaling? Antioxid. Redox Signal. 2007, 9, 1535–1540. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Kim, Y.S.; Liu, Z. Lipid rafts and oxidative stress-induced cell death. Antioxid. Redox Signal. 2007, 9, 1471–1483. [Google Scholar] [CrossRef] [PubMed]
- Schlegel, A.; Volonte, D.; Engelman, J.A.; Galbiati, F.; Mehta, P.; Zhang, X.L.; Scherer, P.E.; Lisanti, M.P. Crowded little caves: Structure and function of caveolae. Cell Signal. 1998, 10, 457–463. [Google Scholar] [CrossRef]
- Parton, R.G. Caveolae and caveolins. Curr. Opin. Cell Biol. 1996, 8, 542–548. [Google Scholar] [CrossRef]
- Sanguinetti, A.R.; Mastick, C.C. c-Abl is required for oxidative stress-induced phosphorylation of caveolin-1 on tyrosine 14. Cell Signal. 2003, 15, 289–298. [Google Scholar] [CrossRef]
- Li, S.; Seitz, R.; Lisanti, M.P. Phosphorylation of caveolin by src tyrosine kinases. The alpha-isoform of caveolin is selectively phosphorylated by v-Src in vivo. J. Biol. Chem. 1996, 271, 3863–3868. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.B.; Li, S.M.; Qian, X.X.; Moon, C.; Zheng, J. Tyrosine phosphorylation of caveolin 1 by oxidative stress is reversible and dependent on the c-src tyrosine kinase but not mitogen-activated protein kinase pathways in placental artery endothelial cells. Biol. Reprod. 2005, 73, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.H.; Insel, P.A. Lipid rafts and caveolae and their role in compartmentation of redox signaling. Antioxid. Redox Signal. 2009, 11, 1357–1372. [Google Scholar] [CrossRef] [PubMed]
- Grande-García, A.; Echarri, A.; de Rooij, J.; Alderson, N.B.; Waterman-Storer, C.M.; Valdivielso, J.M.; del Pozo, M.A. Caveolin-1 regulates cell polarization and directional migration through Src kinase and Rho GTPases. J. Cell Biol. 2007, 177, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Volonte, D.; Zhang, K.; Lisanti, M.P.; Galbiati, F. Expression of caveolin-1 induces premature cellular senescence in primary cultures of murine fibroblasts. Mol. Biol. Cell 2002, 13, 2502–2517. [Google Scholar] [CrossRef] [PubMed]
- Rungtabnapa, P.; Nimmannit, U.; Halim, H.; Rojanasakul, Y.; Chanvorachote, P. Hydrogen peroxide inhibits non-small cell lung cancer cell anoikis through the inhibition of caveolin-1 degradation. Am. J. Physiol. Cell Physiol. 2011, 300, C235–C245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Y.S.; Ko, Y.G.; Seo, J.S. Caveolin internalization by heat shock or hyperosmotic shock. Exp. Cell Res. 2000, 255, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Zhou, F.; Katirai, F.; Li, P.L. Lipid raft redox signaling: Molecular mechanisms in health and disease. Antioxid. Redox Signal. 2011, 15, 1043–1083. [Google Scholar] [CrossRef] [PubMed]
- Oakley, F.D.; Abbott, D.; Li, Q.; Engelhardt, J.F. Signaling components of redox active endosomes: The redoxosomes. Antioxid. Redox Signal. 2009, 11, 1313–1333. [Google Scholar] [CrossRef] [PubMed]
- Miller, F.J., Jr.; Filali, M.; Huss, G.J.; Stanic, B.; Chamseddine, A.; Barna, T.J.; Lamb, F.S. Cytokine activation of nuclear factor ĸB in vascular smooth muscle cells requires signaling endosomes containing Nox1 and ClC-3. Circ. Res. 2007, 101, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhang, Y.; Marden, J.J.; Banfi, B.; Engelhardt, J.F. Endosomal NADPH oxidase regulates c-Src activation following hypoxia/reoxygenation injury. Biochem. J. 2008, 411, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Puri, C.; Tosoni, D.; Comai, R.; Rabellino, A.; Segat, D.; Caneva, F.; Luzzi, P.; Di Fiore, P.P.; Tacchetti, C. Relationships between EGFR signaling-competent and endocytosis-competent membrane microdomains. Mol. Biol. Cell 2005, 16, 2704–2718. [Google Scholar] [CrossRef] [PubMed]
- Vieira, A.V.; Lamaze, C.; Schmid, S.L. Control of EGF receptor signaling by clathrin-mediated endocytosis. Science 1996, 274, 2086–2089. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Pennock, S.; Chen, X.; Wang, Z. Endosomal signaling of epidermal growth factor receptor stimulates signal transduction pathways leading to cell survival. Mol. Cell. Biol. 2002, 22, 7279–7290. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Pennock, S.D.; Chen, X.; Kazlauskas, A.; Wang, Z. Platelet-derived growth factor receptor-mediated signal transduction from endosomes. J. Biol. Chem. 2004, 279, 8038–8046. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.B.; Harris, R.C. Autocrine, paracrine and juxtacrine signaling by EGFR ligands. Cell Signal. 2005, 17, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Farrow, M.A.; Chumbler, N.M.; Lapierre, L.A.; Franklin, J.L.; Rutherford, S.A.; Goldenring, J.R.; Lacy, D.B. Clostridium difficile toxin B-induced necrosis is mediated by the host epithelial cell NADPH oxidase complex. Proc. Natl. Acad. Sci. USA 2013, 110, 18674–18679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, H.; Fan, L.; Chen, T.; Li, R.; Li, X.; He, Q.; Botella, M.A.; Lin, J. Clathrin and membrane microdomains cooperatively regulate RbohD dynamics and activity in Arabidopsis. Plant. Cell 2014, 26, 1729–1745. [Google Scholar] [CrossRef] [PubMed]
- Balla, T. Phosphoinositides: Tiny lipids with giant impact on cell regulation. Physiol. Rev. 2013, 93, 1019–1137. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A. Phosphoinositide recognition domains. Traffic 2003, 4, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Hammond, G.R.; Balla, T. Polyphosphoinositide binding domains: Key to inositol lipid biology. Biochim. Biophys. Acta 2015, 1851, 746–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, G.; Lambeth, J.D. NOXO1, regulation of lipid binding, localization, and activation of Nox1 by the Phox homology (PX) domain. J. Biol. Chem. 2004, 279, 4737–4742. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Virbasius, J.V.; Song, X.; Pomerleau, D.P.; Zhou, G.W. The p40phox and p47phox PX domains of NADPH oxidase target cell membranes via direct and indirect recruitment by phosphoinositides. J. Biol. Chem. 2002, 277, 4512–4518. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, T.; Lambeth, J.D. Phosphatidylinositol (4,5)-bisphosphate modulates Nox5 localization via an N-terminal polybasic region. Mol. Biol. Cell 2008, 19, 4020–4031. [Google Scholar] [CrossRef] [PubMed]
- Fulton, D.J. Nox5 and the regulation of cellular function. Antioxid. Redox Signal. 2009, 11, 2443–2452. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Song, L.; Jope, R.S. Cholinergic stimulation of AP-1 and NF kappa B transcription factors is differentially sensitive to oxidative stress in SH-SY5Y neuroblastoma: Relationship to phosphoinositide hydrolysis. J. Neurosci. Off. J. Soc. Neurosci. 1996, 16, 5914–5922. [Google Scholar] [CrossRef]
- van den Bout, I.; Jones, D.R.; Shah, Z.H.; Halstead, J.R.; Keune, W.J.; Mohammed, S.; D’Santos, C.S.; Divecha, N. Collaboration of AMPK and PKC to induce phosphorylation of Ser413 on PIP5K1B resulting in decreased kinase activity and reduced PtdIns(4,5)P2 synthesis in response to oxidative stress and energy restriction. Biochem. J. 2013, 455, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.P. Hydrogen peroxide stimulates the epithelial sodium channel through a phosphatidylinositide 3-kinase-dependent pathway. J. Biol. Chem. 2011, 286, 32444–32453. [Google Scholar] [CrossRef] [PubMed]
- Müller, G.; Wied, S.; Walz, N.; Jung, C. Translocation of glycosylphosphatidylinositol-anchored proteins from plasma membrane microdomains to lipid droplets in rat adipocytes is induced by palmitate, H2O2, and the sulfonylurea drug glimepiride. Mol. Pharmacol. 2008, 73, 1513–1529. [Google Scholar] [CrossRef] [PubMed]
- Kwon, J.; Lee, S.R.; Yang, K.S.; Ahn, Y.; Kim, Y.J.; Stadtman, E.R.; Rhee, S.G. Reversible oxidation and inactivation of the tumor suppressor PTEN in cells stimulated with peptide growth factors. Proc. Natl. Acad. Sci. USA 2004, 101, 16419–16424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finegold, A.A.; Shatwell, K.P.; Segal, A.W.; Klausner, R.D.; Dancis, A. Intramembrane bis-heme motif for transmembrane electron transport conserved in a yeast iron reductase and the human NADPH oxidase. J. Biol. Chem. 1996, 271, 31021–31024. [Google Scholar] [CrossRef] [PubMed]
- Stasia, M.J. CYBA encoding p22phox, the cytochrome b558 alpha polypeptide: Gene structure, expression, role and physiopathology. Gene 2016, 586, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Sugawara, M.; Sugawara, Y.; Wen, K.; Giulivi, C. Generation of oxygen free radicals in thyroid cells and inhibition of thyroid peroxidase. Exp. Biol. Med. 2002, 227, 141–146. [Google Scholar] [CrossRef]
- Lambeth, J.D.; Neish, A.S. Nox enzymes and new thinking on reactive oxygen: A double-edged sword revisited. Ann. Rev.Pathol. 2014, 9, 119–145. [Google Scholar] [CrossRef] [PubMed]
- Nisimoto, Y.; Jackson, H.M.; Ogawa, H.; Kawahara, T.; Lambeth, J.D. Constitutive NADPH-dependent electron transferase activity of the Nox4 dehydrogenase domain. Biochemistry 2010, 49, 2433–2442. [Google Scholar] [CrossRef] [PubMed]
- Laurindo, F.R.; Araujo, T.L.; Abrahao, T.B. Nox NADPH oxidases and the endoplasmic reticulum. Antioxid. Redox Signal. 2014, 20, 2755–2775. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, J.; Nakagawa, K.; Yamasaki, T.; Nakamura, K.; Takeya, R.; Kuribayashi, F.; Imajoh-Ohmi, S.; Igarashi, K.; Shibata, Y.; Sueishi, K.; et al. The superoxide-producing NAD(P)H oxidase Nox4 in the nucleus of human vascular endothelial cells. Genes Cells Devoted Mol. Cell. Mech. 2005, 10, 1139–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilenski, L.L.; Clempus, R.E.; Quinn, M.T.; Lambeth, J.D.; Griendling, K.K. Distinct subcellular localizations of Nox1 and Nox4 in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Clempus, R.E.; Griendling, K.K. Reactive oxygen species signaling in vascular smooth muscle cells. Cardiovasc. Res. 2006, 71, 216–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gianni, D.; Diaz, B.; Taulet, N.; Fowler, B.; Courtneidge, S.A.; Bokoch, G.M. Novel p47phox-related organizers regulate NADPH oxidase 1 (Nox1) activity and localization. Sci. Signal. 2010, 2, ra54. [Google Scholar]
- Arbiser, J.L.; Petros, J.; Klafter, R.; Govindajaran, B.; McLaughlin, E.R.; Brown, L.F.; Cohen, C.; Moses, M.; Kilroy, S.; Arnold, R.S.; et al. Reactive oxygen generated by Nox1 triggers the angiogenic switch. Proc. Natl. Acad. Sci. USA 2002, 99, 715–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montezano, A.C.; Touyz, R.M. Reactive oxygen species and endothelial function—Role of nitric oxide synthase uncoupling and Nox family nicotinamide adenine dinucleotide phosphate oxidases. Basic Clin. Pharmacol. Toxicol. 2012, 110, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.L.; Brockman, D.; Campos, B.; Myatt, L. Expression of NADPH oxidase isoform 1 (Nox1) in human placenta: Involvement in preeclampsia. Placenta 2006, 27, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Murillo, I.; Henderson, L.M. Expression of GP91phox/Nox2 in COS-7 cells: Cellular localization of the protein and the detection of outward proton currents. Biochem. J. 2005, 385, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Byrne, J.A.; Grieve, D.J.; Bendall, J.K.; Li, J.M.; Gove, C.; Lambeth, J.D.; Cave, A.C.; Shah, A.M. Contrasting roles of NADPH oxidase isoforms in pressure-overload versus angiotensin II-induced cardiac hypertrophy. Circ. Res. 2003, 93, 802–805. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Cruz, A.; Vilchis-Landeros, M.M.; Guinzberg, R.; Villalobos-Molina, R.; Pina, E. Nox2 activated by alpha1-adrenoceptors modulates hepatic metabolic routes stimulated by beta-adrenoceptors. Free Radic. Res. 2011, 45, 1366–1378. [Google Scholar] [CrossRef] [PubMed]
- Piccoli, C.; Ria, R.; Scrima, R.; Cela, O.; D’Aprile, A.; Boffoli, D.; Falzetti, F.; Tabilio, A.; Capitanio, N. Characterization of mitochondrial and extra-mitochondrial oxygen consuming reactions in human hematopoietic stem cells. Novel evidence of the occurrence of NAD(P)H oxidase activity. J. Biol. Chem. 2005, 280, 26467–26476. [Google Scholar] [CrossRef] [PubMed]
- Paffenholz, R.; Bergstrom, R.A.; Pasutto, F.; Wabnitz, P.; Munroe, R.J.; Jagla, W.; Heinzmann, U.; Marquardt, A.; Bareiss, A.; Laufs, J.; et al. Vestibular defects in head-tilt mice result from mutations in Nox3, encoding an NADPH oxidase. Genes Dev. 2004, 18, 486–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, Y.; Banfi, B.; Jesaitis, A.J.; Dinauer, M.C.; Allen, L.A.; Nauseef, W.M. Critical roles for p22phox in the structural maturation and subcellular targeting of Nox3. Biochem. J. 2007, 403, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Chun, Y.S.; Park, K.S.; Kim, S.J.; Choi, S.O.; Kim, H.L.; Park, J.W. Identification of subdomains in NADPH oxidase-4 critical for the oxygen-dependent regulation of TASK-1 K+ channels. Am. J. Physiol. Cell Physiol. 2009, 297, C855–C864. [Google Scholar] [CrossRef] [PubMed]
- Fischer, H. Mechanisms and function of DUOX in epithelia of the lung. Antioxid. Redox Signal. 2009, 11, 2453–2465. [Google Scholar] [CrossRef] [PubMed]
- Dupuy, C.; Ohayon, R.; Valent, A.; Noel-Hudson, M.S.; Deme, D.; Virion, A. Purification of a novel flavoprotein involved in the thyroid NADPH oxidase. Cloning of the porcine and human cdnas. J. Biol. Chem. 1999, 274, 37265–37269. [Google Scholar] [CrossRef] [PubMed]
- De Deken, X.; Wang, D.; Many, M.C.; Costagliola, S.; Libert, F.; Vassart, G.; Dumont, J.E.; Miot, F. Cloning of two human thyroid cDNAs encoding new members of the NADPH oxidase family. J. Biol. Chem. 2000, 275, 23227–23233. [Google Scholar] [CrossRef] [PubMed]
- Forteza, R.; Salathe, M.; Miot, F.; Conner, G.E. Regulated hydrogen peroxide production by Duox in human airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2005, 32, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Rada, B.; Leto, T.L. Oxidative innate immune defenses by Nox/Duox family NADPH oxidases. Contrib. Microbiol. 2008, 15, 164–187. [Google Scholar] [PubMed]
- Sumimoto, H. Structure, regulation and evolution of Nox-family NADPH oxidases that produce reactive oxygen species. FEBS J. 2008, 275, 3249–3277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lambeth, J.D. Nox enzymes, ROS, and chronic disease: An example of antagonistic pleiotropy. Free Radic. Biol. Med. 2007, 43, 332–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilhardt, F.; van Deurs, B. The phagocyte NADPH oxidase depends on cholesterol-enriched membrane microdomains for assembly. EMBO J. 2004, 23, 739–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Rizzo, V. TNF-alpha potentiates protein-tyrosine nitration through activation of NADPH oxidase and eNOS localized in membrane rafts and caveolae of bovine aortic endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H954–H962. [Google Scholar] [CrossRef] [PubMed]
- Bokoch, G.M.; Diebold, B.A. Current molecular models for NADPH oxidase regulation by Rac GTPase. Blood 2002, 100, 2692–2696. [Google Scholar] [CrossRef] [PubMed]
- Allen, L.A.; DeLeo, F.R.; Gallois, A.; Toyoshima, S.; Suzuki, K.; Nauseef, W.M. Transient association of the nicotinamide adenine dinucleotide phosphate oxidase subunits p47phox and p67phox with phagosomes in neutrophils from patients with X-linked chronic granulomatous disease. Blood 1999, 93, 3521–3530. [Google Scholar] [PubMed]
- Pierini, L.M.; Eddy, R.J.; Fuortes, M.; Seveau, S.; Casulo, C.; Maxfield, F.R. Membrane lipid organization is critical for human neutrophil polarization. J. Biol. Chem. 2003, 278, 10831–10841. [Google Scholar] [CrossRef] [PubMed]
- Tall, E.G.; Spector, I.; Pentyala, S.N.; Bitter, I.; Rebecchi, M.J. Dynamics of phosphatidylinositol 4,5-bisphosphate in actin-rich structures. Curr. Biol. 2000, 10, 743–746. [Google Scholar] [CrossRef]
- Grasberger, H.; De Deken, X.; Miot, F.; Pohlenz, J.; Refetoff, S. Missense mutations of dual oxidase 2 (DUOX2) implicated in congenital hypothyroidism have impaired trafficking in cells reconstituted with DUOX2 maturation factor. Mol. Endocrinol. 2007, 21, 1408–1421. [Google Scholar] [CrossRef] [PubMed]
- Grasberger, H.; Refetoff, S. Identification of the maturation factor for dual oxidase. Evolution of an eukaryotic operon equivalent. J. Biol. Chem. 2006, 281, 18269–18272. [Google Scholar] [CrossRef] [PubMed]
- Isshiki, M.; Anderson, R.G. Calcium signal transduction from caveolae. Cell Calcium 1999, 26, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Weerth, S.H.; Holtzclaw, L.A.; Russell, J.T. Signaling proteins in raft-like microdomains are essential for Ca2+ wave propagation in glial cells. Cell Calcium 2007, 41, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.A.; Kim, B.; You, H.; Lee, W.J. Uracil-induced signaling pathways for DUOX-dependent gut immunity. Fly 2015, 9, 115–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heppner, D.E.; van der Vliet, A. Redox-dependent regulation of epidermal growth factor receptor signaling. Redox Biol. 2016, 8, 24–27. [Google Scholar] [CrossRef] [PubMed]
- Mistry, R.K.; Brewer, A.C. Redox regulation of gasotransmission in the vascular system: A focus on angiogenesis. Free Radic. Biol. Med. 2017, 108, 500–516. [Google Scholar] [CrossRef] [PubMed]
- Ogier-Denis, E.; Mkaddem, S.B.; Vandewalle, A. NOX enzymes and Toll-like receptor signaling. Sem. Immunopathol. 2008, 30, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.; Smith, S.W.; Anderson, B.D. Kinetics and mechanism of the reaction of cysteine and hydrogen peroxide in aqueous solution. J. Pharm. Sci. 2005, 94, 304–316. [Google Scholar] [CrossRef] [PubMed]
- Wadley, A.J.; Aldred, S.; Coles, S.J. An unexplored role for Peroxiredoxin in exercise-induced redox signalling? Redox Biol. 2016, 8, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Antunes, F.; Brito, P.M. Quantitative biology of hydrogen peroxide signaling. Redox Biol. 2017, 13, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Bienert, G.P.; Chaumont, F. Aquaporin-facilitated transmembrane diffusion of hydrogen peroxide. Biochim. Biophys. Acta 2014, 1840, 1596–1604. [Google Scholar] [CrossRef] [PubMed]
- Verkman, A.S. Aquaporins at a glance. J. Cell Sci. 2011, 124, 2107–2112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bienert, G.P.; Schjoerring, J.K.; Jahn, T.P. Membrane transport of hydrogen peroxide. Biochim. Biophys. Acta 2006, 1758, 994–1003. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.W.; Dickinson, B.C.; Chang, C.J. Aquaporin-3 mediates hydrogen peroxide uptake to regulate downstream intracellular signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 15681–15686. [Google Scholar] [CrossRef] [PubMed]
- Bertolotti, M.; Bestetti, S.; Garcia-Manteiga, J.M.; Medrano-Fernandez, I.; Dal Mas, A.; Malosio, M.L.; Sitia, R. Tyrosine kinase signal modulation: A matter of H2O2 membrane permeability? Antioxid. Redox Signal. 2013, 19, 1447–1451. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Moniaga, C.S.; Nielsen, S.; Hara-Chikuma, M. Aquaporin-9 facilitates membrane transport of hydrogen peroxide in mammalian cells. Biochem. Biophys. Res. Commun. 2016, 471, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Appenzeller-Herzog, C.; Bánhegyi, G.; Bogeski, I.; Davies, K.J.; Delaunay-Moisan, A.; Forman, H.J.; Görlach, A.; Kietzmann, T.; Laurindo, F.; Margittai, E.; et al. Transit of H2O2 across the endoplasmic reticulum membrane is not sluggish. Free Radic. Biol. Med. 2016, 94, 157–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, Q.; Steudle, E. Oxidative gating of water channels (aquaporins) in corn roots. Plant Cell Environ. 2006, 29, 459–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ampilogova, Y.N.; Zhestkova, I.M.; Trofimova, M.S. Redox modulation of osmotic water permeability in plasma membranes isolated from roots and shoots of pea seedlings. Russ. J. Plant. Physiol. 2006, 53, 622–628. [Google Scholar] [CrossRef]
- Henzler, T.; Ye, Q.; Steudle, E. Oxidative gating of water channels (aquaporins) in Chara by hydroxyl radicals. Plant. Cell Environ. 2004, 27, 1184–1195. [Google Scholar] [CrossRef]
- Dynowski, M.; Schaaf, G.; Loque, D.; Moran, O.; Ludewig, U. Plant plasma membrane water channels conduct the signalling molecule H2O2. Biochem. J. 2008, 414, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Bienert, G.P.; Cavez, D.; Besserer, A.; Berny, M.C.; Gilis, D.; Rooman, M.; Chaumont, F. A conserved cysteine residue is involved in disulfide bond formation between plant plasma membrane aquaporin monomers. Biochem. J. 2012, 445, 101–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boursiac, Y.; Boudet, J.; Postaire, O.; Luu, D.T.; Tournaire-Roux, C.; Maurel, C. Stimulus-induced downregulation of root water transport involves reactive oxygen species-activated cell signalling and plasma membrane intrinsic protein internalization. Plant J. Cell Mol. Biol. 2008, 56, 207–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bestetti, S.; Medraño-Fernandez, I.; Galli, M.; Ghitti, M.; Bienert, G.P.; Musco, G.; Orsi, A.; Rubartelli, A.; Sitia, R. A persulfidation-based mechanism controls aquaporin-8 conductance. Sci. Adv. 2018, 4, eaar5770. [Google Scholar] [CrossRef] [PubMed]
- Hara-Chikuma, M.; Satooka, H.; Watanabe, S.; Honda, T.; Miyachi, Y.; Watanabe, T.; Verkman, A.S. Aquaporin-3-mediated hydrogen peroxide transport is required for NF-κB signalling in keratinocytes and development of psoriasis. Nat. Commun. 2015, 6, 7454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertolotti, M.; Farinelli, G.; Galli, M.; Aiuti, A.; Sitia, R. AQP8 transports Nox2-generated H2O2 across the plasma membrane to promote signaling in B cells. J. Leukocyte Biol. 2016, 100, 1071–1079. [Google Scholar] [CrossRef] [PubMed]
- Bocharov, E.V.; Sharonov, G.V.; Bocharova, O.V.; Pavlov, K.V. Conformational transitions and interactions underlying the function of membrane embedded receptor protein kinases. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1417–1429. [Google Scholar] [CrossRef] [PubMed]
- Oshikawa, J.; Urao, N.; Kim, H.W.; Kaplan, N.; Razvi, M.; McKinney, R.; Poole, L.B.; Fukai, T.; Ushio-Fukai, M. Extracellular SOD-derived H2O2 promotes VEGF signaling in caveolae/lipid rafts and post-ischemic angiogenesis in mice. PLoS ONE 2010, 5, e10189. [Google Scholar] [CrossRef] [PubMed]
- Mazzone, A.; Tietz, P.; Jefferson, J.; Pagano, R.; LaRusso, N.F. Isolation and characterization of lipid microdomains from apical and basolateral plasma membranes of rat hepatocytes. Hepatology 2006, 43, 287–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shenoy-Scaria, A.M.; Dietzen, D.J.; Kwong, J.; Link, D.C.; Lublin, D.M. Cysteine3 of Src family protein tyrosine kinase determines palmitoylation and localization in caveolae. J. Cell Biol. 1994, 126, 353–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Leu, S.; Cheong, A.; Zhang, H.; Baibakov, B.; Shih, C.; Birnbaum, M.J.; Donowitz, M. Akt2, phosphatidylinositol 3-kinase, and PTEN are in lipid rafts of intestinal cells: Role in absorption and differentiation. Gastroenterology 2004, 126, 122–135. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.Y.; Park, S.J.; Joe, E.H.; Jou, I. Raft-mediated Src homology 2 domain-containing proteintyrosine phosphatase 2 (SHP-2) regulation in microglia. J. Biol. Chem. 2006, 281, 11872–11878. [Google Scholar] [CrossRef] [PubMed]
- Caselli, A.; Mazzinghi, B.; Camici, G.; Manao, G.; Ramponi, G. Some protein tyrosine phosphatases target in part to lipid rafts and interact with caveolin-1. Biochem. Biophys. Res. Commun. 2002, 296, 692–697. [Google Scholar] [CrossRef] [Green Version]
- Yi, F.; Jin, S.; Zhang, F.; Xia, M.; Bao, J.X.; Hu, J.; Poklis, J.L.; Li, P.L. Formation of lipid raft redox signalling platforms in glomerular endothelial cells: An early event of homocysteine-induced glomerular injury. J. Cell. Mol. Med. 2009, 13, 3303–3314. [Google Scholar] [CrossRef] [PubMed]
- Brinckmann, R.; Schnurr, K.; Heydeck, D.; Rosenbach, T.; Kolde, G.; Kuhn, H. Membrane translocation of 15-lipoxygenase in hematopoietic cells is calcium-dependent and activates the oxygenase activity of the enzyme. Blood 1998, 91, 64–74. [Google Scholar] [PubMed]
- Perrone, G.; Zagami, M.; Altomare, V.; Battista, C.; Morini, S.; Rabitti, C. Cox-2 localization within plasma membrane caveolae-like structures in human lobular intraepithelial neoplasia of the breast. Virchows Arch. 2007, 451, 1039–1045. [Google Scholar] [CrossRef] [PubMed]
- Michel, T.; Feron, O. Nitric oxide synthases: Which, where, how, and why? J. Clin. Investig. 1997, 100, 2146–2152. [Google Scholar] [CrossRef] [PubMed]
- Braam, B.; Verhaar, M.C. Understanding eNOS for pharmacological modulation of endothelial function: A translational view. Curr. Pharm. Des. 2007, 13, 1727–1740. [Google Scholar] [CrossRef] [PubMed]
- Dudzinski, D.M.; Igarashi, J.; Greif, D.; Michel, T. The regulation and pharmacology of endothelial nitric oxide synthase. Ann. Rev. Pharmacol. Toxicol. 2006, 46, 235–276. [Google Scholar] [CrossRef] [PubMed]
- Bouloumie, A.; Bauersachs, J.; Linz, W.; Scholkens, B.A.; Wiemer, G.; Fleming, I.; Busse, R. Endothelial dysfunction coincides with an enhanced nitric oxide synthase expression and superoxide anion production. Hypertension 1997, 30, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Heinzel, B.; John, M.; Klatt, P.; Bohme, E.; Mayer, B. Ca2+/calmodulin-dependent formation of hydrogen peroxide by brain nitric oxide synthase. Biochem. J. 1992, 281 Pt 3, 627–630. [Google Scholar] [CrossRef]
- Stuehr, D.; Pou, S.; Rosen, G.M. Oxygen reduction by nitric-oxide synthases. J. Biol. Chem. 2001, 276, 14533–14536. [Google Scholar] [CrossRef] [PubMed]
- Roe, N.D.; Ren, J. Nitric oxide synthase uncoupling: A therapeutic target in cardiovascular diseases. Vasc. Pharmacol. 2012, 57, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C.; Xie, Q.W. Nitric oxide synthases: Roles, tolls, and controls. Cell 1994, 78, 915–918. [Google Scholar] [CrossRef]
- Gonzalez, E.; Kou, R.; Lin, A.J.; Golan, D.E.; Michel, T. Subcellular targeting and agonist-induced site-specific phosphorylation of endothelial nitric-oxide synthase. J. Biol. Chem. 2002, 277, 39554–39560. [Google Scholar] [CrossRef] [PubMed]
- Shaul, P.W. Regulation of endothelial nitric oxide synthase: Location, location, location. Ann. Rev. Physiol. 2002, 64, 749–774. [Google Scholar] [CrossRef] [PubMed]
- Yeh, D.C.; Duncan, J.A.; Yamashita, S.; Michel, T. Depalmitoylation of endothelial nitric-oxide synthase by acyl-protein thioesterase 1 is potentiated by Ca2+-calmodulin. J. Biol. Chem. 1999, 274, 33148–33154. [Google Scholar] [CrossRef] [PubMed]
- Michel, J.B.; Feron, O.; Sase, K.; Prabhakar, P.; Michel, T. Caveolin versus calmodulin. Counterbalancing allosteric modulators of endothelial nitric oxide synthase. J. Biol. Chem. 1997, 272, 25907–25912. [Google Scholar] [CrossRef] [PubMed]
- Drab, M.; Verkade, P.; Elger, M.; Kasper, M.; Lohn, M.; Lauterbach, B.; Menne, J.; Lindschau, C.; Mende, F.; Luft, F.C.; et al. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science 2001, 293, 2449–2452. [Google Scholar] [CrossRef] [PubMed]
- Prabhakar, P.; Cheng, V.; Michel, T. A chimeric transmembrane domain directs endothelial nitric-oxide synthase palmitoylation and targeting to plasmalemmal caveolae. J. Biol. Chem. 2000, 275, 19416–19421. [Google Scholar] [CrossRef] [PubMed]
- Felley-Bosco, E.; Bender, F.C.; Courjault-Gautier, F.; Bron, C.; Quest, A.F. Caveolin-1 down-regulates inducible nitric oxide synthase via the proteasome pathway in human colon carcinoma cells. Proc. Natl. Acad. Sci. USA 2000, 97, 14334–14339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gharavi, N.M.; Baker, N.A.; Mouillesseaux, K.P.; Yeung, W.; Honda, H.M.; Hsieh, X.; Yeh, M.; Smart, E.J.; Berliner, J.A. Role of endothelial nitric oxide synthase in the regulation of SREBP activation by oxidized phospholipids. Circ. Res. 2006, 98, 768–776. [Google Scholar] [CrossRef] [PubMed]
- Espey, M.G.; Thomas, D.D.; Miranda, K.M.; Wink, D.A. Focusing of nitric oxide mediated nitrosation and oxidative nitrosylation as a consequence of reaction with superoxide. Proc. Natl. Acad. Sci. USA 2002, 99, 11127–11132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrett, D.M.; Black, S.M.; Todor, H.; Schmidt-Ullrich, R.K.; Dawson, K.S.; Mikkelsen, R.B. Inhibition of protein-tyrosine phosphatases by mild oxidative stresses is dependent on S-nitrosylation. J. Biol. Chem. 2005, 280, 14453–14461. [Google Scholar] [CrossRef] [PubMed]
- Hess, D.T.; Matsumoto, A.; Kim, S.O.; Marshall, H.E.; Stamler, J.S. Protein S-nitrosylation: Purview and parameters. Nat. Rev. Mol. Cell Biol. 2005, 6, 150–166. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: Oxidative eustress. Redox Biol. 2017, 11, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.H.; Griffiths, H.R. The dual role of ROS in autoimmune and inflammatory diseases: Evidence from preclinical models. Free Radic. Biol. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Nauseef, W.M. Assembly of the phagocyte NADPH oxidase. Histochem. Cell Biol. 2004, 122, 277–291. [Google Scholar] [CrossRef] [PubMed]
- Bylund, J.; Brown, K.L.; Movitz, C.; Dahlgren, C.; Karlsson, A. Intracellular generation of superoxide by the phagocyte NADPH oxidase: How, where, and what for? Free Radic. Biol. Med. 2010, 49, 1834–1845. [Google Scholar] [CrossRef] [PubMed]
- Decoursey, T.E.; Ligeti, E. Regulation and termination of NADPH oxidase activity. Cell. Mol. Life Sci. 2005, 62, 2173–2193. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C.; Kettle, A.J.; Hampton, M.B. Reactive Oxygen Species and Neutrophil Function. Ann. Rev. Biochem. 2016, 85, 765–792. [Google Scholar] [CrossRef] [PubMed]
- Roos, D.; Kuhns, D.B.; Maddalena, A.; Bustamante, J.; Kannengiesser, C.; de Boer, M.; van Leeuwen, K.; Koker, M.Y.; Wolach, B.; Roesler, J.; et al. Hematologically important mutations: The autosomal recessive forms of chronic granulomatous disease (second update). Blood Cells Mol. Dis. 2010, 44, 291–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roos, D.; Kuhns, D.B.; Maddalena, A.; Roesler, J.; Lopez, J.A.; Ariga, T.; Avcin, T.; de Boer, M.; Bustamante, J.; Condino-Neto, A.; et al. Hematologically important mutations: X-linked chronic granulomatous disease (third update). Blood Cells Mol. Dis. 2010, 45, 246–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambruso, D.R.; Knall, C.; Abell, A.N.; Panepinto, J.; Kurkchubasche, A.; Thurman, G.; Gonzalez-Aller, C.; Hiester, A.; deBoer, M.; Harbeck, R.J.; et al. Human neutrophil immunodeficiency syndrome is associated with an inhibitory Rac2 mutation. Proc. Natl. Acad. Sci. USA 2000, 97, 4654–4659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, D.A.; Tao, W.; Yang, F.; Kim, C.; Gu, Y.; Mansfield, P.; Levine, J.E.; Petryniak, B.; Derrow, C.W.; Harris, C.; et al. Dominant negative mutation of the hematopoietic-specific Rho GTPase, Rac2, is associated with a human phagocyte immunodeficiency. Blood 2000, 96, 1646–1654. [Google Scholar] [PubMed]
- van den Berg, J.M.; van Koppen, E.; Ahlin, A.; Belohradsky, B.H.; Bernatowska, E.; Corbeel, L.; Espanol, T.; Fischer, A.; Kurenko-Deptuch, M.; Mouy, R.; et al. Chronic granulomatous disease: The European experience. PLoS ONE 2009, 4, e5234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holland, S.M. Chronic granulomatous disease. Hematol./Oncol. Clin. N. Am. 2013, 27, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Baehner, R.L.; Karnovsky, M.L. Deficiency of reduced nicotinamide-adenine dinucleotide oxidase in chronic granulomatous disease. Science 1968, 162, 1277–1279. [Google Scholar] [CrossRef] [PubMed]
- Winkelstein, J.A.; Marino, M.C.; Johnston, R.B., Jr.; Boyle, J.; Curnutte, J.; Gallin, J.I.; Malech, H.L.; Holland, S.M.; Ochs, H.; Quie, P.; et al. Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine 2000, 79, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Rieber, N.; Hector, A.; Kuijpers, T.; Roos, D.; Hartl, D. Current concepts of hyperinflammation in chronic granulomatous disease. Clin. Dev. Immunol. 2012, 2012, 252460. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.R.; Goldblatt, D.; Buddle, J.; Morton, L.; Thrasher, A.J. Diminished production of anti-inflammatory mediators during neutrophil apoptosis and macrophage phagocytosis in chronic granulomatous disease (CGD). J. Leukocyte Biol. 2003, 73, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Bylund, J.; MacDonald, K.L.; Brown, K.L.; Mydel, P.; Collins, L.V.; Hancock, R.E.; Speert, D.P. Enhanced inflammatory responses of chronic granulomatous disease leukocytes involve ROS-independent activation of NF-κB. Eur. J. Immunol. 2007, 37, 1087–1096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, K.L.; Bylund, J.; MacDonald, K.L.; Song-Zhao, G.X.; Elliott, M.R.; Falsafi, R.; Hancock, R.E.; Speert, D.P. ROS-deficient monocytes have aberrant gene expression that correlates with inflammatory disorders of chronic granulomatous disease. Clin. Immunol. 2008, 129, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Bjorkman, L.; Dahlgren, C.; Karlsson, A.; Brown, K.L.; Bylund, J. Phagocyte-derived reactive oxygen species as suppressors of inflammatory disease. Arthritis Rheum. 2008, 58, 2931–2935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartl, D.; Lehmann, N.; Hoffmann, F.; Jansson, A.; Hector, A.; Notheis, G.; Roos, D.; Belohradsky, B.H.; Wintergerst, U. Dysregulation of innate immune receptors on neutrophils in chronic granulomatous disease. J. Allergy Clin. Immunol. 2008, 121, 375.e379–382.e379. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, S.J.; Kettle, A.J.; Rosen, H.; Winterbourn, C.C.; Nauseef, W.M. Myeloperoxidase: A front-line defender against phagocytosed microorganisms. J. Leukocyte Biol. 2013, 93, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Campbell, A.M.; Kashgarian, M.; Shlomchik, M.J. NADPH oxidase inhibits the pathogenesis of systemic lupus erythematosus. Sci. Transl. Med. 2012, 4, 157ra141. [Google Scholar] [CrossRef] [PubMed]
- Hultqvist, M.; Bäcklund, J.; Bauer, K.; Gelderman, K.A.; Holmdahl, R. Lack of reactive oxygen species breaks T cell tolerance to collagen type II and allows development of arthritis in mice. J. Immunol. 2007, 179, 1431–1437. [Google Scholar] [CrossRef] [PubMed]
- Kelkka, T.; Kienhöfer, D.; Hoffmann, M.; Linja, M.; Wing, K.; Sareila, O.; Hultqvist, M.; Laajala, E.; Chen, Z.; Vasconcelos, J.; et al. Reactive oxygen species deficiency induces autoimmunity with type 1 interferon signature. Antioxid. Redox Signal. 2014, 21, 2231–2245. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, G.J.; Deffit, S.N.; McLetchie, S.; Perez, L.; Walline, C.C.; Blum, J.S. A role for NADPH oxidase in antigen presentation. Front. Immunol. 2013, 4, 295. [Google Scholar] [CrossRef] [PubMed]
- Schappi, M.G.; Jaquet, V.; Belli, D.C.; Krause, K.H. Hyperinflammation in chronic granulomatous disease and anti-inflammatory role of the phagocyte NADPH oxidase. Sem. Immunopathol. 2008, 30, 255–271. [Google Scholar] [CrossRef] [PubMed]
- Jancic, C.; Savina, A.; Wasmeier, C.; Tolmachova, T.; El-Benna, J.; Dang, P.M.; Pascolo, S.; Gougerot-Pocidalo, M.A.; Raposo, G.; Seabra, M.C.; et al. Rab27a regulates phagosomal pH and NADPH oxidase recruitment to dendritic cell phagosomes. Nat. Cell Biol. 2007, 9, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Savina, A.; Jancic, C.; Hugues, S.; Guermonprez, P.; Vargas, P.; Moura, I.C.; Lennon-Dumenil, A.M.; Seabra, M.C.; Raposo, G.; Amigorena, S. Nox2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell 2006, 126, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Kotsias, F.; Hoffmann, E.; Amigorena, S.; Savina, A. Reactive oxygen species production in the phagosome: Impact on antigen presentation in dendritic cells. Antioxid. Redox Signal. 2013, 18, 714–729. [Google Scholar] [CrossRef] [PubMed]
- Mantegazza, A.R.; Savina, A.; Vermeulen, M.; Perez, L.; Geffner, J.; Hermine, O.; Rosenzweig, S.D.; Faure, F.; Amigorena, S. NADPH oxidase controls phagosomal pH and antigen cross-presentation in human dendritic cells. Blood 2008, 112, 4712–4722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hari, A.; Ganguly, A.; Mu, L.; Davis, S.P.; Stenner, M.D.; Lam, R.; Munro, F.; Namet, I.; Alghamdi, E.; Furstenhaupt, T.; et al. Redirecting soluble antigen for MHC class I cross-presentation during phagocytosis. Eur. J. Immunol. 2015, 45, 383–395. [Google Scholar] [CrossRef] [PubMed]
- Rybicka, J.M.; Balce, D.R.; Chaudhuri, S.; Allan, E.R.; Yates, R.M. Phagosomal proteolysis in dendritic cells is modulated by NADPH oxidase in a pH-independent manner. EMBO J. 2012, 31, 932–944. [Google Scholar] [CrossRef] [PubMed]
- Allan, E.R.; Tailor, P.; Balce, D.R.; Pirzadeh, P.; McKenna, N.T.; Renaux, B.; Warren, A.L.; Jirik, F.R.; Yates, R.M. NADPH oxidase modifies patterns of MHC class II-restricted epitopic repertoires through redox control of antigen processing. J. Immunol. 2014, 192, 4989–5001. [Google Scholar] [CrossRef] [PubMed]
- Cotugno, N.; Finocchi, A.; Cagigi, A.; Di Matteo, G.; Chiriaco, M.; Di Cesare, S.; Rossi, P.; Aiuti, A.; Palma, P.; Douagi, I. Defective B-cell proliferation and maintenance of long-term memory in patients with chronic granulomatous disease. J. Allergy Clin. Immunol. 2015, 135, 753–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bleesing, J.J.; Souto-Carneiro, M.M.; Savage, W.J.; Brown, M.R.; Martinez, C.; Yavuz, S.; Brenner, S.; Siegel, R.M.; Horwitz, M.E.; Lipsky, P.E.; et al. Patients with chronic granulomatous disease have a reduced peripheral blood memory B cell compartment. J. Immunol. 2006, 176, 7096–7103. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Feng, X.; He, C.; Gao, H.; Yang, L.; Ma, Q.; Guo, L.; Qiao, Y.; Yang, H.; Ma, T. Defective macrophage function in aquaporin-3 deficiency. FASEB J. 2011, 25, 4233–4239. [Google Scholar] [CrossRef] [PubMed]
- Lindskog, C.; Asplund, A.; Catrina, A.; Nielsen, S.; Rutzler, M. A Systematic Characterization of Aquaporin-9 Expression in Human Normal and Pathological Tissues. J. Histochem. Cytochem. Off. J. Histochem. Soc. 2016, 64, 287–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moon, C.; Rousseau, R.; Soria, J.C.; Hoque, M.O.; Lee, J.; Jang, S.J.; Trink, B.; Sidransky, D.; Mao, L. Aquaporin expression in human lymphocytes and dendritic cells. Am. J. Hematol. 2004, 75, 128–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara-Chikuma, M.; Chikuma, S.; Sugiyama, Y.; Kabashima, K.; Verkman, A.S.; Inoue, S.; Miyachi, Y. Chemokine-dependent T cell migration requires aquaporin-3-mediated hydrogen peroxide uptake. J. Exp. Med. 2012, 209, 1743–1752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loitto, V.M.; Forslund, T.; Sundqvist, T.; Magnusson, K.E.; Gustafsson, M. Neutrophil leukocyte motility requires directed water influx. J. Leukocyte Biol. 2002, 71, 212–222. [Google Scholar] [PubMed]
- Moniaga, C.S.; Watanabe, S.; Honda, T.; Nielsen, S.; Hara-Chikuma, M. Aquaporin-9-expressing neutrophils are required for the establishment of contact hypersensitivity. Sci. Rep. 2015, 5, 15319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, C.; Cho, J.H. Inflammatory bowel disease. N. Engl. J. Med. 2009, 361, 2066–2078. [Google Scholar] [CrossRef] [PubMed]
- Biasi, F.; Leonarduzzi, G.; Oteiza, P.I.; Poli, G. Inflammatory bowel disease: Mechanisms, redox considerations, and therapeutic targets. Antioxid. Redox Signal. 2013, 19, 1711–1747. [Google Scholar] [CrossRef] [PubMed]
- Corridoni, D.; Arseneau, K.O.; Cominelli, F. Inflammatory bowel disease. Immunol. Lett. 2014, 161, 231–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gevers, D.; Kugathasan, S.; Denson, L.A.; Vazquez-Baeza, Y.; Van Treuren, W.; Ren, B.; Schwager, E.; Knights, D.; Song, S.J.; Yassour, M.; et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 2014, 15, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Haberman, Y.; Tickle, T.L.; Dexheimer, P.J.; Kim, M.O.; Tang, D.; Karns, R.; Baldassano, R.N.; Noe, J.D.; Rosh, J.; Markowitz, J.; et al. Pediatric Crohn disease patients exhibit specific ileal transcriptome and microbiome signature. J. Clin. Investig. 2014, 124, 3617–3633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goyette, P.; Labbé, C.; Trinh, T.T.; Xavier, R.J.; Rioux, J.D. Molecular pathogenesis of inflammatory bowel disease: Genotypes, phenotypes and personalized medicine. Ann. Med. 2007, 39, 177–199. [Google Scholar] [CrossRef] [PubMed]
- Cheung, E.C.; Lee, P.; Ceteci, F.; Nixon, C.; Blyth, K.; Sansom, O.J.; Vousden, K.H. Opposing effects of TIGAR- and RAC1-derived ROS on Wnt-driven proliferation in the mouse intestine. Genes Dev. 2016, 30, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Alzoghaibi, M.A. Concepts of oxidative stress and antioxidant defense in Crohn’s disease. World J. Gastroenterol. 2013, 19, 6540–6547. [Google Scholar] [CrossRef] [PubMed]
- Pravda, J. Radical induction theory of ulcerative colitis. World J. Gastroenterol. 2005, 11, 2371–2384. [Google Scholar] [CrossRef] [PubMed]
- Geiszt, M.; Lekstrom, K.; Brenner, S.; Hewitt, S.M.; Dana, R.; Malech, H.L.; Leto, T.L. NAD(P)H oxidase 1, a product of differentiated colon epithelial cells, can partially replace glycoprotein 91phox in the regulated production of superoxide by phagocytes. J. Immunol. 2003, 171, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Sommer, F.; Bäckhed, F. The gut microbiota engages different signaling pathways to induce Duox2 expression in the ileum and colon epithelium. Mucosal Immunol. 2015, 8, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Coant, N.; Ben Mkaddem, S.; Pedruzzi, E.; Guichard, C.; Treton, X.; Ducroc, R.; Freund, J.N.; Cazals-Hatem, D.; Bouhnik, Y.; Woerther, P.L.; et al. NADPH oxidase 1 modulates WNT and NOTCH1 signaling to control the fate of proliferative progenitor cells in the colon. Mol. Cell. Biol. 2010, 30, 2636–2650. [Google Scholar] [CrossRef] [PubMed]
- van der Flier, L.G.; Clevers, H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Ann. Rev. Physiol. 2009, 71, 241–260. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.C.; Tan, X.Y.; Luxenberg, D.P.; Karim, R.; Dunussi-Joannopoulos, K.; Collins, M.; Fouser, L.A. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J. Exp. Med. 2006, 203, 2271–2279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, E.M.; Oh, C.T.; Bae, Y.S.; Lee, W.J. A direct role for dual oxidase in Drosophila gut immunity. Science 2005, 310, 847–850. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Misaka, T.; Matsumoto, I.; Watanabe, H.; Abe, K. Aquaporin-9 is expressed in a mucus-secreting goblet cell subset in the small intestine. FEBS Lett. 2003, 540, 157–162. [Google Scholar] [CrossRef] [Green Version]
- Fischer, H.; Stenling, R.; Rubio, C.; Lindblom, A. Differential expression of aquaporin 8 in human colonic epithelial cells and colorectal tumors. BMC Physiol. 2001, 1, 1. [Google Scholar] [CrossRef]
- Yang, B.; Song, Y.; Zhao, D.; Verkman, A.S. Phenotype analysis of aquaporin-8 null mice. Am. J. Physiol. Cell Physiol. 2005, 288, C1161–C1170. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Li, J.; Wang, J.; Shen, X.; Sun, J. Aquaporin 3 and 8 are down-regulated in TNBS-induced rat colitis. Biochem. Biophys. Res. Commun. 2014, 443, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Hardin, J.A.; Wallace, L.E.; Wong, J.F.; O’Loughlin, E.V.; Urbanski, S.J.; Gall, D.G.; MacNaughton, W.K.; Beck, P.L. Aquaporin expression is downregulated in a murine model of colitis and in patients with ulcerative colitis, Crohn’s disease and infectious colitis. Cell Tissue Res. 2004, 318, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Zahn, A.; Moehle, C.; Langmann, T.; Ehehalt, R.; Autschbach, F.; Stremmel, W.; Schmitz, G. Aquaporin-8 expression is reduced in ileum and induced in colon of patients with ulcerative colitis. World J. Gastroenterol. 2007, 13, 1687–1695. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.X.; Dong, P.P.; Peng, R.; Li, J.; Zhang, D.Y.; Wang, J.Y.; Shen, X.Z.; Dong, L.; Sun, J.Y. Expression, localization and possible functions of aquaporins 3 and 8 in rat digestive system. Biotech. Histochem. Off. Publ. Biol. Stain Comm. 2016, 91, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Ikarashi, N.; Kon, R.; Sugiyama, K. Aquaporins in the Colon as a New Therapeutic Target in Diarrhea and Constipation. Int. J. Mol. Sci. 2016, 17, 1172. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, T.; Suzuki, T.; Koyama, H.; Tanaka, S.; Takata, K. Water channel protein AQP3 is present in epithelia exposed to the environment of possible water loss. J. Histochem. Cytochem. Off. J. Histochem. Soc. 1999, 47, 1275–1286. [Google Scholar] [CrossRef] [PubMed]
- Thiagarajah, J.R.; Zhao, D.; Verkman, A.S. Impaired enterocyte proliferation in aquaporin-3 deficiency in mouse models of colitis. Gut 2007, 56, 1529–1535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Xu, Y.; Chen, Z.; Xu, Z.; Xu, H. Knockdown of aquaporin 3 is involved in intestinal barrier integrity impairment. FEBS Lett. 2011, 585, 3113–3119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, S.; Brault, J.; Stasia, M.J.; Knaus, U.G. Genetic disorders coupled to ROS deficiency. Redox Biol. 2015, 6, 135–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorce, S.; Krause, K.H. NOX enzymes in the central nervous system: From signaling to disease. Antioxid. Redox Signal. 2009, 11, 2481–2504. [Google Scholar] [CrossRef] [PubMed]
- Choi, D.H.; Cristóvão, A.C.; Guhathakurta, S.; Lee, J.; Joh, T.H.; Beal, M.F.; Kim, Y.S. NADPH oxidase 1-mediated oxidative stress leads to dopamine neuron death in Parkinson’s disease. Antioxid. Redox Signal. 2012, 16, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Dugan, L.L.; Ali, S.S.; Shekhtman, G.; Roberts, A.J.; Lucero, J.; Quick, K.L.; Behrens, M.M. IL-6 mediated degeneration of forebrain GABAergic interneurons and cognitive impairment in aged mice through activation of neuronal NADPH oxidase. PLoS ONE 2009, 4, e5518. [Google Scholar] [CrossRef] [PubMed]
- Brennan, A.M.; Suh, S.W.; Won, S.J.; Narasimhan, P.; Kauppinen, T.M.; Lee, H.; Edling, Y.; Chan, P.H.; Swanson, R.A. NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat. Neurosci. 2009, 12, 857–863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiavone, S.; Jaquet, V.; Sorce, S.; Dubois-Dauphin, M.; Hultqvist, M.; Backdahl, L.; Holmdahl, R.; Colaianna, M.; Cuomo, V.; Trabace, L.; et al. NADPH oxidase elevations in pyramidal neurons drive psychosocial stress-induced neuropathology. Transl. Psychiatry 2012, 2, e111. [Google Scholar] [CrossRef] [PubMed]
- Kallenborn-Gerhardt, W.; Schröder, K.; Del Turco, D.; Lu, R.; Kynast, K.; Kosowski, J.; Niederberger, E.; Shah, A.M.; Brandes, R.P.; Geisslinger, G.; et al. NADPH oxidase-4 maintains neuropathic pain after peripheral nerve injury. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 10136–10145. [Google Scholar] [CrossRef] [PubMed]
- Kleinschnitz, C.; Grund, H.; Wingler, K.; Armitage, M.E.; Jones, E.; Mittal, M.; Barit, D.; Schwarz, T.; Geis, C.; Kraft, P.; et al. Post-stroke inhibition of induced NADPH oxidase type 4 prevents oxidative stress and neurodegeneration. PLoS Biol. 2010, 8, e1000479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, M.T.; Sharma, R.; Lim, J.L.; Haider, L.; Frischer, J.M.; Drexhage, J.; Mahad, D.; Bradl, M.; van Horssen, J.; Lassmann, H. NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain J. Neurol. 2012, 135, 886–899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chéret, C.; Gervais, A.; Lelli, A.; Colin, C.; Amar, L.; Ravassard, P.; Mallet, J.; Cumano, A.; Krause, K.H.; Mallat, M. Neurotoxic activation of microglia is promoted by a Nox1-dependent NADPH oxidase. J. Neurosci. Off. J. Soc. Neurosci. 2008, 28, 12039–12051. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Bedard, K.; Sorce, S.; Hinz, B.; Dubois-Dauphin, M.; Krause, K.H. NOX4 expression in human microglia leads to constitutive generation of reactive oxygen species and to constitutive IL-6 expression. J. Innate Immun. 2009, 1, 570–581. [Google Scholar] [CrossRef] [PubMed]
- Nayernia, Z.; Jaquet, V.; Krause, K.H. New insights on NOX enzymes in the central nervous system. Antioxid. Redox Signal. 2014, 20, 2815–2837. [Google Scholar] [CrossRef] [PubMed]
- MacFarlane, P.M.; Satriotomo, I.; Windelborn, J.A.; Mitchell, G.S. NADPH oxidase activity is necessary for acute intermittent hypoxia-induced phrenic long-term facilitation. J. Physiol. 2009, 587, 1931–1942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tejada-Simon, M.V.; Serrano, F.; Villasana, L.E.; Kanterewicz, B.I.; Wu, G.Y.; Quinn, M.T.; Klann, E. Synaptic localization of a functional NADPH oxidase in the mouse hippocampus. Mol. Cell. Neurosci. 2005, 29, 97–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reyes, R.C.; Brennan, A.M.; Shen, Y.; Baldwin, Y.; Swanson, R.A. Activation of neuronal NMDA receptors induces superoxide-mediated oxidative stress in neighboring neurons and astrocytes. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 12973–12978. [Google Scholar] [CrossRef] [PubMed]
- Kishida, K.T.; Pao, M.; Holland, S.M.; Klann, E. NADPH oxidase is required for NMDA receptor-dependent activation of ERK in hippocampal area CA1. J. Neurochem. 2005, 94, 299–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, N.; Yoneda, K.; Asai, K.; Sobue, K.; Tada, T.; Fujita, Y.; Katsuya, H.; Fujita, M.; Aihara, N.; Mase, M.; et al. Alterations in the expression of the AQP family in cultured rat astrocytes during hypoxia and reoxygenation. Brain Res. Mol. Brain Res. 2001, 90, 26–38. [Google Scholar] [CrossRef]
- Yang, M.; Gao, F.; Liu, H.; Yu, W.H.; Sun, S.Q. Temporal changes in expression of aquaporin-3, -4, -5 and -8 in rat brains after permanent focal cerebral ischemia. Brain Res. 2009, 1290, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.J.; Wang, K.J.; Gan, S.W.; Xu, J.; Xu, S.Y.; Sun, S.Q. Expression of aquaporin8 in human astrocytomas: Correlation with pathologic grade. Biochem. Biophys. Res. Commun. 2013, 440, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Badaut, J.; Hirt, L.; Granziera, C.; Bogousslavsky, J.; Magistretti, P.J.; Regli, L. Astrocyte-specific expression of aquaporin-9 in mouse brain is increased after transient focal cerebral ischemia. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2001, 21, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Gorelick, D.A.; Praetorius, J.; Tsunenari, T.; Nielsen, S.; Agre, P. Aquaporin-11: A channel protein lacking apparent transport function expressed in brain. BMC Biochem. 2006, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Koike, S.; Tanaka, Y.; Matsuzaki, T.; Morishita, Y.; Ishibashi, K. Aquaporin-11 (AQP11) Expression in the Mouse Brain. Int. J. Mol. Sci. 2016, 17, 861. [Google Scholar] [CrossRef] [PubMed]
- Dong, R.; Tao, S.; Liu, Z.; Zheng, W.; Yu, D. Down-regulation of AQP8 suppresses glioma cells growth and invasion/migration via cell cycle pathway. Int. J. Clin Exp. Pathol. 2016, 9, 1240–1248. [Google Scholar]
- Jelen, S.; Parm Ulhøi, B.; Larsen, A.; Frøkiaer, J.; Nielsen, S.; Rützler, M. AQP9 expression in glioblastoma multiforme tumors is limited to a small population of astrocytic cells and CD15+/CalB+ leukocytes. PLoS ONE 2013, 8, e75764. [Google Scholar] [CrossRef] [PubMed]
- Kishida, K.T.; Hoeffer, C.A.; Hu, D.; Pao, M.; Holland, S.M.; Klann, E. Synaptic plasticity deficits and mild memory impairments in mouse models of chronic granulomatous disease. Mol. Cell. Biol. 2006, 26, 5908–5920. [Google Scholar] [CrossRef] [PubMed]
- Pao, M.; Wiggs, E.A.; Anastacio, M.M.; Hyun, J.; DeCarlo, E.S.; Miller, J.T.; Anderson, V.L.; Malech, H.L.; Gallin, J.I.; Holland, S.M. Cognitive function in patients with chronic granulomatous disease: A preliminary report. Psychosomatics 2004, 45, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C. Mechanisms of inflammatory neurodegeneration: INOS and NADPH oxidase. Biochem. Soc. Trans. 2007, 35, 1119–1121. [Google Scholar] [CrossRef] [PubMed]
- Bamberger, M.E.; Harris, M.E.; McDonald, D.R.; Husemann, J.; Landreth, G.E. A cell surface receptor complex for fibrillar beta-amyloid mediates microglial activation. J. Neurosci. Off. J. Soc. Neurosci. 2003, 23, 2665–2674. [Google Scholar] [CrossRef]
- Bianca, V.D.; Dusi, S.; Bianchini, E.; Dal Pra, I.; Rossi, F. beta-amyloid activates the O−2 forming NADPH oxidase in microglia, monocytes, and neutrophils. A possible inflammatory mechanism of neuronal damage in Alzheimer’s disease. J. Biol. Chem. 1999, 274, 15493–15499. [Google Scholar] [CrossRef] [PubMed]
- Shelat, P.B.; Chalimoniuk, M.; Wang, J.H.; Strosznajder, J.B.; Lee, J.C.; Sun, A.Y.; Simonyi, A.; Sun, G.Y. Amyloid beta peptide and NMDA induce ROS from NADPH oxidase and AA release from cytosolic phospholipase A2 in cortical neurons. J. Neurochem. 2008, 106, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, E.; Hashimoto, Y.; Kanekura, K.; Niikura, T.; Aiso, S.; Nishimoto, I. Characterization of the toxic mechanism triggered by Alzheimer’s amyloid-β peptides via p75 neurotrophin receptor in neuronal hybrid cells. J. Neurosci. Res. 2003, 73, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Liu, Y.; Cooper, C.; Liu, B.; Wilson, B.; Hong, J.S. Microglia enhance β-amyloid peptide-induced toxicity in cortical and mesencephalic neurons by producing reactive oxygen species. J. Neurochem. 2002, 83, 973–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, L.; Anrather, J.; Zhou, P.; Frys, K.; Pitstick, R.; Younkin, S.; Carlson, G.A.; Iadecola, C. NADPH-oxidase-derived reactive oxygen species mediate the cerebrovascular dysfunction induced by the amyloid β peptide. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 1769–1777. [Google Scholar] [CrossRef] [PubMed]
- Park, L.; Zhou, P.; Pitstick, R.; Capone, C.; Anrather, J.; Norris, E.H.; Younkin, L.; Younkin, S.; Carlson, G.; McEwen, B.S.; et al. Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc. Natl. Acad. Sci. USA 2008, 105, 1347–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, L.; Zheng, W.; Xin, N.; Xie, J.W.; Wang, T.; Wang, Z.Y. Ebselen inhibits iron-induced tau phosphorylation by attenuating DMT1 up-regulation and cellular iron uptake. Neurochem. Int. 2012, 61, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Henchcliffe, C.; Beal, M.F. Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat. Clin. Pract. Neurol. 2008, 4, 600–609. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.C.; Jackson-Lewis, V.; Vila, M.; Tieu, K.; Teismann, P.; Vadseth, C.; Choi, D.K.; Ischiropoulos, H.; Przedborski, S. Blockade of microglial activation is neuroprotective in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson disease. J. Neurosci. Off. J. Soc. Neurosci. 2002, 22, 1763–1771. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, T.; Qin, L.; Gao, H.M.; Wilson, B.; Ali, S.F.; Hong, J.S.; Liu, B. Neuroprotective effect of dextromethorphan in the MPTP Parkinson’s disease model: Role of NADPH oxidase. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2004, 18, 589–591. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L.; Li, G.; Qin, L.; Wu, X.; Pei, Z.; Wang, T.; Wilson, B.; Yang, J.; Hong, J.S. Potent regulation of microglia-derived oxidative stress and dopaminergic neuron survival: Substance P vs. dynorphin. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2006, 20, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhou, Y.; Hong, J.S.; Zhang, J. Aggregated alpha-synuclein activates microglia: A process leading to disease progression in Parkinson’s disease. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2005, 19, 533–542. [Google Scholar]
- Hu, X.; Zhang, D.; Pang, H.; Caudle, W.M.; Li, Y.; Gao, H.; Liu, Y.; Qian, L.; Wilson, B.; Di Monte, D.A.; et al. Macrophage antigen complex-1 mediates reactive microgliosis and progressive dopaminergic neurodegeneration in the MPTP model of Parkinson’s disease. J. Immunol. 2008, 181, 7194–7204. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Choi, D.H.; Block, M.L.; Lorenzl, S.; Yang, L.; Kim, Y.J.; Sugama, S.; Cho, B.P.; Hwang, O.; Browne, S.E.; et al. A pivotal role of matrix metalloproteinase-3 activity in dopaminergic neuronal degeneration via microglial activation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2007, 21, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.C.; Re, D.B.; Nagai, M.; Ischiropoulos, H.; Przedborski, S. The inflammatory NADPH oxidase enzyme modulates motor neuron degeneration in amyotrophic lateral sclerosis mice. Proc. Natl. Acad. Sci. USA 2006, 103, 12132–12137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beers, D.R.; Henkel, J.S.; Zhao, W.; Wang, J.; Appel, S.H. CD4+ T cells support glial neuroprotection, slow disease progression, and modify glial morphology in an animal model of inherited ALS. Proc. Natl. Acad. Sci. USA 2008, 105, 15558–15563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marden, J.J.; Harraz, M.M.; Williams, A.J.; Nelson, K.; Luo, M.; Paulson, H.; Engelhardt, J.F. Redox modifier genes in amyotrophic lateral sclerosis in mice. J. Clin. Investig. 2007, 117, 2913–2919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cristóvão, A.C.; Choi, D.H.; Baltazar, G.; Beal, M.F.; Kim, Y.S. The role of NADPH oxidase 1-derived reactive oxygen species in paraquat-mediated dopaminergic cell death. Antioxid. Redox Signal. 2009, 11, 2105–2118. [Google Scholar] [CrossRef] [PubMed]
- Cristovao, A.C.; Guhathakurta, S.; Bok, E.; Je, G.; Yoo, S.D.; Choi, D.H.; Kim, Y.S. NADPH oxidase 1 mediates α-synucleinopathy in Parkinson’s disease. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 14465–14477. [Google Scholar] [CrossRef] [PubMed]
- Boillee, S.; Vande Velde, C.; Cleveland, D.W. ALS: A disease of motor neurons and their nonneuronal neighbors. Neuron 2006, 52, 39–59. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.J.; Wu, P.F.; Long, L.H.; Yu, D.F.; Wu, W.N.; Hu, Z.L.; Fu, H.; Xie, N.; Jin, Y.; Ni, L.; et al. Reversal of aging-associated hippocampal synaptic plasticity deficits by reductants via regulation of thiol redox and NMDA receptor function. Aging Cell 2010, 9, 709–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Muñoz, M.; Garzón, J. Nitric oxide and zinc-mediated protein assemblies involved in mu opioid receptor signaling. Mol. Neurobiol. 2013, 48, 769–782. [Google Scholar] [CrossRef] [PubMed]
- Calero, C.I.; Vickers, E.; Moraga Cid, G.; Aguayo, L.G.; von Gersdorff, H.; Calvo, D.J. Allosteric modulation of retinal GABA receptors by ascorbic acid. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 9672–9682. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Major producing and removal pathways for reactive oxygen species (ROS). The sequential steps of the univalent reduction of molecular oxygen (O2) to water (H2O) leading to the generation of several ROS intermediates are shown. Diverse redox enzymatic systems, mainly mitochondrial respiration complexes and membrane-residing NADPH oxidases (NOXes), can convert O2 into superoxide (O2•−). Superoxide dismutases (SOD) catalyze the dismutation of superoxide (O2•−) into H2O2 and O2. H2O2 can be reduced directly to water by peroxiredoxins (Prx), glutathione peroxidases (GPX), or catalases (CAT). Alternatively, hydroxyl radicals (OH•) are generated from H2O2 in the presence of reduced transition metals, such as Fe2+ (Fenton reaction). Red squares and green squares represent paired and unpaired electrons, respectively, in the oxygen atom. White squares represent the electron provided by hydrogen atoms.
Figure 1.
Major producing and removal pathways for reactive oxygen species (ROS). The sequential steps of the univalent reduction of molecular oxygen (O2) to water (H2O) leading to the generation of several ROS intermediates are shown. Diverse redox enzymatic systems, mainly mitochondrial respiration complexes and membrane-residing NADPH oxidases (NOXes), can convert O2 into superoxide (O2•−). Superoxide dismutases (SOD) catalyze the dismutation of superoxide (O2•−) into H2O2 and O2. H2O2 can be reduced directly to water by peroxiredoxins (Prx), glutathione peroxidases (GPX), or catalases (CAT). Alternatively, hydroxyl radicals (OH•) are generated from H2O2 in the presence of reduced transition metals, such as Fe2+ (Fenton reaction). Red squares and green squares represent paired and unpaired electrons, respectively, in the oxygen atom. White squares represent the electron provided by hydrogen atoms.

Figure 2.
Main models for ROS signal transmission to specific cysteines. (A) The direct model presumes that redox targets (depicted as a blue teardrop) are constituted by proteins whose rate constant of oxidation is higher than that of the reaction of ROS with its major cellular scavengers, particularly peroxiredoxins. Direct chemical reaction of ROS with those targets is thus kinetically possible and leads to their oxidation. (B) The binding hypothesis proposes that H2O2 sources and targets are bound or in close proximity, allowing for site-localized oxidation to occur. Also in this case, ROS will directly oxidize targets but as a function of their relative proximity to the source instead of depending on their rate constant of reaction. (C) The floodgate model overcomes the apparent kinetic limitation of target oxidation by suggesting that overoxidation of peroxiredoxins (represented by green hexagons) permits further oxidation of other proteins with slower reaction rates. (D) Peroxiredoxins have been already shown to behave as relays, transmitting redox equivalents to targets during their oxidation–reduction cycle, and thus allowing for signal transduction. This model implies an indirect ROS effect on targets, as ROS will oxidize peroxiredoxins and will not chemically react with targets. In all cases, a plasma-membrane-bound NADPH oxidase (NOX, in dark pink) has been chosen as a representative source of ROS acting at the cellular surface. Note that the four possibilities are not mutually excluding.
Figure 2.
Main models for ROS signal transmission to specific cysteines. (A) The direct model presumes that redox targets (depicted as a blue teardrop) are constituted by proteins whose rate constant of oxidation is higher than that of the reaction of ROS with its major cellular scavengers, particularly peroxiredoxins. Direct chemical reaction of ROS with those targets is thus kinetically possible and leads to their oxidation. (B) The binding hypothesis proposes that H2O2 sources and targets are bound or in close proximity, allowing for site-localized oxidation to occur. Also in this case, ROS will directly oxidize targets but as a function of their relative proximity to the source instead of depending on their rate constant of reaction. (C) The floodgate model overcomes the apparent kinetic limitation of target oxidation by suggesting that overoxidation of peroxiredoxins (represented by green hexagons) permits further oxidation of other proteins with slower reaction rates. (D) Peroxiredoxins have been already shown to behave as relays, transmitting redox equivalents to targets during their oxidation–reduction cycle, and thus allowing for signal transduction. This model implies an indirect ROS effect on targets, as ROS will oxidize peroxiredoxins and will not chemically react with targets. In all cases, a plasma-membrane-bound NADPH oxidase (NOX, in dark pink) has been chosen as a representative source of ROS acting at the cellular surface. Note that the four possibilities are not mutually excluding.

Figure 3.
Cellular factors of plasma membrane redox signaling. As integral membrane proteins, NADPH oxidases (NOX, dark pink) are present either on lipid rafts (dashed blue line) or in caveolae, releasing their product superoxide (O2•−) into the extracellular space. Upon activation of NOXes, these enzymes can be also internalized into forming redoxosomes where they are still functional. Besides, extracellular O2•− can be generated by the activity of an extracellularly linked xanthine oxidase (XO, blue). In its uncoupled state, NOS enzymes also produce O2•− in the vicinity of caveolae, but since there is a certain controversy on whether this happens in non-pathological conditions, the fact is indicated with a question mark. Nevertheless, extracellular (SODex, yellow) or intracellular (SODin, brown) superoxide dismutases convert O2•− into hydrogen peroxide (H2O2) depending on the localization of the sources. H2O2 is transported inside or outside the cell by specialized aquaporins (AQP, orange channel), following cellular needs.
Figure 3.
Cellular factors of plasma membrane redox signaling. As integral membrane proteins, NADPH oxidases (NOX, dark pink) are present either on lipid rafts (dashed blue line) or in caveolae, releasing their product superoxide (O2•−) into the extracellular space. Upon activation of NOXes, these enzymes can be also internalized into forming redoxosomes where they are still functional. Besides, extracellular O2•− can be generated by the activity of an extracellularly linked xanthine oxidase (XO, blue). In its uncoupled state, NOS enzymes also produce O2•− in the vicinity of caveolae, but since there is a certain controversy on whether this happens in non-pathological conditions, the fact is indicated with a question mark. Nevertheless, extracellular (SODex, yellow) or intracellular (SODin, brown) superoxide dismutases convert O2•− into hydrogen peroxide (H2O2) depending on the localization of the sources. H2O2 is transported inside or outside the cell by specialized aquaporins (AQP, orange channel), following cellular needs.

Figure 4.
Putative mechanisms adopted by cells to achieve redox signal transmission after peroxiporin-mediated routing of ROS through the plasma membrane. The NOX/AQP system, situated in privileged lipid platforms for signaling (blue dashed line), offers a new perspective to re-interpret the models for redox signal transmission to intracellular targets (depicted as blue teardrops): site-localized oxidation of vicinal proteins will be favored by strategical positioning near the cytosolic mouth of the channel, possibly by localizing them to lipid raft domains, while further proteins would be reached either directly by overoxidation of peroxiredoxins (represented as green hexagons) or indirectly via peroxiredoxin-mediated relays. The particular disposition of NOXes releasing ROS to the extracellular space will also induce similar pathways in a peroxiporin-equipped adjacent cell (upper part of the scheme), thus allowing for a coordinated response. The figure has been organized as a cascade for clarity. However, all modes of signal transduction may be not hierarchical and concur simultaneously.
Figure 4.
Putative mechanisms adopted by cells to achieve redox signal transmission after peroxiporin-mediated routing of ROS through the plasma membrane. The NOX/AQP system, situated in privileged lipid platforms for signaling (blue dashed line), offers a new perspective to re-interpret the models for redox signal transmission to intracellular targets (depicted as blue teardrops): site-localized oxidation of vicinal proteins will be favored by strategical positioning near the cytosolic mouth of the channel, possibly by localizing them to lipid raft domains, while further proteins would be reached either directly by overoxidation of peroxiredoxins (represented as green hexagons) or indirectly via peroxiredoxin-mediated relays. The particular disposition of NOXes releasing ROS to the extracellular space will also induce similar pathways in a peroxiporin-equipped adjacent cell (upper part of the scheme), thus allowing for a coordinated response. The figure has been organized as a cascade for clarity. However, all modes of signal transduction may be not hierarchical and concur simultaneously.


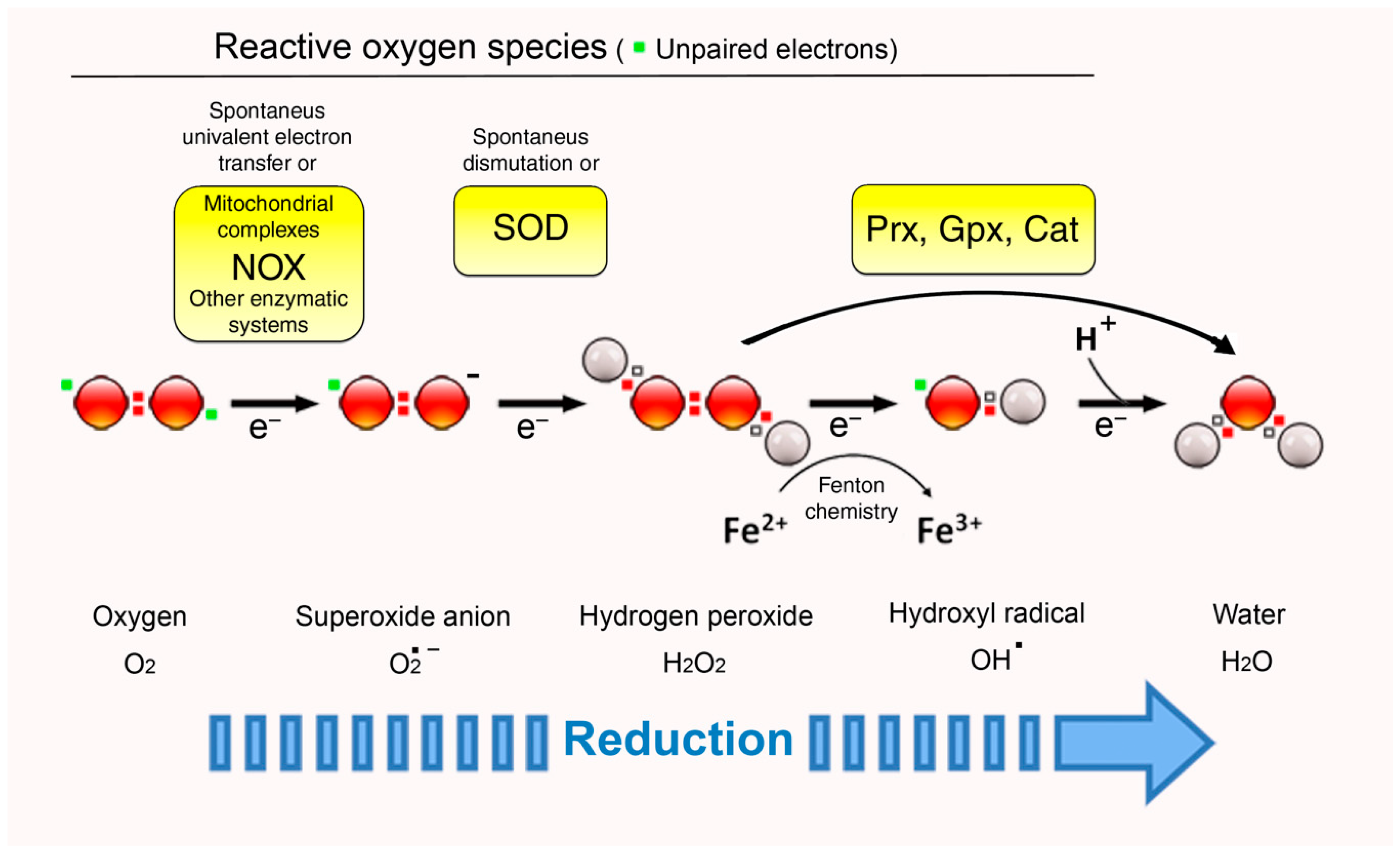{kind=link}
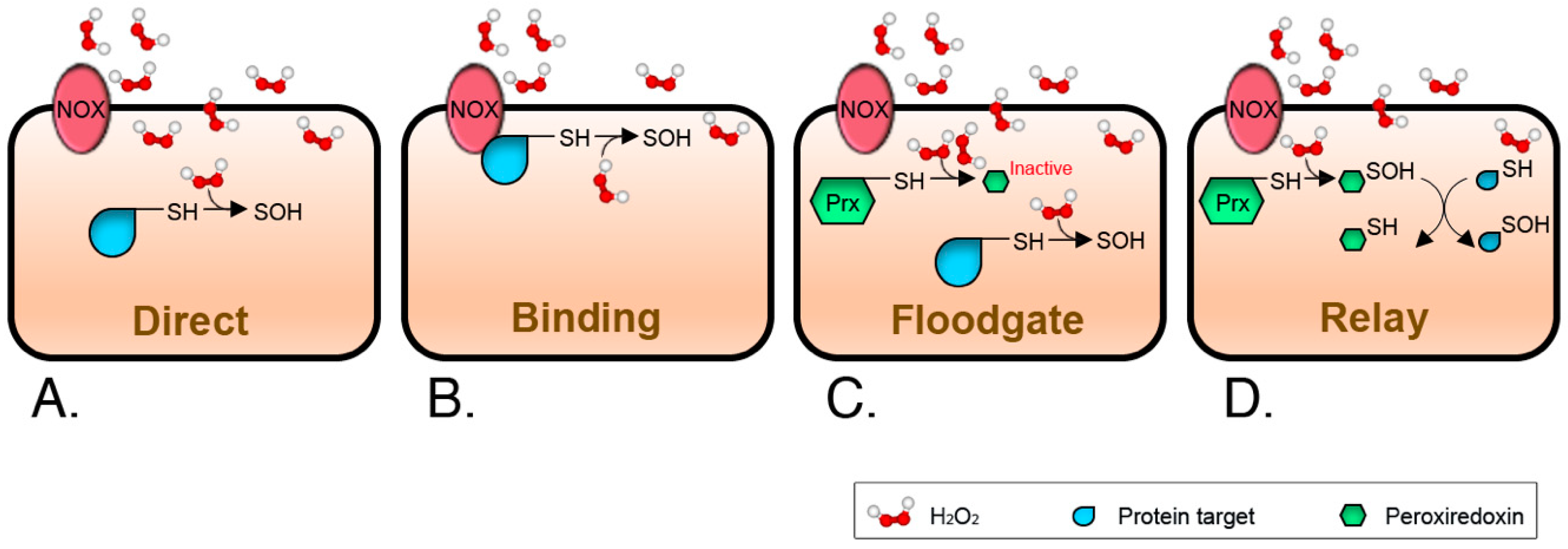{kind=link}
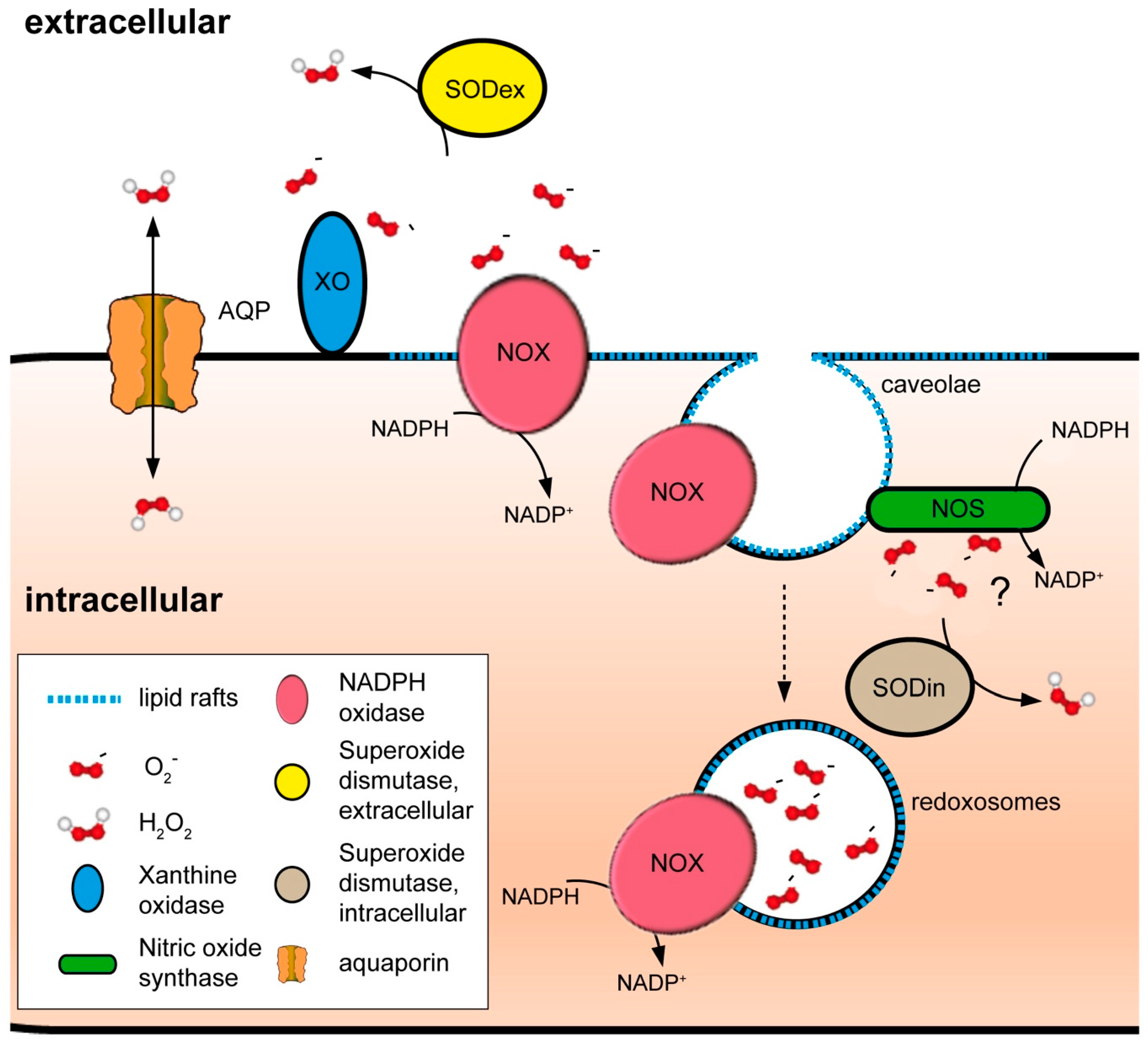{kind=link}
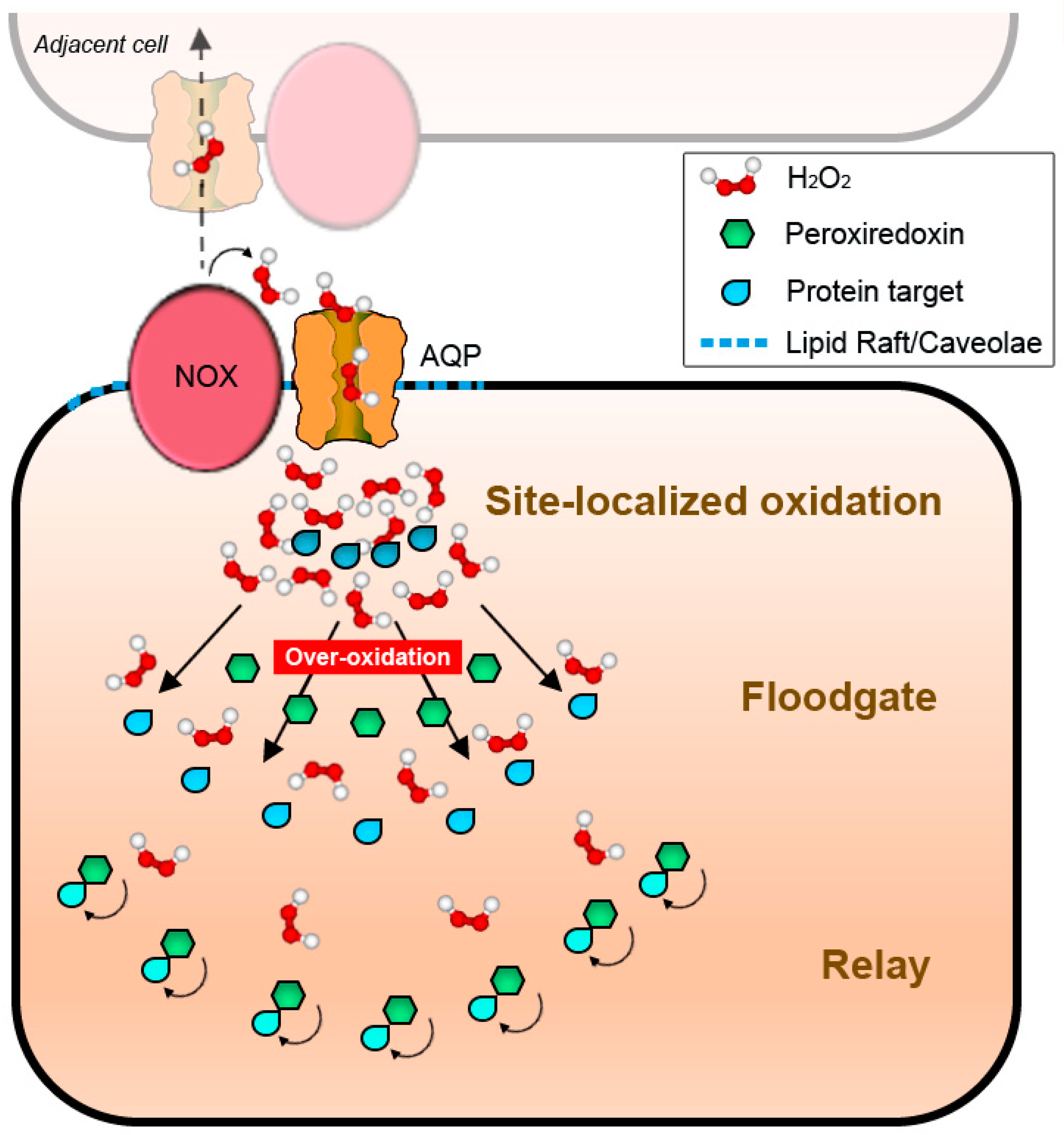{kind=link}
Table 1.
The main tissue distribution of NOX isoforms exhibiting plasma membrane localization.
| Isoform | Regulatory Subunits | Major Distribution Sites Reported | References |
|---|---|---|---|
| Nox1 | p22phox NOXA1 NOXO1 Rac | Vascular smooth muscle cells (VSMC), colon, endothelium, placenta, central nervous system (CNS) | [115,116,117,118,119] |
| Nox2 | p22phox p67phox p47phox p40phox Rac | Phagocytes, ovary, central nervous system (CNS), cardiomyocytes, hepatocytes, endothelium, gastrointestinal tract, hematopoietic stem cells | [13,118,120,121,122,123] |
| Nox3 | p22phox NOXA1 NOXO1 Rac? | Inner ear | [124,125] |
| Nox4 | p22phox | Ubiquitous, including: Endothelial cells, vascular smooth muscle cells (VSMC), kidney, lungs, hematopoietic stem cells, placenta, central nervous system (CNS) | [123,126] |
| Nox5 | Ca2+ (as activator) | Lymphoid tissue, testis, ovary, spleen | [100,101] |
| DUOX1 | Ca2+ (as activator) DUOXA1 (as maturation factor) | Respiratory epithelium, thyroid, gastrointestinal epithelia, salivary and rectal glands | [127,128,129,130,131] |
| DUOX2 | Ca2+ (as activator) DUOXA2 (as maturation factor) | Respiratory epithelium, thyroid, gastrointestinal epithelia, salivary and rectal glands | [127,128,129,130,131] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Nordzieke, D.E.; Medraño-Fernandez, I. The Plasma Membrane: A Platform for Intra- and Intercellular Redox Signaling. Antioxidants 2018, 7, 168. https://doi.org/10.3390/antiox7110168
AMA Style
Nordzieke DE, Medraño-Fernandez I. The Plasma Membrane: A Platform for Intra- and Intercellular Redox Signaling. Antioxidants. 2018; 7(11):168. https://doi.org/10.3390/antiox7110168
Chicago/Turabian StyleNordzieke, Daniela E., and Iria Medraño-Fernandez. 2018. "The Plasma Membrane: A Platform for Intra- and Intercellular Redox Signaling" Antioxidants 7, no. 11: 168. https://doi.org/10.3390/antiox7110168
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.