Redox for Repair: Cold Physical Plasmas and Nrf2 Signaling Promoting Wound Healing


1
Plasma Life Science, Leibniz Institute for Plasma Science and Technology (INP Greifswald), Felix-Hausdorff-Str. 2, 17489 Greifswald, Germany
2
ZIK-PRE, Leibniz Institute for Plasma Science and Technology (INP Greifswald), Felix-Hausdorff-Str. 2, 17489 Greifswald, Germany
*
Author to whom correspondence should be addressed.
Antioxidants 2018, 7(10), 146; https://doi.org/10.3390/antiox7100146
Submission received: 28 September 2018
/
Revised: 12 October 2018
/
Accepted: 18 October 2018
/
Published: 19 October 2018
(This article belongs to the Special Issue Nrf2 in Dermatological Pathologies)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Chronic wounds and ulcers are major public health threats. Being a substantial burden for patients and health care systems alike, better understanding of wound pathophysiology and new avenues in the therapy of chronic wounds are urgently needed. Cold physical plasmas are particularly effective in promoting wound closure, irrespective of its etiology. These partially ionized gases deliver a therapeutic cocktail of reactive oxygen and nitrogen species safely at body temperature and without genotoxic side effects. This field of plasma medicine reanimates the idea of redox repair in physiological healing. This review compiles previous findings of plasma effects in wound healing. It discusses new links between plasma treatment of cells and tissues, and the perception and intracellular translation of plasma-derived reactive species via redox signaling pathways. Specifically, (i) molecular switches governing redox-mediated tissue response; (ii) the activation of the nuclear E2-related factor (Nrf2) signaling, together with antioxidative and immunomodulatory responses; and (iii) the stabilization of the scaffolding function and actin network in dermal fibroblasts are emphasized in the light of wound healing.
1. Introduction
Defective wound healing affects millions of people in the western world, and is a strong burden to patients and health care systems [1]. Although there is a plethora of wound therapies available [2], challenges remain in the treatment of problematic wounds [3]. Phases of wound healing are complex and dynamically regulated during physiological tissue repair; they can co-exist in different wound areas and tend to blend together depending on the degree of wound closure [4,5,6]. Moreover, several phases such as blood coagulation, thrombosis, fibrosis, migration, proliferation, and programmed cell death are dependent on reactive species-mediated signaling, redox level, and wound oxygenation [7]. Endogenous generation of oxidants leads to reversible oxidative modifications, including redox regulation, sensing, and redox signaling, which contribute to physiological or pathological stages of wound healing [8].
In comparison to normal wound healing, several studies have shown pathophysiological mechanisms counteract healing processes in chronic wounds. One parameter that has been described to aggravate wound healing is diabetes mellitus (DM), which is ranked as the 10th leading cause of death in 2012 [9], affecting 24 million people in the U.S. [10] or 382 million people worldwide (International Diabetes Federation). The incidence of diabetes continues to rise rapidly. In Germany, 3.8% of the insured population have been diagnosed with diabetic foot ulcers [11]. Extensive differences were found regarding the cellular redox level in non-healing (diabetic) wounds [8,12]. Accordingly, they display an imbalance in redox processes and are often linked to chronic inflammation [13], ultimately disturbing the sequence of events necessary for healing. For example, low-grade inflammation is involved in diabetic micro-angiopathy [14]. In type 1 diabetes, the level of IL-2 is reduced, and hence, wound healing is in a prolonged inflammatory phase [15,16]. A modulated apoptosis [17] or an increased apoptosis of T lymphocytes [18], a reduced number and activation of macrophages, which results in reduced lymphatic vessel formation [19], leads to impaired wound healing in diabetic patients [20]. In the granulation phase, DM disturbs re-epithelialization by affecting multiple proteins and genes such as angiopoietin-4 [21]. Moreover, DM has a negative influence on signaling intermediates responsible for coordination and regulation of wound healing, angiogenesis, and vasculogenesis [22], leading to a decreased expression of angiogenetic growth factors [23] and an impaired neovascularization [24]. As a result, diabetic patients are prone to the development of chronic wounds due to deficiencies in either endothelial progenitor cells or peripheral tissue homing and in engraftment of bone marrow [25]. In the matrix recovering phase, dysfunction of fibroblasts mediated by heat shock proteins (HSPs) together with aberrant collagen homeostasis contribute to impaired healing [26]. In addition, gap junctional connexins form channels between two adjacent cells and their expression is highly regulated after wound formation at transcriptional, translational, and post translational levels [27]. Upregulation of connexin proteins during diabetes leads to improper formation of gap junctions and attributes to the passage of apoptotic and inflammatory signals, thereby resulting in delayed healing of chronic diabetic ulcers [28]. In addition, dysregulation of apoptosis in response to hyperglycemia causes impaired wound healing, which can further worsen ischemia and coronary artery disease [14]. Thus, alternative therapeutic strategies are required for treatment of non-healing wounds.
Since inflammation and wound healing are subject to redox control [29], there is an obvious link to plasma medicine. By using cold physical plasmas generating or mimicking physiologically active reactive species, a beneficial strategy of tissue repair in chronic (diabetic) wounds seems possible. However, the application of cold plasma as a new medical alternative and/or supporting therapy requires the identification of the molecular switches governing redox-regulated tissue responses and key molecules as well as signaling pathways. Over the past 10 years, a huge number of in vitro studies have demonstrated plasma-triggered wound healing based on the stimulation of cell proliferation and survival [30,31], extracellular matrix (ECM) protein synthesis [32,33,34], changes of junctional proteins and cytoskeletal architecture [35], as well as apoptosis [36,37,38]. Moreover, several studies have showed that cold plasma treatment favors the redox regulation of targets known to be important in wound healing [39,40,41]. Specifically, this includes redox pathways and the regulation of redox proteins, ultimately resetting inflammation to reinitialize the sequence of events necessary for healing [42].
This article summarizes state-of-the-art of plasma medical research to understand the biological effects of plasma-generated reactive species, as well as plasma-induced signaling pathways and to further elucidate the relevance and significance of redox regulation in different stages and events of (diabetic) wound healing.
2. Cold Physical Plasma-Derived Reactive Species and Their Biological Effects
2.1. Plasma-Derived Reactive Species
Cold physical plasmas are multicomponent and complex systems that have unique features, but also differences [33,43]. Most of them are a significant source of highly reactive oxygen species (ROS) like ozone (O3), hydroxyl radicals (OH−), superoxide (O2−), and singlet oxygen (1O2), as well as nitrogen species (RNS) such as nitric oxide (NO), peroxynitrite (ONOO−), and nitrogen dioxide (NO2−) [44]. Some of these reactive components are being considered second messengers in the field of redox biology [45]. Additionally, unique characteristics of cold plasmas are charged particles, ions, thermal and ultraviolet radiation, as well as electrical fields [46,47]. A detailed overview about physicochemical and biological parameters plus their biological application along with a detailed risk estimation of plasma sources was recently given [48]. The most striking parameter provided by plasma in tissues is the superficial penetration depth of approximately 10 µm (especially for short-living reactive species) to 60 µm [49]. However, due to its rapid reactions with biomolecules (proteins, lipids, DNA), plasma-generated stable species have oxidizing properties, e.g., protein oxidation of redox-sensitive cysteine residues and thiol groups [50,51] evoking paracrine effects and changes of microenvironment in deeper skin layers [34]. Nevertheless, a lack of histological damage was observed after 10 min of single-spot plasma treatment of human skin biopsies [30].
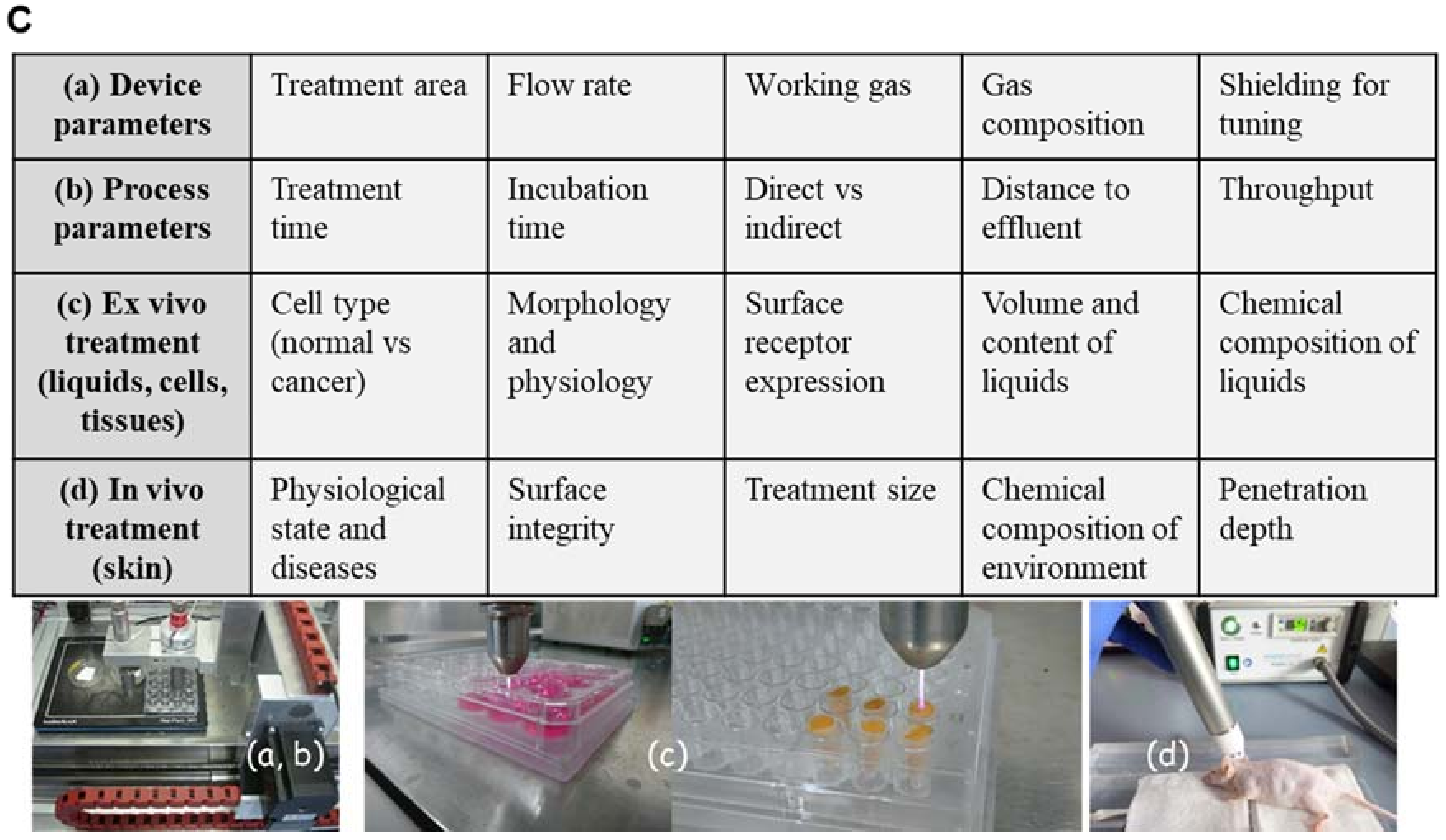
Besides other plasma sources (e.g., dielectric barrier or microwave-based) [52], the kINPen plasma jet (neoplas tools GmbH, Greifswald, Germany) composition including power supply, electrode, dielectric and plasma effluent was depicted to show the general structure of a jet-based plasma device (Figure 1A). The pen-sized hand-held unit generates an atmospheric argon plasma primarily for spot-like treatments of target cells [53]. Plasma composition varied through the way from gaseous over liquid phase to the target [54,55]. Additionally, biological plasma effects are largely dependent on plasma-generated reactive species in the gas phase, which subsequently diffuse or react with proteins and lipids in cells or tissues. Hence, monitoring of the gas via optical emission spectroscopy phase allows monitoring of the reactive species output and biological effect. For example, biological effects of argon-oxygen-generated plasmas are mainly explained by the formation of ROS [56,57]. Based on this analytical technique, it was possible to compare spectra in the ultraviolet (UV) and visible range regarding the present plasma-excited species produced by kINPen and to standardize plasma treatment for biological applications (Figure 1B) [58]. In addition to device parameters such as treatment area, flow rate, working gas, components of gas and their tuning, process parameters can also be modulated including treatment and incubation time, direct vs. indirect treatment, distance to the target from effluents and throughput [59]. One favorable advantage of cold plasma’s use is the generation of highly active species at the site of interest (liquids, cells and tissues), where they can directly function as signaling or redox-reactive molecules (Figure 1C). In this context, the general applicability of cold plasmas to treat the skin or infected wounds has been shown in vivo [60,61,62].
2.2. Nrf2 Signaling in Plasma-Assisted Wound Healing
While plasma-induced wound healing revealed differences of a huge number of factors, selected targets were introduced. Beyond that, changes in ROS levels trigger a coordinated action of redox-sensitive transcription factors [63] underlining the importance of controlled redox signaling during wound healing. The nuclear factor erythroid 2-related factor 2 (Nrf2), a basic leucine zipper (bZIP) transcription factor, is a crucial translator for redox signaling and functions in cellular defense against imbalances in redox homeostasis. Such imbalance between the production and the detoxification of reactive intermediates affects the cellular stress level. Generally, Nrf2 activates cellular rescue pathways against oxidative injury [64,65,66], inflammation [67,68] as well as apoptosis [69,70] and plays a key role in regulation of genes, which encode detoxifying enzymes and non-enzymatic proteins [71]. Under basal conditions, Nrf2 is associated with an actin-binding protein, Kelch-like ECH-associated protein 1 (Keap1), a vital factor in Nrf2 signaling cascade, which retains Nrf2 in the cytoplasm, where it is targeted for ubiquitin-mediated degradation [72,73]. As a key regulator of oxidative and electrophilic stress, Keap1 is constitutively expressed in cytoplasmic regions of cells [74,75] as shown in dermal fibroblasts isolated from mouse skin (Figure 2A). Keap1 acts not only as an oxidative stress sensor but also functions as a regulator of F-actin filament architecture [75], supporting the assumption that Keap1 also possesses Nrf2-independent functions [76,77]. Colocalization of Keap1 (red) with actin filaments (green) promotes scaffolding functions and a controlled Nrf2 regulation in dermal fibroblasts (Figure 2B). Furthermore, plasma-mediated activation of Keap1 supports the reorganization of architecture of actin cytoskeleton and focal adhesion [35].
After release of Nrf2 from Keap1 by oxidation events at cysteine, Nrf2 translocates to the nucleus (Figure 2C), binds to specific DNA recognition sites, namely, antioxidant responsive elements (AREs) in the promoters of its target genes, and activates their transcription [78]. Most skin cells are equipped with mechanisms to detoxify reactive species via expression of antioxidant enzymes such as heme oxygenase 1 (HMOX-1), NADPH quinone oxidoreductase 1 (NQO1), glutathione S-transferase (GST), cytochrome P450, γ-glutamylcysteine ligase catalytic (GCLC) and modifier subunit (GCLM), superoxide dismutases 1-3 (SOD1-3), glutathione reductase (GSR), thioredoxin reductase (TRxR), thioredoxin (TXN), catalase (CAT), glutathione peroxidase (GPx), and non-enzymatic antioxidants like glutathione, thioredoxin and ferritin [63,71]. Plasma-generated reactive species were similarly translated via the redox-sensitive Nrf2 signaling, leading to an activation of Nrf2-ARE targets, in particular GPx, CAT, SODs, HMOX1, NQO1 and enhanced antioxidant defense [42]. In an in vivo wound model, a more robust and accelerated physiological response was obtained by activating of Nrf2 [33]. ROS production was profoundly elevated in HMOX1 knockout mice [79], showing that HMOX1 is able to metabolize high levels of reactive intermediates. Furthermore, a pharmacological activation of Nrf2 or a knockdown of Keap1 supports wound closure in diabetic mice [80,81].
2.3. Plasma-Induced Angiogenesis and Immunomodulation
In addition to its cyto-protective effects, an increasing number of studies support the pivotal role of Nrf2 in angiogenesis [82,83]. The activation of Nrf2 reduces oxidative stress in endothelial cells and suppresses inflammatory responses that may lead to several diseases [84]. Upregulation of Nrf2 by a chemical activator prevents diabetes-induced Erk activation and insulin-signaling downregulation in diabetic patients [85]. Although Nrf2 may promote vascular development via protection of retina from hyperoxia-induced oxidative stress [86], Nrf2 dysfunction may be a potential mechanism underlying impaired angiogenesis and microvascular rarefaction in aging [87]. Plasma-induced regulation of Nrf2 and down-stream targets coincides with the physiological features of wound healing, such as re-epithelialization [35,88], angiogenesis [33], oxidation of lipid layer, and TGFβ signaling [89]. TGFβ signaling is crucially involved in inflammation, angiogenesis, granulation for tissue formation and overall epidermal maintenance [90]. The beneficial activity of plasma is promoted by the production of a plasma-specific signature of inflammatory, growth and angiogenetic factors (e.g., fibroblast growth factor, FGF; vascular endothelial growth factor, VEGF; keratinocyte growth factor, KGF; cyclooxygenase 2,COX2; cluster of differentiation 31, CD31; protein kinase B; Akt etc.) in vitro [39] and in vivo. Regeneration of full-thickness wounds occurred after an early infiltration of neutrophils and macrophages concomitant with a balanced expression of pro- (e.g., IL-1β, IL-6, TNFα) and anti-inflammatory (TGFβ) mediators [33].
2.4. Estimations of Risks after Plasma-Induced Activation of Nrf2 Signaling
Some studies suggest an oncogenic characteristic of Nrf2 causing a constitutive activation during early phases of skin tumorigenesis [91], resistance to chemotherapy [92], and an induction of a cancer-associated fibroblast phenotype [93]. Although small amounts of constitutively nuclear localized Nrf2 maintain cellular redox homeostasis through regulation of basal expression of antioxidant genes, after plasma treatment, a transient or persistent activation of Nrf2 does not further increase the cytoplasmic-to-nuclear translocation, thereby preventing cancer formation [42]. Moreover, our data highlight the pivotal role played by Nrf2 in regulating cellular adaptions in response to a repeated cold plasma-induced redox change [94]. Risk estimation studies permitted a clinical application of plasma due to the fail of mutagenicity, genotoxicity [95,96,97], excessive inflammation, and tumor formation [88]. As such, anatomical magnetic-resonance imaging, positron emission tomography-computed tomography, histological and immune histochemical analysis as well as quantitative PCR of several tumor markers showed no apparent signs of tumor manifestation in any of the investigated structures and organs, suggesting that cold plasma treatment is tissue compatible and not damaging to the skin per se. Moreover, other pathways (e.g., Nrf1/3) can partially compensate functions of Nrf2 as shown by Nrf2 knockdown experiments in plasma-treated epidermal keratinocytes (manuscript submitted). Nrf2 in myeloid cells being dispensable for wound healing also indicates the presence of additional antioxidant defense strategies of these cells that compensate for the loss of Nrf2 even in skin wounds [98].
2.5. p53 and MAPK Signaling, Directly Influence Nrf2 Activity
The primary event in down-stream signaling of Nrf2 is the recognition of plasma-generated reactive species by specific ROS sensors, e.g., Keap1. Recently, an important new paradigm was introduced by which p53 can mediate a two-phase Nrf2 response; for example, to determine cell fate [99] as well as the cross-talk between oxidative stress (e.g., Nrf2 signaling) and DNA-damage (p53 activation) to define the tipping points where cell injury may switch from adaptation to injury [100]. The nuclear transcription factor and tumor suppressor protein p53 is a key coordinator of oxidative stress [101]. p53 acts as an antioxidant at low levels of oxidative stress to ensure cell survival as well as cellular protection mechanisms and, conversely, p53 has pro-oxidative activities to further increase stress levels to trigger cell death, e.g., apoptosis [102]. Moreover, the redox balance influences the maintenance of cell proliferation rhythms like the cell cycle [103]. p53 signaling can suppress the Nrf2-dependent transcription of antioxidant responsive genes to promote cell survival, apoptosis or cell cycle arrest in a p21-dependent manner [104]. These findings are consistent with the early cell proliferation supporting effect required for rapid tissue repair observed after a transient inhibition of p53 [105], which was also found in a dermal full-thickness wound model upon plasma treatment (manuscript submitted). Several downstream targets of p53 participate in regulation of cellular ROS levels [106], which further influence the activation of all three major mitogen-activated protein (MAP) kinases: p38, the extracellular signal-regulated kinase (Erk), and the stress-activated c-Jun N-terminal kinase (Jnk) [107]. These molecules are also central in cellular Nrf2 signaling processes such as proliferation, differentiation, general stress response [108,109,110,111], and even influencing cell migration [112,113]. The pattern of regulated MAPK expression in different cell settings has also been described after plasma treatment [114,115,116,117]. Moreover, Jnk and p38 modify Nrf2 and p53 activity by phosphorylating it [118,119,120,121] and thus establishing a cross-talk between Nrf2, p53 and MAP kinase signaling [122,123,124]. Altogether, targeting Nrf2 through plasma may offer a novel and better alternative for the therapeutic management of wounds in diabetic patients.
2.6. Junctional Proteins in Plasma-Assisted Wound Healing
Interestingly, the gap junction protein connexin 43 (Cx43) might enhance the activation of Nrf2-ARE pathway by means of inhibiting tyrosine kinase c-Src activity to hinder the nuclear export of Nrf2, ultimately attenuating renal fibrosis in diabetes [125]. Cx43, a gap junctional complex component, is a target of interest in dermal wound healing, since it has essential roles in homeostasis and disease [126]. Inhibition of Cx43 by transient blocking of channels with Cx43 mimetic peptides was associated with a significant decrease of ulcer areas, showing that wound healing in diabetic wounds is enhanced when Cx43 levels are reduced [127,128,129]. After plasma treatment, a significant reduction of Cx43 expression was observed in epithelial cells, suggesting a wound closure promoting feature of plasma, especially in chronic and slowly-healing wounds [35]. Indeed, in deep skin layers, Cx43 was transiently upregulated in fibroblasts in the first hours after wounding, as well as during granulation tissue formation and maturity, indicating a localized modulation of Cx43 levels [129].
However, elevated levels of Cx43 had a negative effect on the migratory ability of skin cells [130]; they inhibit the ability of fibroblasts and keratinocytes to migrate into the wound bed and to heal wounds [131]. Skin cells such as keratinocytes and fibroblasts appear to play an important role in migration, differentiation, and re-epithelialization in the final stage of wound closure. After single treatment, plasma had a modulating effect by facilitating cell progression as well as cell motility [33,34,35,42]. For acute redox stress, plasma notably repressed cell migration, which is in agreement with previous results using a carcinoma cell line [132]. Interestingly, cell migration activity partly recovered afterwards, pointing to an adaption of keratinocytes to periodic redox challenges [94]. Moreover, in wound healing, cell migration through connective tissue requires adhesive cell-matrix interactions and ECM contraction, and is mediated by surface integrins and other adhesion molecules [133]. One of the main tasks of fibroblasts is the synthesis of new granulation tissue to recover connective tissue and components of the extracellular matrix [134], which could be demonstrated in plasma-treated fibroblasts, particularly at early stages [33,135]. Overall downregulation of several integrins [35] offers another explanation of the observed changes in cell morphology, including increased motility, rearrangement of actin filaments or stabilization of cytoskeletal architecture. Additionally, the findings support the assumption that certain aspects of Cx43 function could be used for beneficial strategies in wound management and that epidermal as well as dermal skin cells may play a central role in plasma-mediated wound healing.
3. Clinical Observations with Cold Physical Plasmas in Dermatology
Although a huge number of plasma sources for biomedical applications are described, only three plasma devices are CE-certified medical plasma devices class II. This includes the argon-driven plasma jet kINPenMed (neoplas tools GmbH, Greifswald, Germany), the microwave-driven Adtec SteriPlas (Adtec Plasma Technology, Adtec Europe, Hunslow, UK), and the dielectric barrier discharge (DBD)-based PlasmaDerm® (CYNOGY GmbH, Duderstadt, Germany). All three plasma devices are based on comprehensive physical, molecular biological, pre-clinical and clinical characterization [33,43,47,136,137]. Generally, standardization of devices is in the focus of the plasma community to compare and identify key process parameters, plasma components, and experimental conditions. First success in translating plasma source protocols and biological application culminated in an unique standardization protocol with DIN-specification 91,315 “General requirements for medical plasma sources”, which was based on the characterization of the plasma jet kINPen MED [53].
In clinical dermatology, cold plasmas are mainly used for the treatment of chronic wounds and pathogen-based skin diseases, in which stimulation of tissue repair and decontamination through killing of microorganisms are combined [38,52,138]. Using DBD, a clinical trial was performed to investigate plasma treatment of chronic wounds [139]. A prospective clinical trial was also conducted using the MicroPlasSter to determine the bacterial load reduction on wounds [62,140], and to show beneficial effects of plasma in patients with infected skin blisters [141]. Similarly, pilot studies or case reports were undertaken using kINPen-generated plasma, showing a decrease of bacterial load [142], a skin recovery after laser-induced skin lesion [143,144,145], a wound healing of ulcer [146,147], as well as a beneficial treatment of psoriasis vulgaris [148].
Nevertheless, the linkage between molecular characterization and clinical protocols will remain a key challenge in plasma medical research in dermatology in order to push healing of chronic wounds in the right direction. To understand mechanisms of wound healing and to deliberate about whether plasma treatment is beneficial, major aims include (i) the modulation of plasma components by tuning of feed gas [149,150,151,152]; (ii) the construction of application-oriented plasma sources, e.g., DBD, jet or microwave devices; (iii) the clinical classification into responder or non-responder; and (iv) the identification of potential biological markers for plasma therapy (currently reviewed in [43]). To answer a broad spectrum of open questions, the prominent aim of plasma medical research consists in the creation of molecular fingerprints of a plasma-triggered wound response.
4. Summary and Highlights in Plasma-Induced Wound Healing
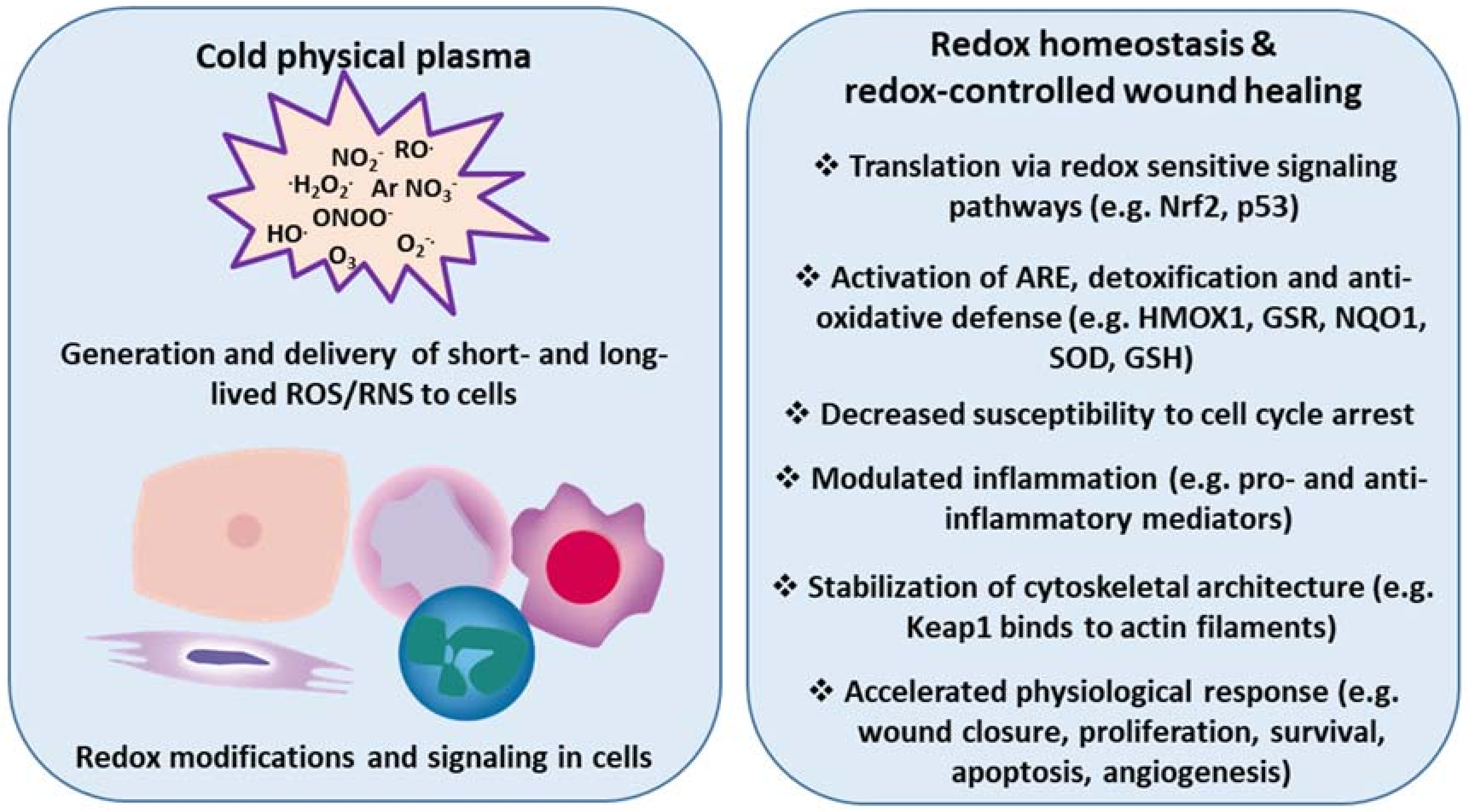
Based on findings, we and others have hypothesized that manipulating the redox state of wounds accelerates wound healing by tuning inflammation via redox signaling, regulation of leukocyte traffic and phenotype, and chemokine/cytokine patterns, ultimately fostering skin cell migration into the wound bed. Given its potential (e.g., to kill microorganisms, stimulate cell proliferation, promote tissue regeneration, and modulate inflammation in tissues), cold physical plasma appears to be a promising biomedical tool for the treatment of chronic wounds. Here, we describe that redox-mediated pathways, particularly Nrf2 signaling, are key modulators, not only in wound healing, but also in their promotion via cold plasma treatment (Figure 3).
Key events are:
- Chronic wounds display subacute inflammation with cellular senescence and bacterial burden
- Plasma accelerates wound healing by different mechanisms of action during the consecutive phases of wound healing
- Plasma controls Nrf2 signaling and inflammatory response in skin cells
- Plasma stabilizes the scaffolding function and actin network in dermal fibroblasts
- Plasma induces changes in Cx43 expression, which could be used for beneficial therapies
- Plasma as a therapeutic option of mild pro-oxidant therapy in chronic (diabetic) wound healing
Funding
This work was supported by the Ministry of Education, Science and Culture of the State of Mecklenburg-Western Pomerania (Germany), the European Union, European Social Fund (grant numbers AU 11 038; ESF/IV-BM-B35-0010/13 and AU 15 001), and the German Federal Ministry of Education and Research (BMBF, grant number 03Z22DN11).
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Abbreviations
| ARE | antioxidant response element |
| Keap1 | Kelch-like ECH-associated protein 1 |
| Nrf2 | nuclear factor-erythroid 2-related factor 2 |
| ROS | reactive oxygen species |
| RNS | reactive nitrogen species |
References
- Sen, C.K.; Gordillo, G.M.; Roy, S.; Kirsner, R.; Lambert, L.; Hunt, T.K.; Gottrup, F.; Gurtner, G.C.; Longaker, M.T. Human skin wounds: A major and snowballing threat to public health and the economy. Wound Repair Regen. 2009, 17, 763–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Koppen, C.J.; Hartmann, R.W. Advances in the treatment of chronic wounds: A patent review. Expert Opin. Ther. Patents 2015, 25, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Frykberg, R.G.; Banks, J. Challenges in the Treatment of Chronic Wounds. Adv. Wound Care (New Rochelle) 2015, 4, 560–582. [Google Scholar] [CrossRef] [PubMed]
- Ghatak, S.; Maytin, E.V.; Mack, J.A.; Hascall, V.C.; Atanelishvili, I.; Moreno Rodriguez, R.; Markwald, R.R.; Misra, S. Roles of Proteoglycans and Glycosaminoglycans in Wound Healing and Fibrosis. Int. J. Cell Biol. 2015, 2015, 834893. [Google Scholar] [CrossRef] [PubMed]
- Shakya, S.; Wang, Y.; Mack, J.A.; Maytin, E.V. Hyperglycemia-Induced Changes in Hyaluronan Contribute to Impaired Skin Wound Healing in Diabetes: Review and Perspective. Int. J. Cell Biol. 2015, 2015, 701738. [Google Scholar] [CrossRef] [PubMed]
- Broughton, G.; Janis, J.E.; Attinger, C.E. The basic science of wound healing. Plast. Reconstruct. Surg. 2006, 117, S12–S34. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Khanna, S.; Nallu, K.; Hunt, T.K.; Sen, C.K. Dermal wound healing is subject to redox control. Mol. Ther. 2006, 13, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Heron, M. Deaths: Leading Causes for 2012. Natl. Vital Stat. Rep. 2015, 64, 1–93. [Google Scholar] [PubMed]
- Cowie, C.C.; Rust, K.F.; Ford, E.S.; Eberhardt, M.S.; Byrd-Holt, D.D.; Li, C.; Williams, D.E.; Gregg, E.W.; Bainbridge, K.E.; Saydah, S.H.; et al. Full accounting of diabetes and pre-diabetes in the U.S. population in 1988–1994 and 2005–2006. Diabetes Care 2009, 32, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Heyer, K.; Herberger, K.; Protz, K.; Glaeske, G.; Augustin, M. Epidemiology of chronic wounds in Germany: Analysis of statutory health insurance data. Wound Repair Regen. 2016, 24, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Sen, C.K. Wound healing essentials: Let there be oxygen. Wound Repair Regen. 2009, 17, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, C.K. The general case for redox control of wound repair. Wound Repair Regen. 2003, 11, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Arya, A.K.; Tripathi, R.; Kumar, S.; Tripathi, K. Recent advances on the association of apoptosis in chronic non healing diabetic wound. World J. Diabetes 2014, 5, 756–762. [Google Scholar] [CrossRef] [PubMed]
- Wetzler, C.; Kampfer, H.; Stallmeyer, B.; Pfeilschifter, J.; Frank, S. Large and sustained induction of chemokines during impaired wound healing in the genetically diabetic mouse: Prolonged persistence of neutrophils and macrophages during the late phase of repair. J. Investig. Dermatol. 2000, 115, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.W. Interleukin 2 Topical Cream for Treatment of Diabetic Foot Ulcer: Experiment Protocol. JMIR Res. Protoc. 2015, 4, e89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jazayeri, L.; Callaghan, M.J.; Grogan, R.H.; Hamou, C.D.; Thanik, V.; Ingraham, C.R.; Capell, B.C.; Pelo, C.R.; Gurtner, G.C. Diabetes increases p53-mediated apoptosis following ischemia. Plast. Reconstruct. Surg. 2008, 121, 1135–1143. [Google Scholar] [CrossRef] [PubMed]
- Arya, A.K.; Pokharia, D.; Tripathi, K. Relationship between oxidative stress and apoptotic markers in lymphocytes of diabetic patients with chronic non healing wound. Diabetes Res. Clin. Pract. 2011, 94, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, K.; Asai, J.; Ii, M.; Thorne, T.; Losordo, D.W.; D’Amore, P.A. Decreased macrophage number and activation lead to reduced lymphatic vessel formation and contribute to impaired diabetic wound healing. Am. J. Pathol. 2007, 170, 1178–1191. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.T.; Seth, A.K.; Hong, S.J.; Geringer, M.R.; Xie, P.; Leung, K.P.; Mustoe, T.A.; Galiano, R.D. Deficient cytokine expression and neutrophil oxidative burst contribute to impaired cutaneous wound healing in diabetic, biofilm-containing chronic wounds. Wound Repair Regen. 2013, 21, 833–841. [Google Scholar] [CrossRef] [PubMed]
- Arya, A.K.; Tripathi, K.; Das, P. Promising role of ANGPTL4 gene in diabetic wound healing. Int. J. Lower Extrem. Wounds 2014, 13, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, K.A.; Liu, Z.J.; Xiao, M.; Chen, H.; Goldstein, L.J.; Buerk, D.G.; Nedeau, A.; Thom, S.R.; Velazquez, O.C. Diabetic impairments in NO-mediated endothelial progenitor cell mobilization and homing are reversed by hyperoxia and SDF-1 alpha. J. Clin. Investig. 2007, 117, 1249–1259. [Google Scholar] [CrossRef] [PubMed]
- Botusan, I.R.; Sunkari, V.G.; Savu, O.; Catrina, A.I.; Grunler, J.; Lindberg, S.; Pereira, T.; Yla-Herttuala, S.; Poellinger, L.; Brismar, K.; et al. Stabilization of HIF-1alpha is critical to improve wound healing in diabetic mice. Proc. Natl. Acad. Sci. USA 2008, 105, 19426–19431. [Google Scholar] [CrossRef] [PubMed]
- Callaghan, M.J.; Ceradini, D.J.; Gurtner, G.C. Hyperglycemia-induced reactive oxygen species and impaired endothelial progenitor cell function. Antioxid. Redox Signal. 2005, 7, 1476–1482. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.J.; Velazquez, O.C. Hyperoxia, endothelial progenitor cell mobilization, and diabetic wound healing. Antioxid. Redox Signal. 2008, 10, 1869–1882. [Google Scholar] [CrossRef] [PubMed]
- Lerman, O.Z.; Galiano, R.D.; Armour, M.; Levine, J.P.; Gurtner, G.C. Cellular Dysfunction in the Diabetic Fibroblast. Am. J. Pathol. 2003, 162, 303–312. [Google Scholar] [CrossRef]
- Wang, C.M.; Lincoln, J.; Cook, J.E.; Becker, D.L. Abnormal connexin expression underlies delayed wound healing in diabetic skin. Diabetes 2007, 56, 2809–2817. [Google Scholar] [CrossRef] [PubMed]
- Bajpai, S.M.M.; Kumar, H.; Tripathi, K.; Singh, S.K.; Srikrishna, S.; Pandey, H.P.; Singh, R.K. Comparative study of connexins expression in diabetic and non-diabetic wounds. Dig. J. Nanomater. Biostruct. 2010, 5, 857–862. [Google Scholar]
- Schafer, M.; Werner, S. Oxidative stress in normal and impaired wound repair. Pharmacol. Res. 2008, 58, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Hasse, S.; Tran, D.T.; Hahn, O.; Kindler, S.; Metelmann, H.R.; von Woedtke, T.; Masur, K. Induction of proliferation of basal epidermal keratinocytes by cold atmospheric-pressure plasma. Clin. Exp. Dermatol. 2016, 41, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.R.; Xu, G.M.; Shi, X.M.; Zhang, G.J. Low temperature plasma promoting fibroblast proliferation by activating the NF-kappaB pathway and increasing cyclinD1 expression. Sci. Rep. 2017, 7, 11698. [Google Scholar] [CrossRef] [PubMed]
- Eisenhauer, P.; Chernets, N.; Song, Y.; Dobrynin, D.; Pleshko, N.; Steinbeck, M.J.; Freeman, T.A. Chemical modification of extracellular matrix by cold atmospheric plasma-generated reactive species affects chondrogenesis and bone formation. J. Tissue Eng. Regen. Med. 2016, 10, 772–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arndt, S.; Schmidt, A.; Karrer, S.; von Woedtke, T. Comparing two different plasma devices kINPen and Adtec SteriPlas regarding their molecular and cellular effects on wound healing. Clin. Plasma Med. 2018, 9, 24–33. [Google Scholar] [CrossRef]
- Arndt, S.; Unger, P.; Wacker, E.; Shimizu, T.; Heinlin, J.; Li, Y.F.; Thomas, H.M.; Morfill, G.E.; Zimmermann, J.L.; Bosserhoff, A.K.; et al. Cold atmospheric plasma (CAP) changes gene expression of key molecules of the wound healing machinery and improves wound healing in vitro and in vivo. PLoS ONE 2013, 8, e79325. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Bekeschus, S.; Wende, K.; Vollmar, B.; von Woedtke, T. A cold plasma jet accelerates wound healing in a murine model of full-thickness skin wounds. Exp. Dermatol. 2017, 26, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Ha, C.S.; Hwang, S.W.; Lee, H.J.; Kim, G.C.; Lee, K.W.; Song, K. Non-thermal atmospheric pressure plasma preferentially induces apoptosis in p53-mutated cancer cells by activating ROS stress-response pathways. PLoS ONE 2014, 9, e91947. [Google Scholar] [CrossRef] [PubMed]
- Laroussi, M.; Mohades, S.; Barekzi, N. Killing adherent and nonadherent cancer cells with the plasma pencil. Biointerphases 2015, 10, 029401. [Google Scholar] [CrossRef] [Green Version]
- Laroussi, M.; Lu, X.; Keidar, M. Perspective: The physics, diagnostics, and applications of atmospheric pressure low temperature plasma sources used in plasma medicine. J. Appl. Phys. 2017, 122, 020901. [Google Scholar] [CrossRef]
- Barton, A.; Wende, K.; Bundscherer, L.; Hasse, S.; Schmidt, A.; Bekeschus, S.; Weltmann, K.-D.; Lindequist, U.; Masur, K. Nonthermal Plasma Increases Expression of Wound Healing Related Genes in a Keratinocyte Cell Line. Plasma Med. 2013, 3, 125–136. [Google Scholar] [CrossRef]
- Dezest, M.; Chavatte, L.; Bourdens, M.; Quinton, D.; Camus, M.; Garrigues, L.; Descargues, P.; Arbault, S.; Burlet-Schiltz, O.; Casteilla, L.; et al. Mechanistic insights into the impact of Cold Atmospheric Pressure Plasma on human epithelial cell lines. Sci. Rep. 2017, 7, 41163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brun, P.; Brun, P.; Vono, M.; Venier, P.; Tarricone, E.; Deligianni, V.; Martines, E.; Zuin, M.; Spagnolo, S.; Cavazzana, R.; et al. Disinfection of ocular cells and tissues by atmospheric-pressure cold plasma. PLoS ONE 2012, 7, e33245. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Dietrich, S.; Steuer, A.; Weltmann, K.D.; von Woedtke, T.; Masur, K.; Wende, K. Non-thermal plasma activates human keratinocytes by stimulation of antioxidant and phase II pathways. J. Biol. Chem. 2015, 290, 6731–6750. [Google Scholar] [CrossRef] [PubMed]
- Bekeschus, S.; Favia, P.; Robert, E.; von Woedtke, T. White paper on plasma for medicine and hygiene: Future in plasma health sciences. Plasma Process. Polym. 2018, 1800033. [Google Scholar] [CrossRef]
- Jablonowski, H.; von Woedtke, T. Research on plasma medicine-relevant plasma–liquid interaction: What happened in the past five years? Clin. Plasma Med. 2015, 3, 42–52. [Google Scholar] [CrossRef]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- Von Woedtke, T.R.S.; Masur, K.; Weltmann, K.-D. Plasmas for medicine. Phys. Rep. 2013, 530, 291–320. [Google Scholar] [CrossRef]
- Weltmann, K.D.; von Woedtke, T. Plasma medicine—Current state of research and medical application. Plasma Phys. Control. Fusion 2017, 59, 014031. [Google Scholar] [CrossRef]
- Bekeschus, S.; Schmidt, A.; Weltmann, K.-D.; von Woedtke, T. The plasma jet kINPen—A powerful tool for wound healing. Clin. Plasma Med. 2016, 4, 19–28. [Google Scholar] [CrossRef]
- Partecke, L.I.; Evert, K.; Haugk, J.; Doering, F.; Normann, L.; Diedrich, S.; Weiss, F.U.; Evert, M.; Huebner, N.O.; Guenther, C.; et al. Tissue tolerable plasma (TTP) induces apoptosis in pancreatic cancer cells in vitro and in vivo. BMC Cancer 2012, 12, 473. [Google Scholar] [CrossRef] [PubMed]
- Forman, H.J.; Fukuto, J.M.; Torres, M. Redox signaling: Thiol chemistry defines which reactive oxygen and nitrogen species can act as second messengers. Am. J. Physiol.-Cell Physiol. 2004, 287, C246–C256. [Google Scholar] [CrossRef] [PubMed]
- Short, J.D.; Downs, K.; Tavakoli, S.; Asmis, R. Protein Thiol Redox Signaling in Monocytes and Macrophages. Antioxid. Redox Signal. 2016, 25, 816–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Woedtke, T.; Metelmann, H.R.; Weltmann, K.D. Clinical Plasma Medicine: State and Perspectives of in Vivo Application of Cold Atmospheric Plasma. Contrib. Plasm Phys. 2014, 54, 104–117. [Google Scholar] [CrossRef]
- Mann, M.S.; Tiede, R.; Gavenis, K.; Daeschlein, G.; Bussiahn, R.; Weltmann, K.-D.; Emmert, S.; Woedtke, T.V.; Ahmed, R. Introduction to DIN-specification 91315 based on the characterization of the plasma jet kINPen® MED. Clin. Plasma Med. 2016, 4, 35–45. [Google Scholar] [CrossRef]
- Darny, T.; Pouvesle, J.M.; Puech, V.; Douat, C.; Dozias, S.; Robert, E. Analysis of conductive target influence in plasma jet experiments through helium metastable and electric field measurements. Plasma Sources Sci. Technol. 2017, 26, 045008. [Google Scholar] [CrossRef]
- Yamada, H.; Sakakita, H.; Kato, S.; Kim, J.; Kiyama, S.; Fujiwara, M.; Itagaki, H.; Okazaki, T.; Ikehara, S.; Nakanishi, H.; et al. Spectroscopy of reactive species produced by low-energy atmospheric-pressure plasma on conductive target material surface. J. Phys. D Appl. Phys. 2016, 49, 394001. [Google Scholar] [CrossRef]
- Jablonowski, H.; Santos Sousa, J.; Weltmann, K.D.; Wende, K.; Reuter, S. Quantification of the ozone and singlet delta oxygen produced in gas and liquid phases by a non-thermal atmospheric plasma with relevance for medical treatment. Sci. Rep. 2018, 8, 12195. [Google Scholar] [CrossRef] [PubMed]
- Graves, D.B. Oxy-nitroso shielding burst model of cold atmospheric plasma therapeutics. Clin. Plasma Med. 2014, 2, 38–49. [Google Scholar] [CrossRef] [Green Version]
- Bruggeman, P.J.; Kushner, M.J.; Locke, B.R.; Gardeniers, J.G.E.; Graham, W.G.; Graves, D.B.; Hofman-Caris, R.C.H.M.; Maric, D.; Reid, J.P.; Ceriani, E.; et al. Plasma–liquid interactions: A review and roadmap. Plasma Sources Sci. Technol. 2016, 25, 053002. [Google Scholar] [CrossRef]
- Bekeschus, S.; Schmidt, A.; Niessner, F.; Gerling, T.; Weltmann, K.D.; Wende, K. Basic Research in Plasma Medicine–A Throughput Approach from Liquids to Cells. J. Vis. Exp. JoVE 2017, e56331. [Google Scholar] [CrossRef] [PubMed]
- Metelmann, H.R.; Sebauer, C.; Rutkowski, R.; Schuster, M.; Bekeschus, S.; Metelmann, P. Treating cancer cells with cold physical plasmas: On the way to evidence-based medicine. Contrib. Plasma Phys. 2018, 58, 415–419. [Google Scholar] [CrossRef]
- Heinlin, J.; Isbary, G.; Stolz, W.; Morfill, G.; Landthaler, M.; Shimizu, T.; Steffes, B.; Nosenko, T.; Zimmermann, J.; Karrer, S. Plasma applications in medicine with a special focus on dermatology. J. Eur. Acad. Dermatol. Venereol. 2011, 25, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Isbary, G.; Morfill, G.; Schmidt, H.U.; Georgi, M.; Ramrath, K.; Heinlin, J.; Karrer, S.; Landthaler, M.; Shimizu, T.; Steffes, B.; et al. A first prospective randomized controlled trial to decrease bacterial load using cold atmospheric argon plasma on chronic wounds in patients. Br. J. Dermatol. 2010, 163, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, A.K. Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic. Biol. Med. 2004, 36, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.Y.; Reddy, S.P.; Debiase, A.; Yamamoto, M.; Kleeberger, S.R. Gene expression profiling of NRF2-mediated protection against oxidative injury. Free Radic. Biol. Med. 2005, 38, 325–343. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Tian, F.; Zheng, H.; Whitman, S.A.; Lin, Y.; Zhang, Z.; Zhang, N.; Zhang, D.D. Nrf2 suppresses lupus nephritis through inhibition of oxidative injury and the NF-kappaB-mediated inflammatory response. Kidney Int. 2014, 85, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.C.; Cui, J.Y.; Klaassen, C.D. Beneficial role of Nrf2 in regulating NADPH generation and consumption. Toxicol. Sci. 2011, 123, 590–600. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.M.; Luo, L.; Namani, A.; Wang, X.J.; Tang, X. Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim. Biophys. Acta 2017, 1863, 585–597. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonay, M.; Deramaudt, T.B. Nrf2: New insight in cell apoptosis. Cell Death Dis. 2015, 6, e1897. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, H.; Yamamoto, M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004, 10, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Schafer, M.; Werner, S. Nrf2—A regulator of keratinocyte redox signaling. Free Radic. Biol. Med. 2015, 88, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, S.B.; Gordan, J.D.; Jin, J.; Harper, J.W.; Diehl, J.A. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: Oxidative stress sensing by a Cul3-Keap1 ligase. Mol. Cell. Biol. 2004, 24, 8477–8486. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Zhang, S.; Chan, J.Y.; Zhang, D.D. Keap1 controls postinduction repression of the Nrf2-mediated antioxidant response by escorting nuclear export of Nrf2. Mol. Cell. Biol. 2007, 27, 6334–6349. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.; Kelso, R.; Cooley, L. The kelch repeat superfamily of proteins: Propellers of cell function. Trends Cell Biol. 2000, 10, 17–24. [Google Scholar] [CrossRef]
- Kang, M.I.; Kobayashi, A.; Wakabayashi, N.; Kim, S.G.; Yamamoto, M. Scaffolding of Keap1 to the actin cytoskeleton controls the function of Nrf2 as key regulator of cytoprotective phase 2 genes. Proc. Natl. Acad. Sci. USA 2004, 101, 2046–2051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinkova-Kostova, A.T.; Kostov, R.V.; Canning, P. Keap1, the cysteine-based mammalian intracellular sensor for electrophiles and oxidants. Arch. Biochem. Biophys. 2017, 617, 84–93. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; You, D.J.; Lee, C.; Ahn, C.; Seong, J.Y.; Hwang, J.I. Suppression of NF-kappaB signaling by KEAP1 regulation of IKKbeta activity through autophagic degradation and inhibition of phosphorylation. Cell Signal. 2010, 22, 1645–1654. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Sherratt, P.J.; Nioi, P.; Yang, C.S.; Pickett, C.B. Nrf2 controls constitutive and inducible expression of ARE-driven genes through a dynamic pathway involving nucleocytoplasmic shuttling by Keap1. J. Biol. Chem. 2005, 280, 32485–32492. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, H.; Ishikawa, K.; Itabe, H.; Maruyama, Y. Carbon monoxide and bilirubin from heme oxygenase-1 suppresses reactive oxygen species generation and plasminogen activator inhibitor-1 induction. Mol. Cell. Biochem. 2006, 291, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Rabbani, P.S.; Zhou, A.; Borab, Z.M.; Frezzo, J.A.; Srivastava, N.; More, H.T.; Rifkin, W.J.; David, J.A.; Berens, S.J.; Chen, R.; et al. Novel lipoproteoplex delivers Keap1 siRNA based gene therapy to accelerate diabetic wound healing. Biomaterials 2017, 132, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Long, M.; Rojo de la Vega, M.; Wen, Q.; Bharara, M.; Jiang, T.; Zhang, R.; Zhou, S.; Wong, P.K.; Wondrak, G.T.; Zheng, H.; et al. An Essential Role of NRF2 in Diabetic Wound Healing. Diabetes 2016, 65, 780–793. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Pan, H.; Wang, H.; Li, X.; Bu, X.; Wang, Q.; Gao, Y.; Wen, G.; Zhou, Y.; Cong, Z.; et al. Interplay between VEGF and Nrf2 regulates angiogenesis due to intracranial venous hypertension. Sci. Rep. 2016, 6, 37338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Florczyk, U.; Jazwa, A.; Maleszewska, M.; Mendel, M.; Szade, K.; Kozakowska, M.; Grochot-Przeczek, A.; Viscardi, M.; Czauderna, S.; Bukowska-Strakova, K.; et al. Nrf2 regulates angiogenesis: Effect on endothelial cells, bone marrow-derived proangiogenic cells and hind limb ischemia. Antioxid. Redox Signal. 2014, 20, 1693–1708. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.L.; Dodd, G.; Thomas, S.; Zhang, X.; Wasserman, M.A.; Rovin, B.H.; Kunsch, C. Activation of Nrf2/ARE pathway protects endothelial cells from oxidant injury and inhibits inflammatory gene expression. Am. J. Physiol. 2006, 290, H1862–H1870. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Ichikawa, T.; Li, J.; Si, Q.; Yang, H.; Chen, X.; Goldblatt, C.S.; Meyer, C.J.; Li, X.; Cai, L.; et al. Diabetic downregulation of Nrf2 activity via ERK contributes to oxidative stress-induced insulin resistance in cardiac cells in vitro and in vivo. Diabetes 2011, 60, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Uno, K.; Prow, T.W.; Bhutto, I.A.; Yerrapureddy, A.; McLeod, D.S.; Yamamoto, M.; Reddy, S.P.; Lutty, G.A. Role of Nrf2 in retinal vascular development and the vaso-obliterative phase of oxygen-induced retinopathy. Exp. Eye Res. 2010, 90, 493–500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valcarcel-Ares, M.N.; Gautam, T.; Warrington, J.P.; Bailey-Downs, L.; Sosnowska, D.; de Cabo, R.; Losonczy, G.; Sonntag, W.E.; Ungvari, Z.; Csiszar, A. Disruption of Nrf2 signaling impairs angiogenic capacity of endothelial cells: Implications for microvascular aging. J. Gerontol. A Biol. Sci. Med. Sci. 2012, 67, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; von Woedtke, T.; Stenzel, J.; Lindner, T.; Polei, S.; Vollmar, B.; Bekeschus, S. One Year Follow-Up Risk Assessment in SKH-1 Mice and Wounds Treated with an Argon Plasma Jet. Int. J. Mol. Sci. 2017, 18, 868. [Google Scholar] [CrossRef] [PubMed]
- Jindam, A.; Yerra, V.G.; Kumar, A. Nrf2: A promising trove for diabetic wound healing. Ann. Transl. Med. 2017, 5, 469. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, H.; Patel, S.B.; Pastar, I. The Role of TGFbeta Signaling in Wound Epithelialization. Adv. Wound Care (New Rochelle) 2014, 3, 482–491. [Google Scholar] [CrossRef] [PubMed]
- Rolfs, F.; Huber, M.; Kuehne, A.; Kramer, S.; Haertel, E.; Muzumdar, S.; Wagner, J.; Tanner, Y.; Bohm, F.; Smola, S.; et al. Nrf2 Activation Promotes Keratinocyte Survival during Early Skin Carcinogenesis via Metabolic Alterations. Cancer Res. 2015, 75, 4817–4829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibata, T.; Kokubu, A.; Gotoh, M.; Ojima, H.; Ohta, T.; Yamamoto, M.; Hirohashi, S. Genetic alteration of Keap1 confers constitutive Nrf2 activation and resistance to chemotherapy in gallbladder cancer. Gastroenterology 2008, 135, 1358–1368. [Google Scholar] [CrossRef] [PubMed]
- Hiebert, P.; Wietecha, M.S.; Cangkrama, M.; Haertel, E.; Mavrogonatou, E.; Stumpe, M.; Steenbock, H.; Grossi, S.; Beer, H.D.; Angel, P.; et al. Nrf2-Mediated Fibroblast Reprogramming Drives Cellular Senescence by Targeting the Matrisome. Dev. Cell 2018, 46, 145–161.e10. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; von Woedtke, T.; Bekeschus, S. Periodic Exposure of Keratinocytes to Cold Physical Plasma: An In Vitro Model for Redox-Related Diseases of the Skin. Oxidative Med. Cell. Longev. 2016, 2016, 9816072. [Google Scholar] [CrossRef] [PubMed]
- Wende, K.; Bekeschus, S.; Schmidt, A.; Jatsch, L.; Hasse, S.; Weltmann, K.D.; Masur, K.; von Woedtke, T. Risk assessment of a cold argon plasma jet in respect to its mutagenicity. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2016, 798–799, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Kluge, S.; Bekeschus, S.; Bender, C.; Benkhai, H.; Sckell, A.; Below, H.; Stope, M.B.; Kramer, A. Investigating the Mutagenicity of a Cold Argon-Plasma Jet in an HET-MN Model. PLoS ONE 2016, 11, e0160667. [Google Scholar] [CrossRef] [PubMed]
- Boxhammer, V.; Li, Y.F.; Koritzer, J.; Shimizu, T.; Maisch, T.; Thomas, H.M.; Schlegel, J.; Morfill, G.E.; Zimmermann, J.L. Investigation of the mutagenic potential of cold atmospheric plasma at bactericidal dosages. Mutat. Res. 2013, 753, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Joshi, N.; Werner, S. Nrf2 is highly expressed in neutrophils, but myeloid cell-derived Nrf2 is dispensable for wound healing in mice. PLoS ONE 2017, 12, e0187162. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Jiang, T.; Wang, H.; Tao, S.; Lau, A.; Fang, D.; Zhang, D.D. Does Nrf2 contribute to p53-mediated control of cell survival and death? Antioxid. Redox Signal. 2012, 17, 1670–1675. [Google Scholar] [CrossRef] [PubMed]
- Hiemstra, S.; Niemeijer, M.; Koedoot, E.; Wink, S.; Klip, J.E.; Vlasveld, M.; de Zeeuw, E.; van Os, B.; White, A.; Water, B.V. Comprehensive Landscape of Nrf2 and p53 Pathway Activation Dynamics by Oxidative Stress and DNA Damage. Chem. Res. Toxicol. 2017, 30, 923–933. [Google Scholar] [CrossRef] [PubMed]
- Budanov, A.V. The role of tumor suppressor p53 in the antioxidant defense and metabolism. Subcell. Biochem. 2014, 85, 337–358. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Xu, Y. Oxidative Stress, and Aging. Antioxid. Redox Signal. 2011, 15, 1669–1678. [Google Scholar] [CrossRef] [PubMed]
- Kondo, N.; Nakamura, H.; Masutani, H.; Yodoi, J. Redox regulation of human thioredoxin network. Antioxid. Redox Signal. 2006, 8, 1881–1890. [Google Scholar] [CrossRef] [PubMed]
- Faraonio, R.; Vergara, P.; Di Marzo, D.; Pierantoni, M.G.; Napolitano, M.; Russo, T.; Cimino, F. p53 suppresses the Nrf2-dependent transcription of antioxidant response genes. J. Biol. Chem. 2006, 281, 39776–39784. [Google Scholar] [CrossRef] [PubMed]
- Vollmar, B.; El-Gibaly, A.M.; Scheuer, C.; Strik, M.W.; Bruch, H.P.; Menger, M.D. Acceleration of cutaneous wound healing by transient p53 inhibition. Lab. Investig. 2002, 82, 1063–1071. [Google Scholar] [CrossRef] [PubMed]
- Olovnikov, I.A.; Kravchenko, J.E.; Chumakov, P.M. Homeostatic functions of the p53 tumor suppressor: Regulation of energy metabolism and antioxidant defense. Semin. Cancer Biol. 2009, 19, 32–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuzawa, A.; Ichijo, H. Redox control of cell fate by MAP kinase: Physiological roles of ASK1-MAP kinase pathway in stress signaling. Biochim. Biophys. Acta 2008, 1780, 1325–1336. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Dickens, M.; Raingeaud, J.; Davis, R.J.; Greenberg, M.E. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 1995, 270, 1326–1331. [Google Scholar] [CrossRef] [PubMed]
- Sakata, N.; Patel, H.R.; Terada, N.; Aruffo, A.; Johnson, G.L.; Gelfand, E.W. Selective activation of c-Jun kinase mitogen-activated protein kinase by CD40 on human B cells. J. Biol. Chem. 1995, 270, 30823–30828. [Google Scholar] [CrossRef] [PubMed]
- Lavoie, J.N.; L’Allemain, G.; Brunet, A.; Muller, R.; Pouyssegur, J. Cyclin D1 expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. J. Biol. Chem. 1996, 271, 20608–20616. [Google Scholar] [CrossRef] [PubMed]
- Naidu, S.; Vijayan, V.; Santoso, S.; Kietzmann, T.; Immenschuh, S. Inhibition and genetic deficiency of p38 MAPK up-regulates heme oxygenase-1 gene expression via Nrf2. J. Immunol. 2009, 182, 7048–7057. [Google Scholar] [CrossRef] [PubMed]
- Roger, L.; Gadea, G.; Roux, P. Control of cell migration: A tumour suppressor function for p53? Biol. Cell 2006, 98, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Pi, J.; Bai, Y.; Reece, J.M.; Williams, J.; Liu, D.; Freeman, M.L.; Fahl, W.E.; Shugar, D.; Liu, J.; Qu, W.; et al. Molecular mechanism of human Nrf2 activation and degradation: Role of sequential phosphorylation by protein kinase CK2. Free Radic. Biol. Med. 2007, 42, 1797–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, A.; Wende, K.; Bekeschus, S.; Bundscherer, L.; Barton, A.; Ottmuller, K.; Weltmann, K.D.; Masur, K. Non-thermal plasma treatment is associated with changes in transcriptome of human epithelial skin cells. Free Radic. Res. 2013, 47, 577–592. [Google Scholar] [CrossRef] [PubMed]
- Bundscherer, L.; Nagel, S.; Hasse, S.; Tresp, H.; Wende, K.; Walther, R.; Reuter, S.; Weltmann, K.D.; Masur, K.; Lindequist, U. Non-thermal plasma treatment induces MAPK signaling in human monocytes. Open Chem. 2015, 13, 606–613. [Google Scholar] [CrossRef]
- Bundscherer, L.; Wende, K.; Ottmuller, K.; Barton, A.; Schmidt, A.; Bekeschus, S.; Hasse, S.; Weltmann, K.D.; Masur, K.; Lindequist, U. Impact of non-thermal plasma treatment on MAPK signaling pathways of human immune cell lines. Immunobiology 2013, 218, 1248–1255. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Bekeschus, S.; Ottmüller, K.; Hasse, S.; von Woedtke, T.; Wende, K. Cold physical plasma modulates p53 and mitogen-activated protein kinase signaling in keratinocytes. Oxidative Med. Cell. Longev. 2018, in press. [Google Scholar]
- Buschmann, T.; Potapova, O.; Bar-Shira, A.; Ivanov, V.N.; Fuchs, S.Y.; Henderson, S.; Fried, V.A.; Minamoto, T.; Alarcon-Vargas, D.; Pincus, M.R.; et al. Jun NH2-terminal kinase phosphorylation of p53 on Thr-81 is important for p53 stabilization and transcriptional activities in response to stress. Mol. Cell. Biol. 2001, 21, 2743–2754. [Google Scholar] [CrossRef] [PubMed]
- Pluquet, O.; North, S.; Bhoumik, A.; Dimas, K.; Ronai, Z.E.; Hainaut, P. The Cytoprotective Aminothiol WR1065 Activates p53 through a Non-genotoxic Signaling Pathway Involving c-Jun N-terminal Kinase. J. Biol. Chem. 2003, 278, 11879–11887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bulavin, D.V.; Higashimoto, Y.; Popoff, I.J.; Gaarde, W.A.; Basrur, V.; Potapova, O.; Appella, E.; Fornace, A.J., Jr. Initiation of a G2/M checkpoint after ultraviolet radiation requires p38 kinase. Nature 2001, 411, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Huang, Z.; Zhang, D.D. Phosphorylation of Nrf2 at multiple sites by MAP kinases has a limited contribution in modulating the Nrf2-dependent antioxidant response. PLoS ONE 2009, 4, e6588. [Google Scholar] [CrossRef] [PubMed]
- Schaeffer, H.J.; Weber, M.J. Mitogen-Activated Protein Kinases: Specific Messages from Ubiquitous Messengers. Mol. Cell. Biol. 1999, 19, 2435–2444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, R.J. Signal Transduction by the JNK Group of MAP Kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef]
- Roux, P.P.; Blenis, J. ERK and p38 MAPK-Activated Protein Kinases: A Family of Protein Kinases with Diverse Biological Functions. Microbiol. Mol. Biol. Rev. 2004, 68, 320–344. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Xie, X.; Huang, J.; Gong, W.; Zhu, X.; Chen, Q.; Huang, J.; Huang, H. Connexin43 regulates high glucose-induced expression of fibronectin, ICAM-1 and TGF-beta1 via Nrf2/ARE pathway in glomerular mesangial cells. Free Radic. Biol. Med. 2017, 102, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, P.; Qiu, C.; Frank, S.; Tamber, K.; Becker, D. Dynamic changes in connexin expression correlate with key events in the wound healing process. Cell Biol. Int. 2003, 27, 525–541. [Google Scholar] [CrossRef]
- Ghatnekar, G.S.; O’Quinn, M.P.; Jourdan, L.J.; Gurjarpadhye, A.A.; Draughn, R.L.; Gourdie, R.G. Connexin43 carboxyl-terminal peptides reduce scar progenitor and promote regenerative healing following skin wounding. Regen. Med. 2009, 4, 205–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grek, C.L.; Montgomery, J.; Sharma, M.; Ravi, A.; Rajkumar, J.S.; Moyer, K.E.; Gourdie, R.G.; Ghatnekar, G.S. A Multicenter Randomized Controlled Trial Evaluating a Cx43-Mimetic Peptide in Cutaneous Scarring. J. Investig. Dermatol. 2017, 137, 620–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montgomery, J.; Ghatnekar, G.S.; Grek, C.L.; Moyer, K.E.; Gourdie, R.G. Connexin 43-Based Therapeutics for Dermal Wound Healing. Int. J. Mol. Sci. 2018, 19, 1778. [Google Scholar] [CrossRef] [PubMed]
- Brandner, J.M.; Houdek, P.; Husing, B.; Kaiser, C.; Moll, I. Connexins 26, 30, and 43: Differences among spontaneous, chronic, and accelerated human wound healing. J. Investig. Dermatol. 2004, 122, 1310–1320. [Google Scholar] [CrossRef] [PubMed]
- Mendoza-Naranjo, A.; Cormie, P.; Serrano, A.E.; Wang, C.M.; Thrasivoulou, C.; Sutcliffe, J.E.; Gilmartin, D.J.; Tsui, J.; Serena, T.E.; Phillips, A.R.; et al. Overexpression of the gap junction protein Cx43 as found in diabetic foot ulcers can retard fibroblast migration. Cell Biol. Int. 2012, 36, 661–667. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Bekeschus, S.; von Woedtke, T.; Hasse, S. Cell migration and adhesion of a human melanoma cell line is decreased by cold plasma treatment. Clin. Plasma Med. 2015, 3, 24–31. [Google Scholar] [CrossRef]
- Reinke, J.M.; Sorg, H. Wound repair and regeneration. Eur. Surg. Res. 2012, 49, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Gabbiani, G. The myofibroblast in wound healing and fibrocontractive diseases. J. Pathol. 2003, 200, 500–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balzer, J.; Heuer, K.; Demir, E.; Hoffmanns, M.A.; Baldus, S.; Fuchs, P.C.; Awakowicz, P.; Suschek, C.V.; Oplander, C. Non-Thermal Dielectric Barrier Discharge (DBD) Effects on Proliferation and Differentiation of Human Fibroblasts Are Primary Mediated by Hydrogen Peroxide. PLoS ONE 2015, 10, e0144968. [Google Scholar] [CrossRef] [PubMed]
- Kuchenbecker, M.; Bibinov, N.; Kaemlimg, A.; Wandke, D.; Awakowicz, P.; Viol, W. Characterization of DBD plasma source for biomedical applications. J. Phys. D Appl. Phys. 2009, 42, 045212. [Google Scholar] [CrossRef]
- Wandke, D. PlasmaDerm®-Based on di_CAP Technology. In Comprehensive Clinical Plasma Medicine; Metelmann, H.-R., von Woedtke, T., Weltmann, K.-D., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 495–502. [Google Scholar]
- Laroussi, M. From Killing Bacteria to Destroying Cancer Cells: 20 Years of Plasma Medicine. Plasma Process. Polym. 2014, 11, 1138–1141. [Google Scholar] [CrossRef]
- Emmert, S.; Brehmer, F.; Hänßle, H.; Helmke, A.; Mertens, N.; Ahmed, R.; Simon, D.; Wandke, D.; Maus-Friedrichs, W.; Däschlein, G. Atmospheric pressure plasma in dermatology: Ulcus treatment and much more. Clin. Plasma Med. 2013, 1, 24–29. [Google Scholar] [CrossRef]
- Isbary, G.; Heinlin, J.; Shimizu, T.; Zimmermann, J.L.; Morfill, G.; Schmidt, H.U.; Monetti, R.; Steffes, B.; Bunk, W.; Li, Y.; et al. Successful and safe use of 2 min cold atmospheric argon plasma in chronic wounds: Results of a randomized controlled trial. Br. J. Dermatol. 2012, 167, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Isbary, G.; Morfill, G.; Zimmermann, J.; Shimizu, T.; Stolz, W. Cold atmospheric plasma: A successful treatment of lesions in Hailey-Hailey disease. Arch. Dermatol. 2011, 147, 388–390. [Google Scholar] [CrossRef] [PubMed]
- Daeschlein, G.; Scholz, S.; Ahmed, R.; von Woedtke, T.; Haase, H.; Niggemeier, M.; Kindel, E.; Brandenburg, R.; Weltmann, K.D.; Juenger, M. Skin decontamination by low-temperature atmospheric pressure plasma jet and dielectric barrier discharge plasma. J. Hosp. Infect. 2012, 81, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Metelmann, H.R.; von Woedtke, T.; Bussiahn, R.; Weltmann, K.D.; Rieck, M.; Khalili, R.; Podmelle, F.; Waite, P.D. Experimental Recovery of CO2-Laser Skin Lesions by Plasma Stimulation. Am. J. Cosmet. Surg. 2012, 29, 52–56. [Google Scholar] [CrossRef]
- Metelmann, H.R.; Podmelle, F.; Waite, P.D.; Muller-Debus, C.F.; Hammes, S.; Funk, W. Conditioning in laser skin resurfacing-betulin emulsion and skin recovery. J. Cranio-Maxillo-Fac. Surg. 2013, 41, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Metelmann, H.-R.; Vu, T.T.; Do, H.T.; Le, T.N.B.; Hoang, T.H.A.; Phi, T.T.T.; Luong, T.M.L.; Doan, V.T.; Nguyen, T.T.H.; Nguyen, T.H.M. Scar formation of laser skin lesions after cold atmospheric pressure plasma (CAP) treatment: A clinical long term observation. Clin. Plasma Med. 2013, 1, 30–35. [Google Scholar] [CrossRef]
- Lademann, J.; Ulrich, C.; Patzelt, A.; Richter, H.; Kluschke, F.; Klebes, M.; Lademann, O.; Kramer, A.; Weltmann, K.D.; Lange-Asschenfeldt, B. Risk assessment of the application of tissue-tolerable plasma on human skin. Clin. Plasma Med. 2013, 1, 5–10. [Google Scholar] [CrossRef]
- Ulrich, C.; Kluschke, F.; Patzelt, A.; Vandersee, S.; Czaika, V.A.; Richter, H.; Bob, A.; Hutten, J.; Painsi, C.; Huge, R.; et al. Clinical use of cold atmospheric pressure argon plasma in chronic leg ulcers: A pilot study. J. Wound Care 2015, 24, 202–193. [Google Scholar] [CrossRef] [PubMed]
- Klebes, M.; Lademann, J.; Philipp, S.; Ulrich, C.; Patzelt, A.; Ulmer, M.; Kluschke, F.; Kramer, A.; Weltmann, K.; Sterry, W. Effects of tissue-tolerable plasma on psoriasis vulgaris treatment compared to conventional local treatment: A pilot study. Clin. Plasma Med. 2014, 2, 22–27. [Google Scholar] [CrossRef]
- Jablonowski, H.; Hansch, M.A.; Dunnbier, M.; Wende, K.; Hammer, M.U.; Weltmann, K.D.; Reuter, S.; Woedtke, T. Plasma jet’s shielding gas impact on bacterial inactivation. Biointerphases 2015, 10, 029506. [Google Scholar] [CrossRef] [PubMed]
- Reuter, S.; Tresp, H.; Wende, K.; Hammer, M.U.; Winter, J.; Masur, K.; Schmidt-Bleker, A.; Weltmann, K.D. From RONS to ROS: Tailoring Plasma Jet Treatment of Skin Cells. IEEE Trans. Plasma Sci. 2012, 40, 2986–2993. [Google Scholar] [CrossRef]
- Reuter, S.; Winter, J.; Schmidt-Bleker, A.; Tresp, H.; Hammer, M.U.; Weltmann, K.D. Controlling the Ambient Air Affected Reactive Species Composition in the Effluent of an Argon Plasma Jet. IEEE Trans. Plasma Sci. 2012, 40, 2788–2794. [Google Scholar] [CrossRef]
- Schmidt, A.; Bekeschus, S.; Jablonowski, H.; Barton, A.; Weltmann, K.D.; Wende, K. Role of Ambient Gas Composition on Cold Physical Plasma-Elicited Cell Signaling in Keratinocytes. Biophys. J. 2017, 112, 2397–2407. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Parameter influencing impact, efficiency and specificity of plasma. (A) Cold physical plasma generates reactive oxygen (ROS) and nitrogen species (RNS), which can be delivered to target cells. (B) Optical emission spectroscopy resolves, for example, OH (~308 nm) and the second positive system of molecular nitrogen (315–380 nm, upper diagram) as well as other excited species such as atomic O (777 nm and 844 nm) and atomic Ar in the range of 696 and 912 nm (lower diagram). (C) Device parameters such as treatment area, flow rate, working gas, components of gas and their tuning (a) as well as process parameter including treatment and incubation time, direct vs. indirect treatment, distance to the effluent and throughput can be modulated. Plasma is used to treat liquids (b), cells or tissues (c) or can be applied in vivo directly to the skin (d).
Figure 1.
Parameter influencing impact, efficiency and specificity of plasma. (A) Cold physical plasma generates reactive oxygen (ROS) and nitrogen species (RNS), which can be delivered to target cells. (B) Optical emission spectroscopy resolves, for example, OH (~308 nm) and the second positive system of molecular nitrogen (315–380 nm, upper diagram) as well as other excited species such as atomic O (777 nm and 844 nm) and atomic Ar in the range of 696 and 912 nm (lower diagram). (C) Device parameters such as treatment area, flow rate, working gas, components of gas and their tuning (a) as well as process parameter including treatment and incubation time, direct vs. indirect treatment, distance to the effluent and throughput can be modulated. Plasma is used to treat liquids (b), cells or tissues (c) or can be applied in vivo directly to the skin (d).


Figure 2.
Cold physical plasma triggers nuclear translocation of Nrf2, and induces co-localization of Keap1 with actin filaments in the cytoplasm. (A) Dermal fibroblasts (bright field image, left) were isolated from SKH1 mouse skin and exposed to cold physical plasma-derived ROS/RNS. Upon nuclear translocation of the nuclear factor erythroid 2-related factor 2 (Nrf2), plasma significantly altered antioxidant and phase II detoxification enzymes and proteins (e.g., heme oxygenase 1 (HMOX-1), NADPH quinone oxidoreductase 1 (NQO1), thioredoxin (TXN), glutathione reductase (GSR), superoxide dismutase (SOD), glutathione S-transferase (GST), glutathione (GSH) etc.). (B) Cytoplasmic localization of Kelch-like ECH-associated protein 1 (Keap1) was detected immunohistochemically by anti-Keap1 antibody (red). Co-localization of Keap1 with actin filaments was visualized by staining with fluorescein isothiocyanate (FITC)-phalloidin (green). (C) Subcellular localization of Keap1 (green) and trans-localization of Nrf2 (red) from the cytoplasm to the nucleus were detected immunohistochemically by anti-Keap1 and anti-Nrf2 antibodies in plasma-treated (right panel), but not control fibroblasts (left panel). Scale bars 100 µm (A), 50 µm (B,C).
Figure 2.
Cold physical plasma triggers nuclear translocation of Nrf2, and induces co-localization of Keap1 with actin filaments in the cytoplasm. (A) Dermal fibroblasts (bright field image, left) were isolated from SKH1 mouse skin and exposed to cold physical plasma-derived ROS/RNS. Upon nuclear translocation of the nuclear factor erythroid 2-related factor 2 (Nrf2), plasma significantly altered antioxidant and phase II detoxification enzymes and proteins (e.g., heme oxygenase 1 (HMOX-1), NADPH quinone oxidoreductase 1 (NQO1), thioredoxin (TXN), glutathione reductase (GSR), superoxide dismutase (SOD), glutathione S-transferase (GST), glutathione (GSH) etc.). (B) Cytoplasmic localization of Kelch-like ECH-associated protein 1 (Keap1) was detected immunohistochemically by anti-Keap1 antibody (red). Co-localization of Keap1 with actin filaments was visualized by staining with fluorescein isothiocyanate (FITC)-phalloidin (green). (C) Subcellular localization of Keap1 (green) and trans-localization of Nrf2 (red) from the cytoplasm to the nucleus were detected immunohistochemically by anti-Keap1 and anti-Nrf2 antibodies in plasma-treated (right panel), but not control fibroblasts (left panel). Scale bars 100 µm (A), 50 µm (B,C).

Figure 3.
Cold physical plasma-derived generation and delivery of ROS/RNS triggers redox modifications and accelerates physiological responses in wound healing. Redox sensitive signaling pathways (e.g., Nrf2, p53) provide protection from excess ROS/RNS to eventually re-establish redox homeostasis. Cold physical plasma-derived ROS/RNS are efficient triggers of such signaling events in skin cells in vitro and in vivo. Accordingly, plasma treatment promotes several cellular and tissue responses linked to molecular signatures accompanying enhanced wound healing.
Figure 3.
Cold physical plasma-derived generation and delivery of ROS/RNS triggers redox modifications and accelerates physiological responses in wound healing. Redox sensitive signaling pathways (e.g., Nrf2, p53) provide protection from excess ROS/RNS to eventually re-establish redox homeostasis. Cold physical plasma-derived ROS/RNS are efficient triggers of such signaling events in skin cells in vitro and in vivo. Accordingly, plasma treatment promotes several cellular and tissue responses linked to molecular signatures accompanying enhanced wound healing.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Schmidt, A.; Bekeschus, S. Redox for Repair: Cold Physical Plasmas and Nrf2 Signaling Promoting Wound Healing. Antioxidants 2018, 7, 146. https://doi.org/10.3390/antiox7100146
AMA Style
Schmidt A, Bekeschus S. Redox for Repair: Cold Physical Plasmas and Nrf2 Signaling Promoting Wound Healing. Antioxidants. 2018; 7(10):146. https://doi.org/10.3390/antiox7100146
Chicago/Turabian StyleSchmidt, Anke, and Sander Bekeschus. 2018. "Redox for Repair: Cold Physical Plasmas and Nrf2 Signaling Promoting Wound Healing" Antioxidants 7, no. 10: 146. https://doi.org/10.3390/antiox7100146
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.