The Loss of α- and β-Tubulin Proteins Are a Pathological Hallmark of Chronic Alcohol Consumption and Natural Brain Ageing

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Brain Samples and Ethics Statement
2.2. Rats
2.3. Brain Tissue Homogenization
2.4. One-Dimensional Polyacrylamide Gel Electrophoresis (1-D PAGE)
2.5. Western (Immuno) Blotting
2.6. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.7. Statistics
3. Results
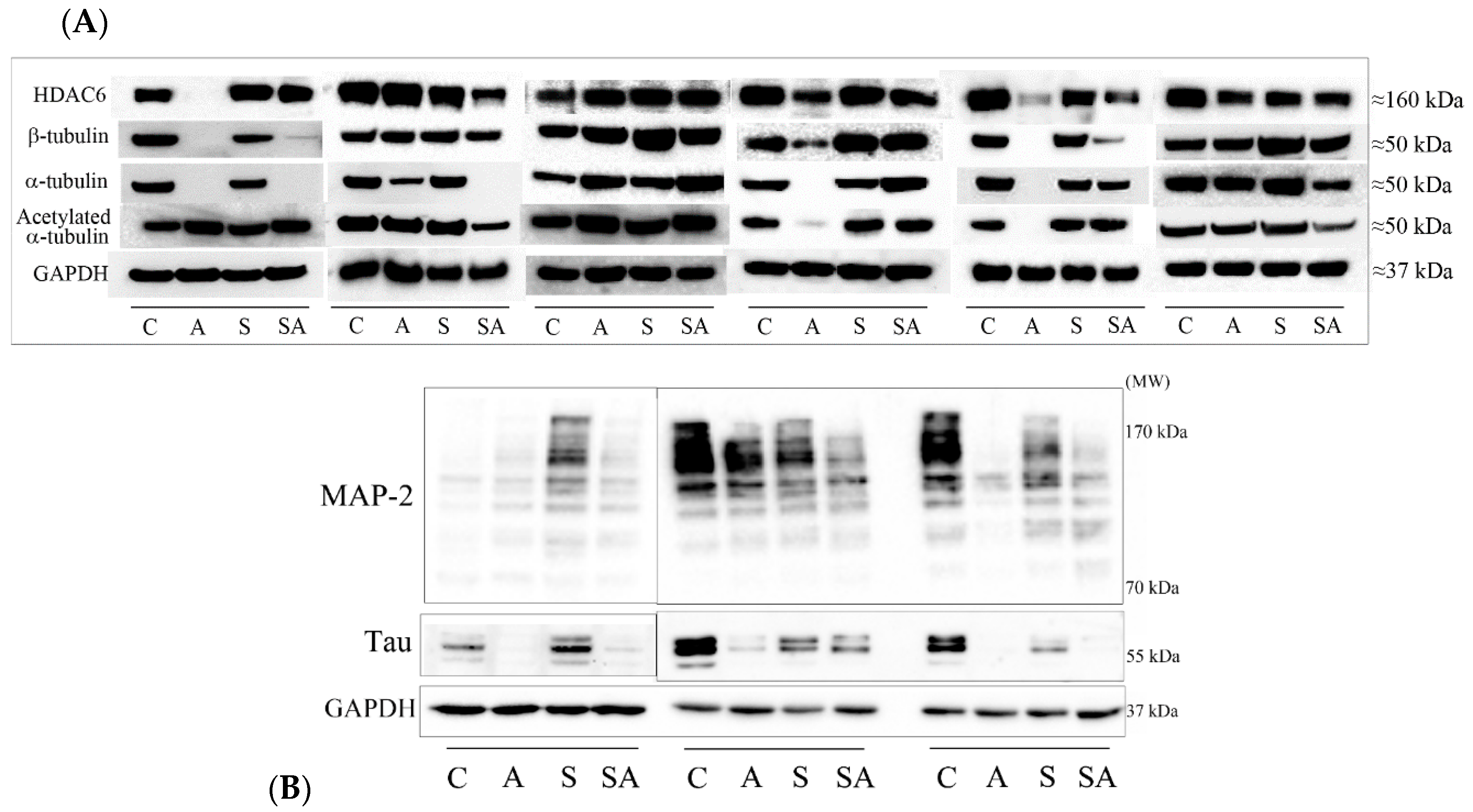
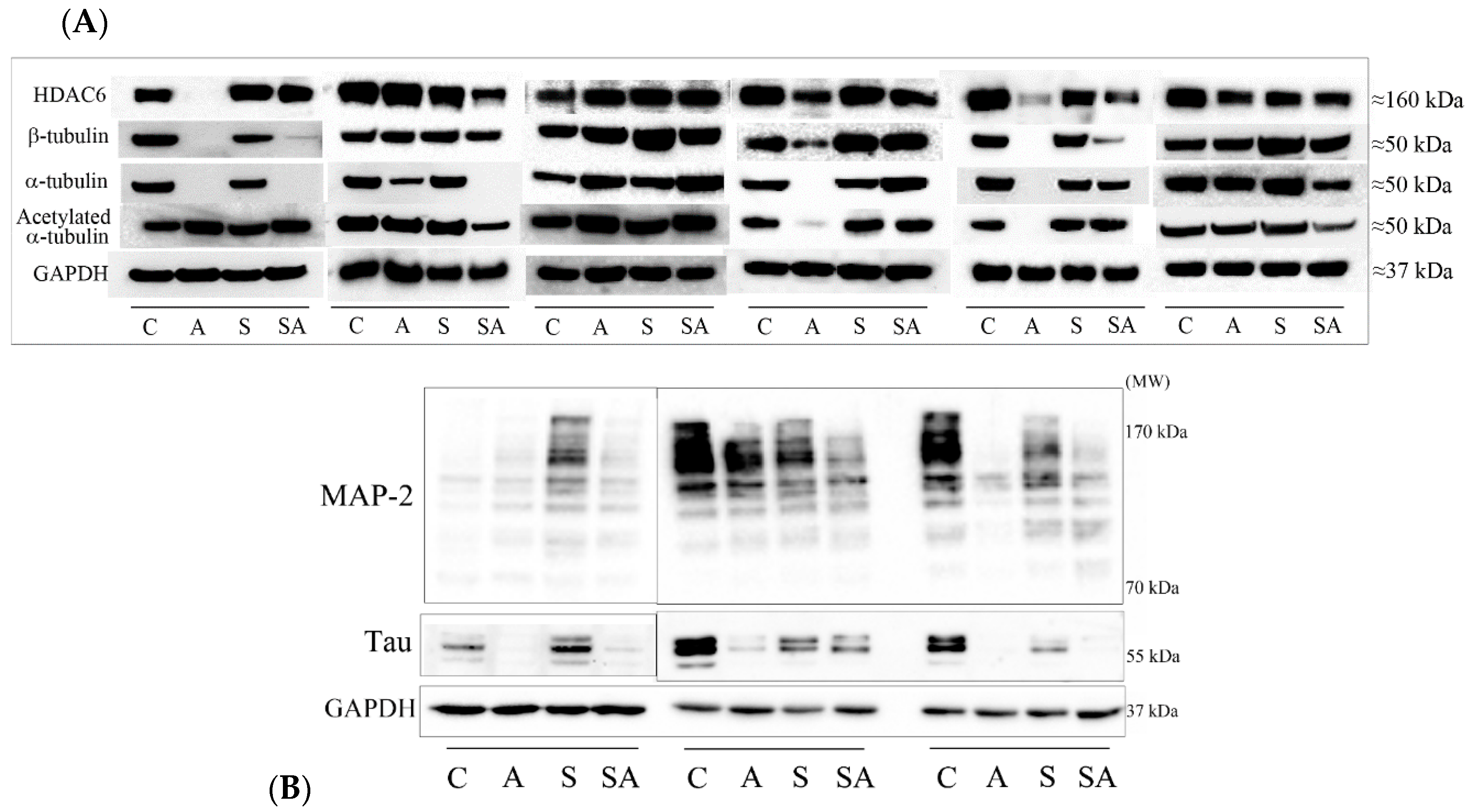
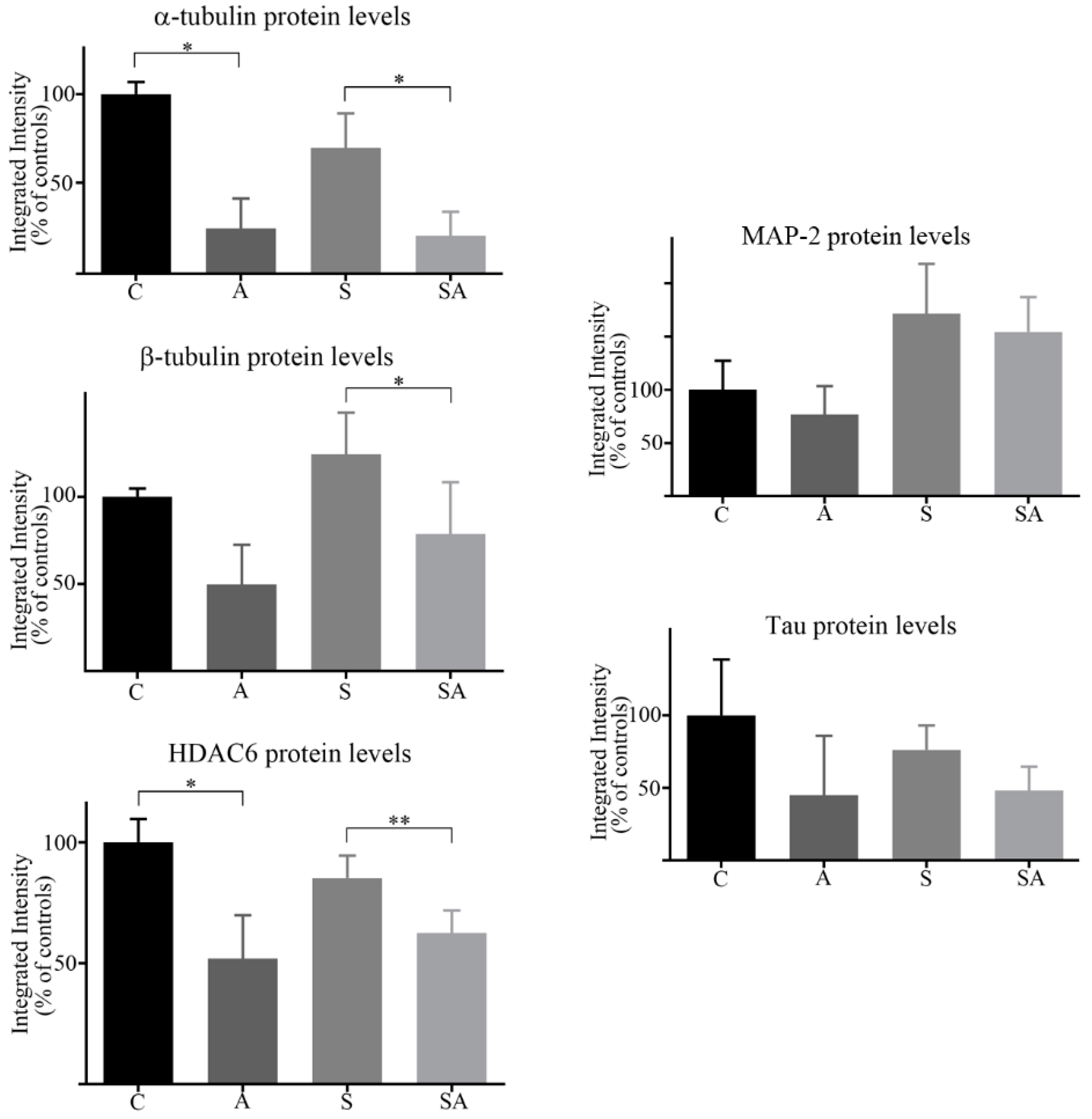
3.1. Reduced α- and β-Tubulins and Microtubule-Associated Proteins in the PFC of Alcoholic Subjects
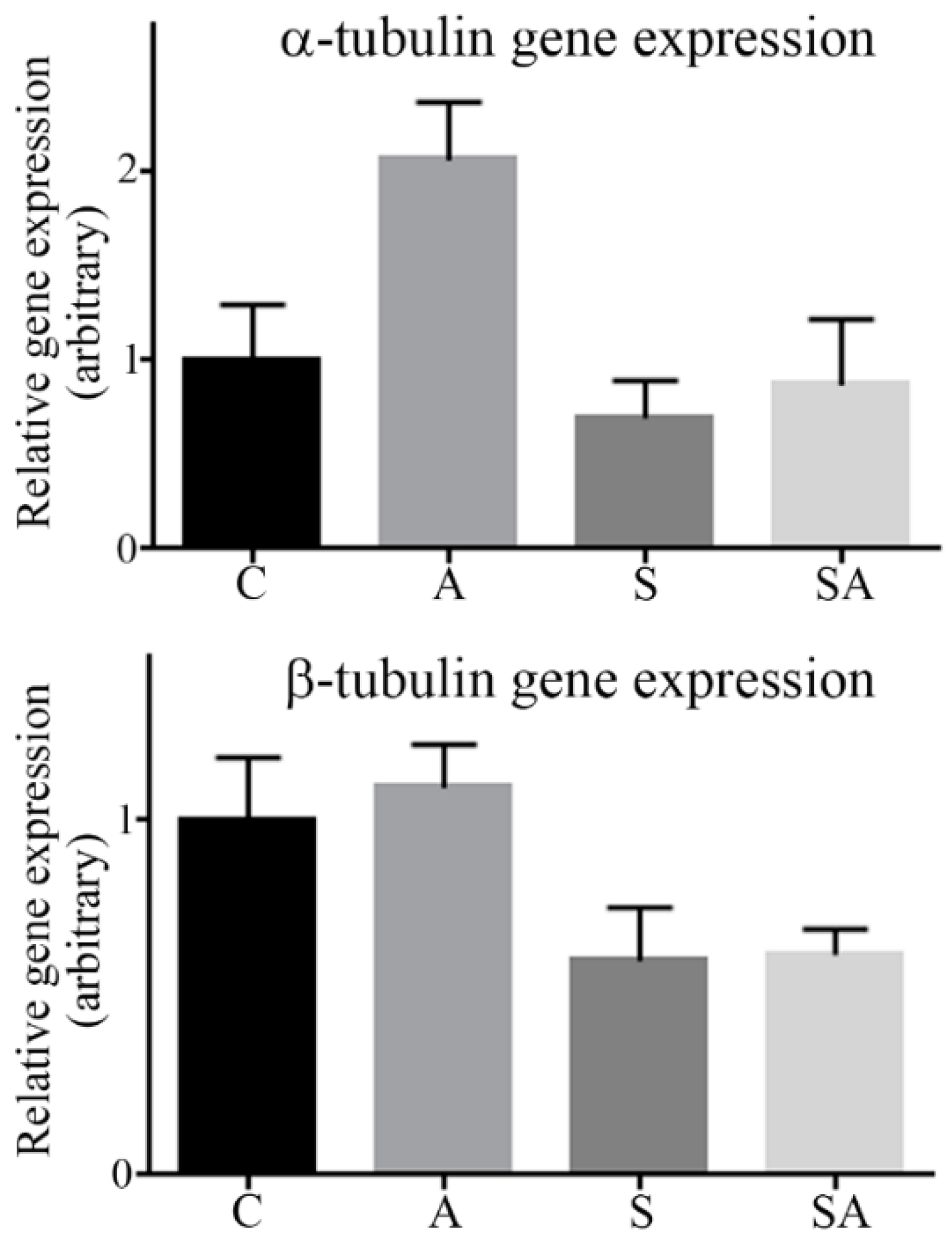
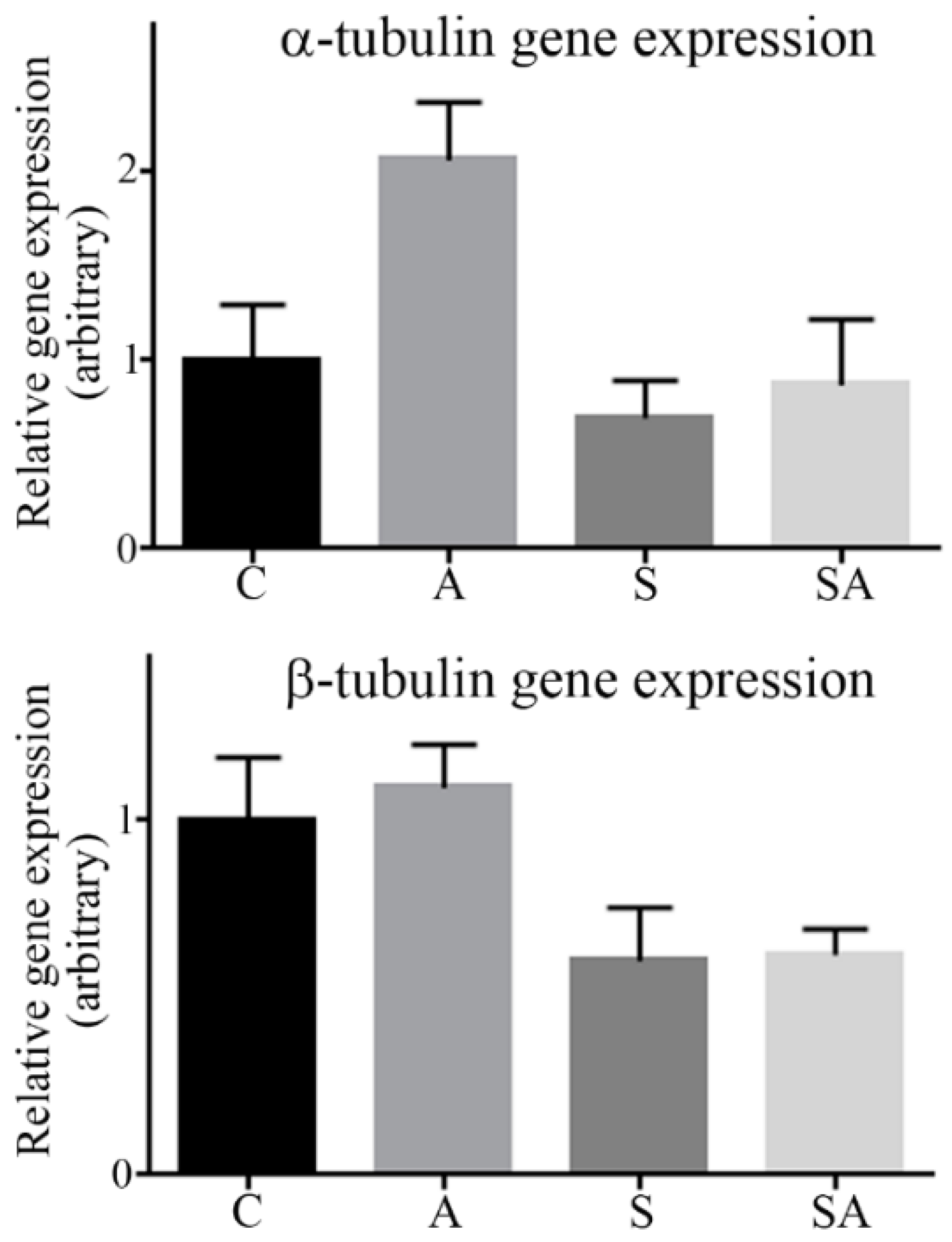
3.2. α- and β-Tubulin Gene Expression Were Moderately Increased in Alcoholic Cohorts
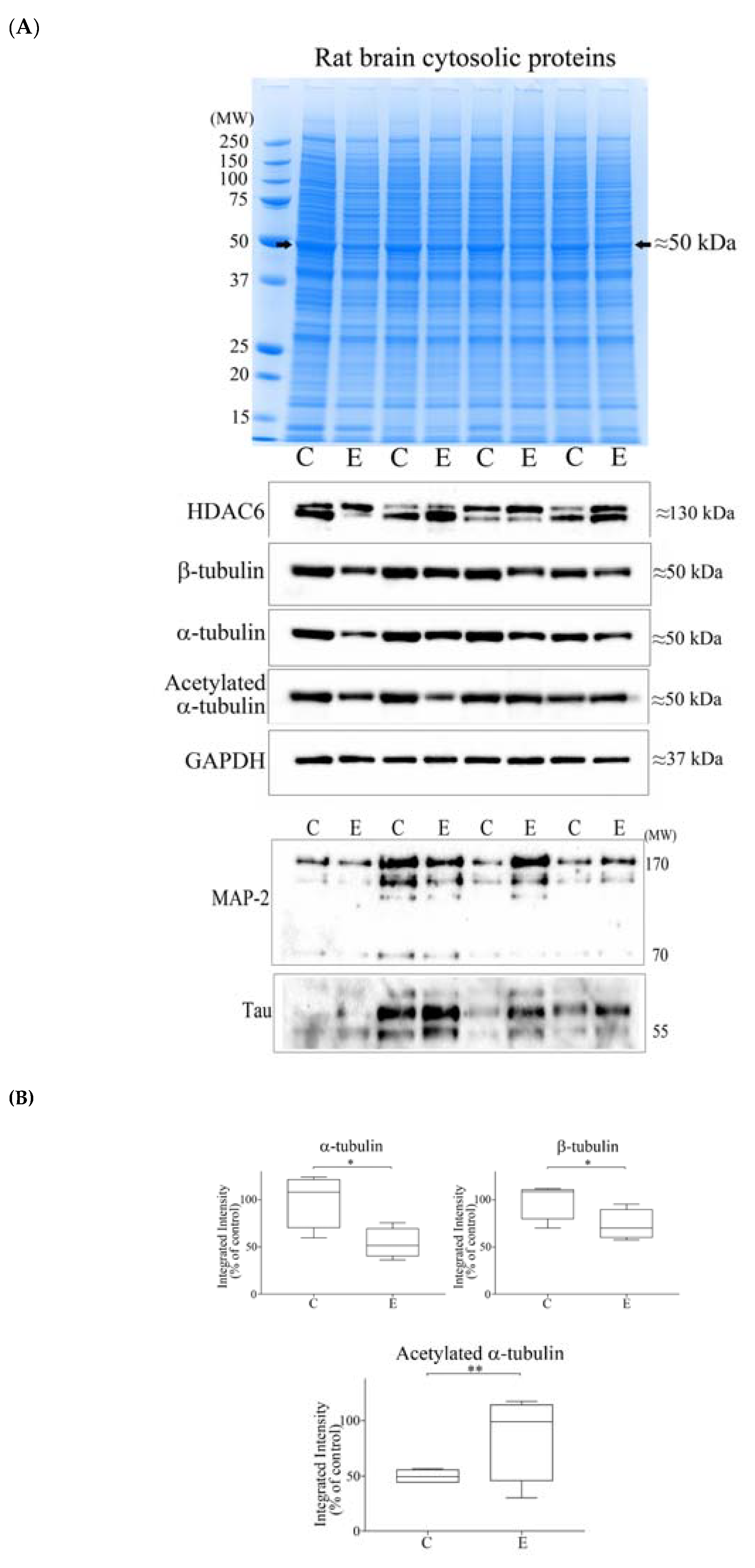
3.3. Alcohol Consumption Modelled In Vivo
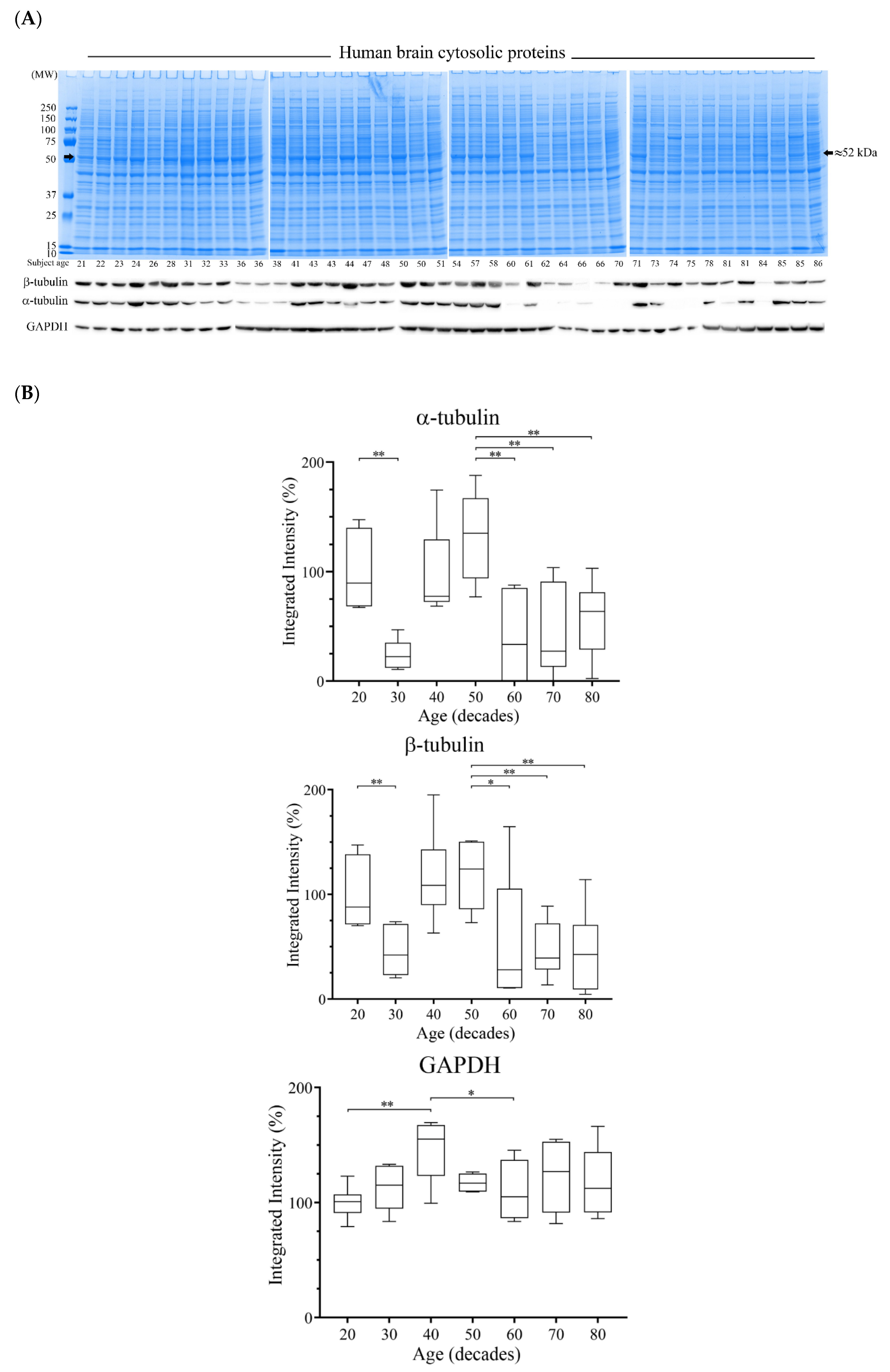
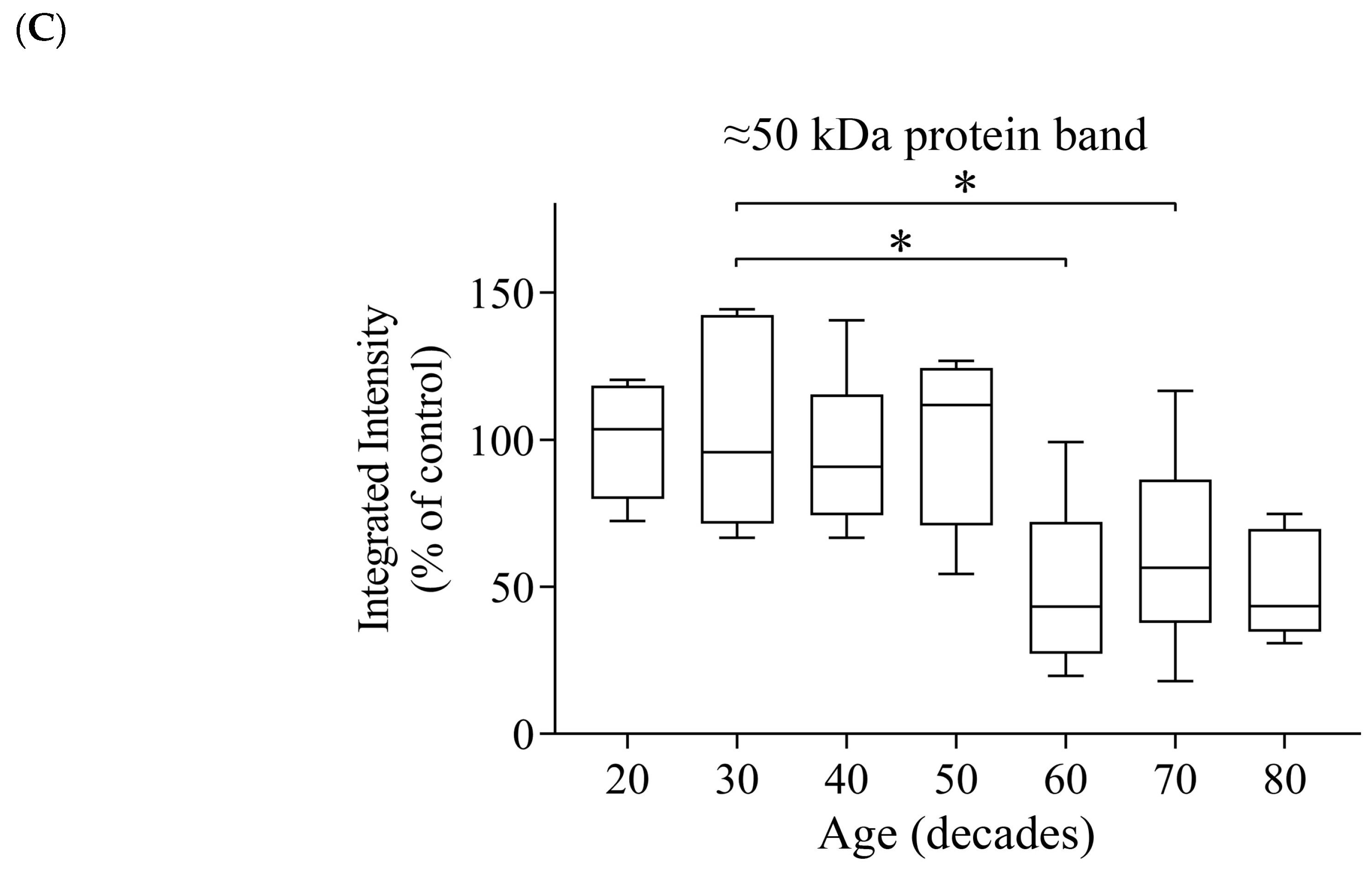
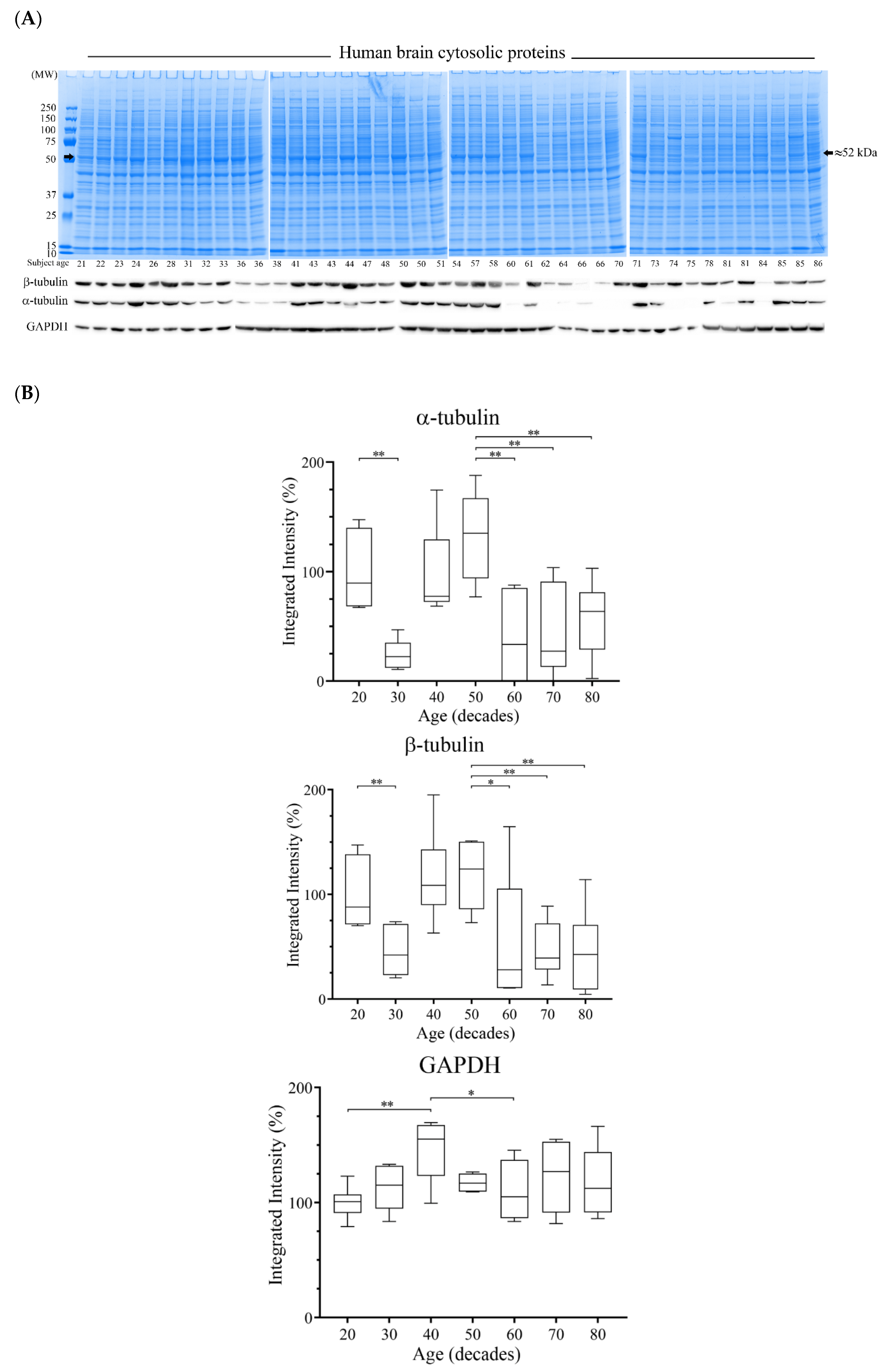
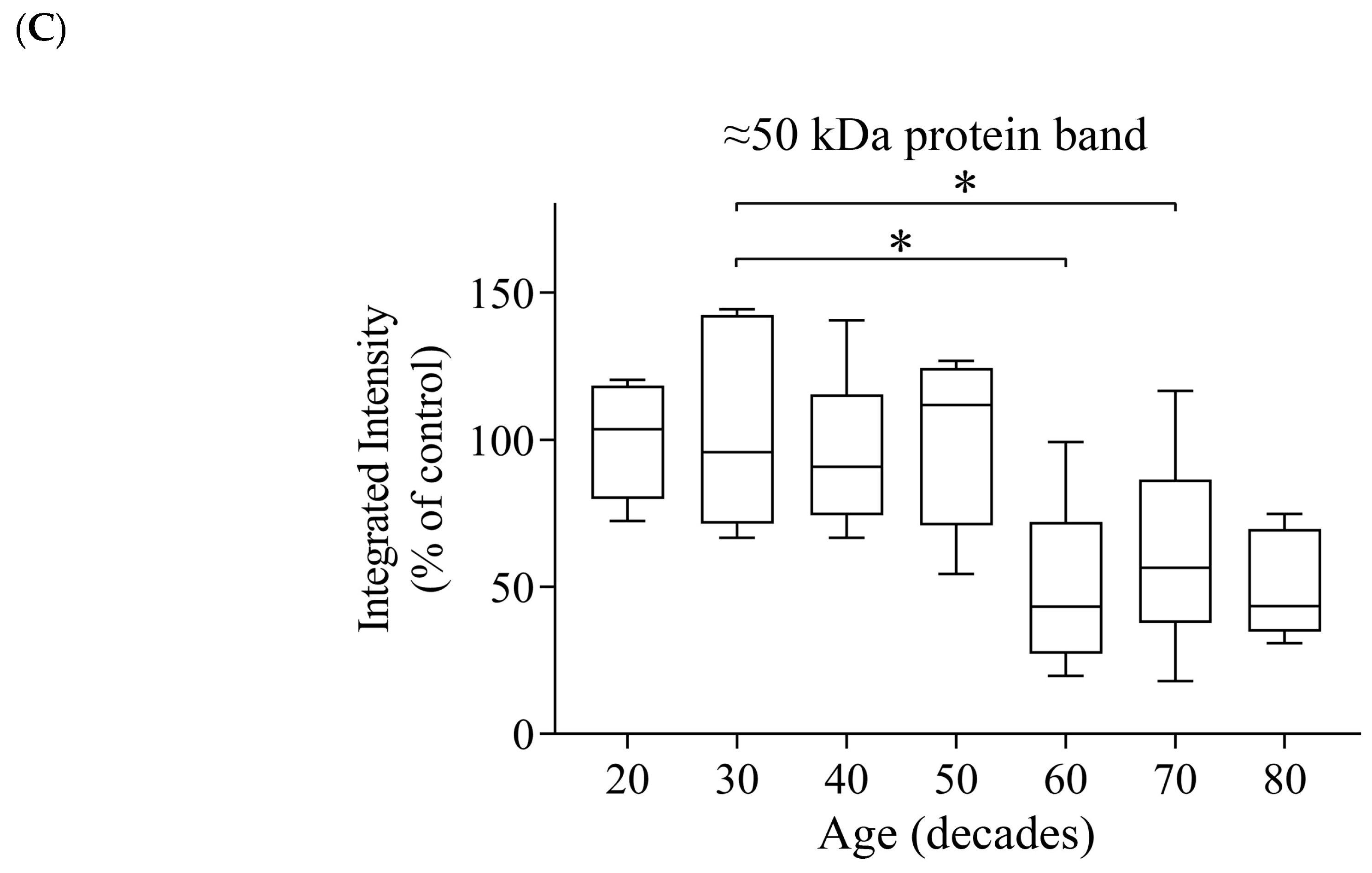
3.4. Reductions of α- and β-Tubulin Protein Levels during Ageing
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Connor, J.P.; Haber, P.S.; Hall, W.D. Alcohol use disorders. Lancet 2016, 387, 988–998. [Google Scholar] [CrossRef] [Green Version]
- Rehm, J.; Mathers, C.; Popova, S.; Thavorncharoensap, M.; Teerawattananon, Y.; Patra, J. Global burden of disease and injury and economic cost attributable to alcohol use and alcohol-use disorders. Lancet 2009, 373, 2223–2233. [Google Scholar] [CrossRef]
- Global Status Report on Alcohol and Health 2014. World Health Organization: Geneva, Switzerland, 2014. Available online: http://www.who.int/substance_abuse/publications/global_alcohol_report/en/ (accessed on 8 May 2014).
- Harper, C. The neurotoxicity of alcohol. Hum. Exp. Toxicol. 2007, 26, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Brust, J.C. Ethanol and cognition: Indirect effects, neurotoxicity and neuroprotection: A review. Int. J. Environ. Res. Public Health 2010, 7, 1540–1557. [Google Scholar] [CrossRef] [PubMed]
- Kandel, D.B.; Huang, F.Y.; Davies, M. Comorbidity between patterns of substance use dependence and psychiatric syndromes. Drug Alcohol Depend. 2001, 64, 233–241. [Google Scholar] [CrossRef]
- Gilpin, N.W.; Koob, G.F. Neurobiology of alcohol dependence: Focus on motivational mechanisms. Alcohol Res. Health 2008, 31, 185–195. [Google Scholar] [PubMed]
- Erdozain, A.M.; Callado, L.F. Neurobiological alterations in alcohol addiction: A review. Adicciones 2014, 26, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Harper, C. The neuropathology of alcohol-related brain damage. Alcohol Alcohol. 2009, 44, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Erdozain, A.M.; Morentin, B.; Bedford, L.; King, E.; Tooth, D.; Brewer, C.; Wayne, D.; Johnson, L.; Gerdes, H.K.; Wigmore, P.; et al. Alcohol-related brain damage in humans. PLoS ONE 2014, 9, e93586. [Google Scholar] [CrossRef] [PubMed]
- De la Monte, S.M.; Kril, J.J. Human alcohol-related neuropathology. Acta Neuropathol. 2014, 127, 71–90. [Google Scholar] [CrossRef] [PubMed]
- Jensen, G.B.; Pakkenberg, B. Do alcoholics drink their neurons away? Lancet 1993, 342, 1201–1204. [Google Scholar] [PubMed]
- Kril, J.J.; Halliday, G.M.; Svoboda, M.D.; Cartwright, H. The cerebral cortex is damaged in chronic alcoholics. Neuroscience 1997, 79, 983–998. [Google Scholar] [CrossRef]
- Harper, C. The neuropathology of alcohol-specific brain damage, or does alcohol damage the brain? J. Neuropathol. Exp. Neurol. 1998, 57, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Kril, J.J.; Halliday, G.M. Brain shrinkage in alcoholics: A decade on and what have we learned? Prog. Neurobiol. 1999, 58, 381–387. [Google Scholar] [CrossRef]
- Skuja, S.; Groma, V.; Smane, L. Alcoholism and cellular vulnerability in different brain regions. Ultrastruct. Pathol. 2012, 36, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Whittom, A.; Villarreal, A.; Soni, M.; Owusu-Duku, B.; Meshram, A.; Rajkowska, G.; Stockmeier, C.A.; Miguel-Hidalgo, J.J. Markers of apoptosis induction and proliferation in the orbitofrontal cortex in alcohol dependence. Alcohol. Clin. Exp. Res. 2014, 38, 2790–2799. [Google Scholar] [CrossRef] [PubMed]
- Harper, C.G.; Kril, J.J. Neuropathology of alcoholism. Alcohol Alcohol. 1990, 25, 207–216. [Google Scholar] [CrossRef]
- Pfefferbaum, A.; Sullivan, E.V.; Mathalon, D.H.; Lim, K.O. Frontal lobe volume loss observed with magnetic resonance imaging in older chronic alcoholics. Alcohol. Clin. Exp. Res. 1997, 21, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Kubota, M.; Nakazaki, S.; Hirai, S.; Saeki, N.; Yamaura, A.; Kusaka, T. Alcohol consumption and frontal lobe shrinkage: Study of 1432 non-alcoholic subjects. J. Neurol. Neurosurg. Psychiatry 2001, 71, 104–106. [Google Scholar] [CrossRef] [PubMed]
- Miguel-Hidalgo, J.J.; Rajkowska, G. Comparison of prefrontal cell pathology between depression alcohol dependence. J. Psychiatr. Res. 2003, 37, 411–420. [Google Scholar] [CrossRef]
- Makris, N.; Oscar-Berman, M.; Jaffin, S.K.; Hodge, S.M.; Kennedy, D.N.; Caviness, V.S.; Marinkovic, K.; Breiter, H.C.; Gasic, G.P.; Harris, G.J. Decreased volume of the brain reward system in alcoholism. Biol. Psychiatry 2008, 64, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Momenan, R.; Steckler, L.E.; Saad, Z.S.; van Rafelghem, S.; Kerich, M.J.; Hommer, D.W. Effects of alcohol dependence on cortical thickness as determined by magnetic resonance imaging. Psychiatry Res. 2012, 204, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Monnig, M.A.; Tonigan, J.S.; Yeo, R.A.; Thoma, R.J.; McCrady, B.S. White matter volume in alcohol use disorders: A meta-analysis. Addict. Biol. 2013, 18, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Xiao, P.; Dai, Z.; Zhong, J.; Zhu, Y.; Shi, H.; Pan, P. Regional gray matter deficits in alcohol dependence: A meta-analysis of voxel-based morphometry studies. Drug Alcohol Depend. 2015, 153, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Tian, F.; Zhang, H.; Zeng, J.; Chen, T.; Wang, S.; Jia, Z.; Gong, Q. Cortical and subcortical gray matter shrinkage in alcohol-use disorders: A voxel-based meta-analysis. Neurosci. Biobehav. Rev. 2016, 66, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Martindale, S.L.; Hurley, R.A.; Taber, K.H. Chronic alcohol use and sleep homeostasis: Risk factors and neuroimaging of recovery. J. Neuropsychiatry Clin. Neurosci. 2017, 29, A6-5. [Google Scholar] [CrossRef] [PubMed]
- Durazzo, T.C.; Gazdzinski, S.; Meyerhoff, D.J. The neurobiological and neurocognitive consequences of chronic cigarette smoking in alcohol use disorders. Alcohol Alcohol. 2007, 42, 174–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fowler, A.K.; Thompson, J.; Chen, L.; Dagda, M.; Dertien, J.; Dossou, K.S.; Moaddel, R.; Bergeson, S.E.; Kruman, I.I. Differential sensitivity of prefrontal cortex and hippocampus to alcohol-induced toxicity. PLoS ONE 2014, 9, e106945. [Google Scholar] [CrossRef] [PubMed]
- Crews, F.T.; Nixon, K. Mechanisms of neurodegeneration and regeneration in alcoholism. Alcohol Alcohol. 2009, 44, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Pfefferbaum, A.; Rosenbloom, M.J.; Chu, W.; Sassoon, S.A.; Rohlfing, T.; Pohl, K.M.; Zahr, N.M.; Sullivan, E.V. White matter microstructural recovery with abstinence and decline with relapse in alcohol dependence interacts with normal ageing: A controlled longitudinal DTI study. Lancet Psychiatry 2014, 1, 202–212. [Google Scholar] [CrossRef]
- Durazzo, T.C.; Mon, A.; Gazdzinski, S.; Yeh, P.H.; Meyerhoff, D.J. Serial longitudinal magnetic resonance imaging data indicate non-linear regional gray matter volume recovery in abstinent alcohol-dependent individuals. Addict. Biol. 2015, 20, 956–967. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.K.; Cohen, J.D. An integrative theory of prefrontal cortex function. Annu. Rev. Neurosci. 2001, 24, 167–202. [Google Scholar] [CrossRef] [PubMed]
- Putzke, J.; De Beun, R.; Schreiber, R.; De Vry, J.; Tölle, T.R.; Zieglgänsberger, W.; Spanagel, R. Long-term alcohol self-administration and alcohol withdrawal differentially modulate microtubule-associated protein 2 (MAP2) gene expression in the rat brain. Brain Res. Mol. Brain Res. 1998, 62, 196–205. [Google Scholar] [CrossRef]
- Evrard, S.G.; Brusco, A. Ethanol effects on the cytoskeleton of nerve tissue cells. In Cytoskeleton of the Nervous System; Springer Science Press: Berlin/Heidelberg, Germany, 2011; Chapter 29; pp. 697–758. [Google Scholar]
- Erdozain, A.M.; Rubio, M.; Valdizan, E.M.; Pazos, A.; Meana, J.J.; Fernández-Ruiz, J.; Alexander, S.P.; Callado, L.F. The endocannabinoid system is altered in the post-mortem prefrontal cortex of alcoholic subjects. Addict. Biol. 2015, 20, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Kharbanda, K.K.; Rogers, D.D., II; Mailliard, M.E.; Siford, G.L.; Barak, A.J.; Beckenhauer, H.C.; Sorrell, M.F.; Tuma, D.J. Role of elevated S-adenosylhomocysteine in rat hepatocyte apoptosis: Protection by betaine. Biochem. Pharmacol. 2005, 70, 1883–1890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leggate, M.; Carter, W.G.; Evans, M.J.; Vennard, R.A.; Sribala-Sundaram, S.; Nimmo, M.A. Determination of inflammatory and prominent proteomic changes in plasma and adipose tissue after high-intensity intermittent training in overweight and obese males. J. Appl. Physiol. 2012, 112, 1353–1360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carter, W.G.; Vigneswara, V.; Newlaczyl, A.; Wayne, D.; Ahmed, B.; Saddington, S.; Brewer, C.; Raut, N.; Gerdes, H.K.; Erdozain, A.M.; et al. Isoaspartate, carbamoyl phosphate synthase-1, and carbonic anhydrase-III as biomarkers of liver injury. Biochem. Biophys. Res. Commun. 2015, 458, 626–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludueña, R.F. A hypothesis on the origin and evolution of tubulin. Int. Rev. Cell Mol. Biol. 2013, 302, 41–185. [Google Scholar] [PubMed]
- Conde, C.; Caceres, A. Microtubule assembly, organization and dynamics in axons and dendrites. Nat. Rev. Neurosci. 2009, 10, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Ludueña, R.F. Multiple forms of tubulin: Different gene products and covalent modifications. Int. Rev. Cytol. 1998, 178, 207–275. [Google Scholar] [PubMed]
- Leandro-García, L.J.; Leskelä, S.; Landa, I.; Montero-Conde, C.; López-Jiménez, E.; Letón, R.; Cascón, A.; Robledo, M.; Rodríguez-Antona, C. Tumoral and tissue-specific expression of the major human beta-tubulin isotypes. Cytoskeleton 2010, 67, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Verhey, K.J.; Gaertig, J. The tubulin code. Cell Cycle 2007, 6, 2152–2160. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Brady, S.T. Post-translational modifications of tubulin: Pathways to functional diversity of microtubules. Trend Cell Biol. 2015, 25, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Dehmelt, L.; Halpain, S. The MAP2/Tau family of microtubule-associated proteins. Genome Biol. 2005, 6, 204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez, C.; Díaz-Nido, J.; Avila, J. Phosphorylation of microtubule-associated protein 2 (MAP2) and its relevance for the regulation of the neuronal cytoskeleton function. Prog. Neurobiol. 2000, 61, 133–168. [Google Scholar] [CrossRef]
- Lee, V.M.; Goedert, M.; Trojanowski, J.Q. Neurodegenerative tauopathies. Annu. Rev. Neurosci. 2001, 24, 1121–1159. [Google Scholar] [CrossRef] [PubMed]
- Noraberg, J.; Zimmer, J. Ethanol induces MAP2 changes in organotypic hippocampal slice cultures. Neuroreport 1998, 9, 3177–3182. [Google Scholar] [CrossRef] [PubMed]
- Tagliaferro, P.; Vega, M.D.; Evrard, S.G.; Ramos, A.J.; Brusco, A. Alcohol exposure during adulthood induces neuronal and astroglial alterations in the hippocampal CA-1 area. Ann. N. Y. Acad. Sci. 2002, 965, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Romero, A.M.; Esteban-Pretel, G.; Marín, M.P.; Ponsoda, X.; Ballestín, R.; Canales, J.J.; Renau-Piqueras, J. Chronic ethanol exposure alters the levels, assembly, and cellular organization of the actin cytoskeleton and microtubules in hippocampal neurons in primary culture. Toxicol. Sci. 2010, 118, 602–612. [Google Scholar] [CrossRef] [PubMed]
- Harada, A.; Oguchi, K.; Okabe, S.; Kuno, J.; Terada, S.; Ohshima, T.; Sato-Yoshitake, R.; Takei, Y.; Noda, T.; Hirokawa, N. Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature 1994, 369, 488–491. [Google Scholar] [CrossRef] [PubMed]
- Harada, A.; Teng, J.; Takei, Y.; Oguchi, K.; Hirokawa, N. MAP2 is required for dendrite elongation, PKA anchoring in dendrites, and proper PKA signal transduction. J. Cell. Biol. 2002, 158, 541–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.F.; Yao, T.P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhang, Y.; Shan, B. Analysis of expression and functions of histone deacetylase 6 (HDAC6). Methods Mol. Biol. 2016, 1436, 85–94. [Google Scholar] [PubMed]
- Skultetyova, L.; Ustinova, K.; Kutil, Z.; Novakova, Z.; Pavlicek, J.; Mikesova, J.; Trapl, D.; Baranova, P.; Havlinova, B.; Hubalek, M.; et al. Human histone deacetylase 6 shows strong preference for tubulin dimers over assembled microtubules. Sci. Rep. 2017, 7, 11547. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.L.; Jennett, R.B.; Sorrell, M.F.; Tuma, D.J. Acetaldehyde substoichiometrically inhibits bovine neurotubulin polymerization. J. Clin. Investig. 1989, 84, 337–341. [Google Scholar] [CrossRef]
- Lee, J.Y.; Koga, H.; Kawaguchi, Y.; Tang, W.; Wong, E.; Gao, Y.S.; Pandey, U.B.; Kaushik, S.; Tresse, E.; Lu, J.; et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 2010, 29, 969–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simões-Pires, C.; Zwick, V.; Nurisso, A.; Schenker, E.; Carrupt, P.; Cuendet, M. HDAC6 as a target for neurodegenerative diseases: What makes it different from the other HDACs? Mol. Neurodegener. 2013, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Lewohl, J.M.; Van Dyk, D.D.; Craft, G.E.; Innes, D.J.; Mayfield, R.D.; Cobon, G.; Harris, R.A.; Dodd, P.R. The application of proteomics to the human alcoholic brain. Ann. N. Y. Acad. Sci. 2004, 1025, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.J.; Butler, T.R.; Prendergast, M.A. Ethanol impairs microtubule formation via interactions at a microtubule associated protein-sensitive site. Alcohol 2013, 47, 539–543. [Google Scholar] [CrossRef] [Green Version]
- Najbauer, J.; Orpiszewski, J.; Aswad, D.W. Molecular aging of tubulin: Accumulation of isoaspartyl sites in vitro and in vivo. Biochemistry 1996, 35, 5183–5190. [Google Scholar] [CrossRef] [PubMed]
- Vigneswara, V.; Lowenson, J.D.; Powell, C.D.; Thakur, M.; Bailey, K.; Clarke, S.; Ray, D.E.; Carter, W.G. Proteomic identification of novel substrates of a protein isoaspartyl methyltransferase repair enzyme. J. Biol. Chem. 2006, 281, 32619–32629. [Google Scholar] [CrossRef] [PubMed]
- Bjork, J.M.; Gilman, J.M. The effects of acute alcohol administration on the human brain: Insights from neuroimaging. Neuropharmacology 2014, 84, 101–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walhovd, K.B.; Fjell, A.M.; Reinvang, I.; Lundervold, A.; Dale, A.M.; Eilertsen, D.E.; Quinn, B.T.; Salat, D.; Makris, N.; Fischl, B. Effects of age on volumes of cortex, white matter and subcortical structures. Neurobiol. Aging 2005, 26, 1261–1270, discussion 1275–1268. [Google Scholar] [CrossRef] [PubMed]
- Raz, N.; Gunning, F.M.; Head, D.; Dupuis, J.H.; McQuain, J.; Briggs, S.D.; Loken, W.J.; Thornton, A.E.; Acker, J.D. Selective aging of the human cerebral cortex observed in vivo: Differential vulnerability of the prefrontal gray matter. Cereb. Cortex 1997, 7, 268–282. [Google Scholar] [CrossRef] [PubMed]
- Tisserand, D.J.; Van Boxtel, M.P.; Pruessner, J.C.; Hofman, P.; Evans, A.C.; Jolles, J. A voxel-based morphometric study to determine individual differences in gray matter density associated with age and cognitive change over time. Cereb. Cortex 2004, 14, 966–973. [Google Scholar] [CrossRef] [PubMed]
- Cowell, P.E.; Sluming, V.A.; Wilkinson, I.D.; Cezayirli, E.; Romanowski, C.A.; Webb, J.A.; Keller, S.S.; Mayes, A.; Roberts, N. Effects of sex and age on regional prefrontal brain volume in two human cohorts. European. J. Neurosci. 2007, 25, 307–318. [Google Scholar]
- Farokhian, F.; Yang, C.; Beheshti, I.; Matsuda, H.; Wu, S. Age-related gray and white matter changes in normal adult brains. Aging Dis. 2017, 8, 899–909. [Google Scholar] [CrossRef] [PubMed]
- Guggenmos, M.; Schmack, K.; Sekutowicz, M.; Garbusow, M.; Sebold, M.; Sommer, C.; Smolka, M.N.; Wittchen, H.U.; Zimmermann, U.S.; Heinz, A.; et al. Quantitative neurobiological evidence for accelerated brain aging in alcohol dependence. Trans. Psychiatry 2017, 7, 1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicholls, C.; Li, H.; Liu, J.P. GAPDH: A common enzyme with uncommon functions. Clin. Exp. Pharmacol. Physiol. 2012, 39, 674–679. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.C.; Berkelman, T.; Yadav, G.; Hammond, M. A defined methodology for reliable quantification of Western blot data. Mol. Biotechnol. 2013, 55, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Giorgio, A.; Santelli, L.; Tomassini, V.; Bosnell, R.; Smith, S.; De Stefano, N.; Johansen-Berg, H. Age-related changes in grey and white matter structure throughout adulthood. Neuroimage 2010, 51, 943–951. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case | Gender (F/M) | Age (Years) | PMD (h) | Psychiatric Diagnose | Etiology of Death | Cause of Death | Ethanol in Blood (mg/mL) | Other Drugs in Blood |
|---|---|---|---|---|---|---|---|---|
| C1 | F | 66 | 15 | Control | Accidental | Run over | 0 | (−) |
| A1 | F | 51 | 8 | Alcoholism | Natural | CRF | 2.98 | Nordiazepam |
| S1 | F | 58 | 48 | Major Depression | Suicidal | Drowning | 0 | (−) |
| SA1 | F | 57 | 24 | Alcoholism | Suicidal | Drug intoxication | 0.23 | Methanol |
| C2 | M | 71 | 19 | Control | Natural | CRF | 0 | Nordiazepam |
| A2 | M | 68 | 15 | Alcoholism | Natural | CRF | 0 | (−) |
| S2 | M | 65 | 19 | Obsessive Disorder | Suicidal | Jumping | 0 | Nordiazepam |
| SA2 | M | 71 | 19 | Alcoholism | Suicidal | Hanging | 0.55 | (−) |
| C3 | M | 48 | 7 | Control | Accidental | Traffic | 0 | (−) |
| A3 | M | 50 | 24 | Alcoholism | Natural | CRF | 0 | (−) |
| S3 | M | 50 | 6 | Bipolar Disorder | Suicidal | Jumping | 0 | Diazepam |
| SA3 | M | 50 | 3 | Alcoholism | Suicidal | Hanging | 0 | (−) |
| C4 | M | 52 | 18 | Control | Accidental | Crushed | 0 | (−) |
| A4 | M | 57 | 12 | Alcoholism | Natural | CRF | 3.37 | (−) |
| S4 | M | 59 | 29 | Major Depression | Suicidal | Hanging | 0 | Citalopram, Nordiazepam |
| SA4 | M | 55 | 24 | Alcoholism | Suicidal | Drug intoxication | 2.4 | (−) |
| C5 | M | 40 | 18 | Control | Accidental | Jumping | 0.56 | (−) |
| A5 | M | 43 | 4 | Alcoholism | Natural | CRF | 1.64 | Chlormethiazole, Metamizole |
| S5 | M | 44 | 15 | Anxiety Disorder | Suicidal | Hanging | 0 | (−) |
| SA5 | M | 40 | 21 | Alcoholism | Suicidal | Run over | 0 | (−) |
| C6 | F | 36 | 9 | Control | Natural | Heart failure | 0 | (−) |
| A6 | F | 43 | 35 | Alcoholism | Natural | Hemorrhage | 0 | Metamizole, Fluoxetine |
| S6 | F | 48 | 9 | Anxiety Disorder | Suicidal | Jumping | 0 | (−) |
| SA6 | F | 50 | 6 | Alcoholism | Suicidal | Jumping | 0 | Hydrochlorothiazide |
| C7 | M | 66 | 50 | Control | Accidental | Traffic | 0 | (−) |
| A7 | M | 71 | 17 | Alcoholism | Accidental | Traffic | 0.35 | (−) |
| S7 | M | 67 | 8 | Major Depression | Suicidal | Sharp weapon | 0 | (−) |
| SA7 | M | 69 | 24 | Alcoholism | Suicidal | Jumping | 0.38 | (−) |
| C8 | M | 37 | 21 | Control | Accidental | Cranoencephalic trauma | 0 | (−) |
| A8 | M | 39 | 19 | Alcoholism | Natural | Hemorrhage | 0.44 | (−) |
| S8 | M | 40 | 15 | Personality Disorder | Suicidal | Jumping | 0 | Diazepam |
| SA8 | M | 37 | 68 | Alcoholism | Suicidal | Hanging | 0.9 | Diazepam |
| C9 | M | 54 | 23 | Control | Accidental | Jumping | 0 | (−) |
| A9 | M | 53 | 12 | Alcoholism | Natural | Suffocation | 0 | (−) |
| S9 | M | 50 | 29 | Major Depression | Suicidal | Hanging | 0 | Diazepam |
| SA9 | M | 50 | 70 | Alcoholism | Suicidal | Hanging | 2.93 | (−) |
| C10 | M | 42 | 27 | Control | Accidental | Traffic | 0 | (−) |
| A10 | M | 42 | 20 | Alcoholism | Natural | Hemorrhage | 0 | (−) |
| S10 | M | 41 | 50 | Major Depression | Suicidal | Hanging | 0 | (−) |
| SA10 | M | 41 | 78 | Alcoholism | Suicidal | Hanging | 3.4 | (−) |
| C11 | M | 47 | 26 | Control | Accidental | Work accident | 0 | (−) |
| A11 | M | 46 | 16 | Alcoholism | Accidental | Suffocation | 0.97 | (−) |
| S11 | M | 46 | 47 | Anxiety Disorder | Suicidal | Hanging | 0 | (−) |
| SA11 | M | 44 | 5 | Alcoholism | Suicidal | Jumping | 2.47 | Diazepam |
| Controls | Alcoholic Subjects | Suicide Subjects | Suicide Alcoholic Subjects | |
|---|---|---|---|---|
| Age (years) | 50 ± 4 | 51 ± 3 | 52 ± 3 | 51 ± 3 |
| PMD (h) | 21 ± 3 | 17 ± 3 | 25 ± 5 | 31 ± 8 |
| Gender (F/M) | 2F/9M | 2F/9M | 2F/9M | 2F/9M |
| Case | Gender (F/M) | Age (Years) | PMD (h) | Cause of Death |
|---|---|---|---|---|
| 1 | M | 21 | 15 | Accident/Traffic |
| 2 | F | 22 | 24 | Accident/Fall from height |
| 3 | M | 23 | 17 | Accident/Work |
| 4 | M | 24 | 20 | Accident/Traffic |
| 5 | F | 26 | 5 | Accident/Traffic |
| 6 | M | 28 | 5 | Accident/Fall from height |
| 7 | M | 31 | 13 | Accident/Traffic |
| 8 | M | 32 | 27 | Accident/Traffic |
| 9 | M | 33 | 13 | Accident/Traffic |
| 10 | M | 36 | 18 | Accident/Work |
| 11 | M | 36 | 23 | Accident/Work |
| 12 | F | 38 | 22 | Accident/Traffic |
| 13 | M | 41 | 14 | Natural/Heart Attack |
| 14 | M | 43 | 10 | Accident/Traffic |
| 15 | F | 43 | 28 | Accident/Train |
| 16 | F | 44 | 9 | Natural/CRF |
| 17 | M | 47 | 15 | Accident/Traffic |
| 18 | F | 48 | 9 | Accident/Fall from height |
| 19 | F | 50 | 11 | Natural/CRF |
| 20 | F | 50 | 11 | Natural/CRF |
| 21 | M | 51 | 18 | Accident/Traffic |
| 22 | M | 54 | 24 | Accident/Traffic |
| 23 | F | 57 | 14 | Natural/CRF |
| 24 | M | 58 | 16 | Accident/Traffic |
| 25 | F | 60 | 27 | Accident/Traffic |
| 26 | M | 61 | 23 | Accident/Traffic |
| 27 | M | 62 | 19 | Accident/Work |
| 28 | F | 64 | 19 | Natural/CRF |
| 29 | F | 66 | 20 | Natural/CRF |
| 30 | M | 66 | 18 | Natural/Tumor |
| 31 | F | 70 | 7 | Accident/Traffic |
| 32 | M | 71 | 21 | Natural/CRF |
| 33 | M | 73 | 19 | Accident/Traffic |
| 34 | F | 74 | 21 | Accident/Traffic |
| 35 | F | 75 | 18 | Accident/Traffic |
| 36 | M | 78 | 29 | Accident/Traffic |
| 37 | F | 81 | 14 | Accident/Traffic |
| 38 | M | 81 | 21 | Accident/Traffic |
| 39 | F | 84 | 18 | Natural/CRF |
| 40 | M | 85 | 19 | Natural/CRF |
| 41 | M | 85 | 10 | Natural/CRF |
| 42 | F | 86 | 18 | Natural/CRF |
| α-Tubulin | β-Tubulin | GAPDH | |
|---|---|---|---|
| All subjects | 42 | 42 | 42 |
| Spearman’s Correlation Coefficient | −0.225 | −0.350 * | 0.068 |
| Significance (two-tailed) | 0.153 | 0.023 | 0.67 |
| Male Subjects | 24 | 24 | 24 |
| Spearman’s Correlation Coefficient | −0.107 | −0.247 | 0.303 |
| Significance (two-tailed) | 0.617 | 0.244 | 0.15 |
| Female Subjects | 18 | 18 | 18 |
| Spearman’s Correlation Coefficient | −0.392 | −0.510 * | −0.274 |
| Significance (two-tailed) | 0.107 | 0.031 | 0.272 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Labisso, W.L.; Raulin, A.-C.; Nwidu, L.L.; Kocon, A.; Wayne, D.; Erdozain, A.M.; Morentin, B.; Schwendener, D.; Allen, G.; Enticott, J.; et al. The Loss of α- and β-Tubulin Proteins Are a Pathological Hallmark of Chronic Alcohol Consumption and Natural Brain Ageing. Brain Sci. 2018, 8, 175. https://doi.org/10.3390/brainsci8090175
Labisso WL, Raulin A-C, Nwidu LL, Kocon A, Wayne D, Erdozain AM, Morentin B, Schwendener D, Allen G, Enticott J, et al. The Loss of α- and β-Tubulin Proteins Are a Pathological Hallmark of Chronic Alcohol Consumption and Natural Brain Ageing. Brain Sciences. 2018; 8(9):175. https://doi.org/10.3390/brainsci8090175
Chicago/Turabian StyleLabisso, Wajana L., Ana-Caroline Raulin, Lucky L. Nwidu, Artur Kocon, Declan Wayne, Amaia M. Erdozain, Benito Morentin, Daniela Schwendener, George Allen, Jack Enticott, and et al. 2018. "The Loss of α- and β-Tubulin Proteins Are a Pathological Hallmark of Chronic Alcohol Consumption and Natural Brain Ageing" Brain Sciences 8, no. 9: 175. https://doi.org/10.3390/brainsci8090175