Clinical and Functional Characterization of the Recurrent TUBA1A p.(Arg2His) Mutation

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Homology Modelling
2.3. Expression Construct Mutagenesis and Cell Culture
2.4. Immunocytochemistry
2.5. Predicting the Probability of TUBA1A Substitutions
3. Results
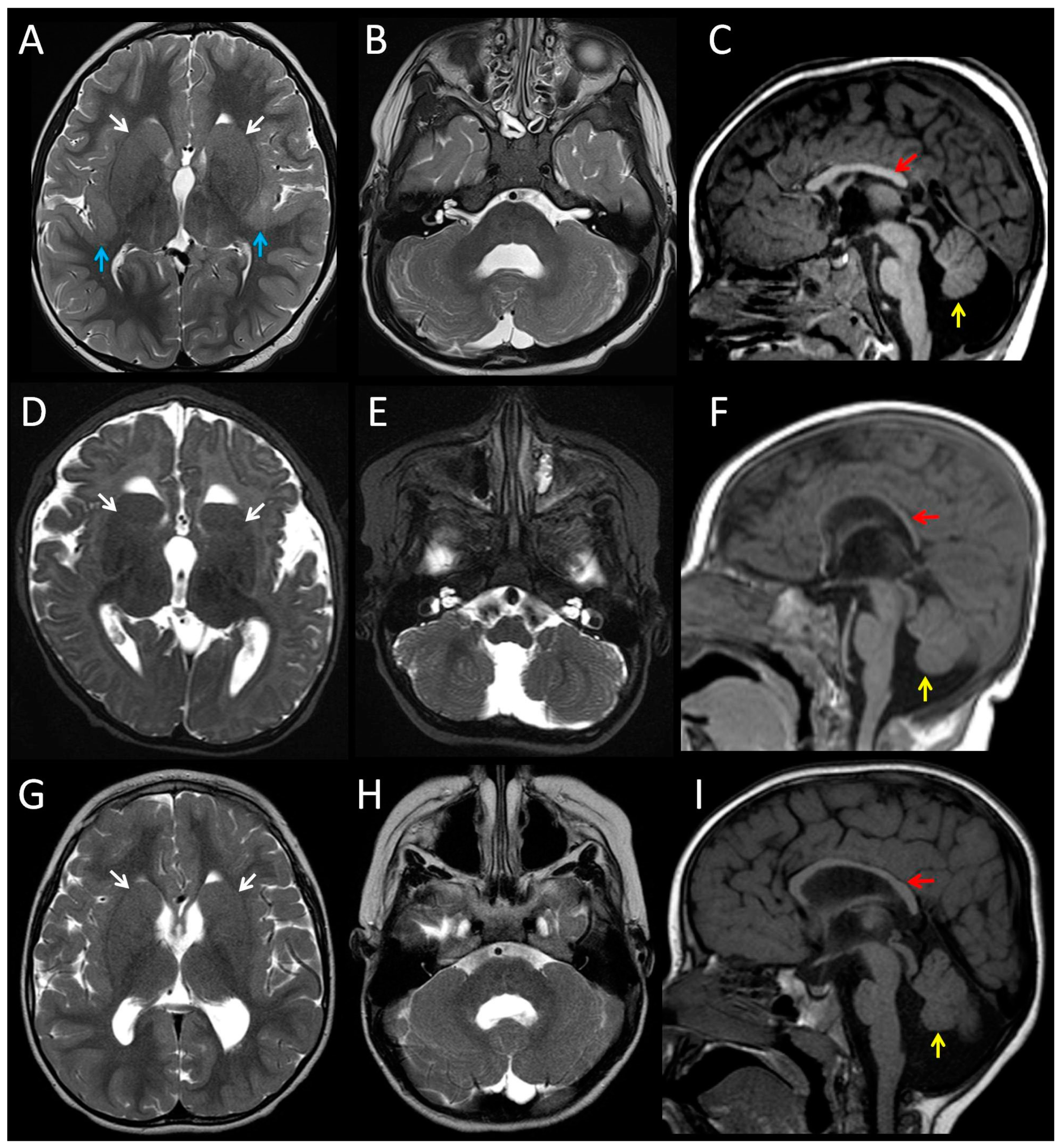
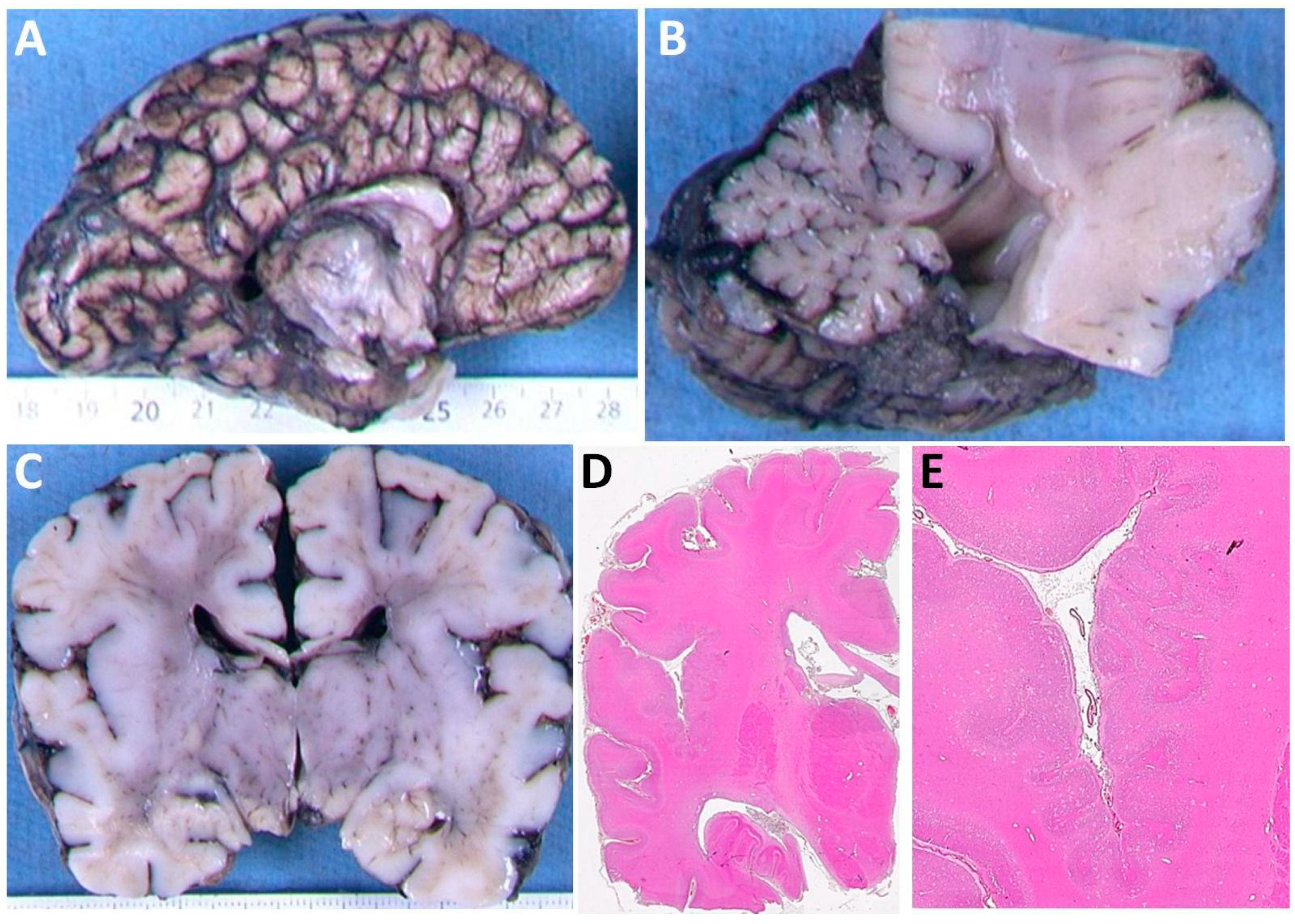
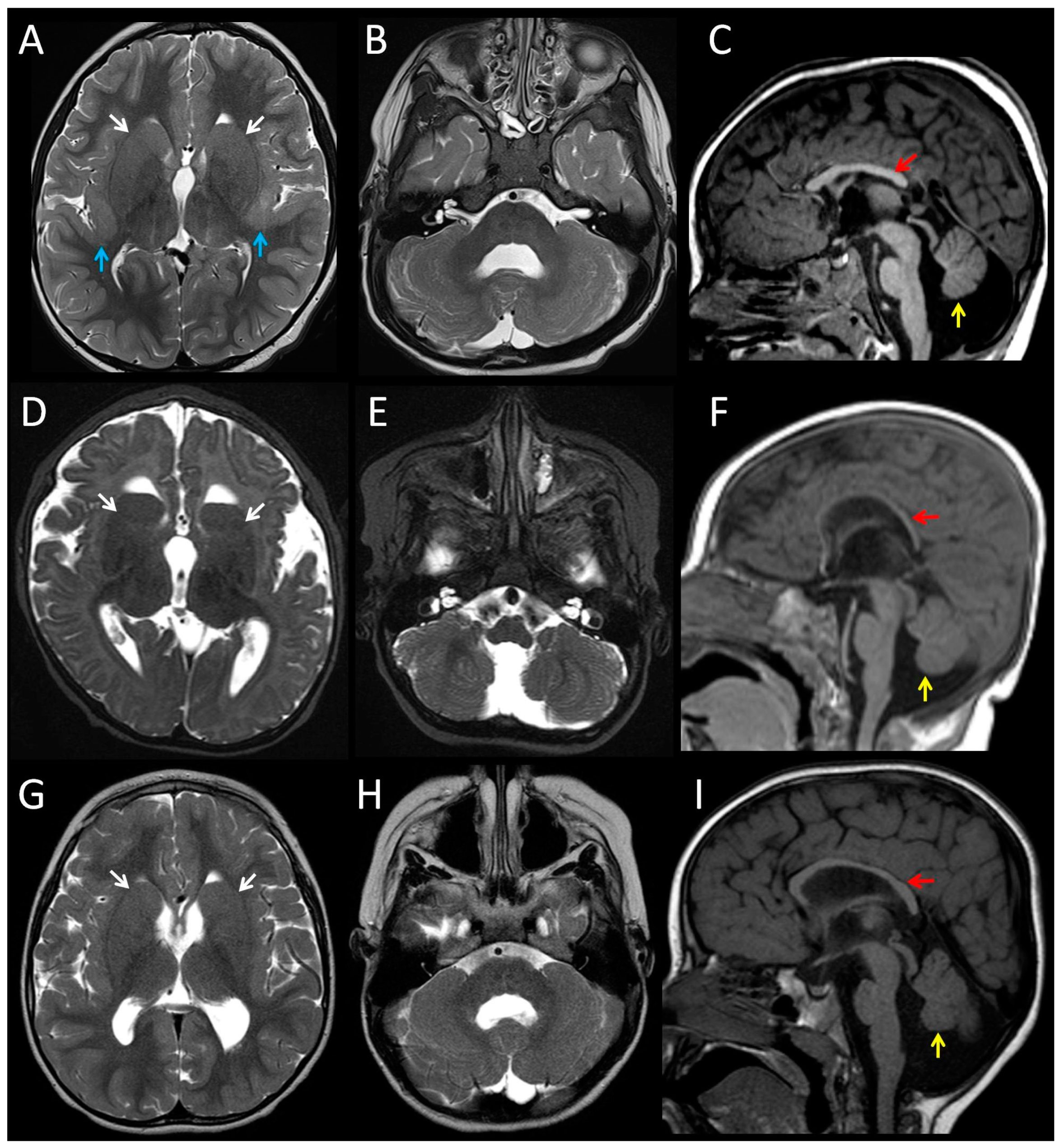
3.1. Clinical Features of Patients with the p.(Arg2His) Mutation
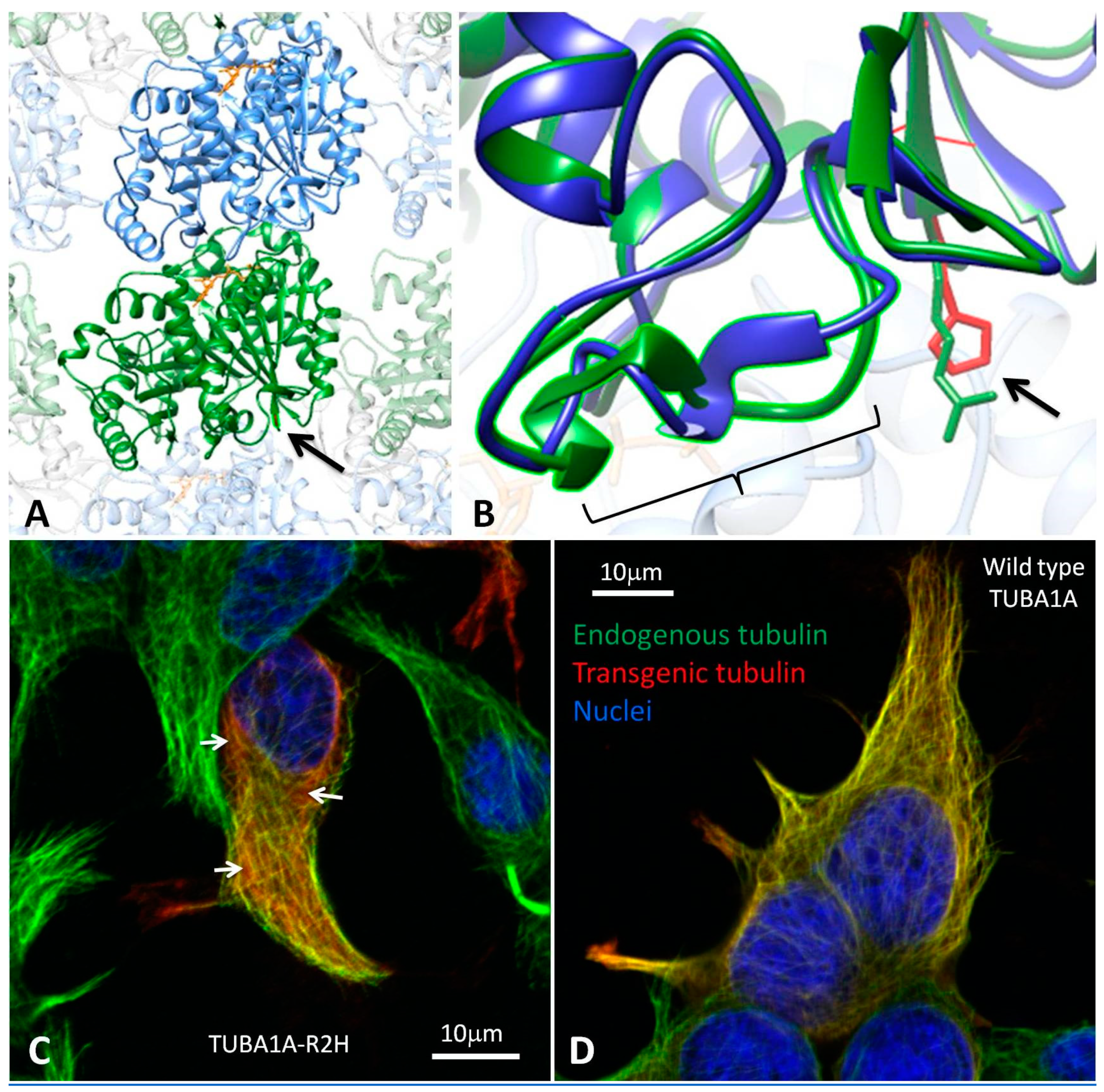
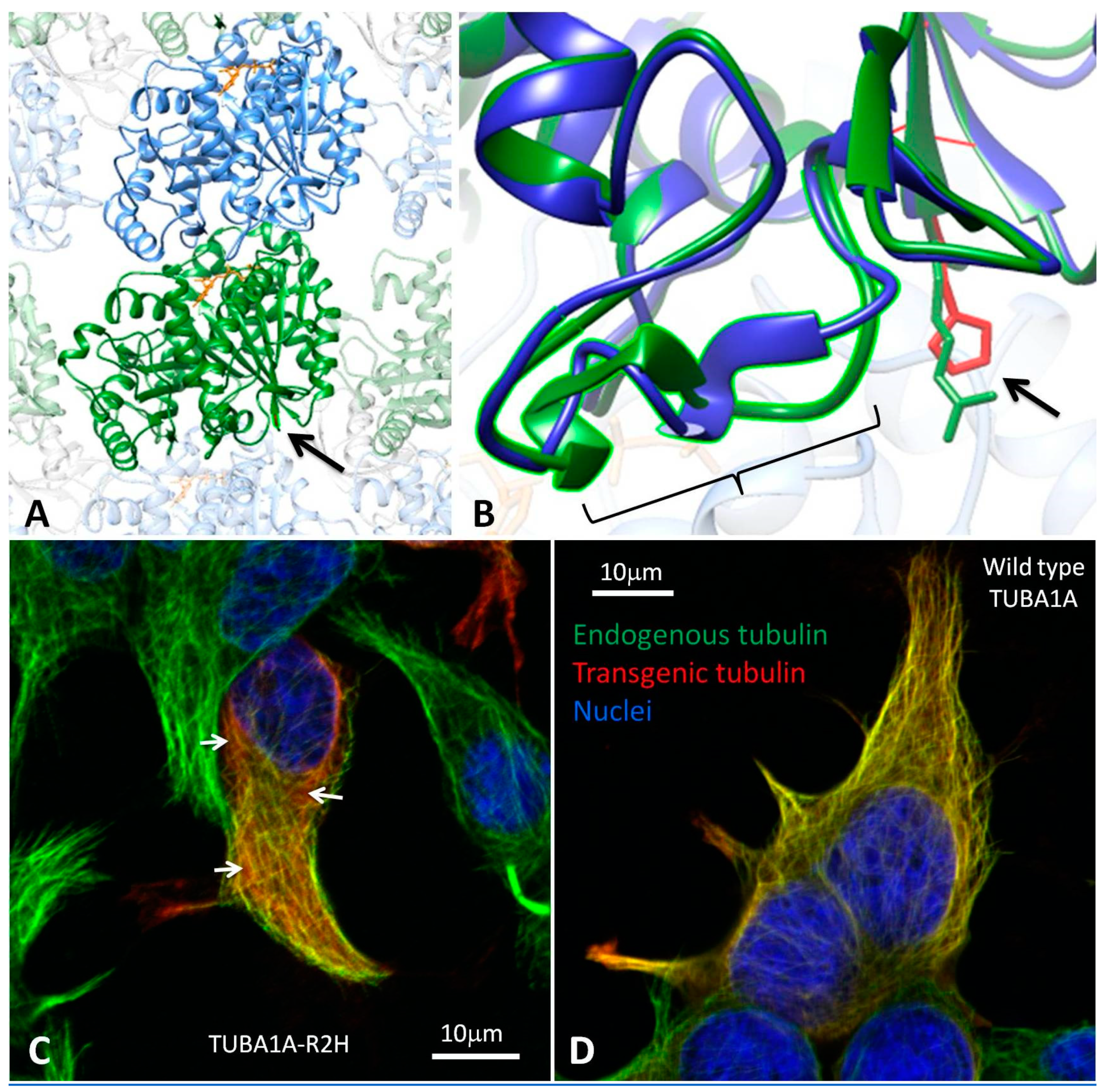
3.2. Modelling the Structural Effects of p.Arg2His
3.3. Heterologous Expression of TUBA1A-R2H in HEK-293 cells
3.4. Substitution Probability of Recurrent TUBA1A Mutations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gloster, A.; Wu, W.; Speelman, A.; Weiss, S.; Causing, C.; Pozniak, C.; Reynolds, B.; Chang, E.; Toma, J.G.; Miller, F.D. The T alpha 1 alpha-tubulin promoter specifies gene expression as a function of neuronal growth and regeneration in transgenic mice. J. Neurosci. 1994, 14, 7319–7330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bamji, S.X.; Miller, F.D. Comparison of the expression of a T alpha 1:nlacZ transgene and T alpha 1 alpha-tubulin mRNA in the mature central nervous system. J. Comp. Neurol. 1996, 374, 52–69. [Google Scholar] [CrossRef]
- Keays, D.A.; Tian, G.; Poirier, K.; Huang, G.-J.; Siebold, C.; Cleak, J.; Oliver, P.L.; Fray, M.; Harvey, R.J.; Molnár, Z.; et al. Mutations in alpha-tubulin cause abnormal neuronal migration in mice and lissencephaly in humans. Cell 2007, 128, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Jansen, A.C.; Oostra, A.; Desprechins, B.; De Vlaeminck, Y.; Verhelst, H.; Régal, L.; Verloo, P.; Bockaert, N.; Keymolen, K.; Seneca, S.; et al. TUBA1A mutations: from isolated lissencephaly to familial polymicrogyria. Neurology 2011, 76, 988–992. [Google Scholar] [CrossRef] [PubMed]
- Cushion, T.D.; Dobyns, W.B.; Mullins, J.G.L.; Stoodley, N.; Chung, S.-K.; Fry, A.E.; Hehr, U.; Gunny, R.; Aylsworth, A.S.; Prabhakar, P.; et al. Overlapping cortical malformations and mutations in TUBB2B and TUBA1A. Brain 2013, 136, 536–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bahi-Buisson, N.; Poirier, K.; Fourniol, F.; Saillour, Y.; Valence, S.; Lebrun, N.; Hully, M.; Bianco, C.F.; Boddaert, N.; Elie, C.; et al. The wide spectrum of tubulinopathies: what are the key features for the diagnosis? Brain 2014, 137, 1676–1700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanders, S.J.; Murtha, M.T.; Gupta, A.R.; Murdoch, J.D.; Raubeson, M.J.; Willsey, A.J.; Ercan-Sencicek, A.G.; DiLullo, N.M.; Parikshak, N.N.; Stein, J.L.; et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature 2012, 485, 237–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMichael, G.; Girirajan, S.; Moreno-De-Luca, A.; Gecz, J.; Shard, C.; Nguyen, L.S.; Nicholl, J.; Gibson, C.; Haan, E.; Eichler, E.; et al. Rare copy number variation in cerebral palsy. Eur. J. Hum. Genet. 2014, 22, 40–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yokoi, S.; Ishihara, N.; Miya, F.; Tsutsumi, M.; Yanagihara, I.; Fujita, N.; Yamamoto, H.; Kato, M.; Okamoto, N.; Tsunoda, T.; et al. TUBA1A mutation can cause a hydranencephaly-like severe form of cortical dysgenesis. Sci. Rep. 2015, 5, 15165. [Google Scholar] [CrossRef] [PubMed]
- Jaglin, X.H.; Poirier, K.; Saillour, Y.; Buhler, E.; Tian, G.; Bahi-Buisson, N.; Fallet-Bianco, C.; Phan-Dinh-Tuy, F.; Kong, X.P.; Bomont, P.; et al. Mutations in the beta-tubulin gene TUBB2B result in asymmetrical polymicrogyria. Nat. Genet. 2009, 41, 746–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tischfield, M.A.; Baris, H.N.; Wu, C.; Rudolph, G.; Van Maldergem, L.; He, W.; Chan, W.-M.; Andrews, C.; Demer, J.L.; Robertson, R.L.; et al. Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance. Cell 2010, 140, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Poirier, K.; Saillour, Y.; Bahi-Buisson, N.; Jaglin, X.H.; Fallet-Bianco, C.; Nabbout, R.; Castelnau-Ptakhine, L.; Roubertie, A.; Attie-Bitach, T.; Desguerre, I.; et al. Mutations in the neuronal β-tubulin subunit TUBB3 result in malformation of cortical development and neuronal migration defects. Hum. Mol. Genet. 2010, 19, 4462–4473. [Google Scholar] [CrossRef] [PubMed]
- Breuss, M.; Heng, J.I.-T.; Poirier, K.; Tian, G.; Jaglin, X.H.; Qu, Z.; Braun, A.; Gstrein, T.; Ngo, L.; Haas, M.; et al. Mutations in the β-tubulin gene TUBB5 cause microcephaly with structural brain abnormalities. Cell Rep. 2012, 2, 1554–1562. [Google Scholar] [CrossRef] [PubMed]
- Cushion, T.D.; Paciorkowski, A.R.; Pilz, D.T.; Mullins, J.G.L.; Seltzer, L.E.; Marion, R.W.; Tuttle, E.; Ghoneim, D.; Christian, S.L.; Chung, S.-K.; et al. De novo mutations in the beta-tubulin gene TUBB2A cause simplified gyral patterning and infantile-onset epilepsy. Am. J. Hum. Genet. 2014, 94, 634–641. [Google Scholar] [CrossRef] [PubMed]
- Poirier, K.; Lebrun, N.; Broix, L.; Tian, G.; Saillour, Y.; Boscheron, C.; Parrini, E.; Valence, S.; Pierre, B.S.; Oger, M.; et al. Mutations in TUBG1, DYNC1H1, KIF5C and KIF2A cause malformations of cortical development and microcephaly. Nat. Genet. 2013, 45, 639–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romaniello, R.; Arrigoni, F.; Panzeri, E.; Poretti, A.; Micalizzi, A.; Citterio, A.; Bedeschi, M.F.; Berardinelli, A.; Cusmai, R.; D’Arrigo, S.; et al. Tubulin-related cerebellar dysplasia: definition of a distinct pattern of cerebellar malformation. Eur. Radiol. 2017, 27, 5080–5092. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.A.; Pilz, D.T.; Babatz, T.D.; Cushion, T.D.; Harvey, K.; Topf, M.; Yates, L.; Robb, S.; Uyanik, G.; Mancini, G.M.S.; et al. TUBA1A mutations cause wide spectrum lissencephaly (smooth brain) and suggest that multiple neuronal migration pathways converge on alpha tubulins. Hum. Mol. Genet. 2010, 19, 2817–2827. [Google Scholar] [CrossRef] [PubMed]
- Bahi-Buisson, N.; Poirier, K.; Boddaert, N.; Saillour, Y.; Castelnau, L.; Philip, N.; Buyse, G.; Villard, L.; Joriot, S.; Marret, S.; et al. Refinement of cortical dysgeneses spectrum associated with TUBA1A mutations. J. Med. Genet. 2008, 45, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Poirier, K.; Keays, D.A.; Francis, F.; Saillour, Y.; Bahi, N.; Manouvrier, S.; Fallet-Bianco, C.; Pasquier, L.; Toutain, A.; Tuy, F.P.D.; et al. Large spectrum of lissencephaly and pachygyria phenotypes resulting from de novo missense mutations in tubulin alpha 1A (TUBA1A). Hum. Mutat. 2007, 28, 1055–1064. [Google Scholar] [CrossRef] [PubMed]
- Tian, G.; Kong, X.-P.; Jaglin, X.H.; Chelly, J.; Keays, D.; Cowan, N.J. A pachygyria-causing alpha-tubulin mutation results in inefficient cycling with CCT and a deficient interaction with TBCB. Mol. Biol. Cell 2008, 19, 1152–1161. [Google Scholar] [CrossRef] [PubMed]
- Farwell, K.D.; Shahmirzadi, L.; El-Khechen, D.; Powis, Z.; Chao, E.C.; Tippin Davis, B.; Baxter, R.M.; Zeng, W.; Mroske, C.; Parra, M.C.; et al. Enhanced utility of family-centered diagnostic exome sequencing with inheritance model-based analysis: results from 500 unselected families with undiagnosed genetic conditions. Genet. Med. 2015, 17, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Alby, C.; Malan, V.; Boutaud, L.; Marangoni, M.A.; Bessières, B.; Bonniere, M.; Ichkou, A.; Elkhartoufi, N.; Bahi-Buisson, N.; Sonigo, P.; et al. Clinical, genetic and neuropathological findings in a series of 138 fetuses with a corpus callosum malformation. Birth Defects Res. Part A Clin. Mol. Teratol. 2016, 106, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, S.; Cohen, J.S.; Vernon, H.; Barañano, K.; McClellan, R.; Jamal, L.; Naidu, S.; Fatemi, A. Clinical whole exome sequencing in child neurology practice. Ann. Neurol. 2014, 76, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Alby, C.; Boutaud, L.; Bessières, B.; Serre, V.; Rio, M.; Cormier-Daire, V.; de Oliveira, J.; Ichkou, A.; Mouthon, L.; Gordon, C.T.; et al. Novel de novo ZBTB20 mutations in three cases with Primrose syndrome and constant corpus callosum anomalies. Am. J. Med. Genet. A 2018, 176, 1091–1098. [Google Scholar] [CrossRef] [PubMed]
- Alby, C.; Boutaud, L.; Bonnière, M.; Collardeau-Frachon, S.; Guibaud, L.; Lopez, E.; Bruel, A.-L.; Aral, B.; Sonigo, P.; Roth, P.; et al. In utero ultrasound diagnosis of corpus callosum agenesis leading to the identification of orofaciodigital type 1 syndrome in female fetuses. Birth Defects Res. 2018, 110, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Mullins, J.G.L. Structural modelling pipelines in next generation sequencing projects. Adv. Protein Chem. Struct. Biol. 2012, 89, 117–167. [Google Scholar] [CrossRef] [PubMed]
- Prota, A.E.; Bargsten, K.; Zurwerra, D.; Field, J.J.; Díaz, J.F.; Altmann, K.-H.; Steinmetz, M.O. Molecular mechanism of action of microtubule-stabilizing anticancer agents. Science 2013, 339, 587–590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fourniol, F.J.; Sindelar, C.V.; Amigues, B.; Clare, D.K.; Thomas, G.; Perderiset, M.; Francis, F.; Houdusse, A.; Moores, C.A. Template-free 13-protofilament microtubule-MAP assembly visualized at 8 A resolution. J. Cell Biol. 2010, 191, 463–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- UCSF Chimera Home Page. Available online: https://www.cgl.ucsf.edu/chimera/ (accessed on 23 July 2018).
- Ensembl Genome Browser 93. Available online: http://www.ensembl.org/index.html (accessed on 23 July 2018).
- Aggarwala, V.; Voight, B.F. An expanded sequence context model broadly explains variability in polymorphism levels across the human genome. Nat. Genet. 2016, 48, 349–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wildeman, M.; van Ophuizen, E.; den Dunnen, J.T.; Taschner, P.E.M. Improving sequence variant descriptions in mutation databases and literature using the Mutalyzer sequence variation nomenclature checker. Hum. Mutat. 2008, 29, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Mutalyzer 2.0.28—Welcome to the Mutalyzer Website. Available online: https://www.mutalyzer.nl/ (accessed on 23 July 2018).
- Oegema, R.; Cushion, T.D.; Phelps, I.G.; Chung, S.-K.; Dempsey, J.C.; Collins, S.; Mullins, J.G.L.; Dudding, T.; Gill, H.; Green, A.J.; et al. Recognizable cerebellar dysplasia associated with mutations in multiple tubulin genes. Hum. Mol. Genet. 2015, 24, 5313–5325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poirier, K.; Saillour, Y.; Fourniol, F.; Francis, F.; Souville, I.; Valence, S.; Desguerre, I.; Marie Lepage, J.; Boddaert, N.; Line Jacquemont, M.; et al. Expanding the spectrum of TUBA1A-related cortical dysgenesis to Polymicrogyria. Eur. J. Hum. Genet. 2013, 21, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Kamiya, K.; Tanaka, F.; Ikeno, M.; Okumura, A.; Aoki, S. DTI tractography of lissencephaly caused by TUBA1A mutation. Neurol. Sci. 2014, 35, 801–803. [Google Scholar] [CrossRef] [PubMed]
- Mokánszki, A.; Körhegyi, I.; Szabó, N.; Bereg, E.; Gergev, G.; Balogh, E.; Bessenyei, B.; Sümegi, A.; Morris-Rosendahl, D.J.; Sztriha, L.; et al. Lissencephaly and band heterotopia: LIS1, TUBA1A, and DCX mutations in Hungary. J. Child Neurol. 2012, 27, 1534–1540. [Google Scholar] [CrossRef] [PubMed]
- Morris-Rosendahl, D.J.; Najm, J.; Lachmeijer, A.M.A.; Sztriha, L.; Martins, M.; Kuechler, A.; Haug, V.; Zeschnigk, C.; Martin, P.; Santos, M.; et al. Refining the phenotype of alpha-1a Tubulin (TUBA1A) mutation in patients with classical lissencephaly. Clin. Genet. 2008, 74, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Feng, R.; Sang, Q.; Kuang, Y.; Sun, X.; Yan, Z.; Zhang, S.; Shi, J.; Tian, G.; Luchniak, A.; Fukuda, Y.; et al. Mutations in TUBB8 and Human Oocyte Meiotic Arrest. N. Engl. J. Med. 2016, 374, 223–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, B.; Li, B.; Li, D.; Yan, Z.; Mao, X.; Xu, Y.; Mu, J.; Li, Q.; Jin, L.; He, L.; et al. Novel mutations and structural deletions in TUBB8: Expanding mutational and phenotypic spectrum of patients with arrest in oocyte maturation, fertilization or early embryonic development. Hum. Reprod. 2017, 32, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Tong, X.; Luo, L.; Zheng, S.; Jin, R.; Fu, Y.; Zhou, G.; Li, D.; Liu, Y. Mutation analysis of the TUBB8 gene in nine infertile women with oocyte maturation arrest. Reprod. Biomed. Online 2017, 35, 305–310. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, K.; Wilcox, R.A.; Winkler, S.; Ramirez, A.; Rakovic, A.; Park, J.-S.; Arns, B.; Lohnau, T.; Groen, J.; Kasten, M.; et al. Whispering dysphonia (DYT4 dystonia) is caused by a mutation in the TUBB4 gene. Ann. Neurol. 2013, 73, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Hersheson, J.; Mencacci, N.E.; Davis, M.; MacDonald, N.; Trabzuni, D.; Ryten, M.; Pittman, A.; Paudel, R.; Kara, E.; Fawcett, K.; et al. Mutations in the autoregulatory domain of β-tubulin 4a cause hereditary dystonia. Ann. Neurol. 2013, 73, 546–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamilton, E.M.; Polder, E.; Vanderver, A.; Naidu, S.; Schiffmann, R.; Fisher, K.; Raguž, A.B.; Blumkin, L.; H-ABC Research Group; van Berkel, C.G.M.; et al. Hypomyelination with atrophy of the basal ganglia and cerebellum: further delineation of the phenotype and genotype-phenotype correlation. Brain 2014, 137, 1921–1930. [Google Scholar] [CrossRef] [PubMed]
- Miyatake, S.; Osaka, H.; Shiina, M.; Sasaki, M.; Takanashi, J.-I.; Haginoya, K.; Wada, T.; Morimoto, M.; Ando, N.; Ikuta, Y.; et al. Expanding the phenotypic spectrum of TUBB4A-associated hypomyelinating leukoencephalopathies. Neurology 2014, 82, 2230–2237. [Google Scholar] [CrossRef] [PubMed]
- Simons, C.; Wolf, N.I.; McNeil, N.; Caldovic, L.; Devaney, J.M.; Takanohashi, A.; Crawford, J.; Ru, K.; Grimmond, S.M.; Miller, D.; et al. A de novo mutation in the β-tubulin gene TUBB4A results in the leukoencephalopathy hypomyelination with atrophy of the basal ganglia and cerebellum. Am. J. Hum. Genet. 2013, 92, 767–773. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Patient | 1 | 2 | 3 | 4 (fetus) |
|---|---|---|---|---|
| Sex | Male | Male | Male | Male |
| OFC at Birth | 30 cm (−3.6 SD) | 34 cm (−0.9 SD) | 33 cm (−1.7 SD) | n/a |
| Age at last review | 4 years | 32 months | 37 months | TOP at 36 weeks gestation |
| Last OFC | 45 cm (−4.9 SD) | 43 cm (−5.9 SD) | 45 cm (−4.6 SD) | n/a |
| Developmental delay | Moderate | Severe | Moderate | n/a |
| Seizures | Yes (onset at 3 years) | Yes (onset at 12 months) | No | n/a |
| Cerebral cortex | Bilateral perisylvian polymicrogyria | Normal | Normal | Bilateral perisylvian polymicrogyria |
| White matter | Reduced | Reduced | Reduced | n/k |
| Corpus callosum | Partial agenesis | Thin | Thin | Short, no rostrum |
| Basal ganglia | Dysmorphic, prominent | Dysmorphic, prominent | Dysmorphic, prominent | n/k |
| Cerebellum | Hypoplasia and dysplasia of vermis | Hypoplasia and dysplasia of vermis | Hypoplasia and dysplasia of vermis | Hypoplasia, Impaired lamination, rare and misaligned Purkinje |
| Brainstem | Small pons | Small pons | Small pons | Neuronal heterotopia of olivary nuclei and hypoplastic pyramids |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gardner, J.F.; Cushion, T.D.; Niotakis, G.; Olson, H.E.; Grant, P.E.; Scott, R.H.; Stoodley, N.; Cohen, J.S.; Naidu, S.; Attie-Bitach, T.; et al. Clinical and Functional Characterization of the Recurrent TUBA1A p.(Arg2His) Mutation. Brain Sci. 2018, 8, 145. https://doi.org/10.3390/brainsci8080145
Gardner JF, Cushion TD, Niotakis G, Olson HE, Grant PE, Scott RH, Stoodley N, Cohen JS, Naidu S, Attie-Bitach T, et al. Clinical and Functional Characterization of the Recurrent TUBA1A p.(Arg2His) Mutation. Brain Sciences. 2018; 8(8):145. https://doi.org/10.3390/brainsci8080145
Chicago/Turabian StyleGardner, Jennifer F., Thomas D. Cushion, Georgios Niotakis, Heather E. Olson, P. Ellen Grant, Richard H. Scott, Neil Stoodley, Julie S. Cohen, Sakkubai Naidu, Tania Attie-Bitach, and et al. 2018. "Clinical and Functional Characterization of the Recurrent TUBA1A p.(Arg2His) Mutation" Brain Sciences 8, no. 8: 145. https://doi.org/10.3390/brainsci8080145