
Nonmechanical Roles of Dystrophin and Associated Proteins in Exercise, Neuromuscular Junctions, and Brains



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
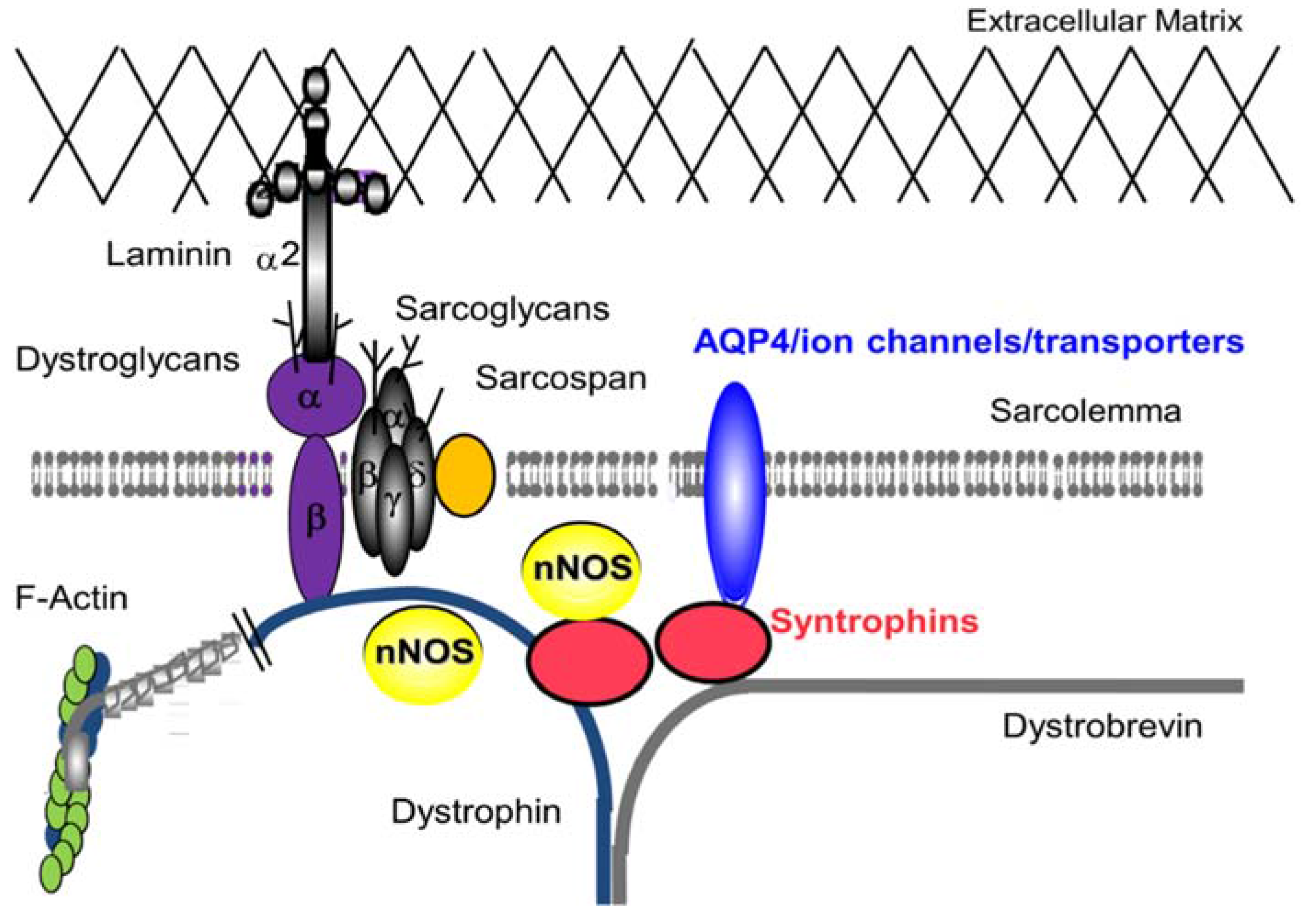
:1. Introduction—Dystrophin-glycoprotein Complex (DGC)
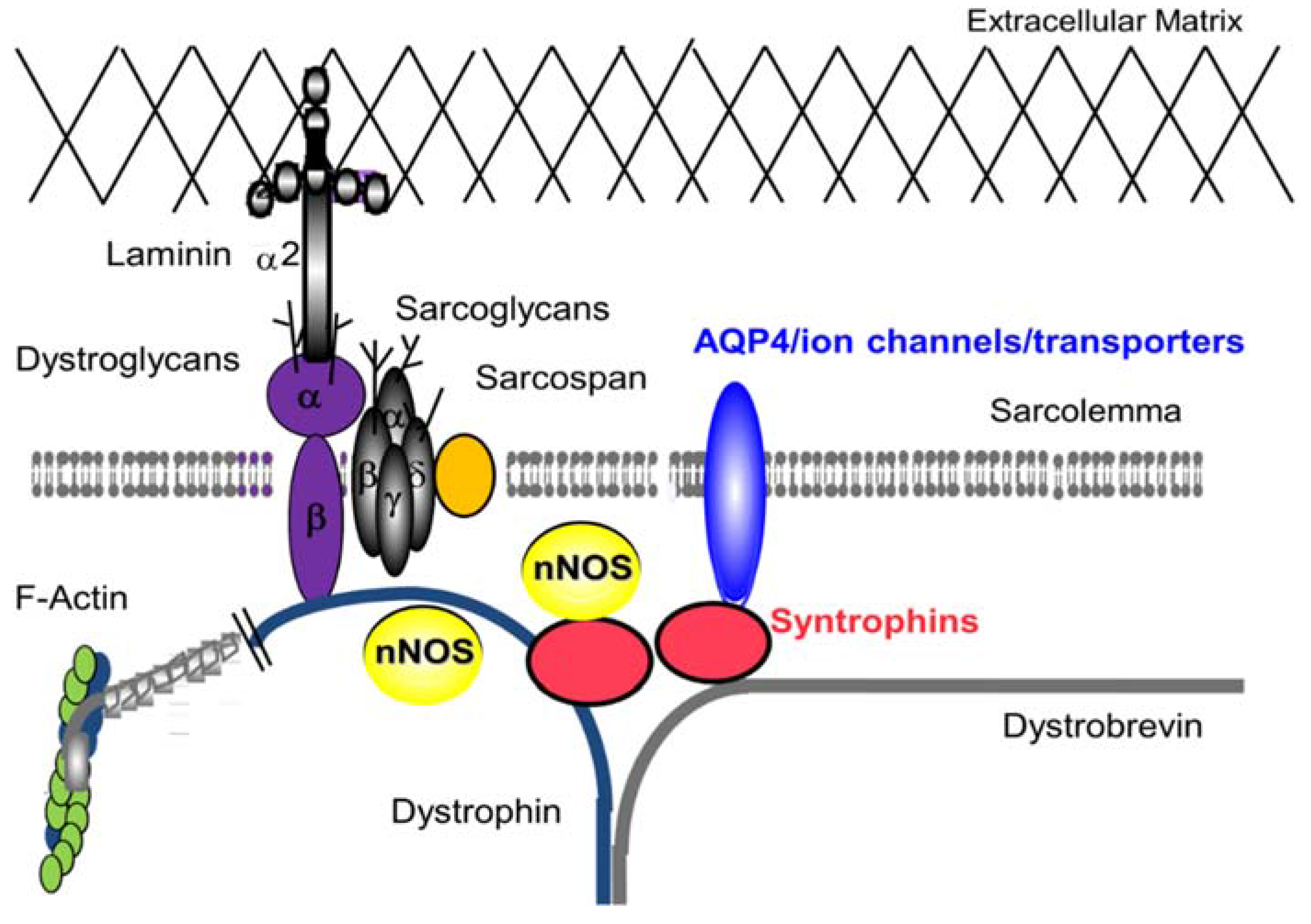
2. DGC—Non Mechanical Roles in Skeletal Muscle
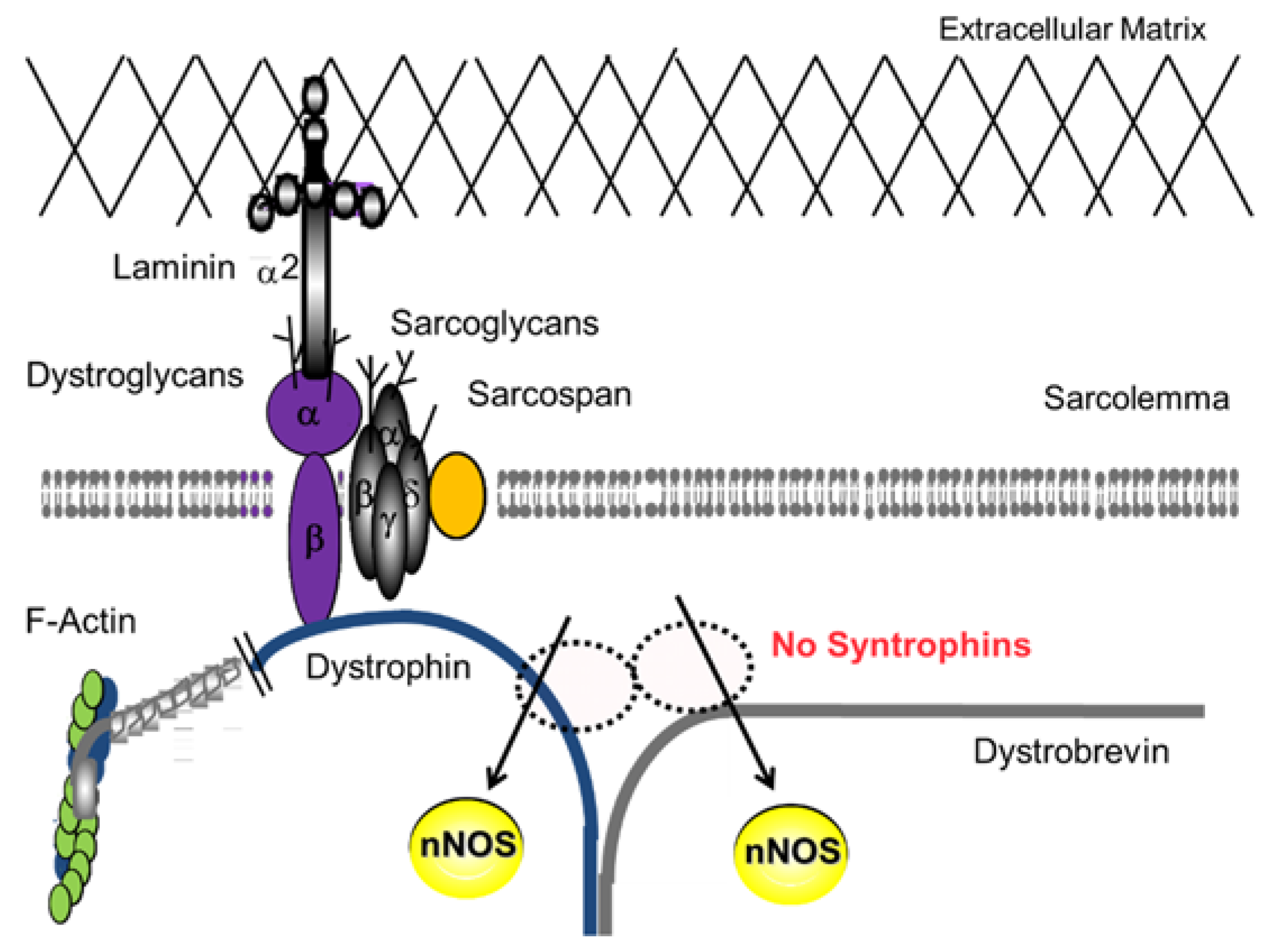
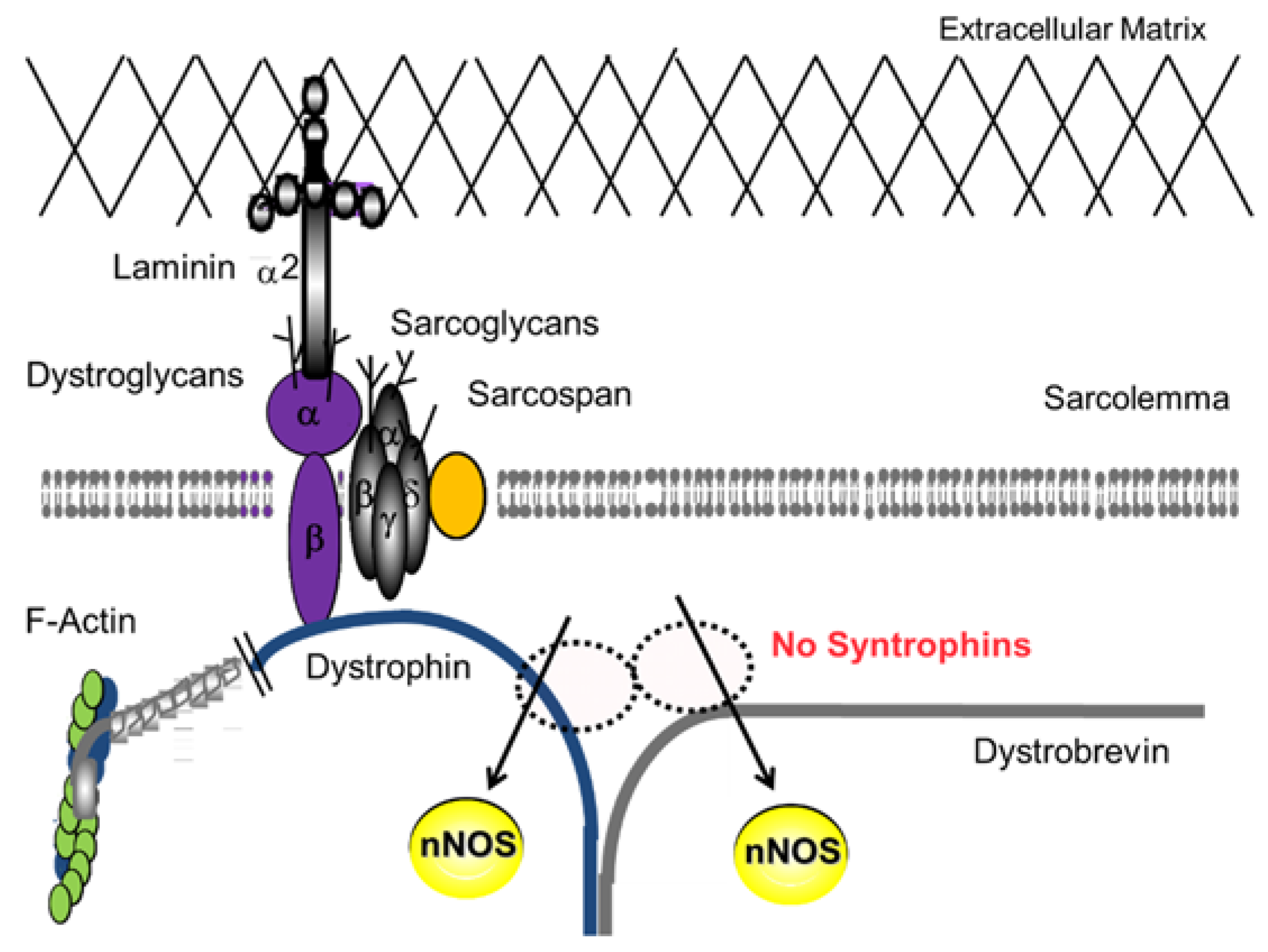
2.1. DGC and Muscle Fatigue

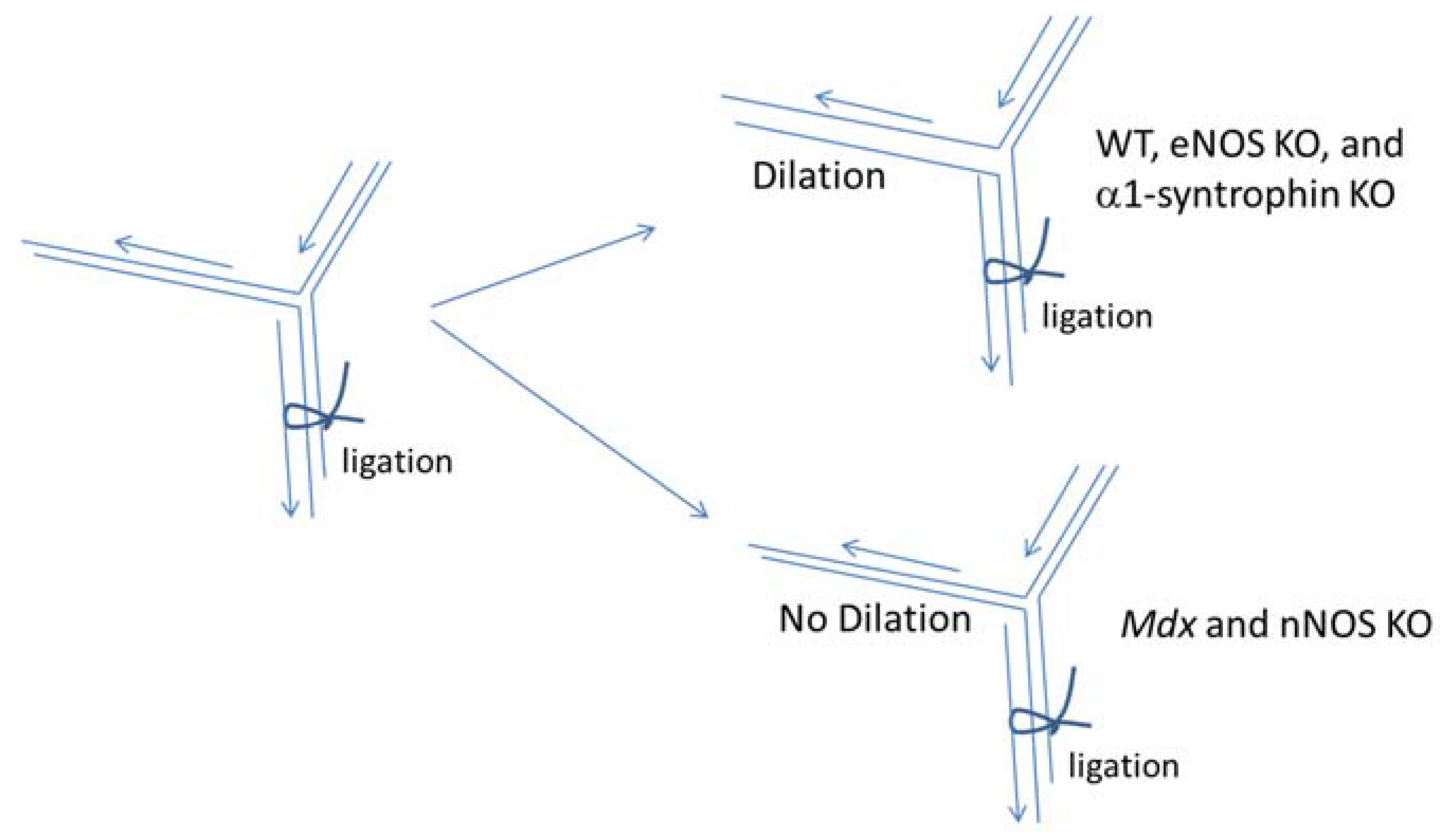
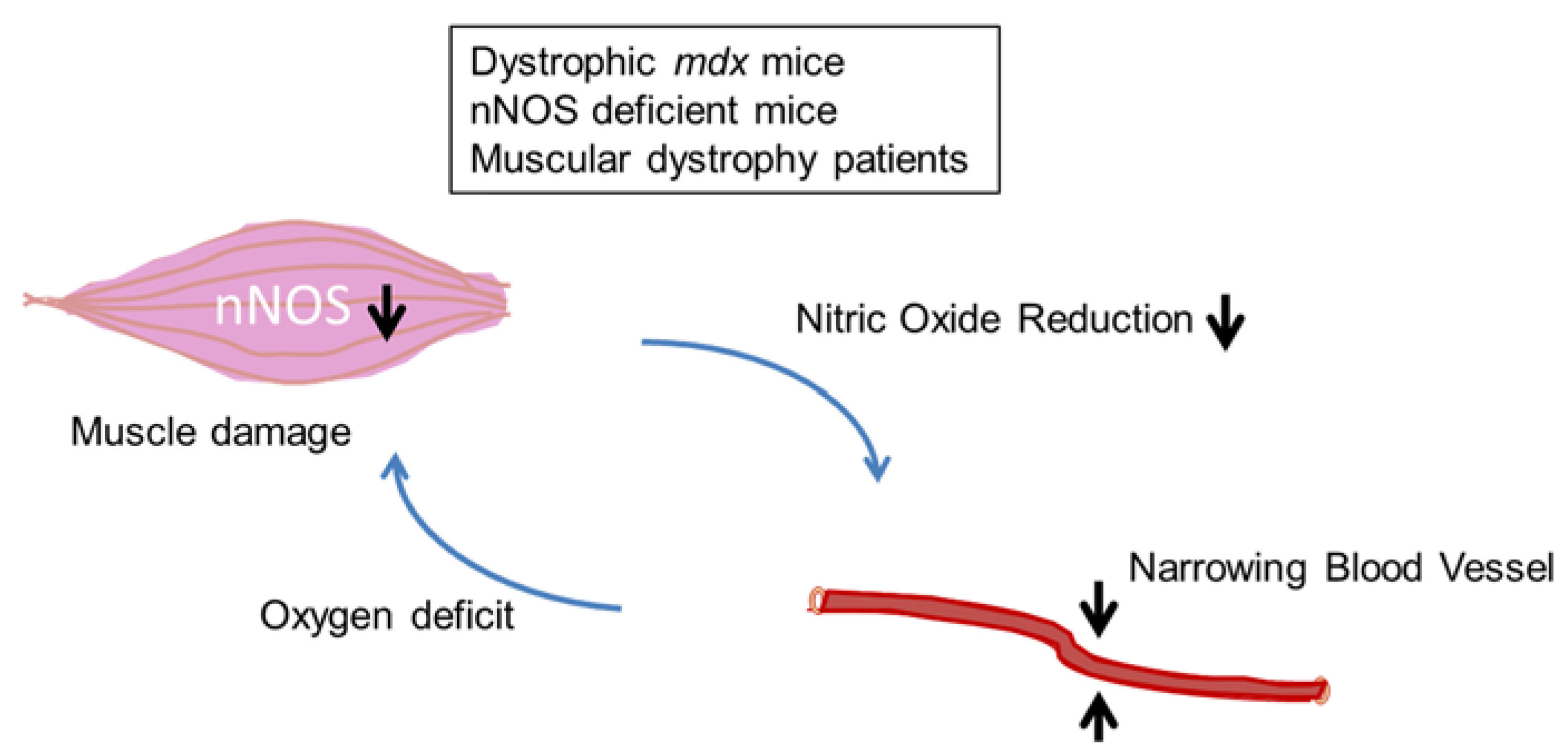
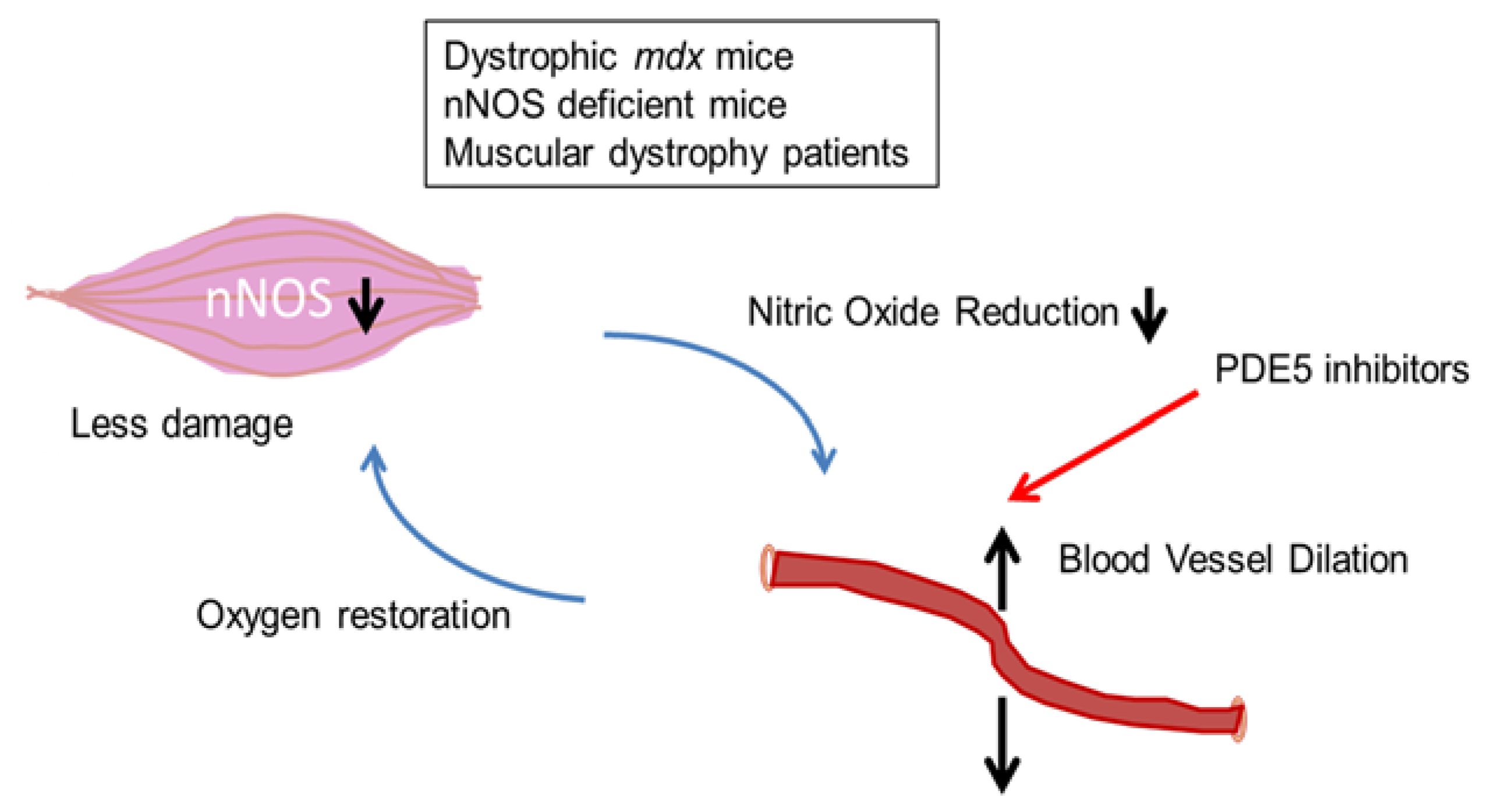
2.2. DGC and Two-Hit Hypothesis
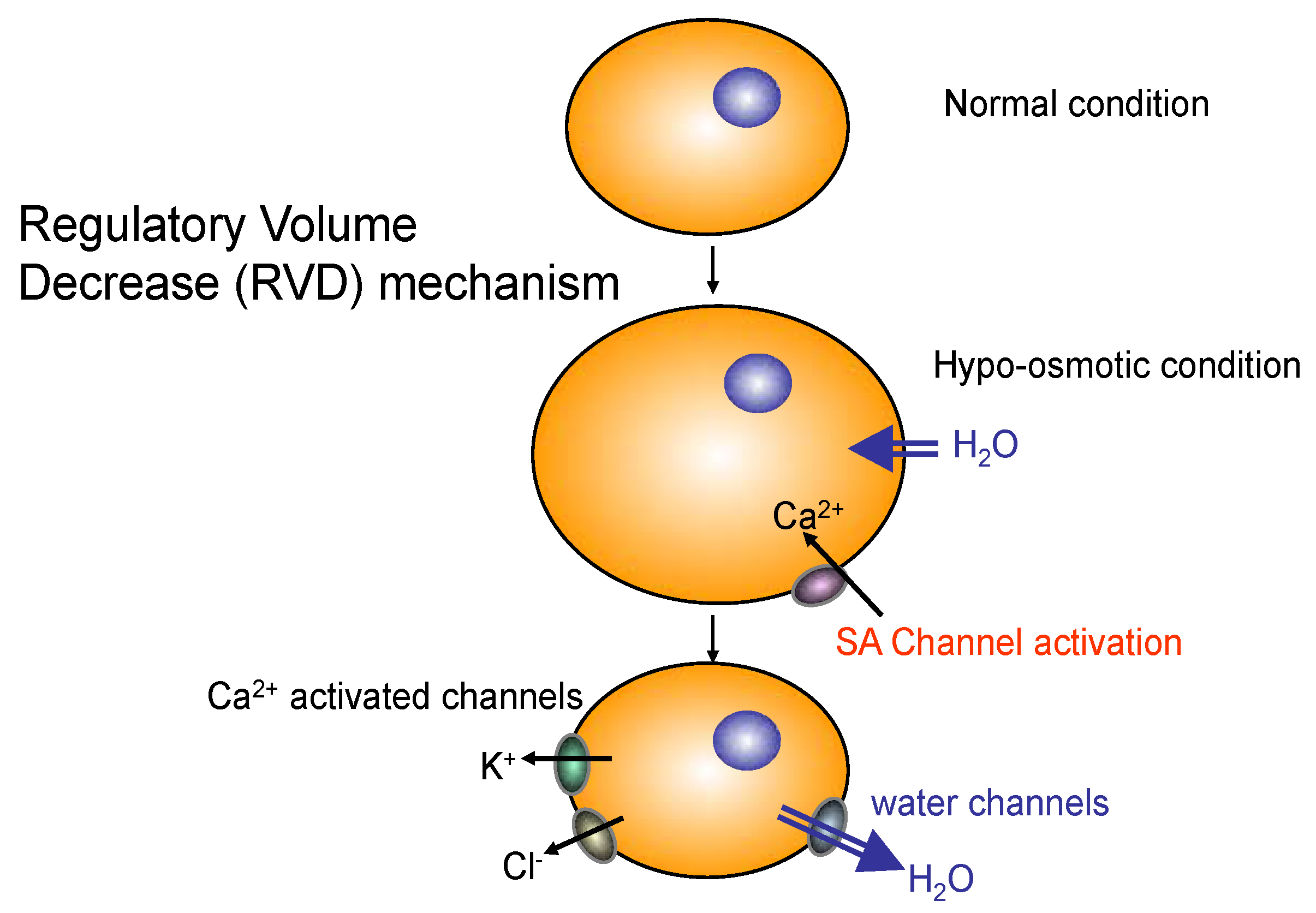
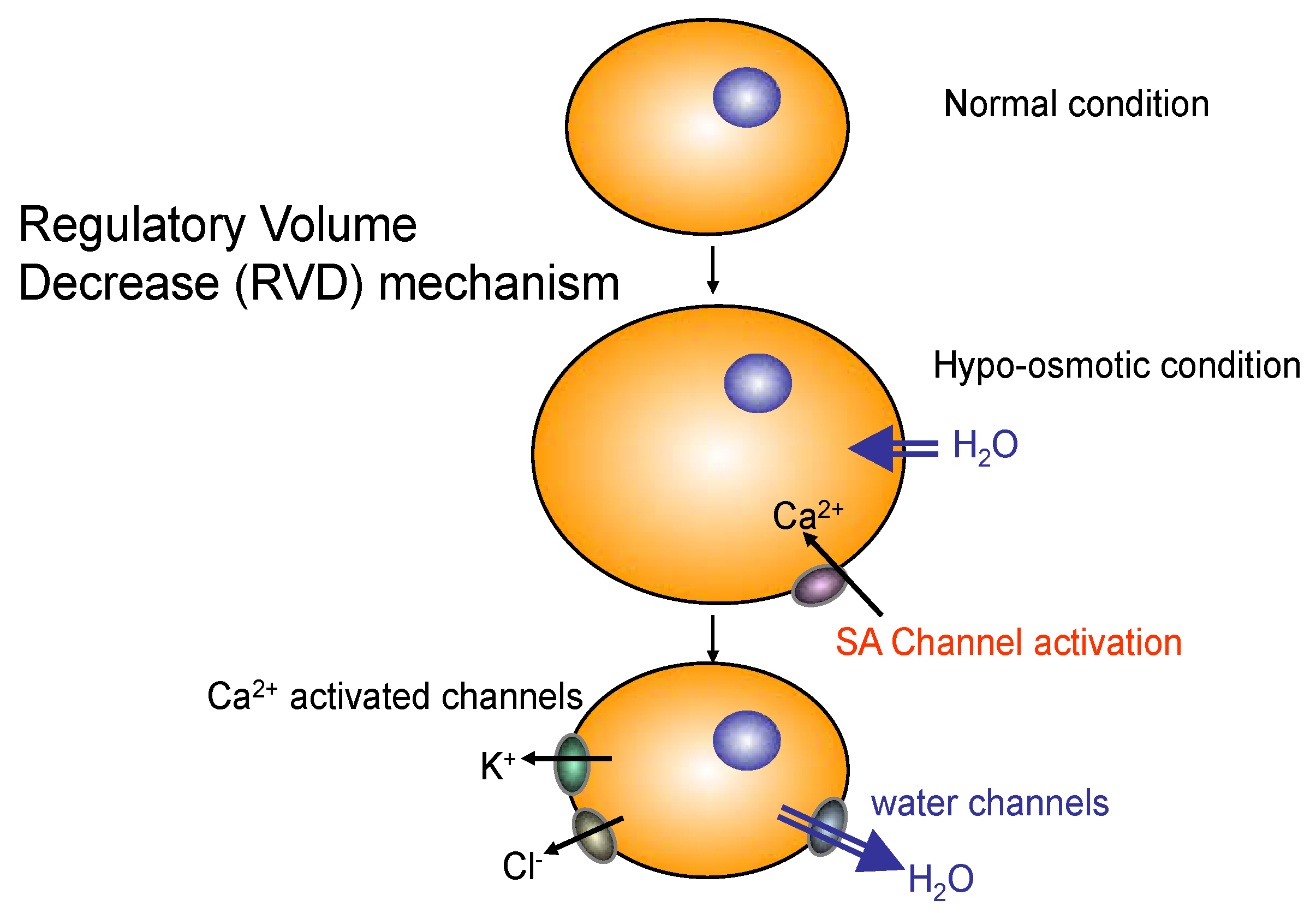
2.3. Role of AQP4 in Skeletal Muscle
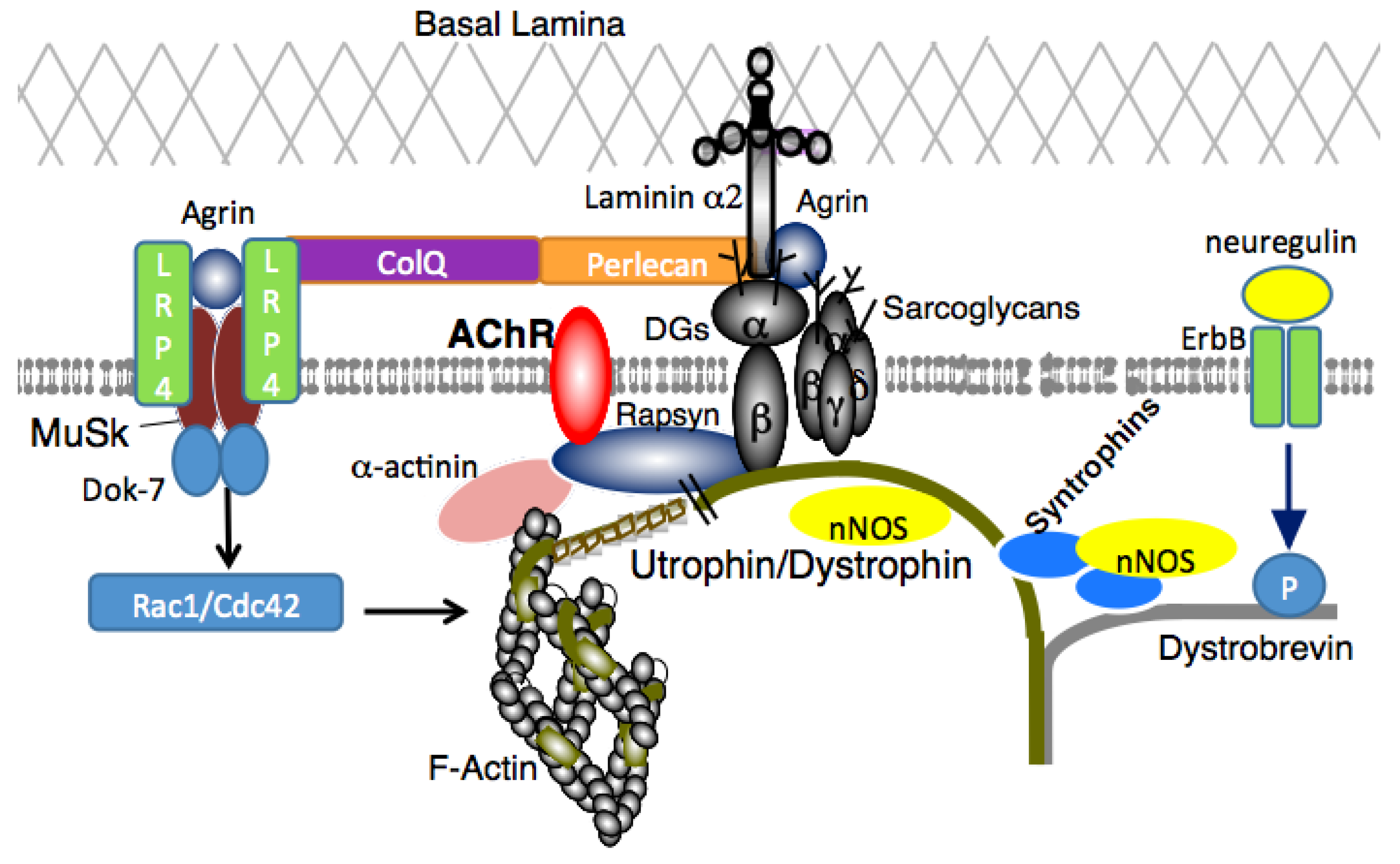
3. DGC Complex at the Neuromuscular Junction

4. DGC Complex in Brain and the Cognitive Impairment in DMD
4.1. Regions of the Brain Affected
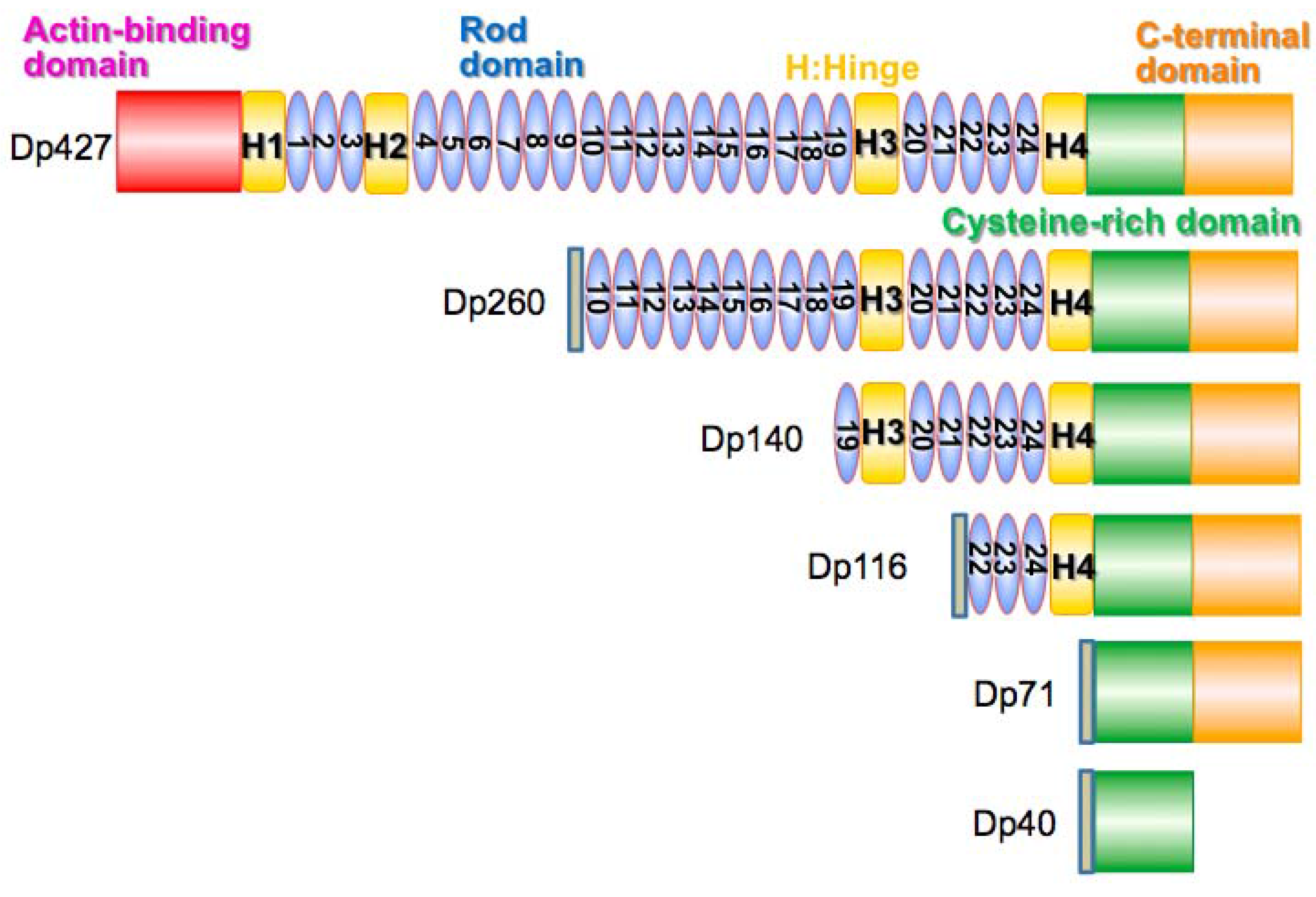
4.2. Dystrophin Protein Isoforms
4.3. Potential Treatments
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hoffman, E.P.; Brown, R.H., Jr.; Kunkel, L.M. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Duchenne. The Pathology of Paralysis with Muscular Degeneration (Paralysie Myosclerotique), or Paralysis with Apparent Hypertrophy. Br. Med. J. 1867, 2, 541–542. [Google Scholar] [CrossRef] [PubMed]
- Koenig, M.; Hoffman, E.P.; Bertelson, C.J.; Monaco, A.P.; Feener, C.; Kunkel, L.M. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 1987, 50, 509–517. [Google Scholar] [CrossRef]
- Ervasti, J.M.; Ohlendieck, K.; Kahl, S.D.; Gaver, M.G.; Campbell, K.P. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature 1990, 345, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Ozawa, E. Glycoprotein complex anchoring dystrophin to sarcolemma. J. Biochem. 1990, 108, 748–752. [Google Scholar] [PubMed]
- Yoshida, M.; Suzuki, A.; Yamamoto, H.; Noguchi, S.; Mizuno, Y.; Ozawa, E. Dissociation of the complex of dystrophin and its associated proteins into several unique groups by N-octyl beta-d-glucoside. Eur. J. Biochem. FEBS 1994, 222, 1055–1061. [Google Scholar] [CrossRef]
- Hewitt, J.E. Abnormal glycosylation of dystroglycan in human genetic disease. Biochim. Biophys. Acta 2009, 1792, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Eagle, M.; Baudouin, S.V.; Chandler, C.; Giddings, D.R.; Bullock, R.; Bushby, K. Survival in Duchenne muscular dystrophy: Improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscul. Disord. 2002, 12, 926–929. [Google Scholar] [CrossRef]
- Heald, A.; Anderson, L.V.; Bushby, K.M.; Shaw, P.J. Becker muscular dystrophy with onset after 60 years. Neurology 1994, 44, 2388–2390. [Google Scholar] [CrossRef] [PubMed]
- Stöllberger, C.; Finsterer, J. Worsening of heart failure in Becker muscular dystrophy after nonsteroidal anti-inflammatory drugs. South Med. J. 2005, 98, 478–480. [Google Scholar] [CrossRef] [PubMed]
- Passamano, L.; Taglia, A.; Palladino, A.; Viggiano, E.; D’Ambrosio, P.; Scutifero, M.; Rosaria Cecio, M.; Torre, V.; DE Luca, F.; Picillo, E.; et al. Improvement of survival in Duchenne Muscular Dystrophy: Retrospective analysis of 835 patients. Acta Myol. 2012, 31, 121–125. [Google Scholar] [PubMed]
- Haginoya, K.; Yamamoto, K.; Iinuma, K.; Yanagisawa, T.; Ichinohasama, Y.; Shimmoto, M.; Suzuki, Y.; Tada, K. Dystrophin immunohistochemistry in a symptomatic carrier of Becker muscular dystrophy. J. Neurol. 1991, 238, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Angelini, C.; Tasca, E.; Nascimbeni, A.C.; Fanin, M. Muscle fatigue, nNOS and muscle fiber atrophy in limb girdle muscular dystrophy. Acta Myol. 2014, 33, 119–126. [Google Scholar] [PubMed]
- Sadoulet-Puccio, H.M.; Khurana, T.S.; Cohen, J.B.; Kunkel, L.M. Cloning and characterization of the human homologue of a dystrophin related phosphoprotein found at the Torpedo electric organ post-synaptic membrane. Hum. Mol. Genet. 1996, 5, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.E.; Butler, M.H.; Dwyer, T.M.; Peters, M.F.; Murnane, A.A.; Froehner, S.C. Two forms of mouse syntrophin, a 58 kd dystrophin-associated protein, differ in primary structure and tissue distribution. Neuron 1993, 11, 531–540. [Google Scholar] [CrossRef]
- Brenman, J.E.; Chao, D.S.; Gee, S.H.; McGee, A.W.; Craven, S.E.; Santillano, D.R.; Wu, Z.; Huang, F.; Xia, H.; Peters, M.F.; et al. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and alpha1-syntrophin mediated by PDZ domains. Cell 1996, 84, 757–767. [Google Scholar] [CrossRef]
- Kameya, S.; Miyagoe, Y.; Nonaka, I.; Ikemoto, T.; Endo, M.; Hanaoka, K.; Nabeshima, Y.; Takeda, S. Alpha1-syntrophin gene disruption results in the absence of neuronal-type nitric-oxide synthase at the sarcolemma but does not induce muscle degeneration. J. Biol. Chem. 1999, 274, 2193–2200. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Hosaka, Y.; Tsukita, K.; Kameya, S.; Shibuya, R.; Matsuda, R.; Wakayama, Y.; Takeda, S. Aquaporin-4 is absent at the sarcolemma and at perivascular astrocyte endfeet in alpha1-syntrophin knockout mice. Proc. Jpn. Acad. 2000, 76B, 22–27. [Google Scholar] [CrossRef]
- Gee, S.H.; Madhavan, R.; Levinson, S.R.; Caldwell, J.H.; Sealock, R.; Froehner, S.C. Interaction of muscle and brain sodium channels with multiple members of the syntrophin family of dystrophin-associated proteins. J. Neurosci. 1998, 18, 128–137. [Google Scholar] [PubMed]
- Hogan, A.; Shepherd, L.; Chabot, J.; Quenneville, S.; Prescott, S.M.; Topham, M.K.; Gee, S.H. Interaction of gamma 1-syntrophin with diacylglycerol kinase-zeta. Regulation of nuclear localization by PDZ interactions. J. Biol. Chem. 2001, 276, 26526–26533. [Google Scholar] [CrossRef] [PubMed]
- Munehira, Y.; Ohnishi, T.; Kawamoto, S.; Furuya, A.; Shitara, K.; Imamura, M.; Yokota, T.; Takeda, S.; Amachi, T.; Matsuo, M.; et al. Alpha1-syntrophin modulates turnover of ABCA1. J. Biol. Chem. 2004, 279, 15091–15095. [Google Scholar] [CrossRef] [PubMed]
- Crosbie, R.H.; Heighway, J.; Venzke, D.P.; Lee, J.C.; Campbell, K.P. Sarcospan, the 25-kDa transmembrane component of the dystrophin-glycoprotein complex. J. Biol. Chem. 1997, 272, 31221–31224. [Google Scholar] [CrossRef] [PubMed]
- Cohn, R.D.; Campbell, K.P. Molecular basis of muscular dystrophies. Muscle Nerve 2000, 23, 1456–1471. [Google Scholar] [CrossRef]
- Asai, A.; Sahani, N.; Kaneki, M.; Ouchi, Y.; Martyn, J.A.; Yasuhara, S.E. Primary role of functional ischemia, quantitative evidence for the two-hit mechanism, and phosphodiesterase-5 inhibitor therapy in mouse muscular dystrophy. PLoS ONE 2007, 2, e806. [Google Scholar] [CrossRef] [PubMed]
- Wust, R.C.; Degens, H. Factors contributing to muscle wasting and dysfunction in COPD patients. Int. J. Chron. Obstruct. Pulmon. Dis. 2007, 2, 289–300. [Google Scholar] [PubMed]
- Kobayashi, Y.M.; Rader, E.P.; Crawford, R.W.; Iyengar, N.K.; Thedens, D.R.; Faulkner, J.A.; Parikh, S.V.; Weiss, R.M.; Chamberlain, J.S.; Moore, S.A.; et al. Sarcolemma-localized nNOS is required to maintain activity after mild exercise. Nature 2008, 456, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Miyagoe-Suzuki, Y.; Ikemoto, T.; Matsuda, R.; Takeda, S. Alpha1-Syntrophin deficient mice exhibit impaired muscle force recovery after osmotic shock. Muscle Nerve 2014, 49, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Angelini, C.; Tasca, E. Fatigue in muscular dystrophies. Neuromuscul. Dis. 2012, 22 (Suppl. S3), 214–220. [Google Scholar] [CrossRef] [PubMed]
- Finanger Hedderick, E.L.; Simmers, J.L.; Soleimani, A.; Andres-Mateos, E.; Marx, R.; Files, D.C.; King, L.; Crawford, T.O.; Corse, A.M.; Cohn, R.D. Loss of sarcolemmal nNOS is common in acquired and inherited neuromuscular disorders. Neurology 2011, 76, 960–967. [Google Scholar] [CrossRef] [PubMed]
- Ehmsen, J.; Poon, E.; Davies, K. The dystrophin-associated protein complex. J. Cell Sci. 2002, 115, 2801–2803. [Google Scholar] [PubMed]
- Meinen, S.; Lin, S.; Ruegg, M.A.; Punga, A.R. Fatigue and muscle atrophy in a mouse model of myasthenia gravis is paralleled by loss of sarcolemmal nNOS. PLoS ONE 2012, 7, e44148. [Google Scholar] [CrossRef] [PubMed]
- Hillier, B.J.; Christopherson, K.S.; Prehoda, K.E.; Bredt, D.S.; Lim, W.A. Unexpected modes of PDZ domain scaffolding revealed by structure of nNOS-syntrophin complex. Science 1999, 284, 812–815. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.E.; Mueller, H.A.; Froehner, S.C. In vivo requirement of the alpha-syntrophin PDZ domain for the sarcolemmal localization of nNOS and aquaporin-4. J. Cell Biol. 2001, 155, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Yokota, T.; Ichioka, S.; Shibata, M.; Takeda, S. Vasodilation of intramuscular arterioles under shear stress in dystrophin-deficient skeletal muscle is impaired through decreased nNOS expression. Acta Myol. 2008, 27, 30–36. [Google Scholar] [PubMed]
- Chang, W.J.; Iannaccone, S.T.; Lau, K.S.; Masters, B.S.; McCabe, T.J.; McMillan, K.; Padre, R.C.; Spencer, M.J.; Tidball, J.G.; Stull, J.T. Neuronal nitric oxide synthase and dystrophin-deficient muscular dystrophy. Proc. Natl. Acad. Sci. USA 1996, 93, 9142–9147. [Google Scholar] [CrossRef] [PubMed]
- Rando, T.A. Role of nitric oxide in the pathogenesis of muscular dystrophies: A “two hit” hypothesis of the cause of muscle necrosis. Microsc. Res. Tech. 2001, 55, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.D.; Rader, F.; Tang, X.; Tavyev, J.; Nelson, S.F.; Miceli, M.C.; Elashoff, R.M.; Sweeney, H.L.; Victor, R.G. PDE5 inhibition alleviates functional muscle ischemia in boys with Duchenne muscular dystrophy. Neurology 2014, 82, 2085–2091. [Google Scholar] [CrossRef] [PubMed]
- Martin, E.A.; Barresi, R.; Byrne, B.J.; Tsimerinov, E.I.; Scott, B.L.; Walker, A.E.; Gurudevan, S.V.; Anene, F.; Elashoff, R.M.; Thomas, G.D.; et al. Tadalafil alleviates muscle ischemia in patients with Becker muscular dystrophy. Sci. Transl. Med. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, M.; Nakatsuji, Y. Where Do AQP4 Antibodies Fit in the Pathogenesis of NMO? Mult. Scler. Int. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Frigeri, A.; Nicchia, G.P.; Balena, R.; Nico, B.; Svelto, M. Aquaporins in skeletal muscle: Reassessment of the functional role of aquaporin-4. FASEB J. 2004, 18, 905–907. [Google Scholar] [CrossRef] [PubMed]
- Devuyst, O.; Nielsen, S.; Cosyns, J.P.; Smith, B.L.; Agre, P.; Squifflet, J.P.; Pouthier, D.; Goffin, E. Aquaporin-1 and endothelial nitric oxide synthase expression in capillary endothelia of human peritoneum. Am. J. Physiol. 1998, 275, 234–242. [Google Scholar]
- Yang, B.; Verbavatz, J.M.; Song, Y.; Vetrivel, L.; Manley, G.; Kao, W.M.; Ma, T.; Verkman, A.S. Skeletal muscle function and water permeability in aquaporin-4 deficient mice. Am. J. Physiol. Cell Physiol. 2000, 278, C1108–C1115. [Google Scholar] [PubMed]
- Ponting, C.P.; Phillips, C. DHR domains in syntrophins, neuronal NO synthases and other intracellular proteins. Trends Biochem. Sci. 1995, 20, 102–103. [Google Scholar] [CrossRef]
- Adams, M.E.; Tesch, Y.; Percival, J.M.; Albrecht, D.E.; Conhaim, J.I.; Anderson, K.; Froehner, S.C. Differential targeting of nNOS and AQP4 to dystrophin-deficient sarcolemma by membrane-directed alpha-dystrobrevin. J. Cell Sci. 2008, 121, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Hosaka, Y.; Yokota, T.; Miyagoe-Suzuki, Y.; Yuasa, K.; Imamura, M.; Matsuda, R.; Ikemoto, T.; Kameya, S.; Takeda, S. Alpha1-syntrophin-deficient skeletal muscle exhibits hypertrophy and aberrant formation of neuromuscular junctions during regeneration. J. Cell Biol. 2002, 158, 1097–1107. [Google Scholar] [CrossRef] [PubMed]
- Crosbie, R.H.; Straub, V.; Yun, H.Y.; Lee, J.C.; Rafael, J.A.; Chamberlain, J.S.; Dawson, V.L.; Dawson, T.M.; Campbell, K.P. mdx muscle pathology is independent of nNOS perturbation. Hum. Mol. Genet. 1998, 7, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Arredondo, J.; Lara, M.; Ng, F.; Gochez, D.A.; Lee, D.C.; Logia, S.P.; Nguyen, J.; Maselli, R.A. COOH-terminal collagen Q (COLQ) mutants causing human deficiency of endplate acetylocholinesterase impair the interaction of ColQ with proteins of the basil lamina. Hum. Genet. 2014, 5, 599–616. [Google Scholar] [CrossRef] [PubMed]
- Sigoillot, S.M.; Bourgeois, F.; Lambergeon, M.; Strochlic, L.; Legay, C. CoIQ controls postsynaptic differentiation at the neuromuscular junction. Neuroscience 2010, 30, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Kibuuka, H.; Guwatudde, D.; Kimutai, R.; Maganga, L.; Maboko, L.; Watyema, C.; Sawe, F.; Shaffer, D.; Matsiko, D.; Millard, M.; et al. Contraceptive use in women enrolled into preventive HIV vaccine trials: Experience from a phase I/II trial in East Africa. PLoS ONE 2009, 4, e5164. [Google Scholar] [CrossRef] [PubMed]
- Deconinck, A.E.; Potter, A.C.; Tinsley, J.M.; Wood, S.J.; Vater, R.; Young, C.; Metzinger, L.; Vincent, A.; Slater, C.R.; Davies, K.E. Postsynaptic abnormalities at the neuromuscular junctions of utrophin-deficient mice. J. Cell Biol. 1997, 136, 883–894. [Google Scholar] [CrossRef] [PubMed]
- Deconinck, A.E.; Rafael, J.A.; Skinner, J.A.; Brown, S.C.; Potter, A.C.; Metzinger, L.; Watt, D.J.; Dickson, J.G.; Tinsley, J.M.; Davies, K.E. Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell 1997, 90, 717–727. [Google Scholar] [CrossRef]
- Grady, R.M.; Merlie, J.P.; Sanes, J.R. Subtle neuromuscular defects in utrophin-deficient mice. J. Cell Biol. 1997, 136, 871–882. [Google Scholar] [CrossRef] [PubMed]
- Akaaboune, M.; Grady, R.M.; Turney, S.; Sanes, J.R.; Lichtman, J.W. Neurotransmitter receptor dynamics studied in vivo by reversible photo-unbinding of fluorescent ligands. Neuron 2002, 34, 865–876. [Google Scholar] [CrossRef]
- Adams, M.E.; Kramarcy, N.; Krall, S.P.; Rossi, S.G.; Rotundo, R.L.; Sealock, R.; Froehner, S.C. Absence of alpha-syntrophin leads to structurally aberrant neuromuscular synapses deficient in utrophin. J. Cell Biol. 2000, 150, 1385–1398. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, C.; Cote, P.D.; Rossi, S.G.; Rotundo, R.L.; Carbonetto, S. The dystroglycan complex is necessary for stabilization of acetylcholine receptor clusters at neuromuscular junctions and formation of the synaptic basement membrane. J. Cell Biol. 2001, 152, 435–450. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, N.; Akaaboune, M.; Gajendran, N.; Martinez-Pena y Valenzuela, I.; Wakefield, S.; Thurnheer, R.; Brenner, H.R. Neuregulin/ErbB regulate neuromuscular junction development by phosphorylation of alpha-dystrobrevin. J. Cell Biol. 2011, 195, 1171–1184. [Google Scholar] [CrossRef] [PubMed]
- Bartoli, M.; Ramarao, M.K.; Cohen, J.B. Interactions of the rapsyn RING-H2 domain with dystroglycan. J. Biol. Chem. 2001, 276, 24911–24917. [Google Scholar] [CrossRef] [PubMed]
- Arimura, S.; Okada, T.; Tezuka, T.; Chiyo, T.; Kasahara, Y.; Yoshimura, T.; Motomura, M.; Yoshida, N.; Beeson, D.; Takeda, S.; et al. Neuromuscular disease. DOK7 gene therapy benefits mouse models of diseases characterized by defects in the neuromuscular junction. Science 2014, 345, 1505–1508. [Google Scholar] [PubMed]
- Hinton, V.J.; De Vivo, D.C.; Nereo, N.E.; Goldstein, E.; Stern, Y. Poor verbal working memory across intellectual level in boys with Duchene dystrophy. Neurology 2000, 54, 2127–2132. [Google Scholar] [CrossRef] [PubMed]
- Billard, C.; Gillet, P.; Signoret, J.L.; Uicaut, E.; Bertrand, P.; Fardeau, M.; Barthez-Carpentier, M.A.; Santini, J.J. Cognitive functions in Duchenne muscular dystrophy: A reappraisal and comparison with spinal muscular atrophy. Neuromuscul. Disord. NMD 1992, 2, 371–378. [Google Scholar] [CrossRef]
- Prosser, E.J.; Murphy, E.G.; Thompson, M.W. Intelligence and the gene for Duchenne muscular dystrophy. Arch. Dis. Child. 1969, 44, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Felisari, G.; Martinelli Boneschi, F.; Bardoni, A.; Sironi, M.; Comi, G.P.; Robotti, M.; Turconi, A.C.; Lai, M.; Corrao, G.; Bresolin, N. Loss of Dp140 dystrophin isoform and intellectual impairment in Duchenne dystrophy. Neurology 2000, 55, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Perronnet, C.; Vaillend, C. Dystrophins, utrophins, and associated scaffolding complexes: Role in mammalian brain and implications for therapeutic strategies. J. Biomed. Biotechnol. 2010, 2010. [Google Scholar] [CrossRef]
- Mento, G.; Tarantino, V.; Bisiacchi, P.S. The neuropsychological profile of infantile Duchene muscular Dystrophy. Clin. Neuropsychol. 2011, 25, 1359–1377. [Google Scholar] [CrossRef] [PubMed]
- Doorenweerd, N.; Straathof, C.S.; Dumas, E.M.; Spitali, P.; Ginjaar, I.B.; Wokke, B.H.; Schrans, D.G.; van den Bergen, J.C.; van Zwet, E.W.; Webb, A.; et al. Reduced cerebral gray matter and altered white matter in boys with Duchenne muscular dystrophy. Ann. Neurol. 2014, 76, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Hinton, V.J.; Nereo, N.E.; Fee, R.J.; Cyrulnik, S.E. Social behavior problems in boys with Duchenne muscular dystrophy. J. Dev. Behav. Pediatr. 2006, 27, 470–476. [Google Scholar] [CrossRef] [PubMed]
- Donders, J.; Taneja, C. Neurobehavioral characteristics of children with Duchenne muscular dystrophy. Child Neuropsychol. 2009, 15, 295–304. [Google Scholar] [CrossRef] [PubMed]
- Hendriksen, J.G.; Vles, J.S. Neuropsychiatric disorders in males with duchenne muscular dystrophy: Frequency rate of attention-deficit hyperactivity disorder (ADHD), autism spectrum disorder, and obsessive- compulsive disorder. J. Child Neurol. 2008, 23, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Roccella, M.; Pace, R.; De Gregorio, M.T. Psychopatho-logical assessment in children affected by Duchenne de Boulogne muscular dystrophy. Minerva Pediatr. 2003, 55, 267–276. [Google Scholar] [PubMed]
- Cyrulnik, S.E.; Hinton, V.J. Duchenne muscular dystrophy: A cerebellar disorder? Neurosci. Biobehav. Rev. 2008, 32, 486–496. [Google Scholar] [CrossRef] [PubMed]
- Culligan, K.; Glover, L.; Dowling, P.; Ohlendieck, K. Brain dystrophin-glycoprotein complex: Persistent expression of beta-dystroglycan, impaired oligomerization of Dp71 and up-regulation of utrophins in animal models of muscular dystrophy. BMC Cell Biol. 2001, 2. [Google Scholar] [CrossRef] [Green Version]
- Lidov, H.G.; Byers, T.J.; Watkins, S.C.; Kunkel, L.M. Localization of dystrophin to postsynaptic regions of central nervous system cortical neurons. Nature 1990, 348, 725–728. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.W.; Wu, K.; Xu, J.L.; Black, I.B. Detection of dystrophin in the postsynaptic density of rat brain and deficiency in a mouse model of Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 1992, 89, 11642–11644. [Google Scholar] [CrossRef] [PubMed]
- Grady, R.M.; Wozniak, D.F.; Ohlemiller, K.K.; Sanes, J.R. Cerebellar synaptic defects and abnormal motor behavior in mice lacking alpha- and beta-dystrobrevin. J. Neurosci. 2006, 26, 2841–2851. [Google Scholar] [CrossRef] [PubMed]
- Vaillend, C.; Billard, J.M.; Laroche, S. Impaired long-term spatial and recognition memory and enhanced CA1 hippocampal LTP in the dystrophin-deficient Dmdmdx mouse. Neurobiol. Dis. 2004, 17, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, M.; Zushida, K.; Yoshida, M.; Maekawa, M.; Kamichi, S.; Yoshida, M.; Sahara, Y.; Yuasa, S.; Takeda, S.; Wada, K. A deficit of brain dystrophin impairs specific amygdala GABAergic transmission and enhances defensive behaviour in mice. Brain J. Neurol. 2009, 132, 124–135. [Google Scholar] [CrossRef] [PubMed]
- Hinton, V.J.; Fee, R.J.; De Vivo, D.C.; Goldstein, E. Poor facial affect recognition among boys with Duchenne muscular dystrophy. J. Autism Dev. Disord. 2007, 37, 1925–1933. [Google Scholar] [CrossRef] [PubMed]
- Vaillend, C.; Rendon, A.; Misslin, R.; Ungerer, A. Influence of dystrophin-gene mutation on mdx mouse behavior. I. Retention deficits at long delays in spontaneous alternation and barpressing tasks. Behav. Genet. 1995, 25, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Vaillend, C.; Ungerer, A. Behavioral characterization of mdx3cv mice deficient in C-terminal dystrophins. Neuromuscul. Disord. 1999, 9, 296–304. [Google Scholar] [CrossRef]
- Hinton, V.J.; De Vivo, D.C.; Nereo, N.E.; Goldstein, E.; Sterm, Y. Selective deficits in verbal working memory associated with a known genetic etiology: The neuropsychological profile of Duchenne muscular dystrophy. J. Int. Neuropsychol. Soc. 2001, 7, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Lidov, H.G. Dystrophin in the nervous system. Brain Pathol. 1996, 6, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Desguerre, I.; Christov, C.; Mayer, M.; Zeller, R.; Becane, H.M.; Bastuji-Garin, S.; Leturcq, F.; Chiron, C.; Chelly, J.; Gherardi, R.K. Clinical heterogeneity of Duchenne muscular dystrophy (DMD): Definition of sub-phenotypes and predictive criteria by long-term follow-up. PLoS ONE 2009, 4, e4347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howard, P.L.; Dally, G.Y.; Ditta, S.D.; Austin, R.C.; Worton, R.G.; Klamut, H.J.; Ray, P.N. Dystrophin isoforms DP71 and DP427 have distinct roles in myogenic cells. Muscle Nerve 1999, 22, 16–27. [Google Scholar] [CrossRef]
- Nudel, U.; Zuk, D.; Einat, P.; Zeelon, E.; Levy, Z.; Neuman, S.; Yaffe, D. Duchenne muscular dystrophy gene product is not identical in muscle and brain. Nature 1989, 337, 76–78. [Google Scholar] [CrossRef] [PubMed]
- Gorecki, D.C.; Monaco, A.P.; Derry, J.M.; Walker, A.P.; Barnard, E.A.; Barnard, P.J. Expression of four alternative dystrophin transcripts in brain regions regulated by different promoters. Hum. Mol. Genet. 1992, 1, 505–510. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, V.N.; Nguyen, T.M.; Morris, G.E.; Karges, W.; Pillers, D.A.; Ray, P.N. A novel dystrophin isoform is required for normal retinal electrophysiology. Hum. Mol. Genet. 1995, 4, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Chamova, T.; Guergueltcheva, V.; Raycheva, M.; Todorov, T.; Genova, J.; Bichev, S.; Bojinova, V.; Mitev, V.; Tournev, I.; Todorova, A. Association between loss of dp140 and cognitive impairment in duchenne and becker dystrophies. Balkan J. Med. Genet. BJMG 2013, 16, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Byers, T.J.; Lidov, H.G.; Kunkel, L.M. An alternative dystrophin transcript specific to peripheral nerve. Nat. Genet. 1993, 4, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Rapaport, D.; Fuchs, O.; Nudel, U.; Yaffe, D. Expression of the Duchenne muscular dystrophy gene products in embryonic stem cells and their differentiated derivatives. J. Biol. Chem. 1992, 267, 21289–21292. [Google Scholar] [PubMed]
- Tozawa, T.; Itoh, K.; Yaoi, T.; Tando, S.; Umekage, M.; Dai, H.; Hosoi, H.; Fushiki, S. The shortest isoform of dystrophin (Dp40) interacts with a group of presynaptic proteins to form a presumptive novel complex in the mouse brain. Mol. Neurobiol. 2012, 45, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, T.; Itoh, K.; Yaoi, T.; Fushiki, S. Somatodendritic and excitatory postsynaptic distribution of neuron-type dystrophin isoform, Dp40, in hippocampal neurons. Biochem. Biophys. Res. Commun. 2014, 452, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Sarig, R.; Mezger-Lallemand, V.; Gitelman, I.; Davis, C.; Fuchs, O.; Yaffe, D.; Nudel, U. Targeted inactivation of Dp71, the major non-muscle product of the DMD gene: Differential activity of the Dp71 promoter during development. Hum. Mol. Genet. 1999, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- El Mathari, B.; Sene, A.; Charles-Messance, H.; Vacca, O.; Guillonneau, X.; Grepin, C.; Sennlaub, F.; Sahel, J.A.; Rendon, A.; Tadayoni, R. Dystrophin Dp71 gene deletion induces retinal vascular inflammation and capillary degeneration. Hum. Mol. Genet. 2015, 24, 3939–3947. [Google Scholar] [CrossRef] [PubMed]
- Hopf, F.W.; Steinhardt, R.A. Regulation of intracellular free calcium in normal and dystrophic mouse cerebellar neurons. Brain Res. 1992, 578, 49–54. [Google Scholar] [CrossRef]
- Daoud, F.; Candelario-Martinez, A.; Billard, J.M.; Avital, A.; Khelfaoui, M.; Rozenvald, Y.; Guegan, M.; Mornet, D.; Jaillard, D.; Nudel, U.; et al. Role of mental retardation-associated dystrophin-gene product Dp71 in excitatory synapse organization, synaptic plasticity and behavioral functions. PLoS ONE 2009, 4, e6574. [Google Scholar] [CrossRef] [PubMed]
- Blake, D.J.; Hawkes, R.; Benson, M.A.; Beesley, P.W. Different dystrophin-like complexes are expressed in neurons and glia. J. Cell Biol. 1999, 147, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Tamma, R.; Annese, T.; Capogrosso, R.F.; Cozzoli, A.; Benagiano, V.; Sblendorio, V.; Ruggieri, S.; Crivellato, E.; Specchia, G.; Ribatti, D.; et al. Effects of prednisolone on the dystrophin-associated proteins in the blood-brain barrier and skeletal muscle of dystrophic mdx mice. Lab. Investig. 2013, 93, 592–610. [Google Scholar] [CrossRef] [PubMed]
- Nico, B.; Frigeri, A.; Nicchia, G.P.; Quondamatteo, F.; Herken, R.; Errede, M.; Ribatti, D.; Svelto, M.; Roncali, L. Role of aquaporin-4 water channel in the development and integrity of the blood-brain barrier. J. Cell Sci. 2001, 114, 1297–1307. [Google Scholar] [PubMed]
- Jancsik, V.; Hajos, F. The demonstration of immunoreactive dystrophin and its developmental expression in perivascular astrocytes. Brain Res. 1999, 831, 200–205. [Google Scholar] [CrossRef]
- Guadagno, E.; Moukhles, H. Laminin-induced aggregation of the inwardly rectifying potassium channel, Kir4.1, and the water-permeable channel, AQP4, via a dystroglycan-containing complex in astrocytes. Glia 2004, 47, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Fort, P.E.; Sene, A.; Pannicke, T.; Roux, M.J.; Forster, V.; Mornet, D.; Nudel, U.; Yaffe, D.; Reichenbach, A.; Sahel, J.A.; et al. Kir4.1 and AQP4 associate with Dp71- and utrophin-DAPs complexes in specific and defined microdomains of Muller retinal glial cell membrane. Glia 2008, 56, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Sene, A.; Tadayoni, R.; Pannicke, T.; Wurm, A.; El Mathari, B.; Benard, R.; Roux, M.J.; Yaffe, D.; Mornet, D.; Reichenbach, A.; et al. Functional implication of Dp71 in osmoregulation and vascular permeability of the retina. PLoS ONE 2009, 4, e7329. [Google Scholar] [CrossRef] [PubMed]
- Górecki, D.C.; Lukasiuk, K.; Szklarczyk, A.; Kaczmarek, L. Kainate-evoked changes in dystrophin messenger RNA levels in the rat hippocampus. Neuroscience 1998, 22, 4274–4285. [Google Scholar] [CrossRef]
- Cerna, J.; Cerecedo, D.; Ortega, A.; García-Sierra, F.; Centeno, F.; Garrido, E.; Mornet, D.; Cisnero, B. Dystrophin Dp71f associates with the β1-integrin adhesion complex to modulate PC12 cell adhesion. J. Mol. Biol. 2006, 362, 954–965. [Google Scholar] [CrossRef] [PubMed]
- Levi, S.; Grady, R.M.; Henry, M.D.; Campbell, K.P.; Sanes, J.R.; Craig, A.M. Dystroglycan is selectively associated with inhibitory GABAergic synapses but is dispensable for their differentiation. J. Neurosci. 2002, 22, 4274–4285. [Google Scholar] [PubMed]
- Benabdesselam, R.; Sene, A.; Raison, D.; Benmessaoud-Mesbah, O.; Ayad, G.; Mornet, D.; Yaffe, D.; Rendon, A.; Hardin-Pouzet, H.; Dorbani-Mamine, L. A deficit of brain dystrophin 71 impairs hypothalamic osmostat. J. Neurosci. Res. 2010, 88, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Benabdesselam, R.; Dorbani-Mamine, L.; Benmessaoud-Mesbah, O.; Rendon, A.; Mhaouty-Kodja, S.; Hardin-Pouzet, H. Dp71 gene disruption alters the composition of the dystrophin-associated protein complex and neuronal nitric oxide synthase expression in the hypothalamic supraoptic and paraventricular nuclei. J. Endocrinol 2012, 213, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Cerna, J.; Osuna-Castro, J.A.; Muñiz, J.; Mornet, D.; García-Sierra, F.; Cisneros, B. Dystrophin Dp71f associates with components of the beta1-integrin adhesion complex in PC12 cell neuritis. Acta Neurol. Belg. 2009, 109, 132–135. [Google Scholar] [PubMed]
- Enriquez-Aragon, J.A.; Cerna-Cortes, J.; Bermudez de Leon, M.; Garcia-Sierra, F.; Gonzalez, E.; Mornet, D.; Cisneros, B. Dystrophin Dp71 in PC12 cell adhesion. Neuroreport 2005, 16, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Villarreal-Silva, M.; Centeno-Cruz, F.; Suarez-Sanchez, R.; Garrido, E.; Cisneros, B. Knockdown of dystrophin Dp71 impairs PC12 cells cycle: Localization in the spindle and cytokinesis structures implies a role for Dp71 in cell division. PLoS ONE 2011, 6, e23504. [Google Scholar] [CrossRef] [PubMed]
- Fuentes-Mera, L.; Rodriguez-Munoz, R.; Gonzalez-Ramirez, R.; Garcia-Sierra, F.; Gonzalez, E.; Mornet, D.; Cisneros, B. Characterization of a novel Dp71 dystrophin-associated protein complex (DAPC) present in the nucleus of HeLa cells: Members of the nuclear DAPC associate with the nuclear matrix. Exp. Cell Res. 2006, 312, 3023–3035. [Google Scholar] [CrossRef] [PubMed]
- González-Ramírez, R.; Morales-Lázaro, S.L.; Tapia-Ramírez, V.; Mornet, D.; Cisneros, B. Nuclear and nuclear envelope localization of dystrophin Dp71 and dystrophin-associated proteins (DAPs) in the C2C12 muscle cells: DAPs nuclear localization is modulated during myogenesis. J. Cell. Biochem. 2008, 105, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Suárez-Sánchez, R.; Aguilar, A.; Wagstaff, K.M.; Velez, G.; Azuara-Medina, P.M.; Gomez, P.; Vásquez-Limeta, A.; Hernández-Hernández, O.; Lieu, K.G.; Jans, D.A.; et al. Nucleocytoplasmic shuttling of the Duchenne muscular dystrophy gene product dystrophin Dp71d is dependent on the importin α/β and CRM1 nuclear transporters and microtubule motor dynein. Biochim. Biophys. Acta 2014, 1843, 985–1001. [Google Scholar] [CrossRef] [PubMed]
- Kreis, R.; Wingeier, K.; Vermathen, P.; Giger, E.; Joncourt, F.; Zwygart, K.; Kaufmann, F.; Boesch, C.; Steinlin, M. Brain metabolite composition in relation to cognitive function and dystrophin mutations in boys with Duchenne muscular dystrophy. NMR Biomed. 2011, 24, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.E.; Simmons, C.; Nguyen, T.M. Apo-Dystrophins (DP140 and DP71) and Dystrophin Splicing Isoforms in Developing Brain. Biochem. Biophys. Res. Commun. 1995, 215, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Sicinski, P.; Geng, Y.; Ryder-Cook, A.S.; Barnard, E.A.; Darlison, M.G.; Barnard, P.J. The molecular basis of muscular dystrophy in the mdx mouse: A point mutation. Science 1989, 244, 1578–1580. [Google Scholar] [CrossRef] [PubMed]
- Dubowitz, V.; Crome, L. The central nervous system in Duchenne muscular dystrophy. Brain 1969, 92, 805–808. [Google Scholar] [CrossRef] [PubMed]
- Mehler, M.F.; Haas, K.Z.; Kessler, J.A.; Stanton, P.K. Enhanced sensitivity of hippocampal pyramidal neurons from mdx mice to hypoxia-induced loss of synaptic trans-mission. Proc. Natl. Acad. Sci. USA 1992, 89, 289–292. [Google Scholar] [CrossRef]
- Anderson, J.L.; Head, S.I.; Morley, J.W. Long- term depression is reduced in cerebellar Purkinje cells of dystrophin-deficient mdx mice. Brain Res. 2004, 1019, 289–292. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.J.; Quarta, E.; Fulgenzi, G.; Minciacchi, D. Acetylcholine, GABA and neuronal networks: A working hypothesis for compensations in the dystrophic brain. Brain Res. Bull. 2015, 110, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Del Tongo, C.; Carretta, D.; Fulgenzi, G.; Catini, C.; Minciacchi, D. Parvalbumin-positive GABAergic interneurons are increased in the dorsal hippocampus of the dystrophic mdx mouse. Acta Neuropathol. 2009, 118, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Graciotti, L.; Minelli, A.; Minciacchi, D.; Procopio, A.; Fulgenzi, G. GABAergic miniature spontaneous activity is increased in the CA1 hippocampal region of dystrophic mdx mice. Neuromuscul. Disord. 2008, 18, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Alkondon, M.; Albuquerque, E.X. Nicotinic acetylcholine receptor alpha7 and alpha4beta2 subtypes differentially control GABAergic input to CA1 neurons in rat hippocampus. J. Neurophysiol. 2001, 86, 3043–3055. [Google Scholar] [PubMed]
- Miranda, R.; Sébrié, C.; Degrouard, J.; Gillet, B.; Jaillard, D.; Laroche, S.; Vaillend, C. Reorganizationof inhibitory synapses and increased PSD length of perforated excitatory synapses in hippocampal area CA1 of dystrophin- deficient mdx mice. Cereb. Cortex 2009, 19, 876–888. [Google Scholar] [CrossRef] [PubMed]
- Ghedini, P.C.; Avellar, M.C.; De Lima, T.C.; Lima-Landman, M.T.; Lapa, A.J.; Souccar, C. Quantitative changes of nicotinic receptors in the hippocampus of dystrophin-deficient mice. Brain Res. 2012, 1483, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Craig, A.M.; Kang, Y. Neurexin-neuroligin signaling in synapse development. Curr. Opin. Neurobiol. 2007, 17, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.L.; Rabinowitz, A.; Chen, Y.C.; Yokota, T.; Yin, H.; Alter, J.; Jadoon, A.; Bou-Gharios, G.; Partridge, T. Systemic delivery of antisense oligoribonucleotide restores dystrophin expression in body-wide skeletal muscles. Proc. Natl. Acad. Sci. USA 2005, 102, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Duddy, W.; Echigoya, Y.; Kolski, H. Exon skipping for nonsense mutations in Duchenne muscular dystrophy: Too many mutations, too few patients? Expert Opin. Biol. Ther. 2012, 12, 1141–1152. [Google Scholar] [CrossRef] [PubMed]
- Touznik, A.; Lee, J.J.; Yokota, T. New developments in exon skipping and splice modulation therapies for neuromuscular diseases. Expert Opin. Biol. Ther. 2014, 14, 809–819. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nichols, B.; Takeda, S.; Yokota, T. Nonmechanical Roles of Dystrophin and Associated Proteins in Exercise, Neuromuscular Junctions, and Brains. Brain Sci. 2015, 5, 275-298. https://doi.org/10.3390/brainsci5030275
Nichols B, Takeda S, Yokota T. Nonmechanical Roles of Dystrophin and Associated Proteins in Exercise, Neuromuscular Junctions, and Brains. Brain Sciences. 2015; 5(3):275-298. https://doi.org/10.3390/brainsci5030275
Chicago/Turabian StyleNichols, Bailey, Shin'ichi Takeda, and Toshifumi Yokota. 2015. "Nonmechanical Roles of Dystrophin and Associated Proteins in Exercise, Neuromuscular Junctions, and Brains" Brain Sciences 5, no. 3: 275-298. https://doi.org/10.3390/brainsci5030275