Sleep Patterns and Homeostatic Mechanisms in Adolescent Mice

Abstract
:1. Introduction
2. Results
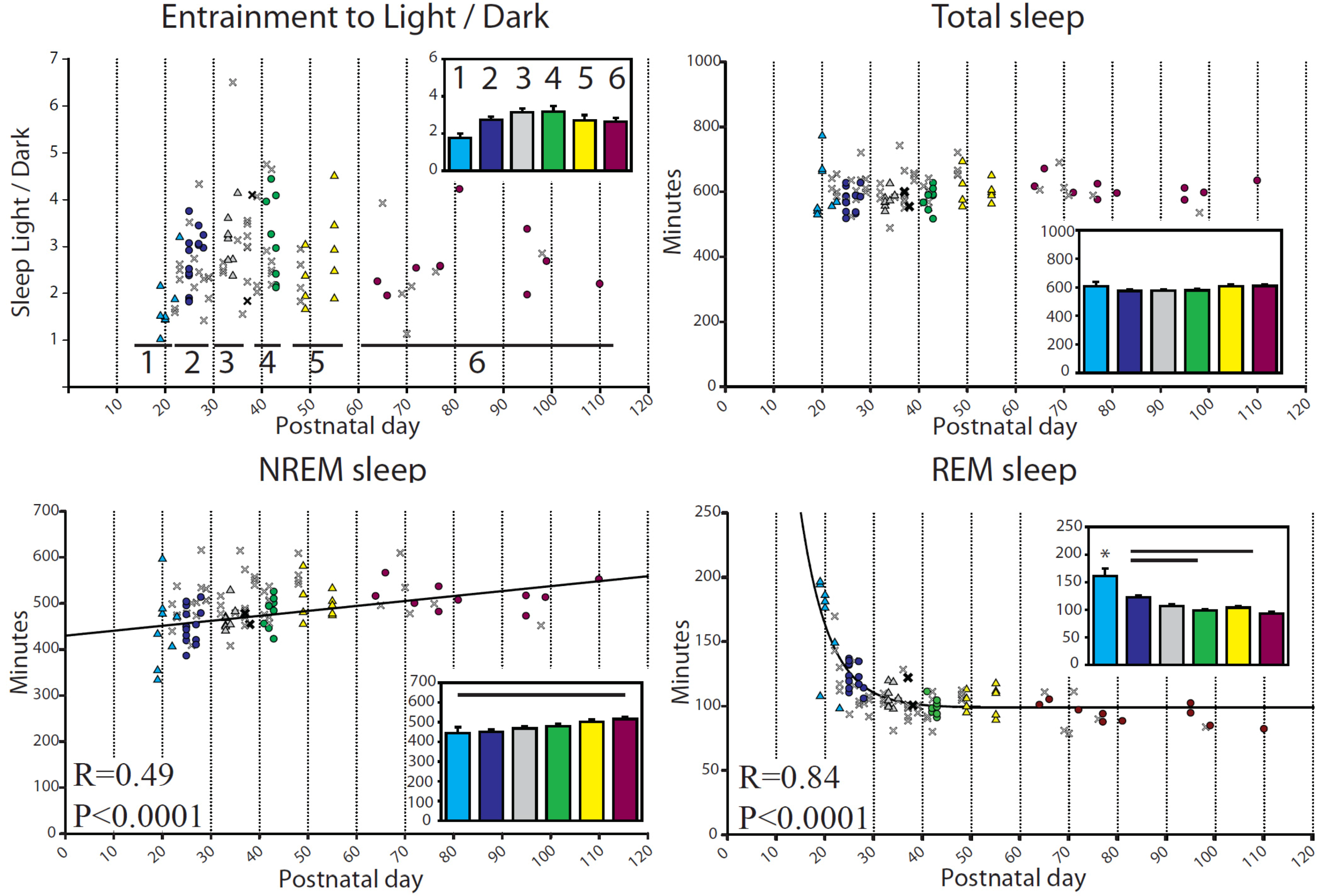
2.1. Sleep in Baseline
2.1.1. Sleep-Wake Patterns during Baseline



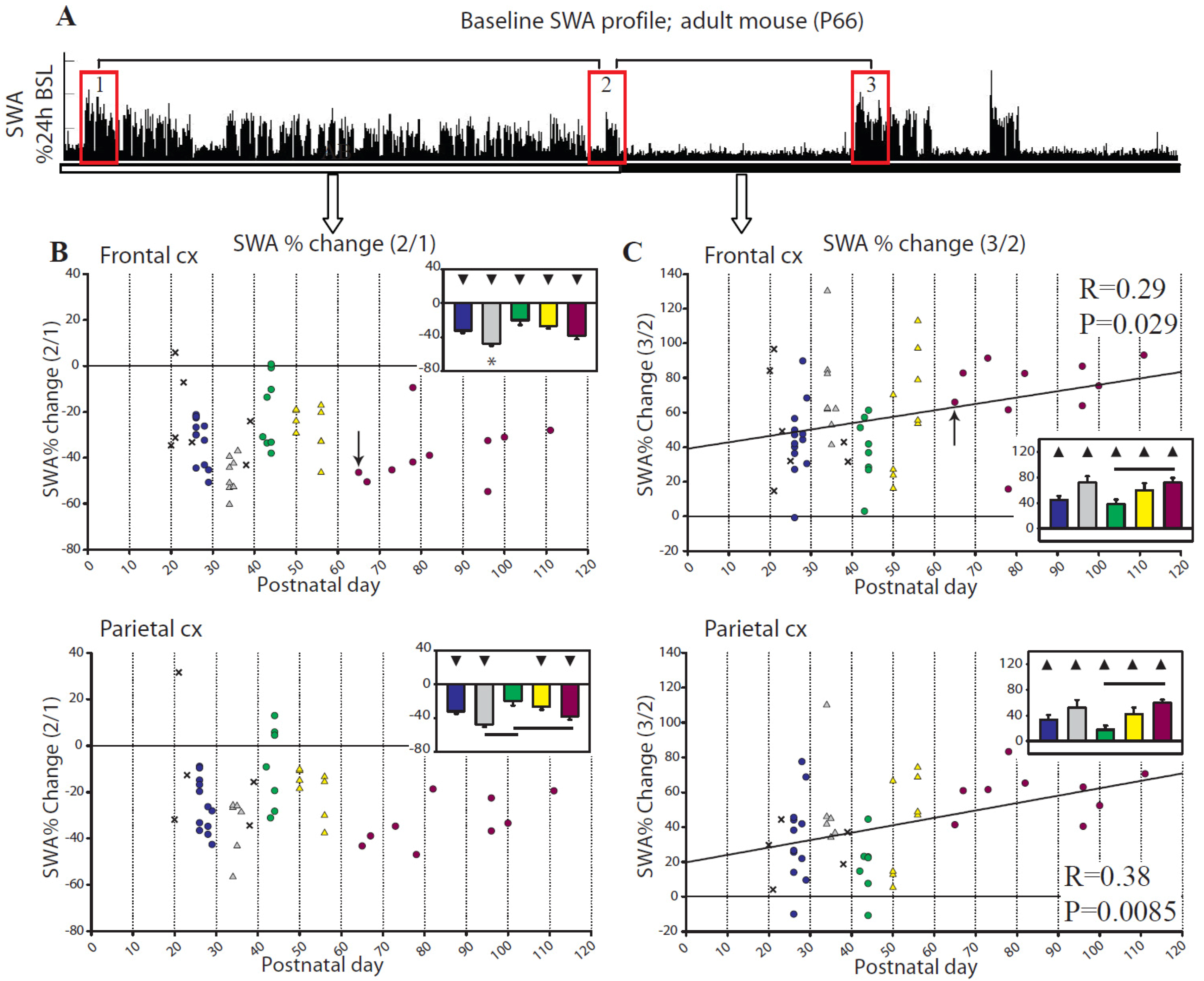
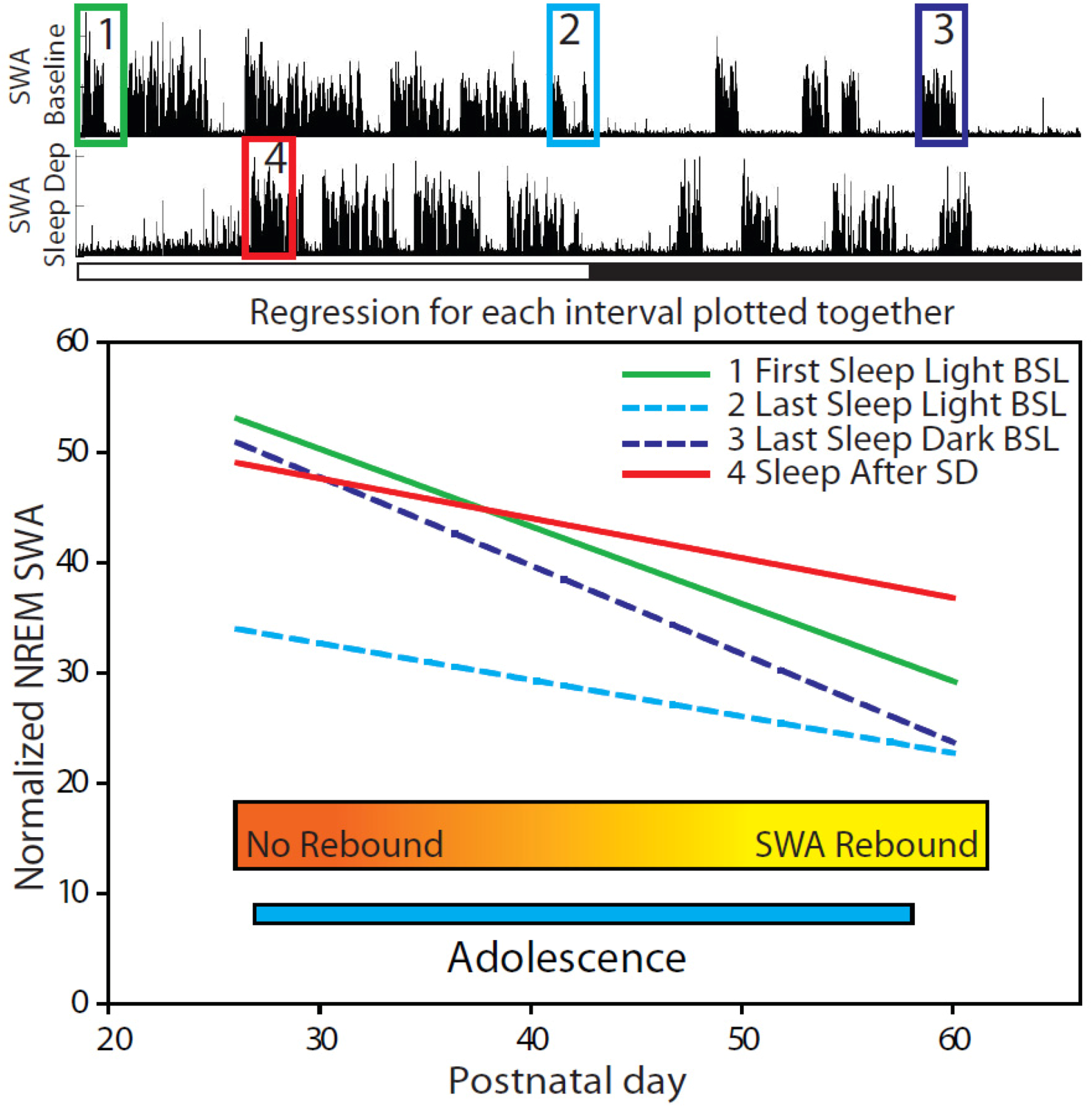
2.1.2. Changes in EEG Power Spectrum during Baseline

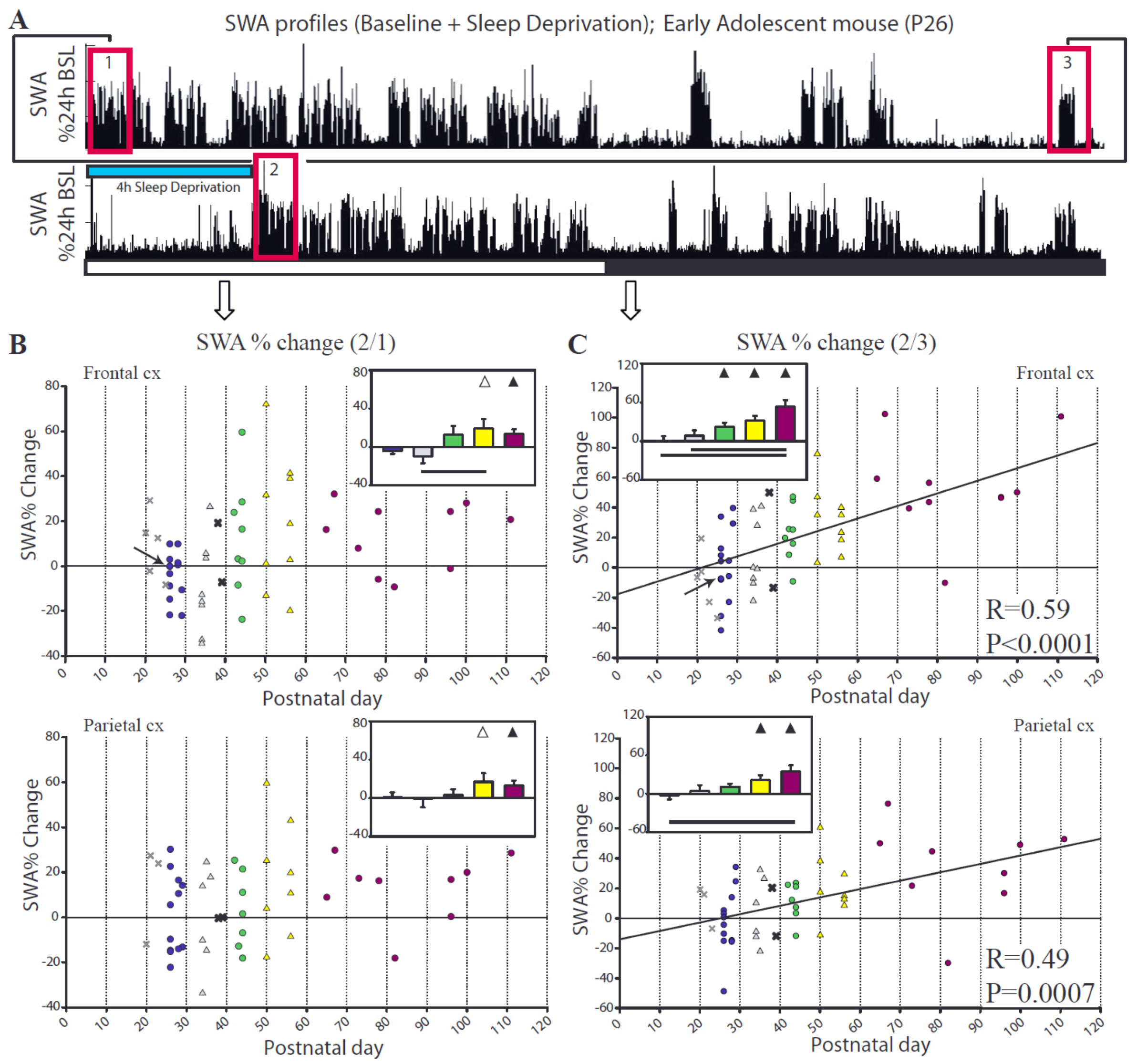
2.2. Sleep after Sleep Deprivation
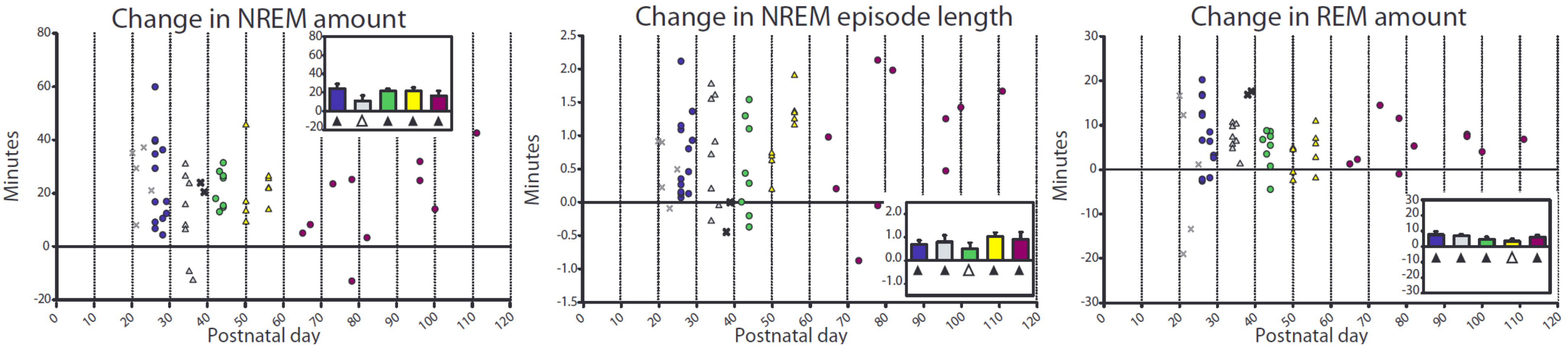
2.2.1. Changes in Sleep duration after Sleep Deprivation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age Group | P26–29 | P34–36 | P41–44 | P50–56 | Adults |
|---|---|---|---|---|---|
| NREM Deficit (min) | −38.13 | −44.60 | −43.00 | −59.36 | −66.91 |
| (11.11) | (13.96) | (15.77) | (10.66) | (11.09) | |
| REM Deficit (min) | −11.68 | −8.32 | −12.75 | −12.58 | −8.72 |
| (3.04) | (2.56) | (3.80) | (3.26) | (3.68) | |
| Change in REM episode length (min) | 0.077 | 0.097 | 0.056 | 0.23 | 0.29 |
| (0.059) | (0.044) | (0.068) | (0.081) | (0.069) | |
| Change in Brief Arousals | −0.0043 | −0.0046 | −0.0017 | −0.0020 | −0.0054 |
| (number per min sleep 1st 4 h) | (0.0014) | (0.0023) | (0.0031) | (0.0021) | (0.0018) |
| Sleep during deprivation (min) | 1.46 | 0.64 | 0.70 | 0.85 | 0.20 |
| (0.16) | (0.26) | (0.29) | (0.34) | (0.16) | |
| Sleep attempts (number during deprivation) | 21.77 | 7.13 | 9.13 | 6.78 | 2.00 |
| (2.33) | (3.78) | (3.34) | (1.76) | (1.57) |
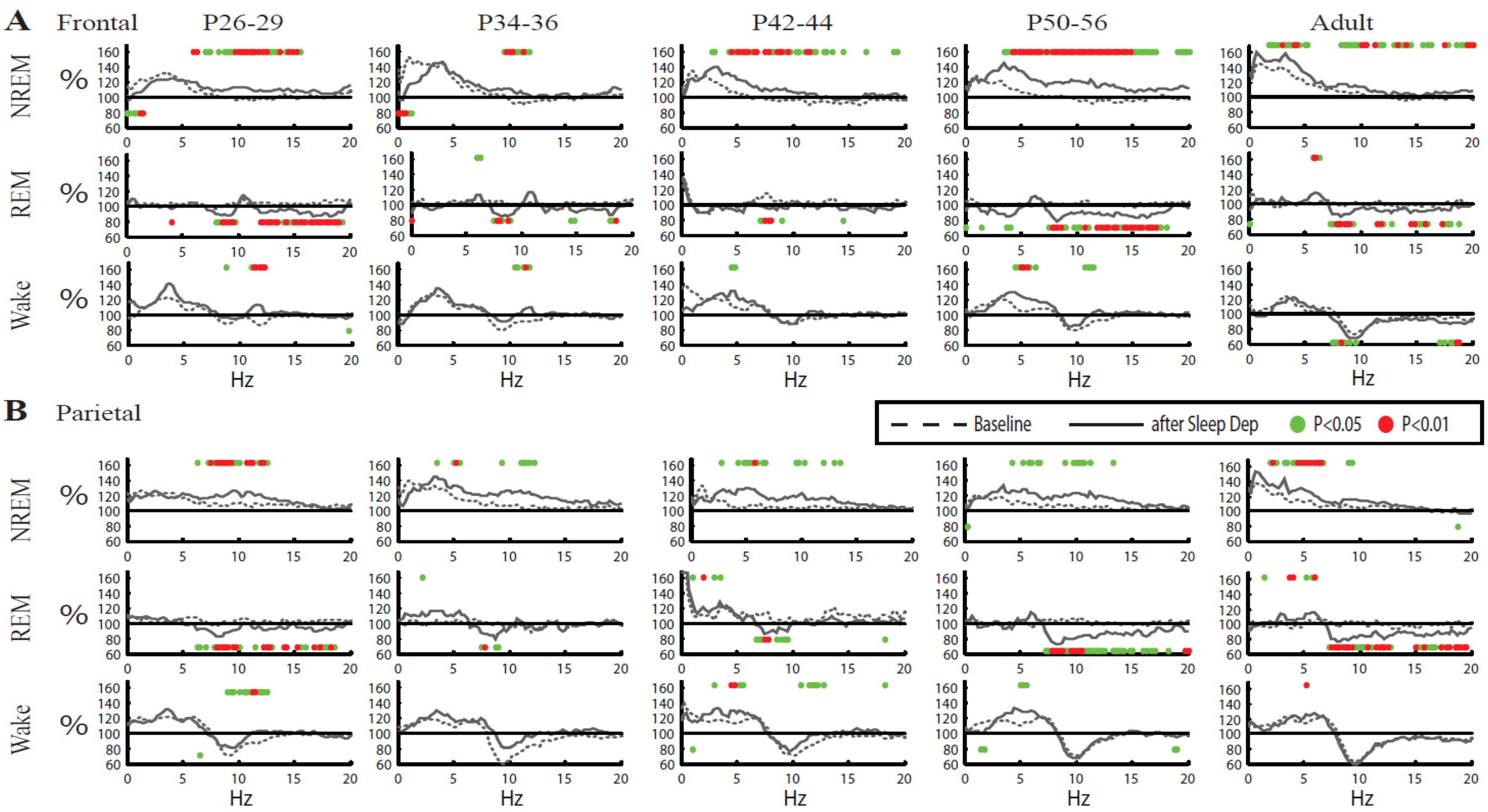
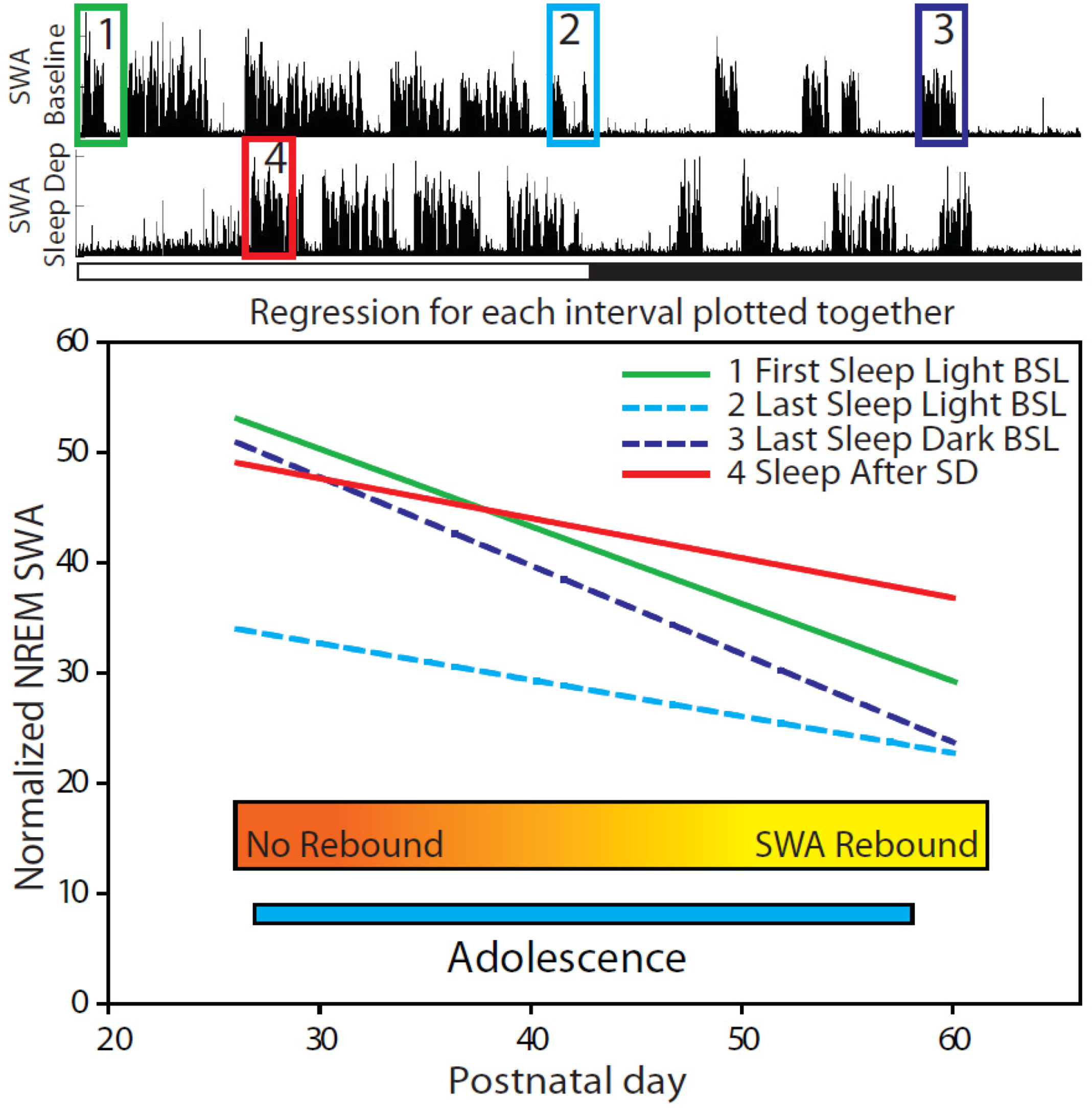
2.2.2. Changes in SWA after Sleep Deprivation

2.2.3. Parameters Affecting SWA Changes after Sleep Deprivation
 | |||
| (A) Frontal: Predictors for SWA rebound (5/1); % change SWA (5/1) = B0 + (predictors) | p | AIC | R2 |
| Weight-adjusted age (using Gompertz fit) | 0.00507 | 362 | 0.189 |
| Recovery days from surgery | 0.43 | 370 | 0.0164 |
| REM/NREM amount (12 h of recovery) | 0.0071 | 363 | 0.175 |
| n of sleep attempts during sleep deprivation | 0.053 | 367 | 0.0953 |
| Sleep during sleep deprivation (min/4 h) | 0.0582 | 367 | 0.0913 |
| Brief arousals (n/min of sleep, first 4 h of recovery) | 0.31 | 369 | 0.0273 |
| Increase in wake SWA (1–4 Hz) during sleep deprivation | 0.76 | 370 | 0.0249 |
| Slow wave energy (SWE = SWA × time) during deprivation (wake and NREM) | 0.84 | 371 | 0.0011 |
| Time spent awake since last sleep | 0.91 | 371 | 0.00033 |
| NREM SWA decline in baseline (1/2) | 0.000154 | 355 | 0.317 |
| NREM SWA decline in baseline (1/2) + | <0.0001 | 352 | 0.395 |
| Increase in wake alpha (8–12 Hz) | 0.0352 | ||
| NREM SWA decline in baseline (1/2) + | 0.0009 | 352 | 0.400 |
| Weight-adjusted age | 0.0297 | ||
| NREM SWA decline in baseline (1/2) + | 0.00043 | 352 | 0.431 |
| Increase in wake alpha (8–12 Hz) + | 0.17293 | ||
| Weight-adjusted age | 0.14348 | ||
| (B) Frontal: Predictors for SWA change from before to after sleep deprivation (5/4); % change SWA (5/4) = B0 + (parameters) | p | AIC | R2 |
| Weight-adjusted age (using Gompertz fit) | 0.00025 | 364 | 0.30 |
| Recovery days from surgery | 0.64 | 378 | 0.00595 |
| REM/NREM amount (12 h of recovery) | 0.0036 | 369 | 0.20 |
| Length of time since last sleep | 0.00462 | 369 | 0.19 |
| Increase in wake alpha (8–12 Hz) | 0.00013 | 362 | 0.324 |
| NREM SWA increase in baseline (4/2) | <0.0001 | 355 | 0.44 |
| NREM SWA (4/2) + | 0.00017 | 350 | 0.53 |
| Weight-adjusted age | 0.01467 | ||
| NREM SWA (4/2) + | 0.000037 | 346 | 0.575 |
| Increase in wake alpha (8–12 Hz) | 0.0015 | ||
| NREM SWA (4/2) + | 0.00044 | 344 | 0.615 |
| Increase in wake alpha (8–12 Hz) + | 0.00618 | ||
| Weight-adjusted age | 0.06158 | ||
| NR SWA (4/2) × Increase wake alpha (8–12 Hz) + | 0.022 | 340 | 0.669 |
| Weight-adjusted age | 0.071 | ||
| (C) Frontal: Models Predicting SWA % change (3/2); % change SWA (3/2) = B0 + (parameters) | p | AIC | R2 |
| Weight-adjusted age (using Gompertz fit) | 0.913 | 383 | 0.0003 |
| Recovery days from surgery | 0.39 | 382 | 0.0197 |
| Length of time since last sleep | 0.016 | 376 | 0.14 |
| Increase in wake alpha (8–12 Hz) | 0.018 | 377 | 0.139 |
| NREM SWA decline in baseline (1/2) | 0.000047 | 365 | 0.357 |
| NREM SWA decline in baseline (1/2) + | 0.00039 | 365 | 0.390 |
| Increase in wake alpha | 0.1654 | ||
| NREM SWA decline in baseline (1/2) + | 0.000095 | 362 | 0.435 |
| Length of time since last sleep | 0.029 | ||
| NREM SWA (1/2) × Length of time since last sleep | 0.029 | 358 | 0.506 |
| (D) Parietal: Predictors for SWA rebound (5/1); % change SWA (5/1) = B0 + (predictors) | p | AIC | R2 |
| Weight-adjusted age (using Gompertz fit) | 0.0045 | 317 | 0.219 |
| Recovery days from surgery | 0.46 | 325 | 0.0165 |
| REM/NREM amount (12 h of recovery) | 0.0024 | 315 | 0.246 |
| N of sleep attempts during sleep deprivation | 0.0100 | 318 | 0.185 |
| Sleep during sleep deprivation (min/4 h) | 0.011 | 318 | 0.180 |
| Brief arousals (N/min of sleep, first 4 h of recovery) | 0.62 | 325 | 0.0076 |
| Increase in wake SWA (1–4 Hz) during sleep deprivation | 0.581 | 325 | 0.0093 |
| Slow wave energy (SWE = SWA × time) during deprivation (wake and NREM) | 0.801 | 325 | 0.0020 |
| Time spent awake since last sleep | 0.967 | 325 | 0.0000 |
| NREM SWA decline in baseline (1/2) | 0.0015 | 314 | 0.266 |
| NREM SWA decline in baseline (1/2) + | 0.00048 | 307 | 0.446 |
| N of sleep attempts during sleep deprivation | 0.00292 | ||
| NREM SWA decline in baseline (1/2) + | 0.012 | 312 | 0.360 |
| Weight-adjusted age | 0.038 | ||
| NREM SWA decline in baseline (1/2) + | 0.0033 | 307 | 0.470 |
| N of sleep attempts during sleep deprivation + | 0.0164 | ||
| Weight-adjusted age | 0.2466 | ||
| (E) Parietal: Predictors for SWA change from before to after sleep deprivation (5/4); % change SWA (5/4) = B0 + (parameters) | p | AIC | R2 |
| Weight-adjusted age (using Gompertz fit) | 0.0004 | 319 | 0.322 |
| Recovery days from surgery | 0.51 | 332 | 0.0135 |
| REM/NREM amount (12 h of recovery) | 0.0004 | 319 | 0.320 |
| Length of time since last sleep | 0.0046 | 324 | 0.219 |
| Increase in wake alpha (8–12 Hz) | 0.00059 | 320 | 0.304 |
| NREM SWA increase in baseline (4/2) | 0.00056 | 320 | 0.306 |
| NREM SWA (4/2) + | 0.034 | 316 | 0.412 |
| Weight-adjusted age | 0.023 | ||
| NREM SWA (4/2) + | 0.0039 | 313 | 0.466 |
| Increase in wake alpha (8–12 Hz) | 0.0041 | ||
| NREM SWA (4/2) + | 0.063 | 311 | 0.526 |
| Increase in wake alpha (8–12 Hz) + | 0.010 | ||
| Weight-adjusted age | 0.057 | ||
| NR SWA (4/2) × Increase wake alpha (8-12 Hz) + | 0.100 | 311 | 0.567 |
| Weight-adjusted age | 0.034 | ||
| (F) Parietal: Models Predicting SWA % change (3/2); % change SWA (3/2) = B0 + (parameters) | p | AIC | R2 |
| Weight-adjusted age (using Gompertz fit) | 0.953 | 334 | 0.000 |
| Recovery days from surgery | 0.47 | 334 | 0.016 |
| Length of time since last sleep | 0.0060 | 326 | 0.207 |
| Increase in wake alpha (8–12 Hz) | 0.133 | 332 | 0.067 |
| NREM SWA decline in baseline (1/2) | 0.0018 | 324 | 0.258 |
| NREM SWA decline in baseline (1/2) + | 0.0061 | 326 | 0.265 |
| Increase in wake alpha | 0.5839 | ||
| NREM SWA decline in baseline (1/2) + | 0.00084 | 316 | 0.444 |
| Length of time since last sleep | 0.00260 | ||
| NREM SWA (1/2) × Length of time since last sleep | 0.026 | 312 | 0.527 |
2.2.4. Other Changes in the EEG Power Spectrum after Sleep Deprivation

3. Discussion
4. Experimental Section
4.1. Recordings of Sleep and Locomotor Activity and Sleep Deprivation
4.2. Weight-Adjusted Age
4.3. Statistical Analysis
5. Conclusions
Acknowledgments
Conflict of Interest
References
- Spear, L.P. The adolescent brain and age-related behavioral manifestations. Neurosci. Biobehav. Rev. 2000, 24, 417–463. [Google Scholar] [CrossRef]
- Laviola, G.; Macri, S.; Morley-Fletcher, S.; Adriani, W. Risk-taking behavior in adolescent mice: Psychobiological determinants and early epigenetic influence. Neurosci. Biobehav. Rev. 2003, 27, 19–31. [Google Scholar] [CrossRef]
- Adriani, W.; Macri, S.; Pacifici, R.; Laviola, G. Peculiar vulnerability to nicotine oral self-administration in mice during early adolescence. Neuropsychopharmacology 2002, 27, 212–224. [Google Scholar] [CrossRef]
- Spear, L.P.; Brake, S.C. Periadolescence: Age-dependent behavior and psychopharmacological responsivity in rats. Dev. Psychobiol. 1983, 16, 83–109. [Google Scholar] [CrossRef]
- Kellogg, C.K.; Awatramani, G.B.; Piekut, D.T. Adolescent development alters stressor-induced Fos immunoreactivity in rat brain. Neuroscience 1998, 83, 681–689. [Google Scholar] [CrossRef]
- Molnar, Z.; Adams, R.; Blakemore, C. Mechanisms underlying the early establishment of thalamocortical connections in the rat. J. Neurosci. 1998, 18, 5723–5745. [Google Scholar]
- Aghajanian, G.K.; Bloom, F.E. The formation of synaptic junctions in developing rat brain: A quantitative electron microscopic study. Brain Res. 1967, 6, 716–727. [Google Scholar] [CrossRef]
- Ashby, M.C.; Isaac, J.T. Maturation of a recurrent excitatory neocortical circuit by experience-dependent unsilencing of newly formed dendritic spines. Neuron 2011, 70, 510–521. [Google Scholar] [CrossRef]
- Caley, D.W.; Maxwell, D.S. An electron microscopic study of neurons during postnatal development of the rat cerebral cortex. J. Comp. Neurol. 1968, 133, 17–44. [Google Scholar] [CrossRef]
- Caley, D.W.; Maxwell, D.S. Development of the blood vessels and extracellular spaces during postnatal maturation of rat cerebral cortex. J. Comp. Neurol. 1970, 138, 31–47. [Google Scholar] [CrossRef]
- Gramsbergen, A. The development of the EEG in the rat. Dev. Psychobiol. 1976, 9, 501–515. [Google Scholar] [CrossRef]
- Mirmiran, M.; Corner, M. Neuronal discharge patterns in the occipital cortex of developing rats during active and quiet sleep. Brain Res. 1982, 255, 37–48. [Google Scholar]
- Frank, M.G.; Heller, H.C. Development of diurnal organization of EEG slow-wave activity and slow-wave sleep in the rat. Am. J. Physiol. 1997, 273, R472–R478. [Google Scholar]
- Jouvet-Mounier, D.; Astic, L.; Lacote, D. Ontogenesis of the states of sleep in rat, cat, and guinea pig during the first postnatal month. Dev. Psychobiol. 1970, 2, 216–239. [Google Scholar] [CrossRef]
- Gramsbergen, A.; Schwartze, P.; Prechtl, H.F. The postnatal development of behavioral states in the rat. Dev. Psychobiol. 1970, 3, 267–280. [Google Scholar] [CrossRef]
- Daszuta, A.; Gambarelli, F. Early postnatal development of EEG and sleep-waking cycle in two inbred mouse strains. Brain Res. 1985, 354, 39–47. [Google Scholar]
- Alfoldi, P.; Tobler, I.; Borbely, A.A. Sleep regulation in rats during early development. Am. J. Physiol. 1990, 258, R634–R644. [Google Scholar]
- Vogel, G.W.; Feng, P.; Kinney, G.G. Ontogeny of REM sleep in rats: Possible implications for endogenous depression. Physiol. Behav. 2000, 68, 453–461. [Google Scholar] [CrossRef]
- Frank, M.G.; Morrissette, R.; Heller, H.C. Effects of sleep deprivation in neonatal rats. Am. J. Physiol. 1998, 275, R148–R157. [Google Scholar]
- Hairston, I.S.; Peyron, C.; Denning, D.P.; Ruby, N.F.; Flores, J.; Sapolsky, R.M.; Heller, H.C.; O’Hara, B.F. Sleep deprivation effects on growth factor expression in neonatal rats: A potential role for BDNF in the mediation of delta power. J. Neurophysiol. 2004, 91, 1586–1595. [Google Scholar] [CrossRef]
- Gvilia, I.; Suntsova, N.; Angara, B.; McGinty, D.; Szymusiak, R. Maturation of sleep homeostasis in developing rats: A role for preoptic area neurons. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R885–R894. [Google Scholar] [CrossRef]
- Huber, R.; Deboer, T.; Tobler, I. Effects of sleep deprivation on sleep and sleep EEG in three mouse strains: Empirical data and simulations. Brain Res. 2000, 857, 8–19. [Google Scholar] [CrossRef]
- Vyazovskiy, V.V.; Ruijgrok, G.; Deboer, T.; Tobler, I. Running wheel accessibility affects the regional electroencephalogram during sleep in mice. Cereb. Cortex 2006, 16, 328–336. [Google Scholar]
- Franken, P.; Chollet, D.; Tafti, M. The homeostatic regulation of sleep need is under genetic control. J. Neurosci. 2001, 21, 2610–2621. [Google Scholar]
- Huber, R.; Deboer, T.; Tobler, I. Topography of EEG dynamics after sleep deprivation in mice. J. Neurophysiol. 2000, 84, 1888–1893. [Google Scholar]
- Tobler, I.; Borbély, A.A. Sleep EEG in the rat as a function of prior waking. Electroencephalogr. Clin. Neurophysiol. 1986, 64, 74–76. [Google Scholar] [CrossRef]
- Deboer, T.; Tobler, I. Sleep regulation in the Djungarian hamster: Comparison of the dynamics leading to the slow-wave activity increase after sleep deprivation and daily torpor. Sleep 2003, 26, 567–572. [Google Scholar]
- Borbely, A.A.; Neuhaus, H.U. Sleep deprivation: Effects on sleep and EEG in the rat. J. Comp. Physiol. 1979, 133, 71–87. [Google Scholar] [CrossRef]
- Torsvall, L.; Akerstedt, T. Sleepiness on the job: Continuously measured EEG changes in train drivers. Electroencephalogr. Clin. Neurophysiol. 1987, 66, 502–511. [Google Scholar] [CrossRef]
- Cajochen, C.; Brunner, D.P.; Krauchi, K.; Graw, P.; Wirz-Justice, A. Power density in theta/alpha frequencies of the waking EEG progressively increases during sustained wakefulness. Sleep 1995, 18, 890–894. [Google Scholar]
- Aeschbach, D.; Matthews, J.R.; Postolache, T.T.; Jackson, M.A.; Giesen, H.A.; Wehr, T.A. Dynamics of the human EEG during prolonged wakefulness: Evidence for frequency-specific circadian and homeostatic influences. Neurosci. Lett. 1997, 239, 121–124. [Google Scholar] [CrossRef]
- Finelli, L.A.; Baumann, H.; Borbely, A.A.; Achermann, P. Dual electroencephalogram markers of human sleep homeostasis: Correlation between theta activity in waking and slow-wave activity in sleep. Neuroscience 2000, 101, 523–529. [Google Scholar] [CrossRef]
- Aeschbach, D.; Matthews, J.R.; Postolache, T.T.; Jackson, M.A.; Giesen, H.A.; Wehr, T.A. Two circadian rhythms in the human electroencephalogram during wakefulness. Am. J. Physiol. 1999, 277, R1771–R1779. [Google Scholar]
- Cajochen, C.; Khalsa, S.B.; Wyatt, J.K.; Czeisler, C.A.; Dijk, D.J. EEG and ocular correlates of circadian melatonin phase and human performance decrements during sleep loss. Am. J. Physiol. 1999, 277, R640–R649. [Google Scholar]
- Cajochen, C.; Wyatt, J.K.; Czeisler, C.A.; Dijk, D.J. Separation of circadian and wake duration-dependent modulation of EEG activation during wakefulness. Neuroscience 2002, 114, 1047–1060. [Google Scholar] [CrossRef]
- Vyazovskiy, V.V.; Tobler, I. Theta-activity in the waking EEG is a marker of sleep propensity in the rat. Brain Res. 2005, 1050, 64–71. [Google Scholar] [CrossRef]
- Retey, J.V.; Adam, M.; Gottselig, J.M.; Khatami, R.; Durr, R.; Achermann, P.; Landolt, H.P. Adenosinergic mechanisms contribute to individual differences in sleep deprivation-induced changes in neurobehavioral function and brain rhythmic activity. J. Neurosci. 2006, 26, 10472–10479. [Google Scholar]
- Tinguely, G.; Finelli, L.A.; Landolt, H.P.; Borbely, A.A.; Achermann, P. Functional EEG topography in sleep and waking: State-dependent and state-independent features. Neuroimage 2006, 32, 283–292. [Google Scholar] [CrossRef]
- Leemburg, S.; Vyazovskiy, V.V.; Olcese, U.; Bassetti, C.L.; Tononi, G.; Cirelli, C. Sleep homeostasis in the rat is preserved during chronic sleep restriction. Proc. Natl. Acad. Sci. USA 2010, 107, 15939–15944. [Google Scholar]
- Ohayon, M.M.; Carskadon, M.A.; Guilleminault, C.; Vitiello, M.V. Meta-analysis of quantitative sleep parameters from childhood to old age in healthy individuals: Developing normative sleep values across the human lifespan. Sleep 2004, 27, 1255–1273. [Google Scholar]
- Carskadon, M.A. Sleep in adolescents: The perfect storm. Pediatr. Clin. North Am. 2011, 58, 637–647. [Google Scholar] [CrossRef]
- Feinberg, I.; Davis, N.M.; de Bie, E.; Grimm, K.J.; Campbell, I.G. The maturational trajectories of NREM and REM sleep durations differ across adolescence on both school-night and extended sleep. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 302, R533–R540. [Google Scholar] [CrossRef]
- Carskadon, M.A.; Harvey, K.; Duke, P.; Anders, T.F.; Litt, I.F.; Dement, W.C. Pubertal changes in daytime sleepiness. Sleep 1980, 2, 453–460. [Google Scholar]
- Gaudreau, H.; Carrier, J.; Montplaisir, J. Age-related modifications of NREM sleep EEG: From childhood to middle age. J. Sleep Res. 2001, 10, 165–172. [Google Scholar] [CrossRef]
- Tarokh, L.; Carskadon, M.A. Developmental changes in the human sleep EEG during early adolescence. Sleep 2010, 33, 801–809. [Google Scholar]
- Tarokh, L.; van Reen, E.; LeBourgeois, M.; Seifer, R.; Carskadon, M.A. Sleep EEG provides evidence that cortical changes persist into late adolescence. Sleep 2011, 34, 1385–1393. [Google Scholar]
- Kurth, S.; Ringli, M.; Geiger, A.; LeBourgeois, M.; Jenni, O.G.; Huber, R. Mapping of cortical activity in the first two decades of life: A high-density sleep electroencephalogram study. J. Neurosci. 2010, 30, 13211–13219. [Google Scholar] [CrossRef] [Green Version]
- Feinberg, I. Changes in sleep cycle patterns with age. J. Psychiatr. Res. 1974, 10, 283–306. [Google Scholar] [CrossRef]
- Todd, W.D.; Gibson, J.L.; Shaw, C.S.; Blumberg, M.S. Brainstem and hypothalamic regulation of sleep pressure and rebound in newborn rats. Behav. Neurosci. 2010, 124, 69–78. [Google Scholar] [CrossRef]
- Yang, S.R.; Sun, H.; Huang, Z.L.; Yao, M.H.; Qu, W.M. Repeated sleep restriction in adolescent rats altered sleep patterns and impaired spatial learning/memory ability. Sleep 2012, 35, 849–859. [Google Scholar]
- Oken, B.S.; Salinsky, M.C.; Elsas, S.M. Vigilance, alertness, or sustained attention: Physiological basis and measurement. Clin. Neurophysiol. 2006, 117, 1885–1901. [Google Scholar] [CrossRef]
- Bachmann, V.; Klaus, F.; Bodenmann, S.; Schafer, N.; Brugger, P.; Huber, S.; Berger, W.; Landolt, H.P. Functional ADA polymorphism increases sleep depth and reduces vigilant attention in humans. Cereb. Cortex 2012, 22, 962–970. [Google Scholar] [CrossRef]
- Ringli, M.; Huber, R. Developmental aspects of sleep slow waves: Linking sleep, brain maturation and behavior. Prog. Brain Res. 2011, 193, 63–82. [Google Scholar] [CrossRef]
- Tarokh, L.; Raffray, T.; van Reen, E.; Carskadon, M.A. Physiology of normal sleep in adolescents. Adolesc. Med. State Art Rev. 2010, 21, 401–417. [Google Scholar]
- Feinberg, I.; Campbell, I.G. Sleep EEG changes during adolescence: An index of a fundamental brain reorganization. Brain Cogn. 2010, 72, 56–65. [Google Scholar] [CrossRef]
- Colrain, I.M.; Baker, F.C. Changes in sleep as a function of adolescent development. Neuropsychol. Rev. 2011, 21, 5–21. [Google Scholar] [CrossRef]
- Jenni, O.G.; Achermann, P.; Carskadon, M.A. Homeostatic sleep regulation in adolescents. Sleep 2005, 28, 1446–1454. [Google Scholar]
- Kurth, S.; Jenni, O.G.; Riedner, B.A.; Tononi, G.; Carskadon, M.A.; Huber, R. Characteristics of sleep slow waves in children and adolescents. Sleep 2010, 33, 475–480. [Google Scholar]
- Maret, S.; Faraguna, U.; Nelson, A.; Cirelli, C.; Tononi, G. Sleep and wake modulate spine turnover in the adolescent mouse cortex. Nat. Neurosci. 2011, 14, 1418–1420. [Google Scholar] [CrossRef]
- Yang, G.; Gan, W.B. Sleep contributes to dendritic spine formation and elimination in the developing mouse somatosensory cortex. Dev. Neurobiol. 2011, 72, 1391–1398. [Google Scholar] [CrossRef]
- Tononi, G.; Cirelli, C. Sleep and synaptic homeostasis: A hypothesis. Brain Res. Bull. 2003, 62, 143–150. [Google Scholar] [CrossRef]
- Tononi, G.; Cirelli, C. Sleep function and synaptic homeostasis. Sleep Med. Rev. 2006, 10, 49–62. [Google Scholar] [CrossRef]
- Tononi, G.; Cirelli, C. Time to be SHY? Some comments on sleep and synaptic homeostasis. Neural Plast. 2012, 2012. [Google Scholar] [CrossRef]
- Borbely, A.A.; Baumann, F.; Brandeis, D.; Strauch, I.; Lehmann, D. Sleep deprivation: Effect on sleep stages and EEG power density in man. Electroencephalogr. Clin. Neurophysiol. 1981, 51, 483–495. [Google Scholar] [CrossRef]
- Dijk, D.J.; Brunner, D.P.; Borbely, A.A. Time course of EEG power density during long sleep in humans. Am. J. Physiol. 1990, 258, R650–R661. [Google Scholar]
- Cajochen, C.; Foy, R.; Dijk, D.J. Frontal predominance of a relative increase in sleep delta and theta eeg activity after sleep loss in humans. Sleep Res. Online 1999, 2, 65–69. [Google Scholar]
- Vyazovskiy, V.V.; Olcese, U.; Hanlon, E.C.; Nir, Y.; Cirelli, C.; Tononi, G. Local sleep in awake rats. Nature 2011, 472, 443–447. [Google Scholar] [CrossRef]
- Hung, C.S.; Sarasso, S.; Ferrarelli, F.; Riedner, B.; Ghilardi, M.F.; Cirelli, C.; Tononi, G. Local, experience-dependent changes in the wake EEG after prolonged wakefulness. Sleep 2013, 36, 59–72. [Google Scholar]
- Jenni, O.G.; Borbely, A.A.; Achermann, P. Development of the nocturnal sleep electroencephalogram in human infants. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 286, R528–R538. [Google Scholar] [CrossRef]
- Franken, P.; Dijk, D.J.; Tobler, I.; Borbely, A.A. Sleep deprivation in rats: Effects on EEG power spectra, vigilance states, and cortical temperature. Am. J. Physiol. 1991, 261, R198–R208. [Google Scholar]
- Deboer, T.; Franken, P.; Tobler, I. Sleep and cortical temperature in the Djungarian hamster under baseline conditions and after sleep deprivation. J. Comp. Physiol. A 1994, 174, 145–155. [Google Scholar]
- Faraguna, U.; Vyazovskiy, V.V.; Nelson, A.B.; Tononi, G.; Cirelli, C. A causal role for brain-derived neurotrophic factor in the homeostatic regulation of sleep. J. Neurosci. 2008, 28, 4088–4095. [Google Scholar] [CrossRef]
- Castellano, C.; Oliverio, A. Early malnutrition and postnatal changes in brain and behavior in the mouse. Brain Res. 1976, 101, 317–325. [Google Scholar] [CrossRef]
- Feinberg, I.; de Bie, E.; Davis, N.M.; Campbell, I.G. Topographic differences in the adolescent maturation of the slow wave EEG during NREM sleep. Sleep 2011, 34, 325–333. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Nelson, A.B.; Faraguna, U.; Zoltan, J.T.; Tononi, G.; Cirelli, C. Sleep Patterns and Homeostatic Mechanisms in Adolescent Mice. Brain Sci. 2013, 3, 318-343. https://doi.org/10.3390/brainsci3010318
Nelson AB, Faraguna U, Zoltan JT, Tononi G, Cirelli C. Sleep Patterns and Homeostatic Mechanisms in Adolescent Mice. Brain Sciences. 2013; 3(1):318-343. https://doi.org/10.3390/brainsci3010318
Chicago/Turabian StyleNelson, Aaron B., Ugo Faraguna, Jeffrey T. Zoltan, Giulio Tononi, and Chiara Cirelli. 2013. "Sleep Patterns and Homeostatic Mechanisms in Adolescent Mice" Brain Sciences 3, no. 1: 318-343. https://doi.org/10.3390/brainsci3010318