NADPH Oxidase and Angiogenesis Following Endothelin-1 Induced Stroke in Rats: Role for Nox2 in Brain Repair

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
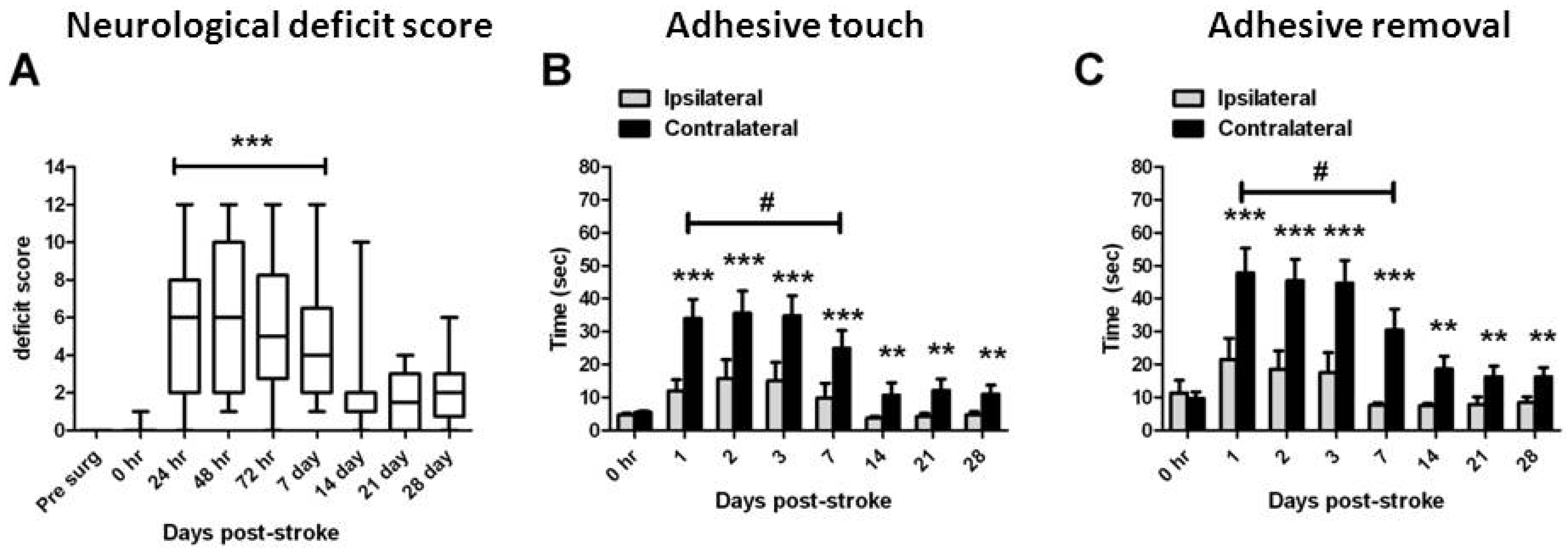
2.1. Stroke Rating
2.2. Functional Assessments

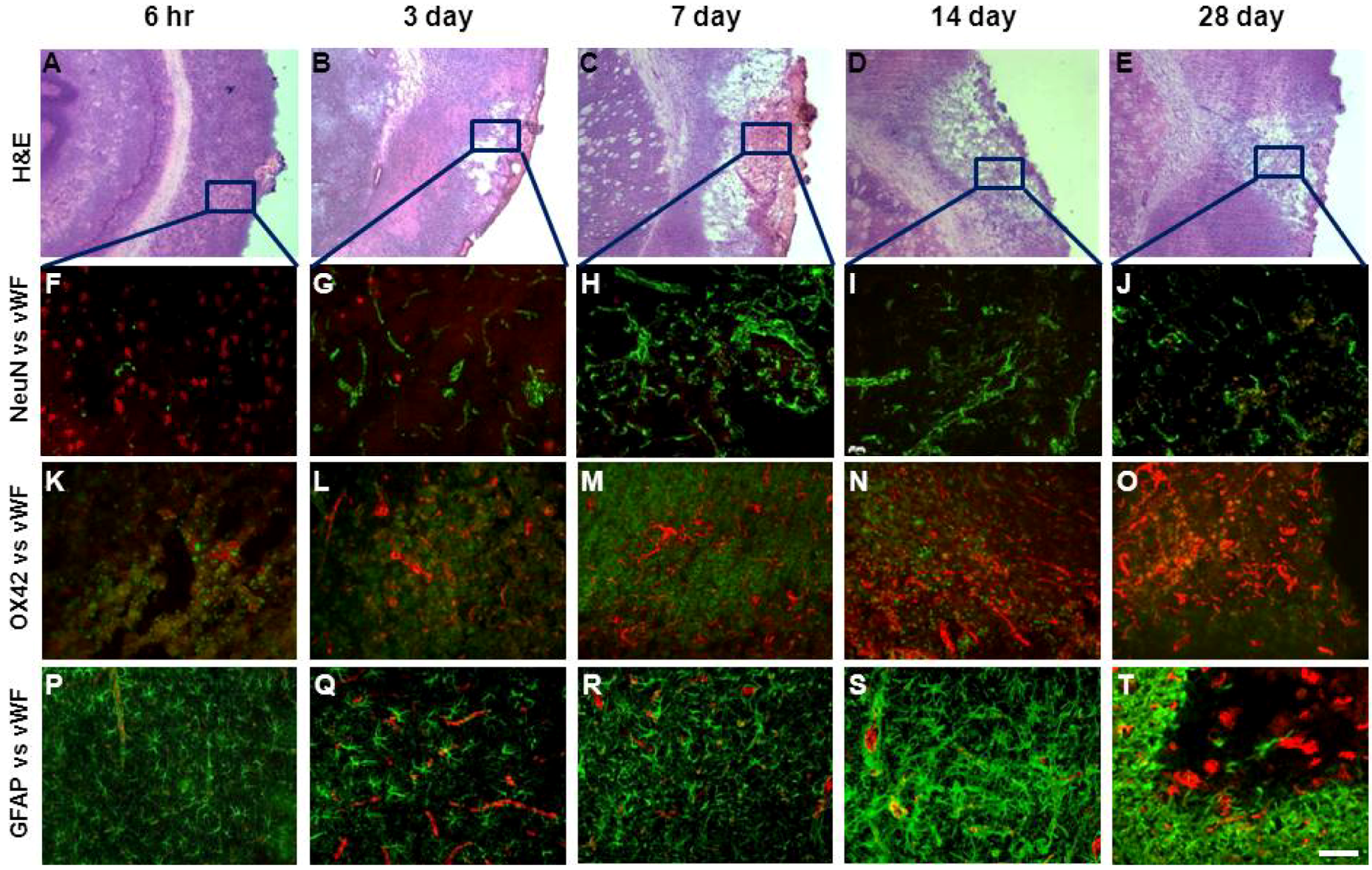
2.3. Assessment of Damage
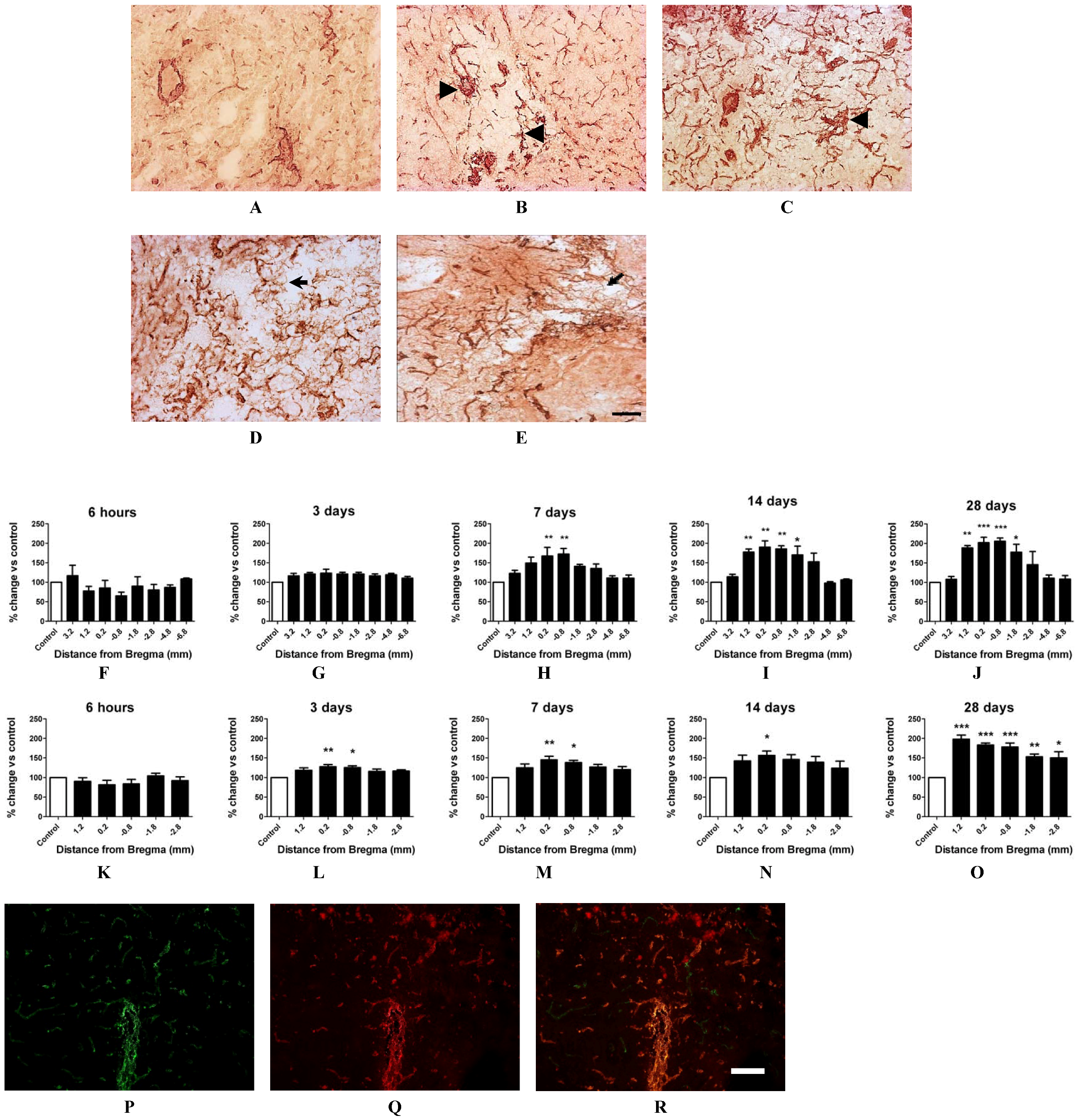
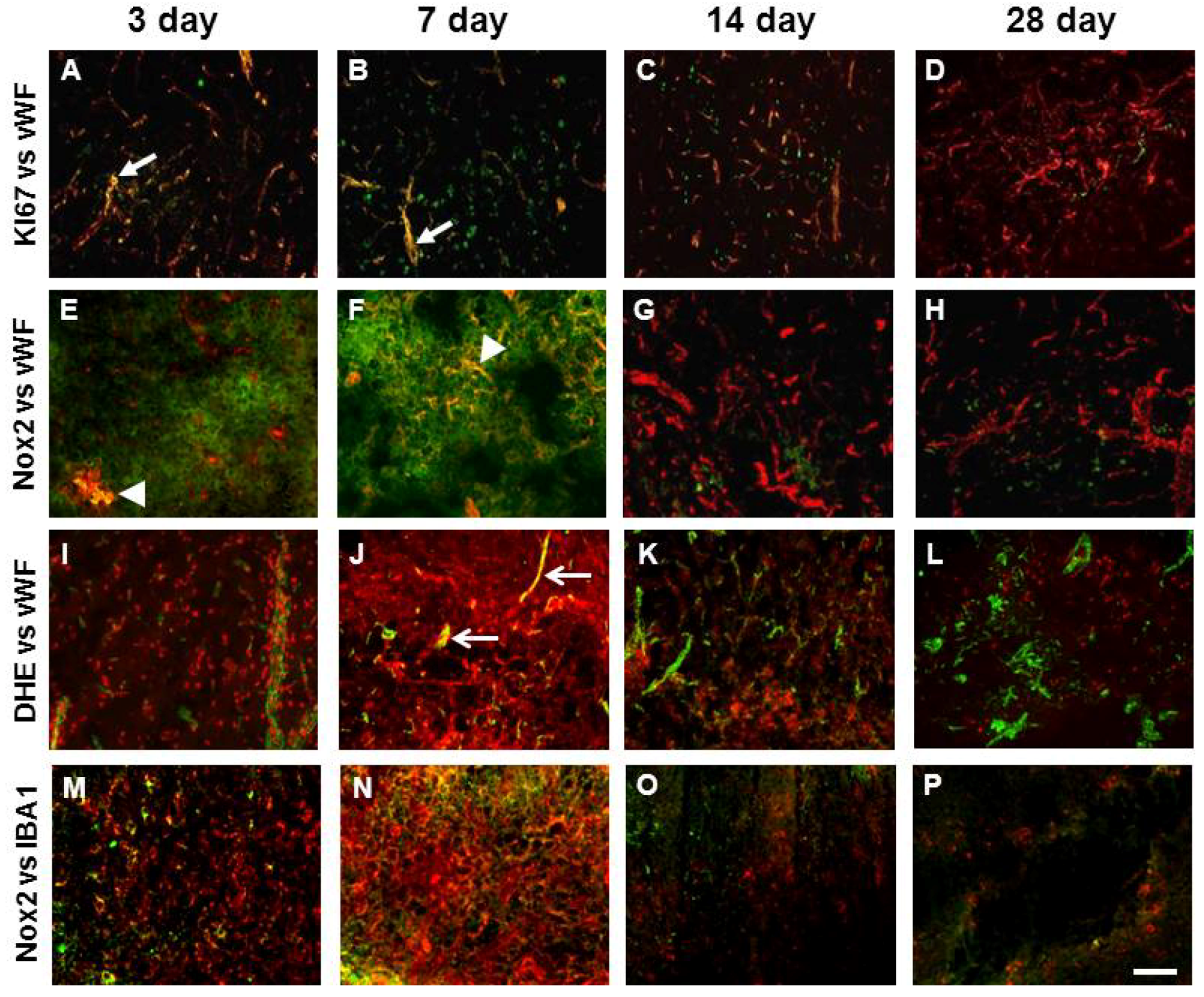
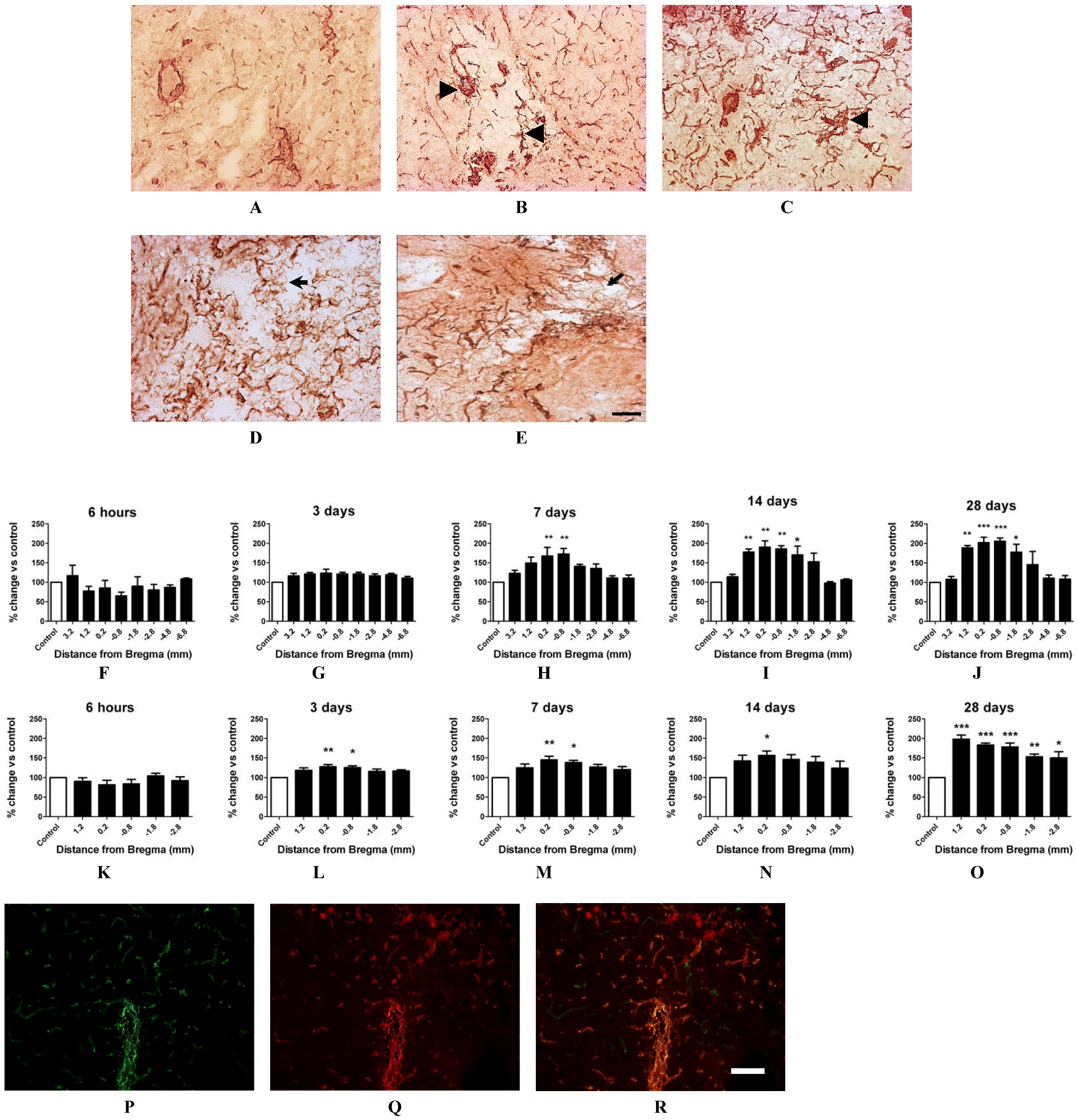
2.4. Angiogenesis after ET-1 Induced Stroke
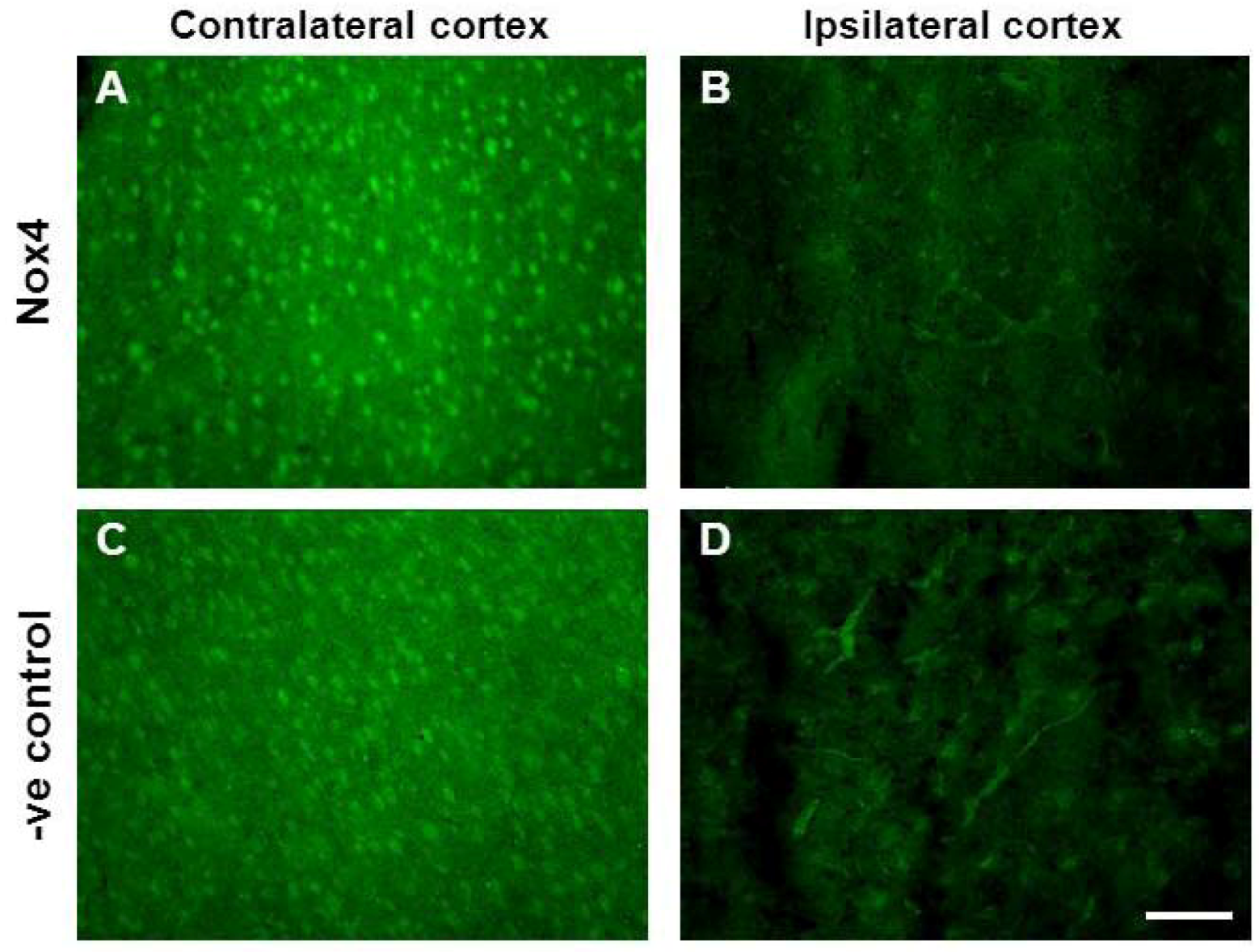
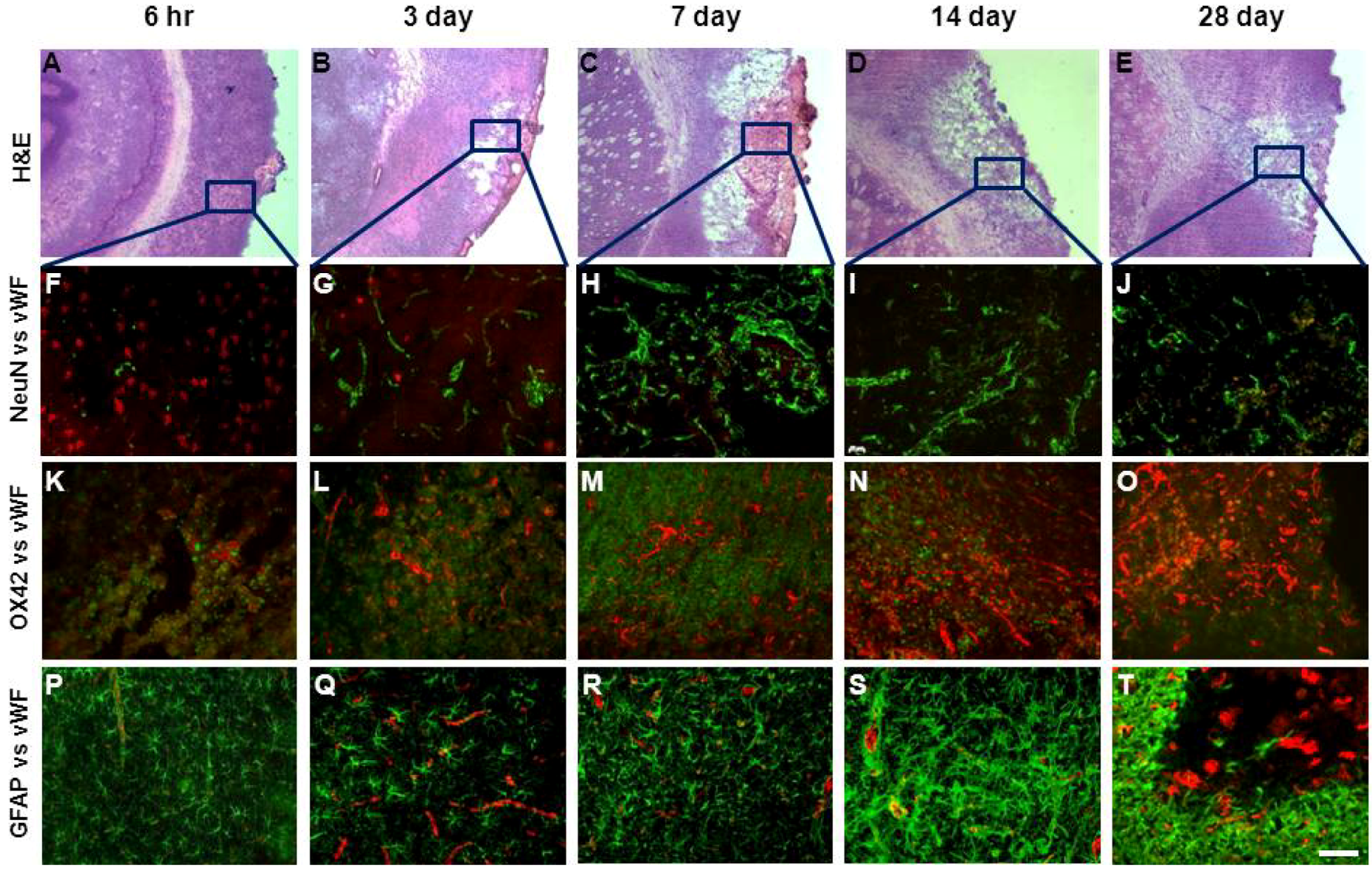
2.5. Changes in Brain Pathology Associated with Increased Angiogenesis
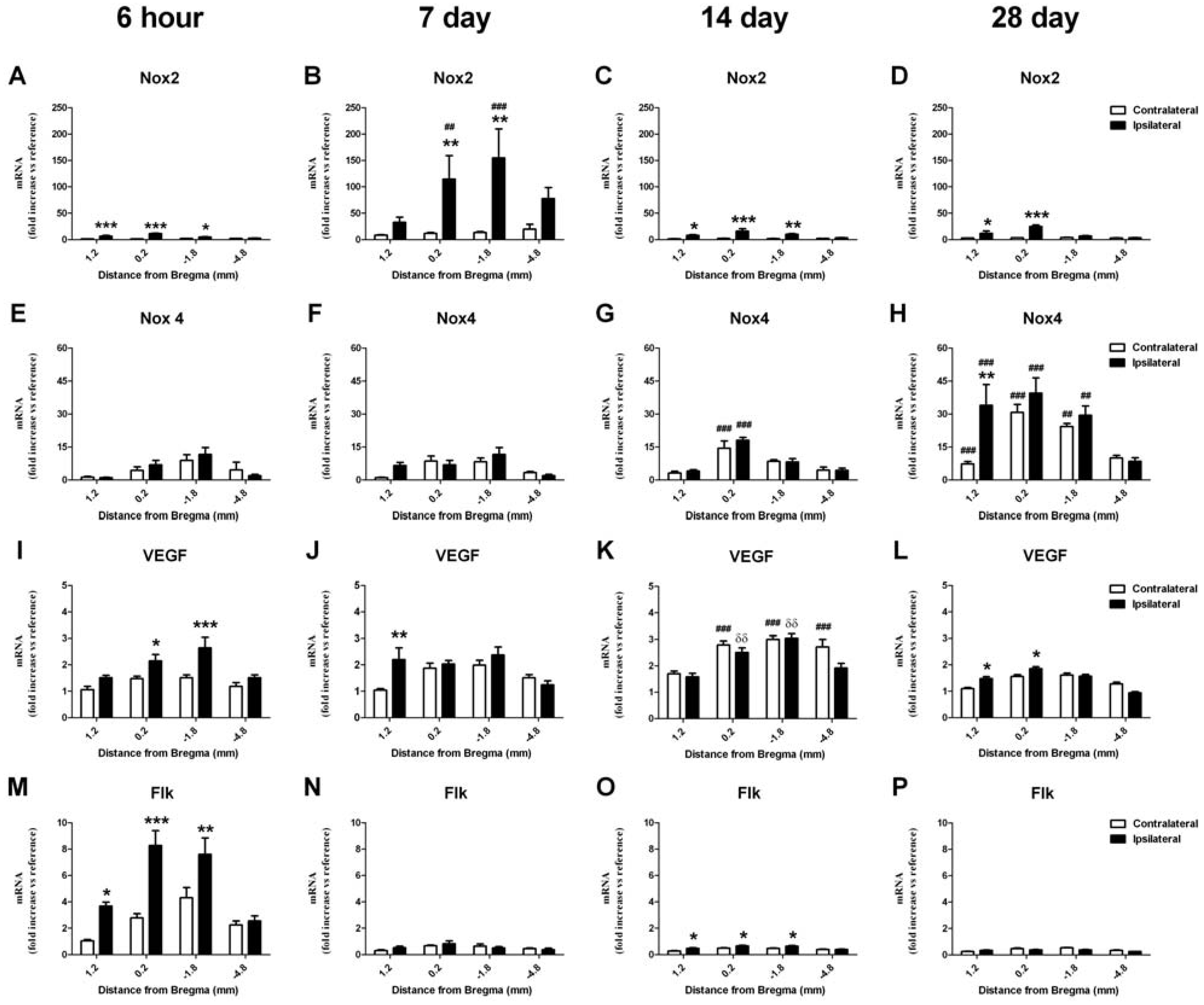
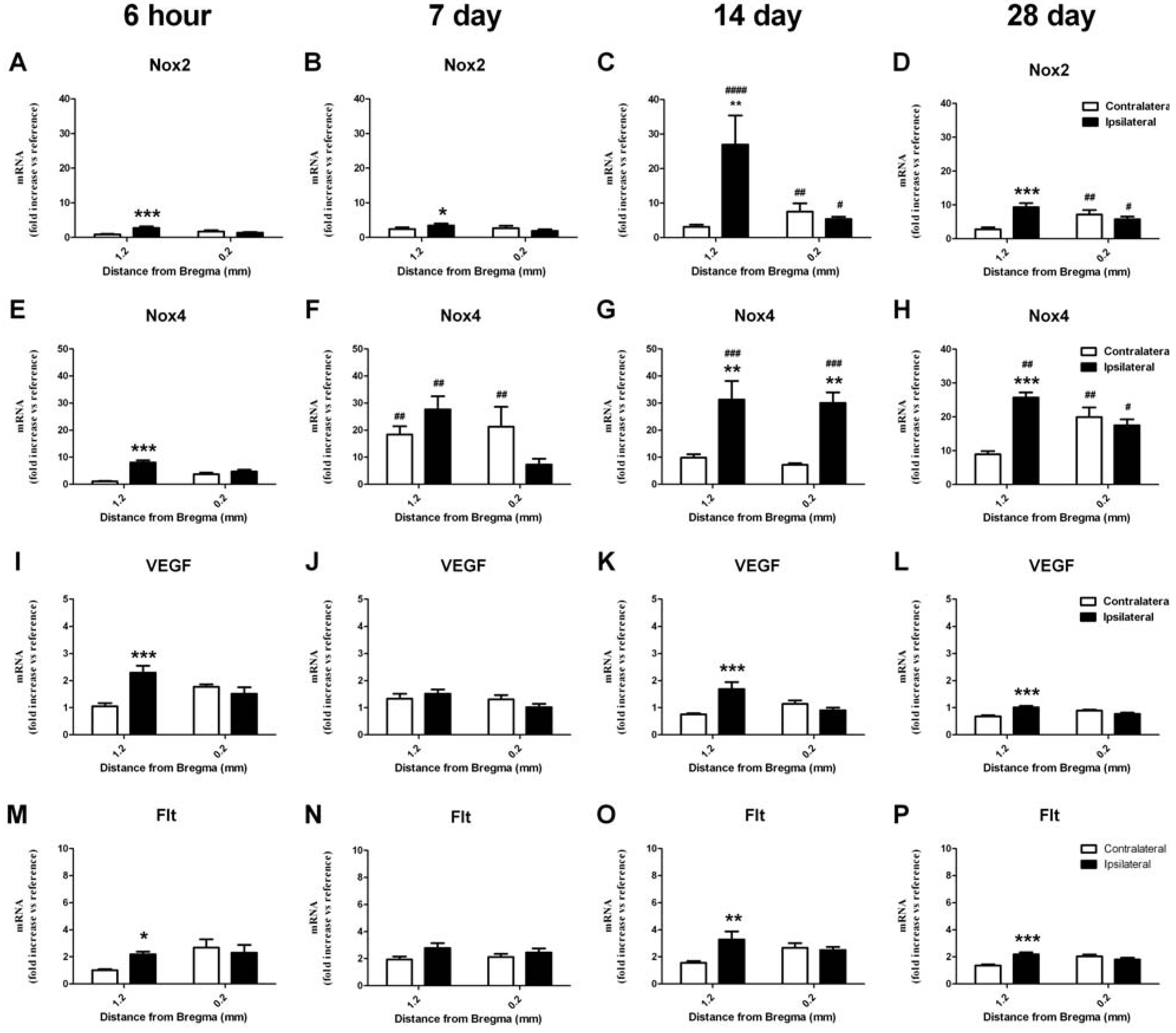
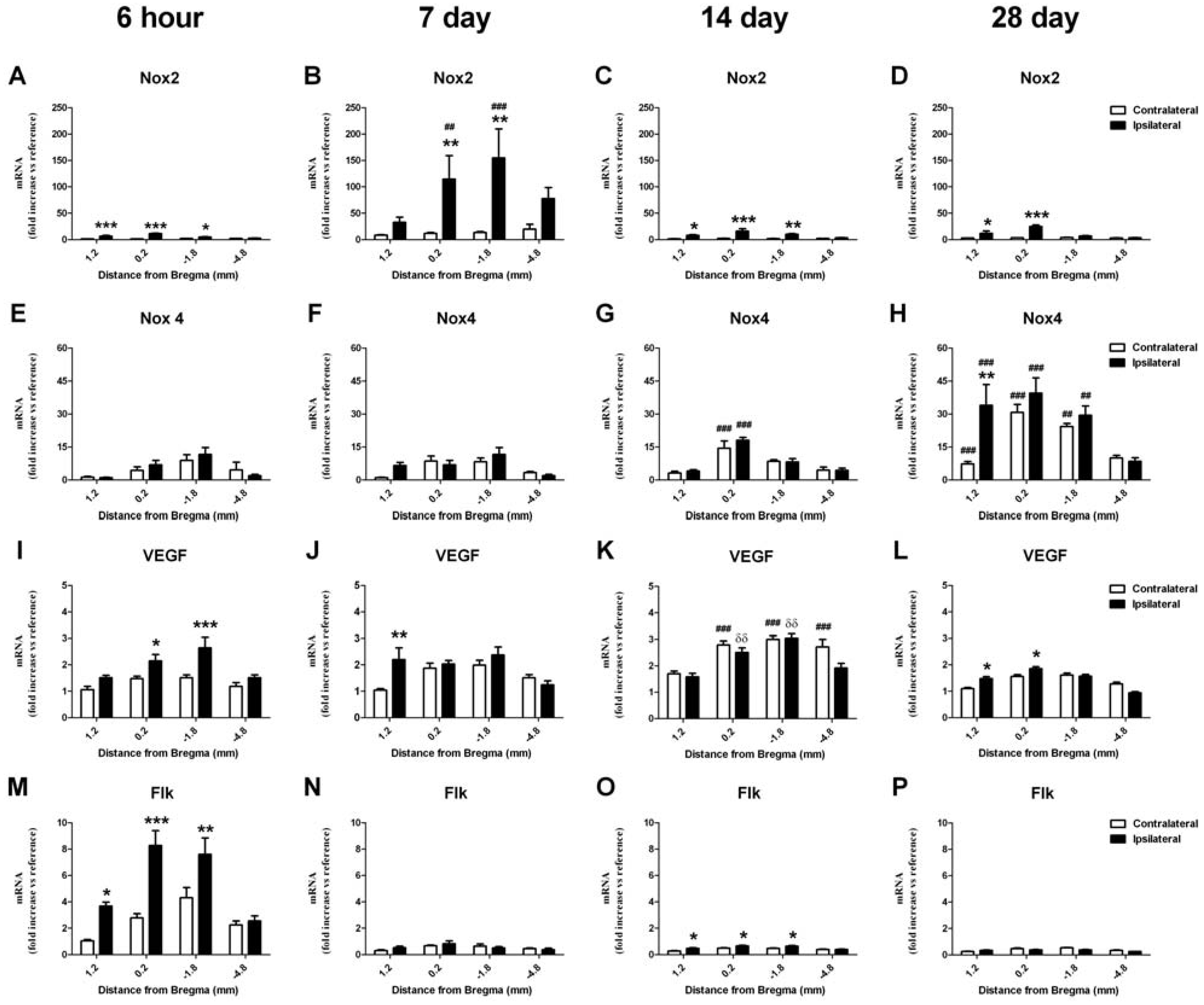
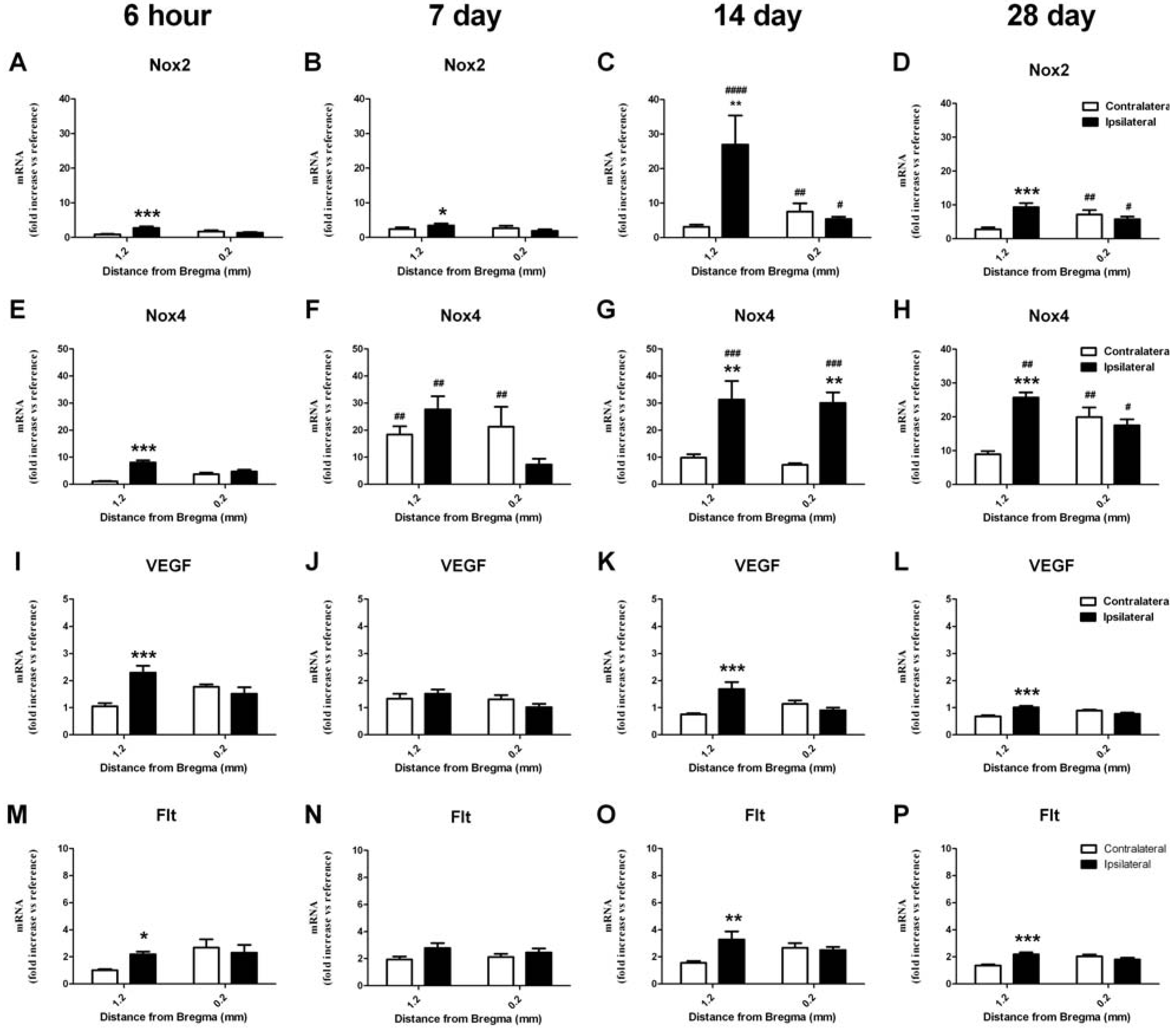
2.6. RT-PCR Detection of NADPH Oxidase Subunits and Angiogenic Factors
2.6.1. Change in mRNA in the Cortex
2.6.2. Change in mRNA in the Striatum
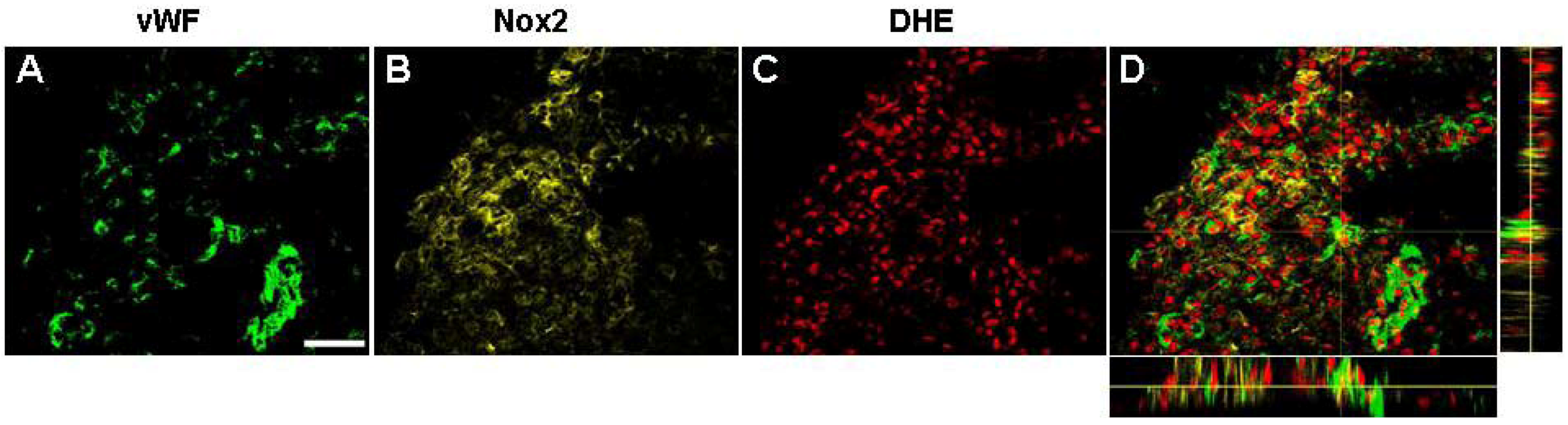
2.7. Vascular Proliferation, Superoxide and Nox2 NADPH Oxidase

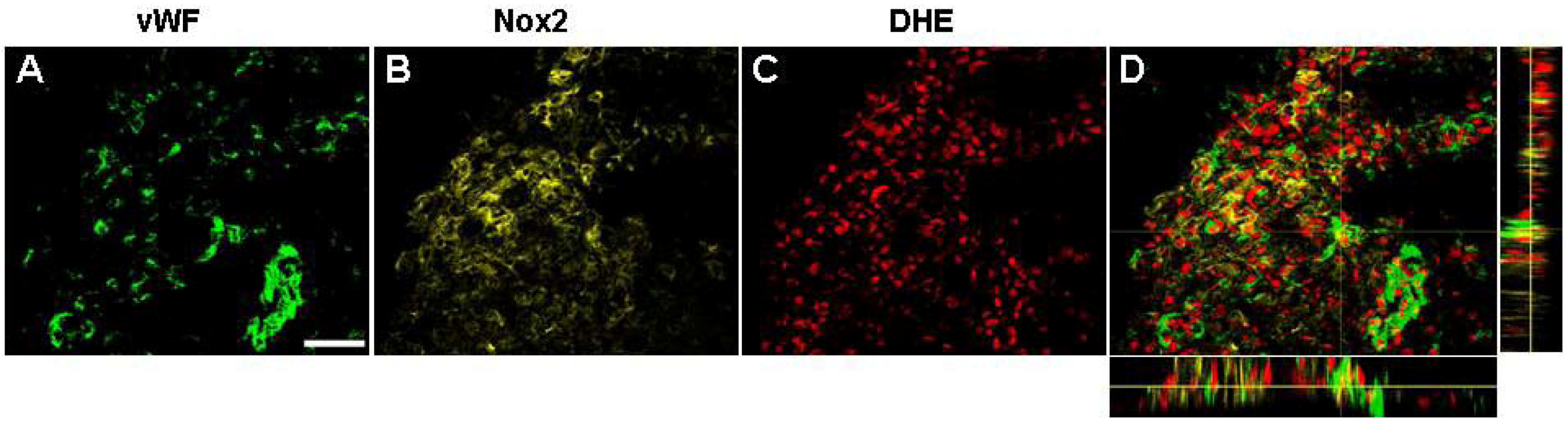
2.8. Discussion
2.8.1. Angiogenesis following ET-1 Induced Stroke
2.8.2. NADPH Oxidase and Angiogenesis
3. Experimental Section
3.1. Ethics Statement
3.2. Endothelin-1 Stroke in Conscious Rats
3.3. Assessment of Functional Outcome
3.4. Confirmation of Ischemic Damage
3.5. Blood Vessel Detection
3.6. Blood Vessel Quantification
3.7. Fluorescent Immunohistochemistry
3.8. Nox Immunohistochemistry

3.9. In Situ Detection of Superoxide Using Dihydroethidium Fluorescence
3.10. RT-PCR Detection of NADPH Oxidase Subunits and Angiogenic Factors
3.11. Statistical Analysis
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Gauthier, L.V.; Taub, E.; Perkins, C.; Ortmann, M.; Mark, V.W.; Uswatte, G. Remodeling the brain: Plastic structural brain changes produced by different motor therapies after stroke. Stroke 2008, 39, 1520–1525. [Google Scholar] [CrossRef]
- Zhang, R.; Zhang, Z.; Wang, L.; Wang, Y.; Gousev, A.; Zhang, L.; Ho, K.L.; Morshead, C.; Chopp, M. Activated neural stem cells contribute to stroke-induced neurogenesis and neuroblast migration toward the infarct boundary in adult rats. J. Cereb. Blood Flow Metab. 2004, 2, 441–448. [Google Scholar]
- Minger, S.L.; Ekonomou, A.; Carta, E.M.; Chinoy, A.; Perry, R.H.; Ballard, C.G. Endogenous neurogenesis in the human brain following cerebral infarction. Regen. Med. 2007, 2, 69–74. [Google Scholar] [CrossRef]
- Ergul, A.; Alhusban, A.; Fagan, S.C. Angiogenesis: A harmonized target for recovery after stroke. Stroke 2012, 43, 2270–2274. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, Z.; Wang, Y.; Zhang, R.; Chopp, M. Treatment of stroke with erythropoietin enhances neurogenesis and angiogenesis and improves neurological function in rats. Stroke 2007, 35, 1732–1737. [Google Scholar]
- Chopp, M.; Zhang, Z.G.; Jiang, Q. Neurogenesis, angiogenesis, and MRI indices of functional recovery from stroke. Stroke 2007, 38, 827–831. [Google Scholar] [CrossRef]
- Krupinski, J.; Kaluza, J.; Kumar, P.; Kumar, S.; Wang, J.M. Role of angiogenesis in patients with cerebral ischemic stroke. Stroke 1994, 25, 1794–1798. [Google Scholar] [CrossRef]
- Henderson, R.D.; Eliasziw, M.; Fox, A.J.; Rothwell, P.M.; Barnett, H.J. Angiographically defined collateral circulation and risk of stroke in patients with severe carotid artery stenosis. North American Symptomatic Carotid Endarterectomy Trial (NASCET) group. Stroke 2000, 31, 128–132. [Google Scholar]
- Nita, D.A.; Nita, V.; Spulber, S.; ldovan, M.; Popa, D.P.; Zagrean, A.M.; Zagrean, L. Oxidative damage following cerebral ischemia depends on reperfusion—A biochemical study in rat. J. Cell. Mol. Med. 2001, 52, 163–170. [Google Scholar]
- Datla, S.R.; Dusting, G.J.; Peshavariya, H.; Mahadev, K.; Goldstein, B.J.; Jiang, F. Important role of Nox4 type NADPH oxidase in angiogenic responses in human microvascular endothelial cells in vitro. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2319–2324. [Google Scholar] [CrossRef]
- Jiang, F.; Zhang, G.; Hashimoto, I.; Kumar, B.S.; Bortolotto, S.; Morrison, W.A.; Dusting, G.J. Neovascularisation in an arterio-venous loop-containing tissue engineering chamber: Role of NADPH oxidase. J. Cell. Mol. Med. 2008, 12, 2062–2072. [Google Scholar] [CrossRef]
- Hachisuka, H.; Dusting, G.J.; Abberton, K.M.; Morrison, W.A.; Jiang, F. Role of NADPH oxidase in tissue growth in a tissue engineering chamber in rats. J. Tissue Eng. Regen. Med. 2008, 2, 430–435. [Google Scholar] [CrossRef]
- Chan, E.C.; Jiang, F.; Peshavariya, H.M.; Dusting, G.J. egulation of cell proliferation by NADPH oxidase-mediated signaling: Potential roles in tissue repair, regenerative medicine and tissue engineering. Pharmacol. Ther. 2009, 122, 97–108. [Google Scholar] [CrossRef]
- Shen, Q.; Goderie, S.K.; Jin, L.; Karanth, N.; Sun, Y.; Abramova, N.; Vincent, P.; Pumiglia, K.; Temple, S. Endothelial cells stimulate self-renewal and expand neurogenesis of neural stem cells. Science 2004, 304, 1338–1340. [Google Scholar] [CrossRef]
- Jiang, F.; Drummond, G.R.; Dusting, G.J. Suppression of oxidative stress in the endothelium and vascular wall. Endothelium 2004, 11, 79–88. [Google Scholar] [CrossRef]
- Griendling, K.K.; Sorescu, D.; Ushio-Fukai, M. NAD(P)H oxidase: Role in cardiovascular biology and disease. Circ. Res. 2000, 86, 494–501. [Google Scholar] [CrossRef]
- Green, S.P.; Cairns, B.; Rae, J.; Errett-Baroncini, C.; Hongo, J.A.; Erickson, R.W.; Curnutte, J.T. Induction of gp91-phox, a component of the phagocyte NADPH oxidase, in microglial cells during central nervous system inflammation. J. Cereb. Blood Flow Metab. 2001, 21, 374–384. [Google Scholar]
- Miller, A.A.; Drummond, G.R.; Schmidt, H.H.; Sobey, C.G. NADPH oxidase activity and function are profoundly greater in cerebral versus systemic arteries. Circ. Res. 2005, 97, 1055–1062. [Google Scholar] [CrossRef]
- Miller, A.A.; Dusting, G.J.; Roulston, C.L.; Sobey, C.G. NADPH-oxidase activity is elevated in penumbral and non-ischemic cerebral arteries following stroke. Brain Res. 2006, 1111, 111–116. [Google Scholar] [CrossRef]
- Vallet, P.; Charnay, Y.; Steger, K.; Ogier-Denis, E.; Kovari, E.; Herrmann, F.; Michel, J.P.; Szanto, I. Neuronal expression of the NADPH oxidase NOX4, and its regulation in mouse experimental brain ischemia. Neuroscience 2005, 132, 233–238. [Google Scholar] [CrossRef]
- McCann, S.K.; Dusting, G.J.; Roulston, C.L. Early increase of Nox4 NADPH oxidase and superoxide generation following endothelin-1-induced stroke in conscious rats. J. Neurosci. Res. 2008, 86, 2524–2534. [Google Scholar] [CrossRef]
- Roulston, C.L.; Callaway, J.K.; Jarrott, B.; Dusting, G.J. Using behaviour to predict stroke severity in conscious rats: Post-stroke treatment with 3′,4′-dihydroxyflavonol improves recovery. Eur. J. Pharm. 2008, 584, 100–110. [Google Scholar] [CrossRef]
- Zhang, R.L.; Zhang, Z.G.; Chopp, M. Ischaemic stroke and neurogenesis in the subventricular zone. Neuropharmacology 2008, 55, 345–352. [Google Scholar] [CrossRef]
- Zhang, R.L.; Zhang, Z.G.; Lu, M.; Wang, Y.; Yang, J.J.; Chopp, M. Reduction of the cell cycle length by decreasing G1 phase and cell cycle re-entry expand neuronal progenitor cells in the subventricular zone of adult rat after stroke. J. Cereb. Blood Flow Metab. 2006, 26, 857–863. [Google Scholar] [CrossRef]
- Nih, L.R.; Deroide, N.; Leré-Déan, C.; Lerouet, D.; Soustrat, M.; Levy, B.I.; Silvestre, J.S.; Merkulova-Rainon, T.; Pocard, M.; Margaill, I.; Kubis, N. Neuroblast survival depends on mature vascular network formation after mouse stroke: Role of endothelial and smooth muscle progenitor cell co-administration. Eur. J. Neurosci. 2012, 35, 1208–1217. [Google Scholar] [CrossRef]
- Yu, S.W.; Friedman, B.; Cheng, Q.; Lyden, P.D. Stroke-evoked angiogenesis results in a transient population of microvessels. J. Cereb. Blood Flow Metab. 2007, 27, 755–763. [Google Scholar]
- Uemura, M.; Kasahara, Y.; Nagatsuka, K.; Taguchi, A. Cell-based therapy to promote angiogenesis in the brain following ischemic damage. Curr. Vasc. Pharmacol. 2012, 10, 285–288. [Google Scholar]
- Peshavariya, H.; Dusting, G.J.; Jiang, F.; Halmos, L.R.; Sobey, C.G.; Drummond, G.R.; Selemidis, S. NADPH oxidase isoform selective regulation of endothelial cell proliferation and survival. Naunyn Schmiedebergs Arch. Pharmacol. 2009, 380, 193–204. [Google Scholar] [CrossRef]
- Ushio-Fukai, M. Redox signaling in angiogenesis: Role of NADPH oxidase. Cardiovasc. Res. 2006, 71, 226–235. [Google Scholar] [CrossRef]
- Tojo, T.; Ushio-Fukai, M.; Yamaoka-Tojo, M.; Ikeda, S.; Patrushev, N.; Alexander, RW. Role of gp91phox (Nox2)-containing NAD(P)H oxidase in angiogenesis in response to hindlimb ischemia. Circulation 2005, 111, 2347–2355. [Google Scholar] [CrossRef]
- Craige, S.M.; Chen, K.; Pei, Y.; Li, C.; Huang, X.; Chen, C.; Shibata, R.; Sato, K.; Walsh, K.; Keaney, J.F., Jr. NADPH oxidase 4 promotes endothelial angiogenesis through endothelial nitric oxide synthase activation. Circulation 2011, 124, 731–740. [Google Scholar] [CrossRef]
- Kleinschnitz, C.; Grund, H.; Wingler, K.; Armitage, M.E.; Jones, E.; Mittal, M.; Barit, D.; Schwarz, T.; Geis, C.; Kraft, P.; et al. Post-stroke inhibition of induced NADPH oxidase type 4 prevents oxidative stress and neurodegeneration. PLoS Biol. 2010, 8, e1000479. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.G.; Zhang, L.; Jiang, Q.; Zhang, R.; Davies, K.; Powers, C.; Bruggen, N.V.; Chopp, M. VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J. Clin. Invest. 2000, 106, 829–838. [Google Scholar] [CrossRef]
- Schröder, K.; Zhang, M.; Benkhoff, S.; Mieth, A.; Pliquett, R.; Kosowski, J.; Kruse, C.; Luedike, P.; Michaelis, U.R.; Weissmann, N.; et al. Nox4 is a protective reactive oxygen species generating vascular NADPH oxidase. Circ. Res. 2012, 110, 1217–1225. [Google Scholar] [CrossRef]
- Krum, J.M.; Mani, N.; Rosenstein, J.M. Roles of endogenous VEGF receptors flt-1 and flk01 in astroglial and vascular remodelling after brain injury. Exp. Neurol. 2008, 212, 108–117. [Google Scholar] [CrossRef]
- Sandoval, K.E.; Witt, K.A. Blood-brain barrier tight junction permeability and ischemic stroke. Neurobiol. Dis. 2008, 32, 200–219. [Google Scholar] [CrossRef]
- Lokmic, Z.; Stillaert, F.; Morrison, W.A.; Thompson, E.W.; Mitchell, G.M. An arteriovenous loop in a protected space generates a permanent, highly vascular, tissue-engineered construct. FASEB J. 2007, 21, 511–522. [Google Scholar] [CrossRef] [Green Version]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Taylor, C.J.; Weston, R.M.; Dusting, G.J.; Roulston, C.L. NADPH Oxidase and Angiogenesis Following Endothelin-1 Induced Stroke in Rats: Role for Nox2 in Brain Repair. Brain Sci. 2013, 3, 294-317. https://doi.org/10.3390/brainsci3010294
Taylor CJ, Weston RM, Dusting GJ, Roulston CL. NADPH Oxidase and Angiogenesis Following Endothelin-1 Induced Stroke in Rats: Role for Nox2 in Brain Repair. Brain Sciences. 2013; 3(1):294-317. https://doi.org/10.3390/brainsci3010294
Chicago/Turabian StyleTaylor, Caroline J., Robert M. Weston, Gregory J. Dusting, and Carli L. Roulston. 2013. "NADPH Oxidase and Angiogenesis Following Endothelin-1 Induced Stroke in Rats: Role for Nox2 in Brain Repair" Brain Sciences 3, no. 1: 294-317. https://doi.org/10.3390/brainsci3010294