Nanostructured Composites of Sodium Montmorillonite Clay and PEO Used in Dissolution Improvement of Aprepitant Drug by Melt Mixing


 , and
, and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
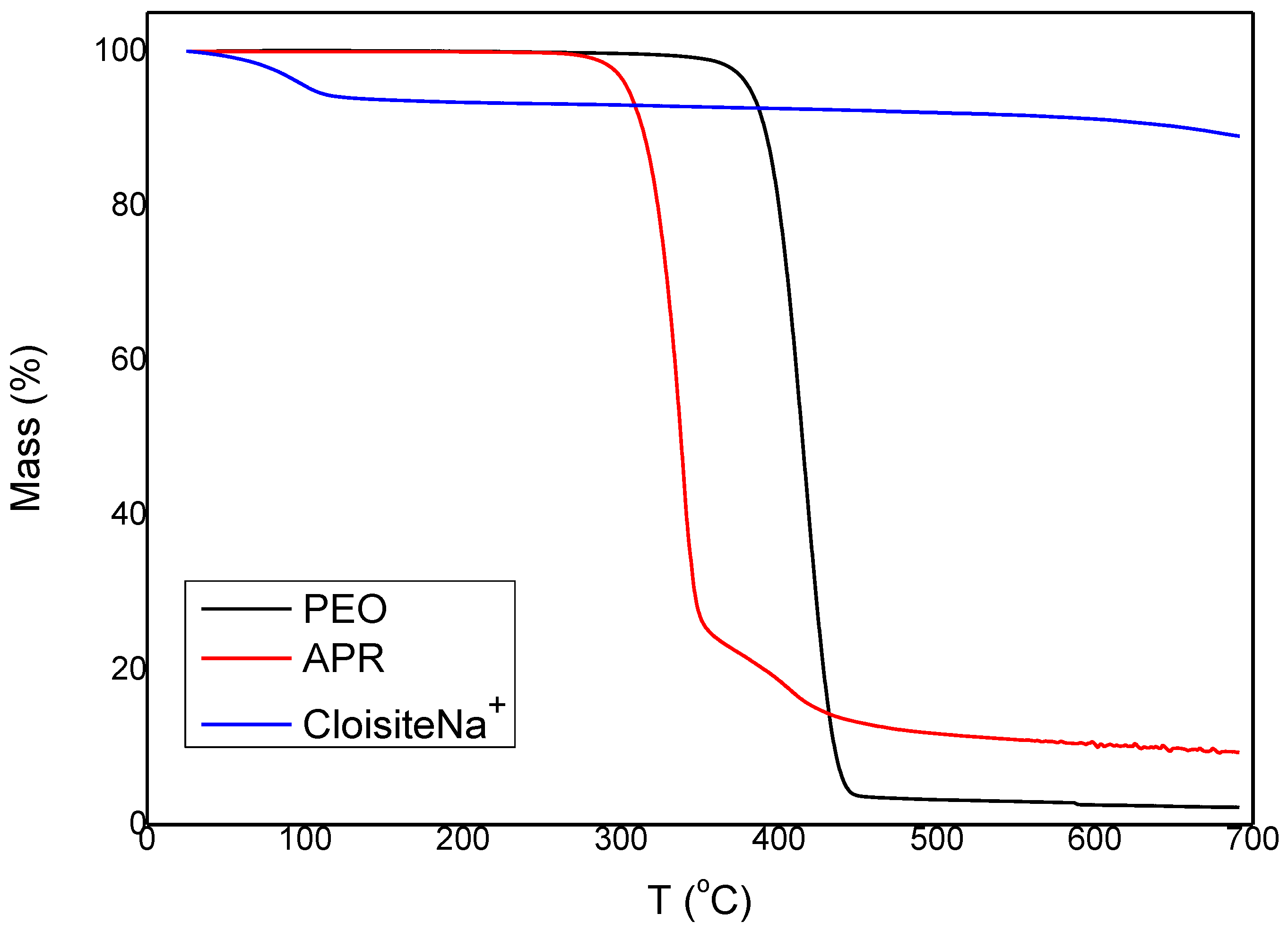
2.2.1. Thermogravimetric Analysis (TGA)
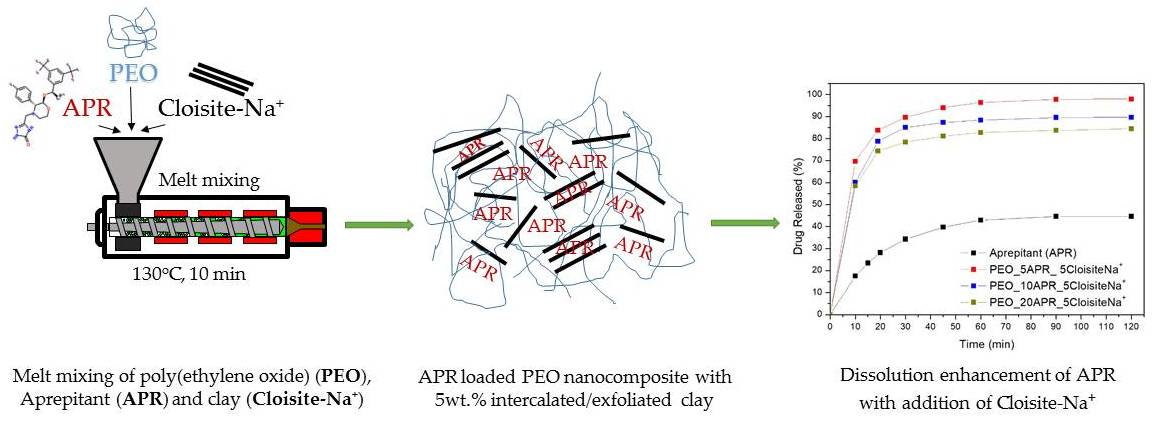
2.2.2. Preparation of Drug-Loaded Polymer and Nanocomposites by Melt Mixing
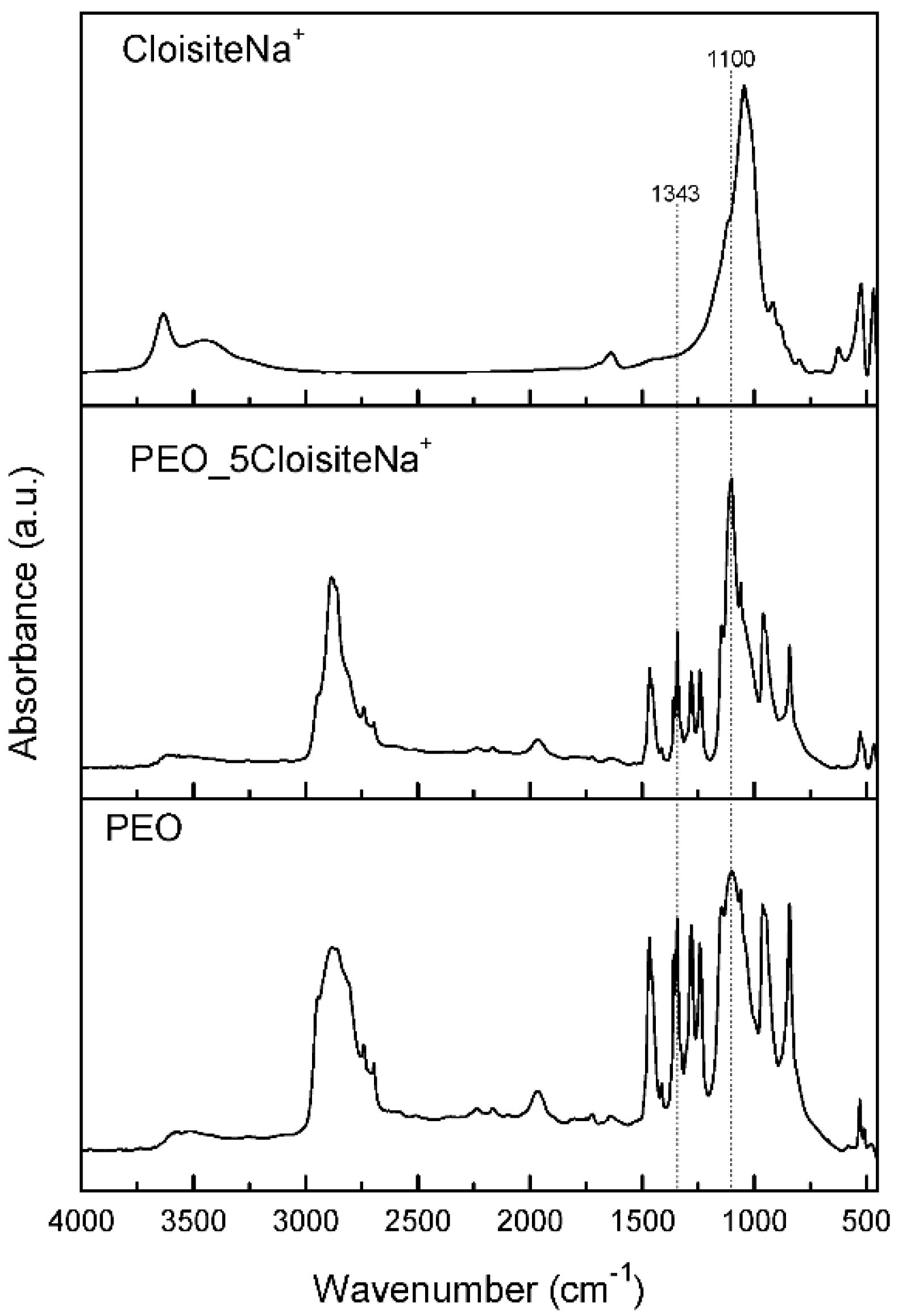
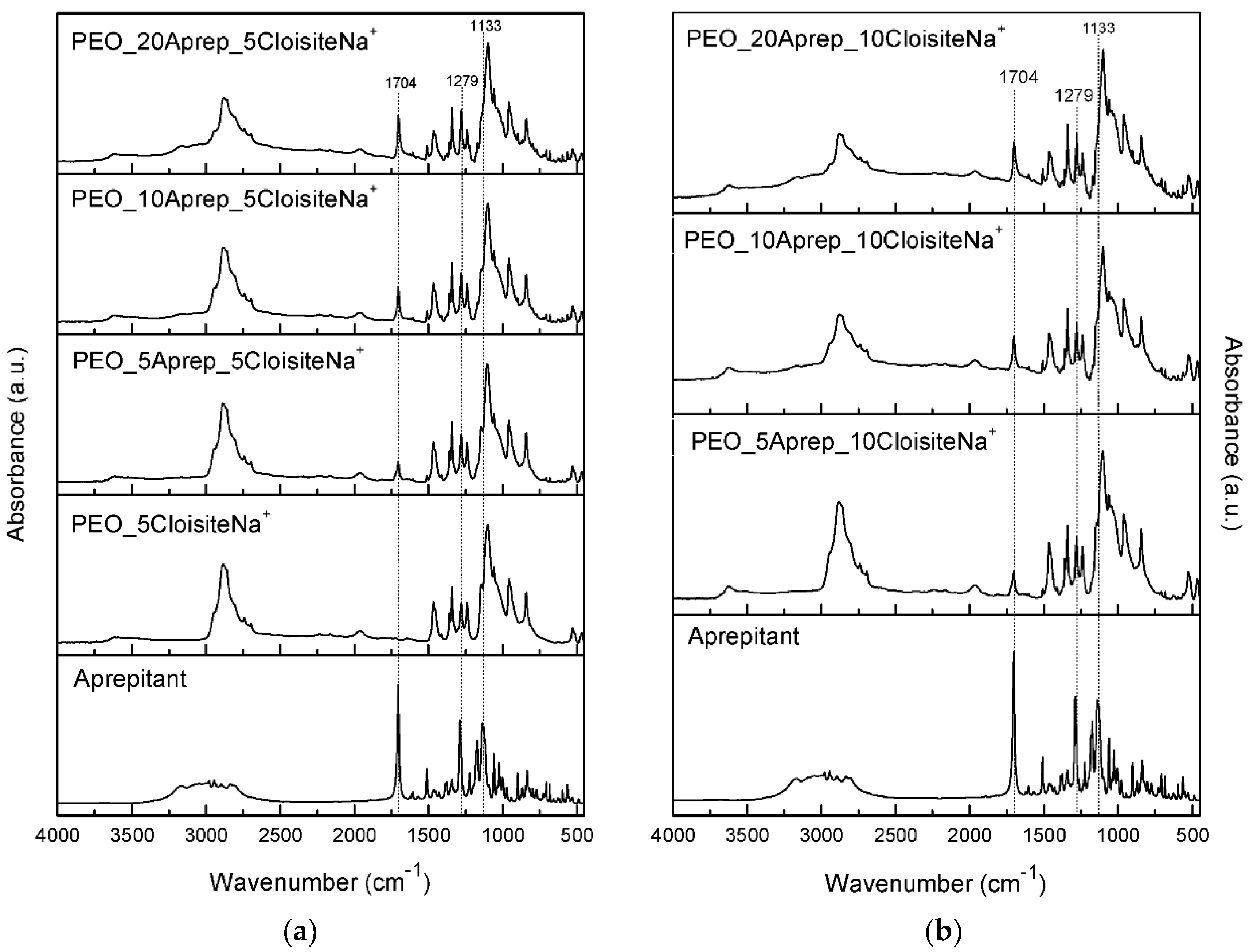
2.2.3. Fourier-Transform Infrared Spectroscopy (FTIR)
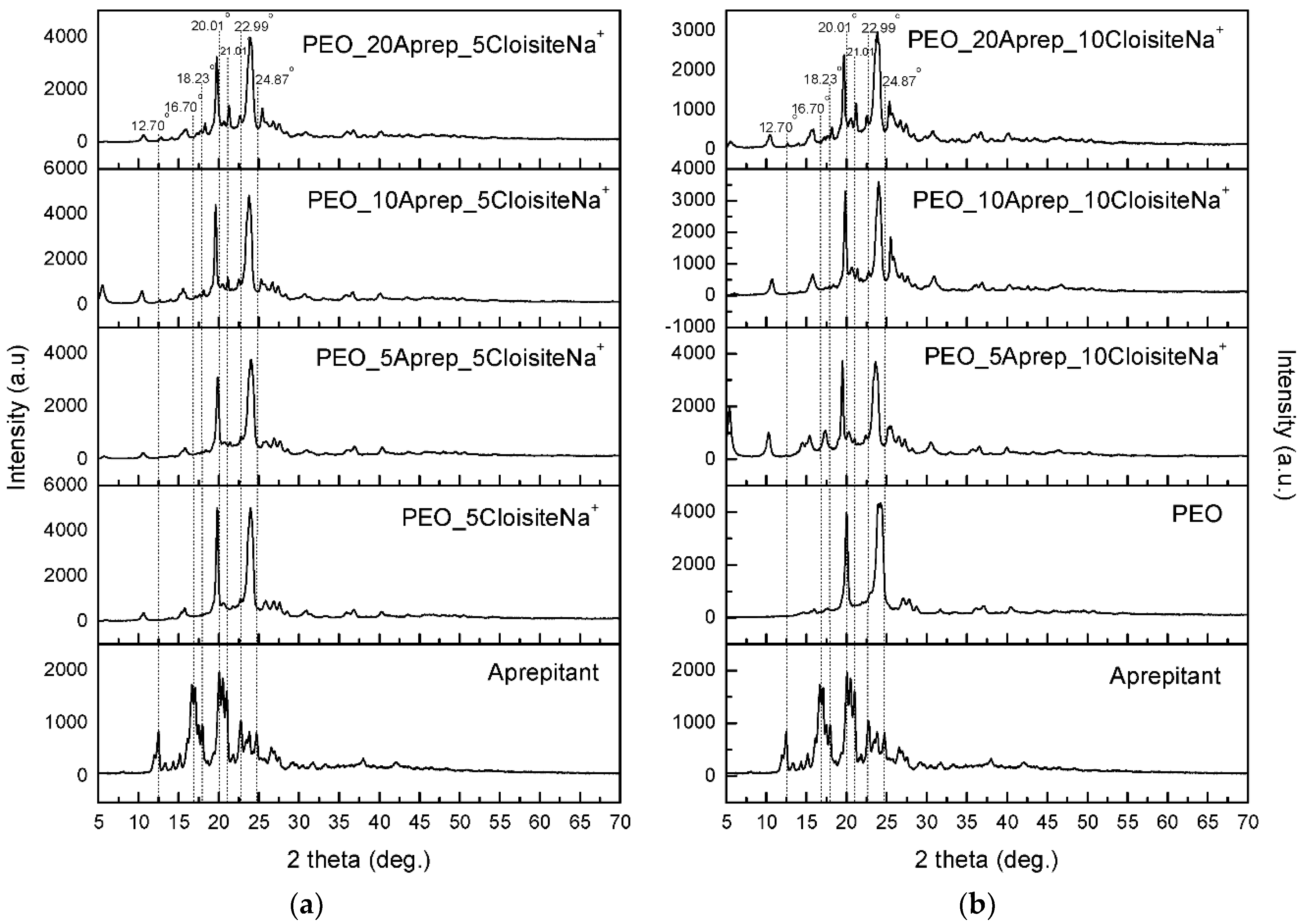
2.2.4. X-Ray Diffraction (XRD)
2.2.5. Differential Scanning Calorimetry (DSC)
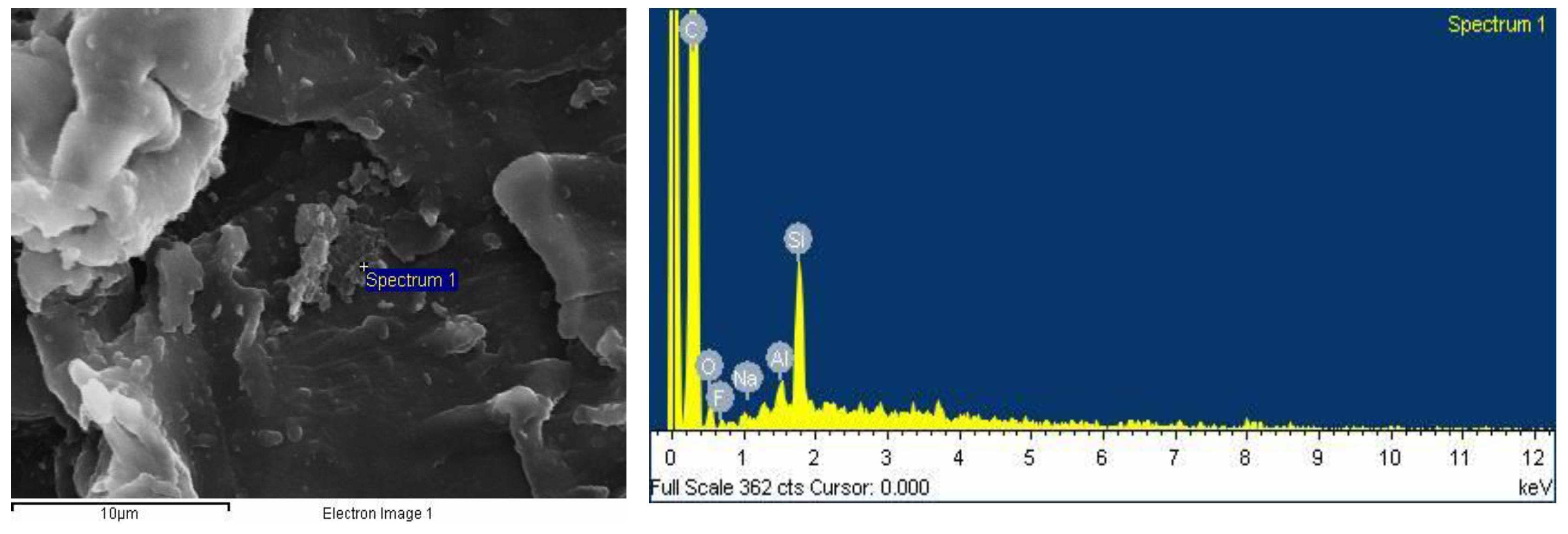
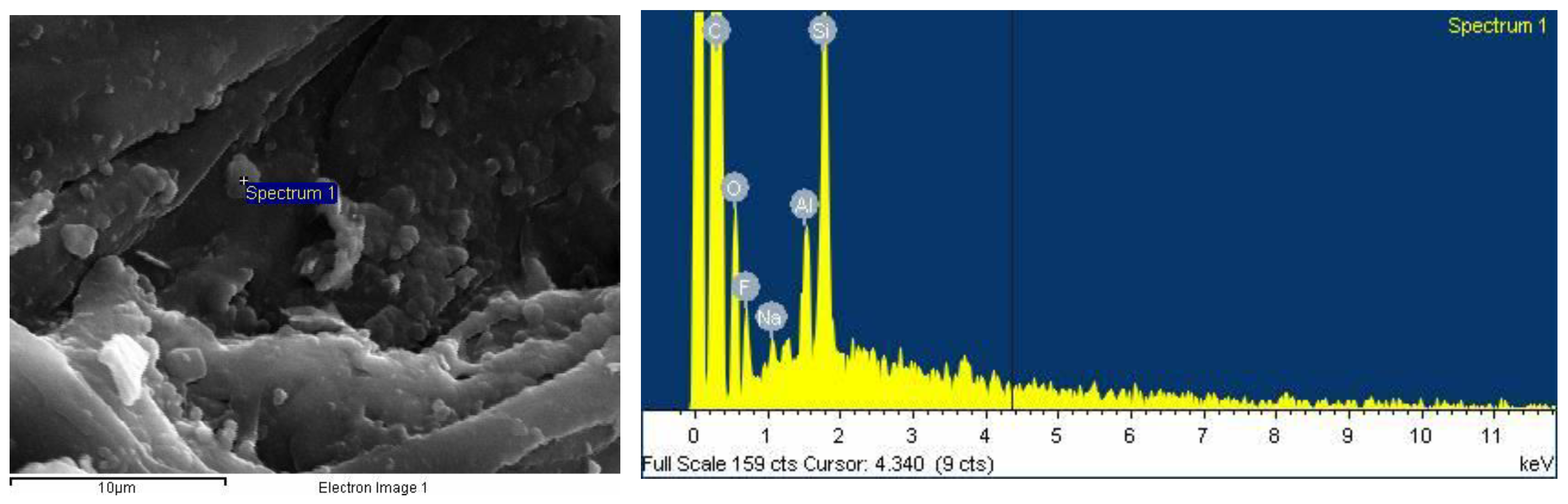
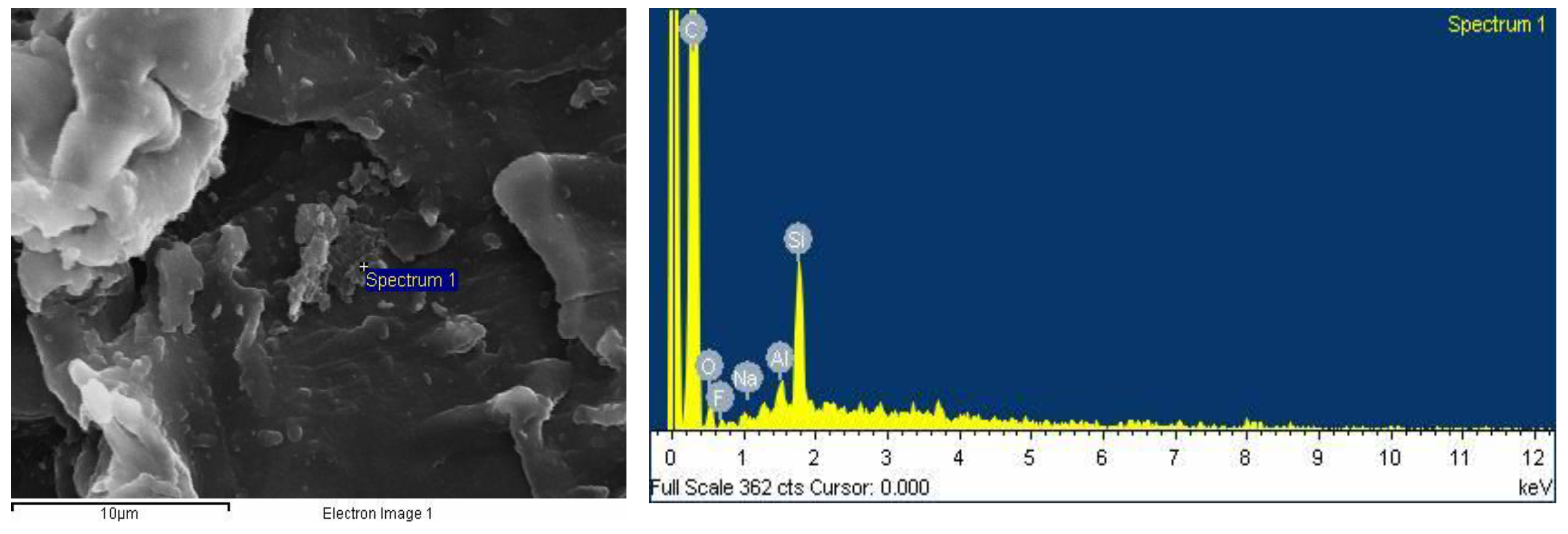
2.2.6. SEM
2.2.7. Dissolution Study/In Vitro Release Profile
2.2.8. HPLC
3. Results and Discussion
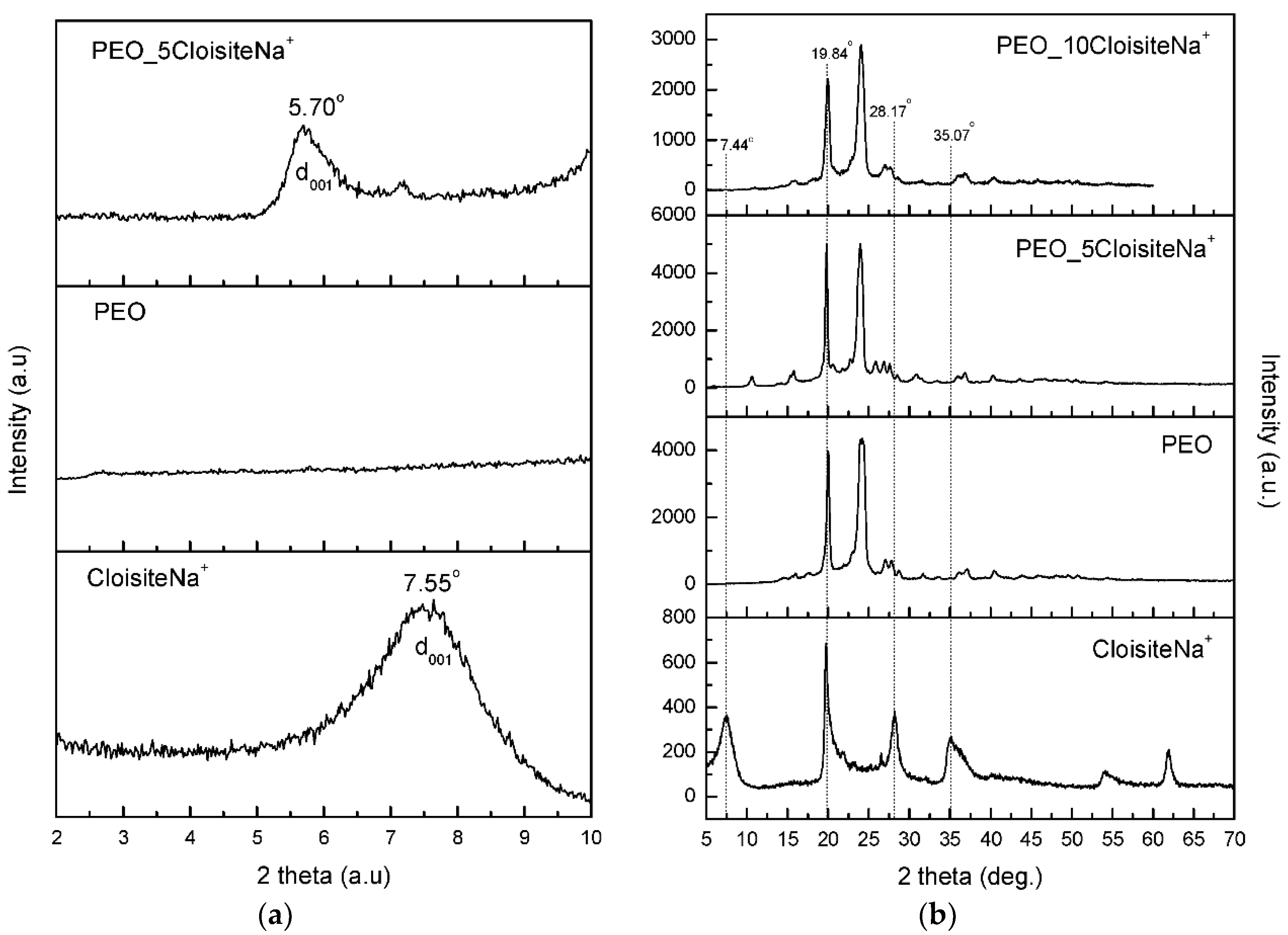
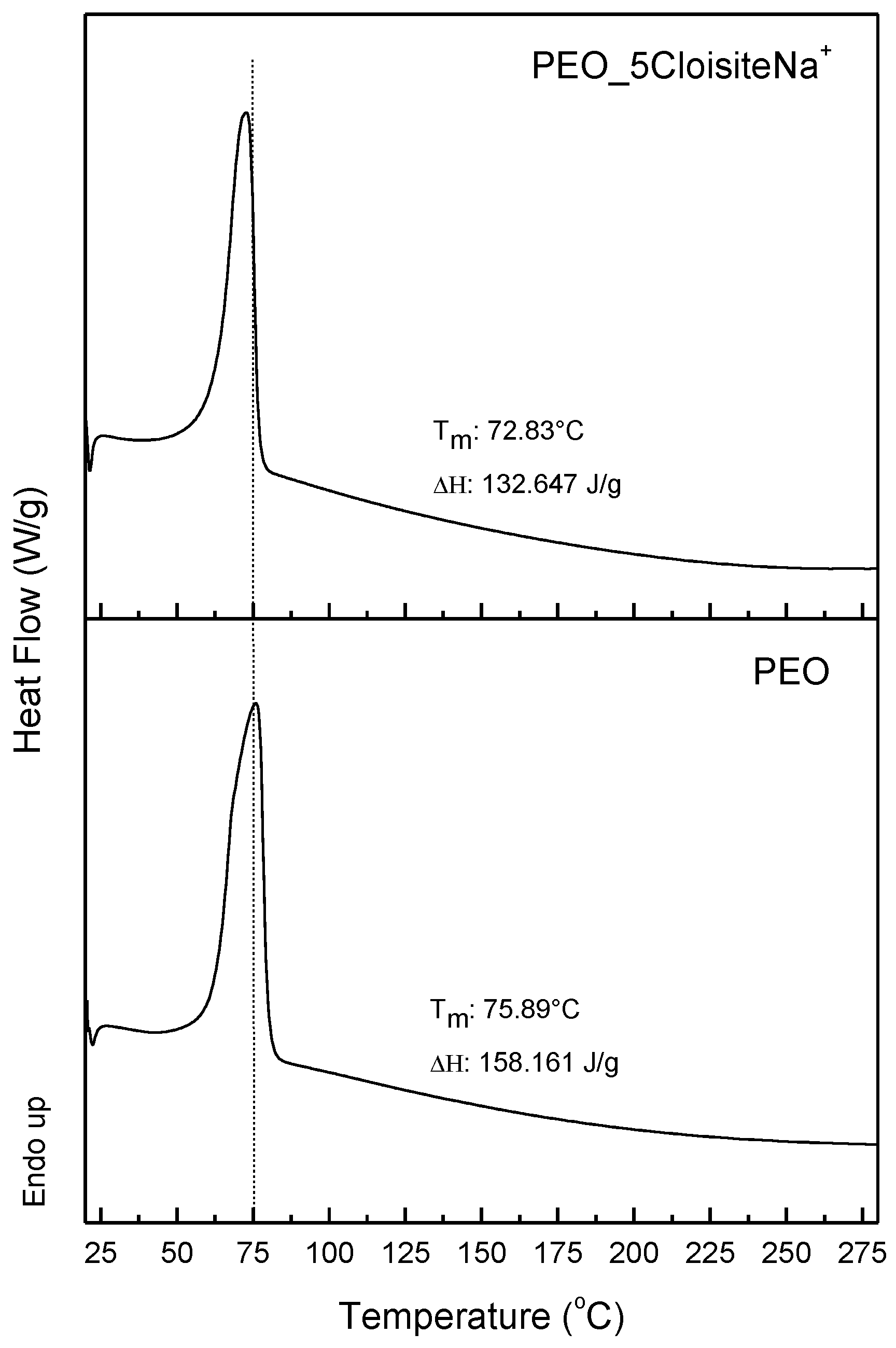
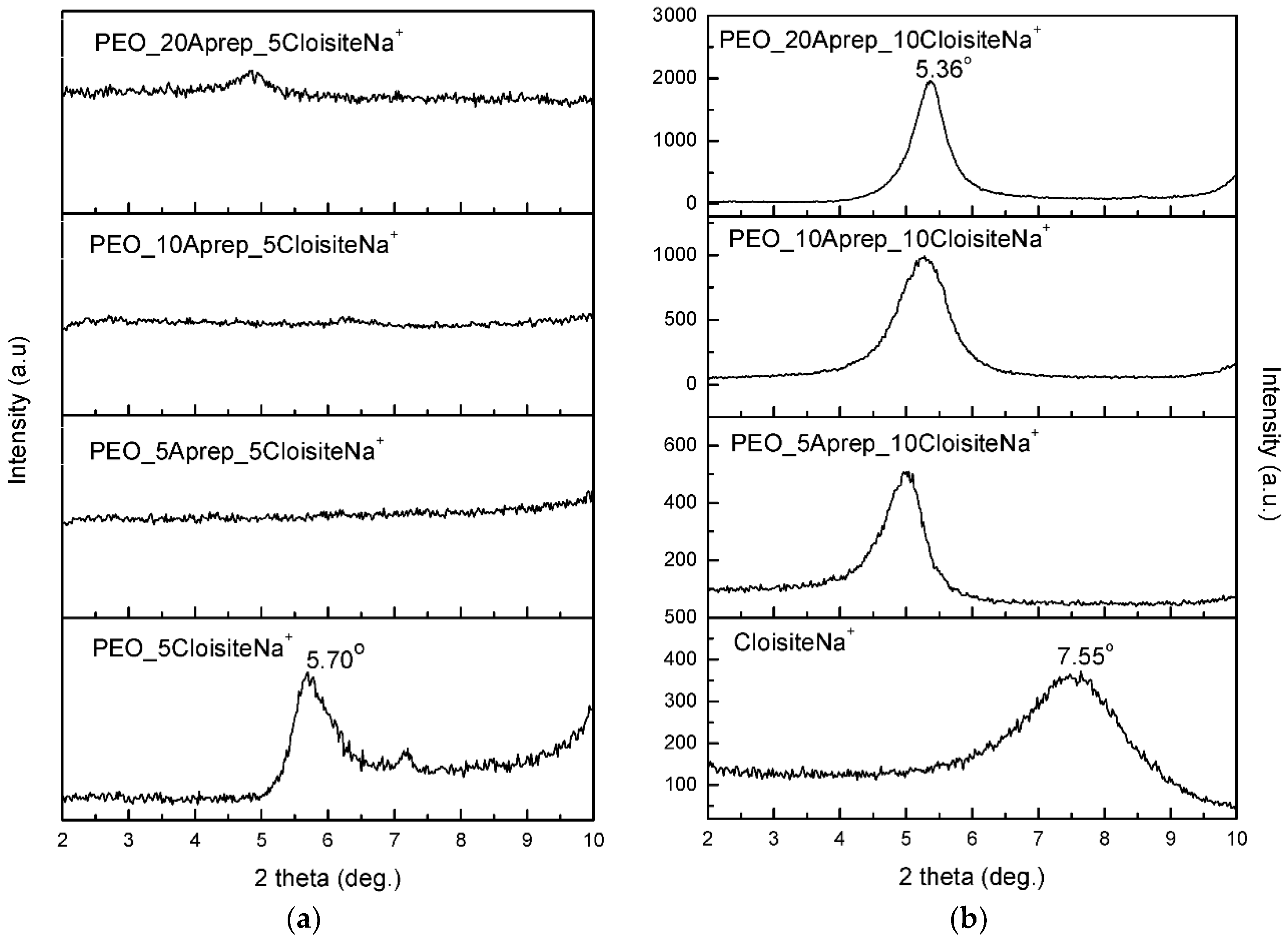
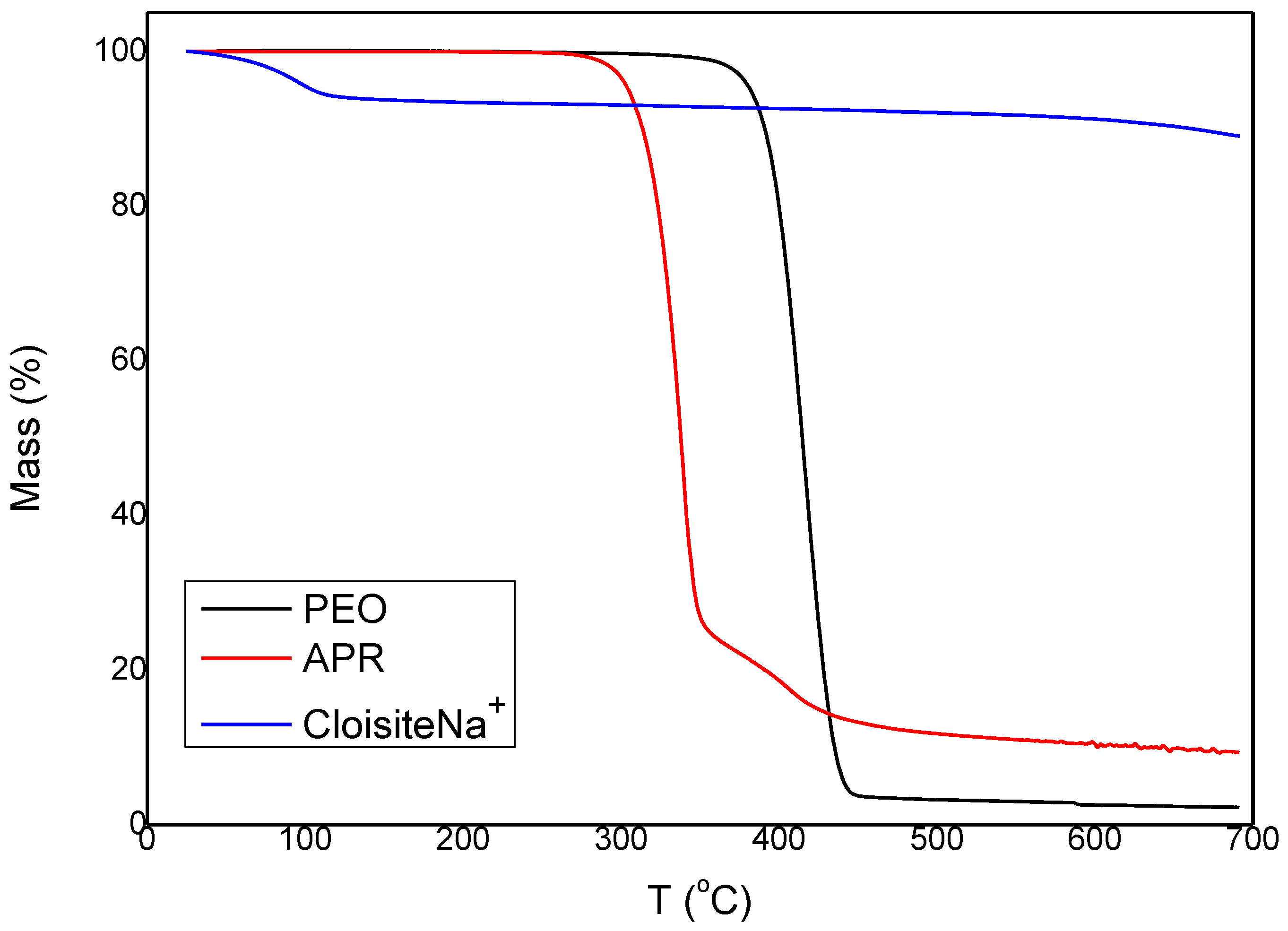
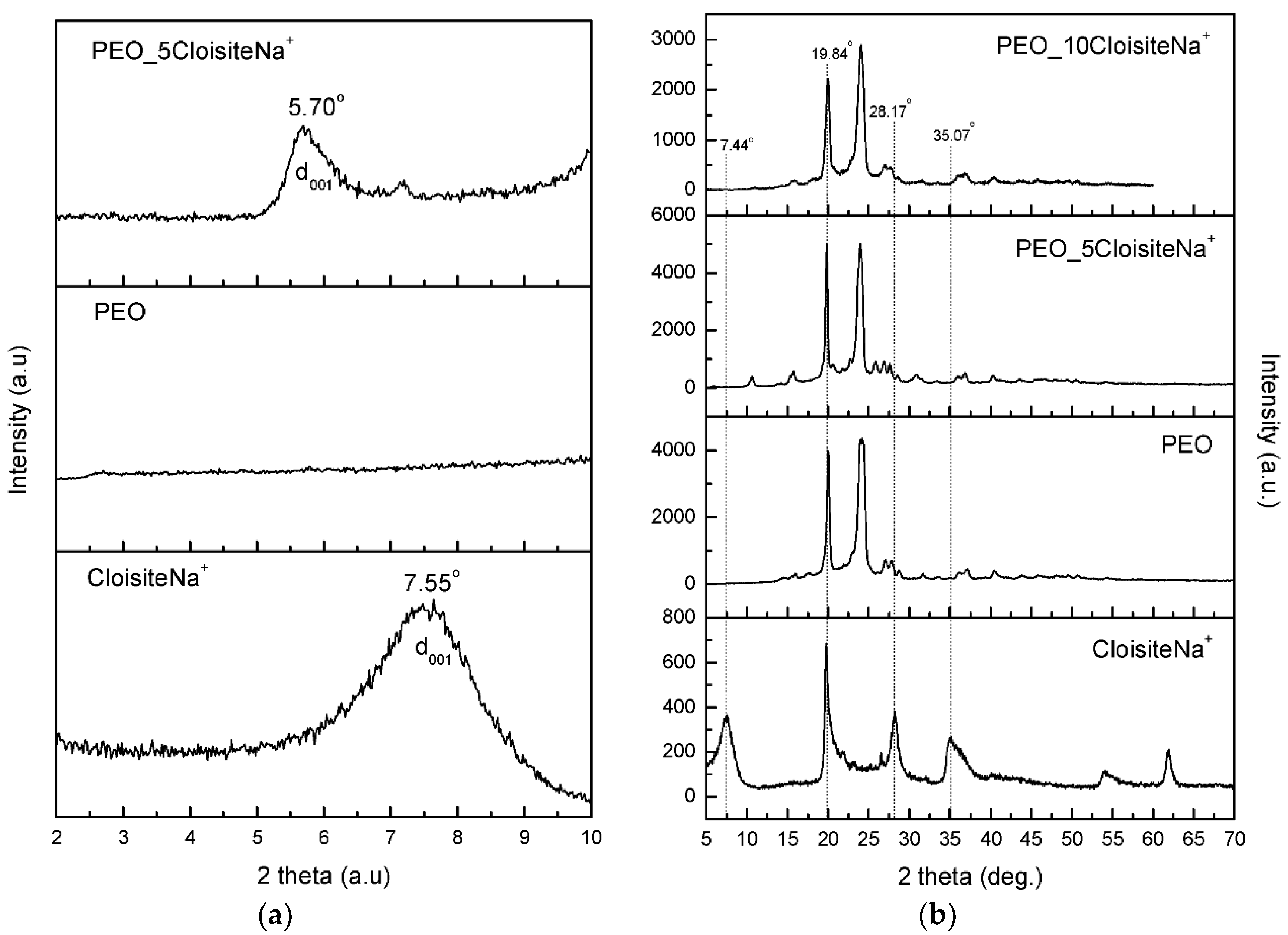
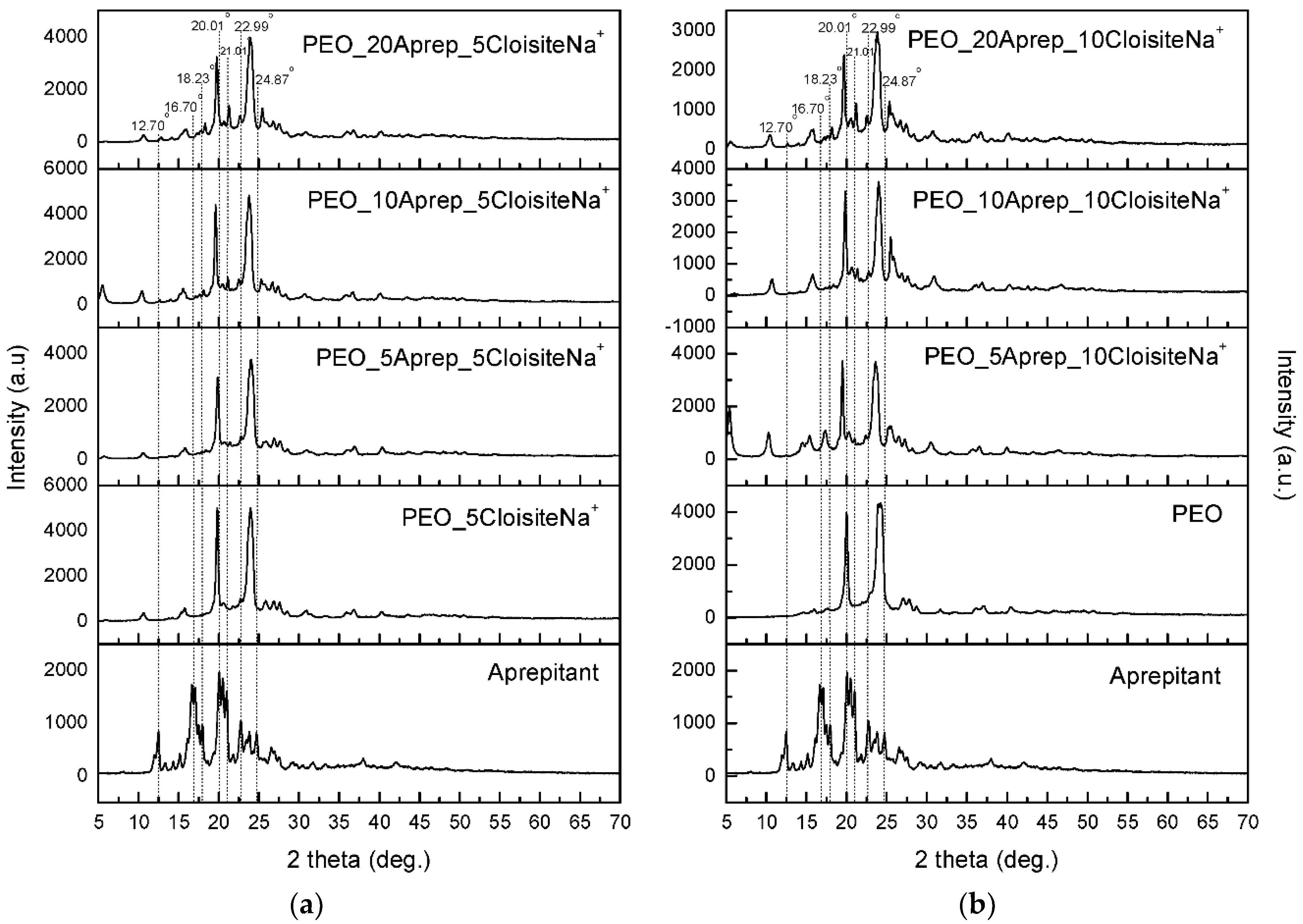
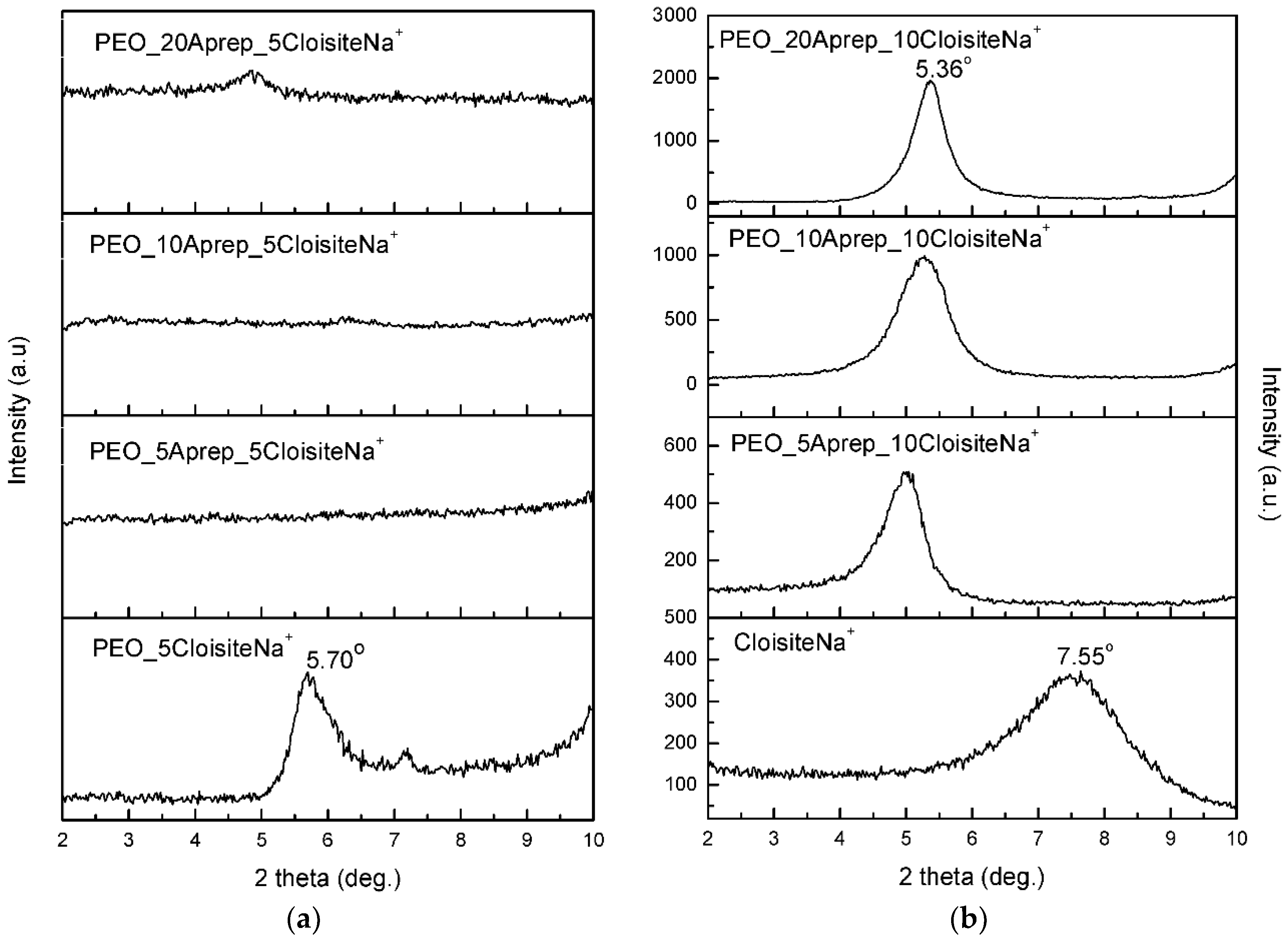
3.1. Characterization and Properties of PEO/Clay Nanocomposites
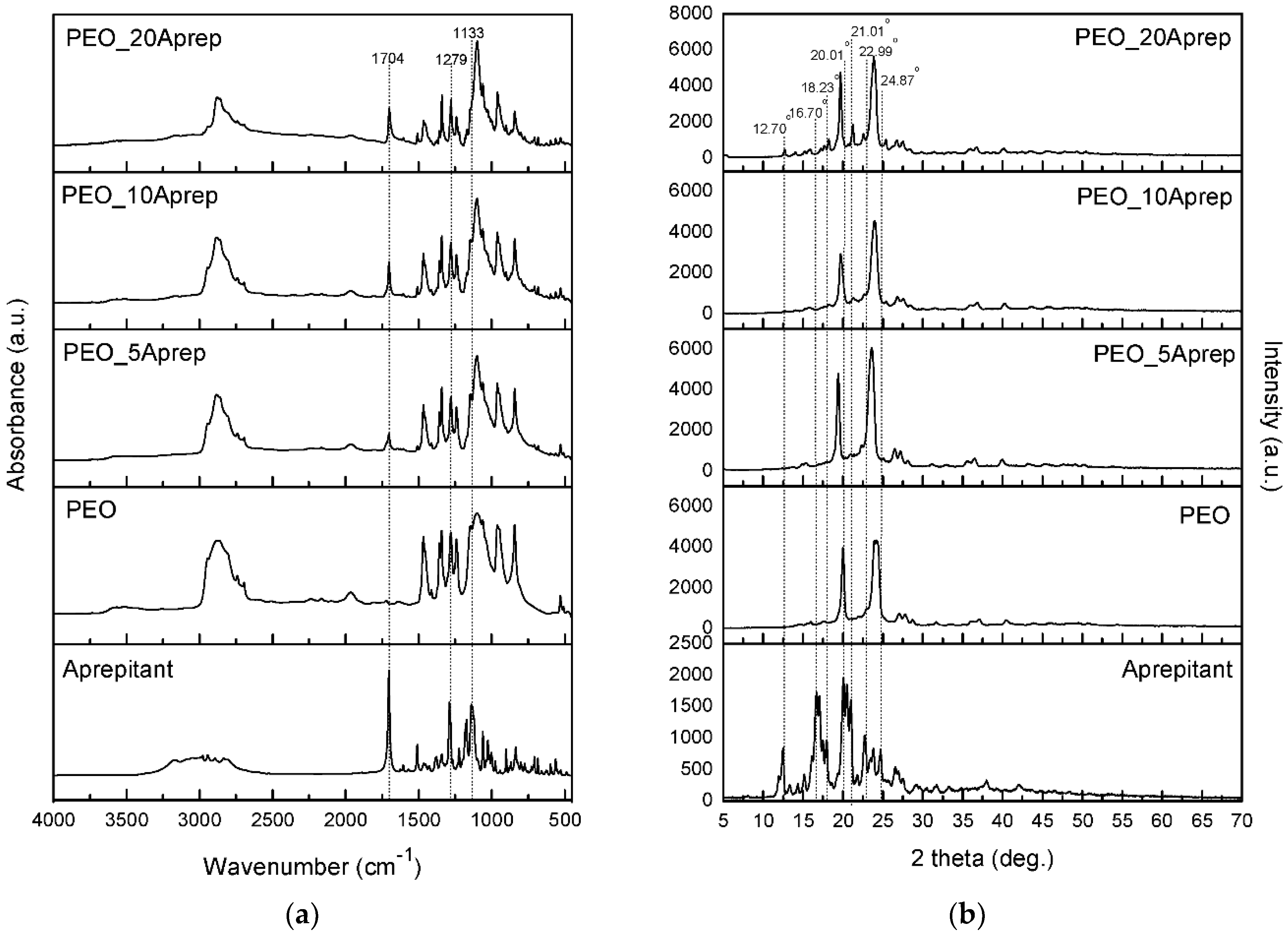
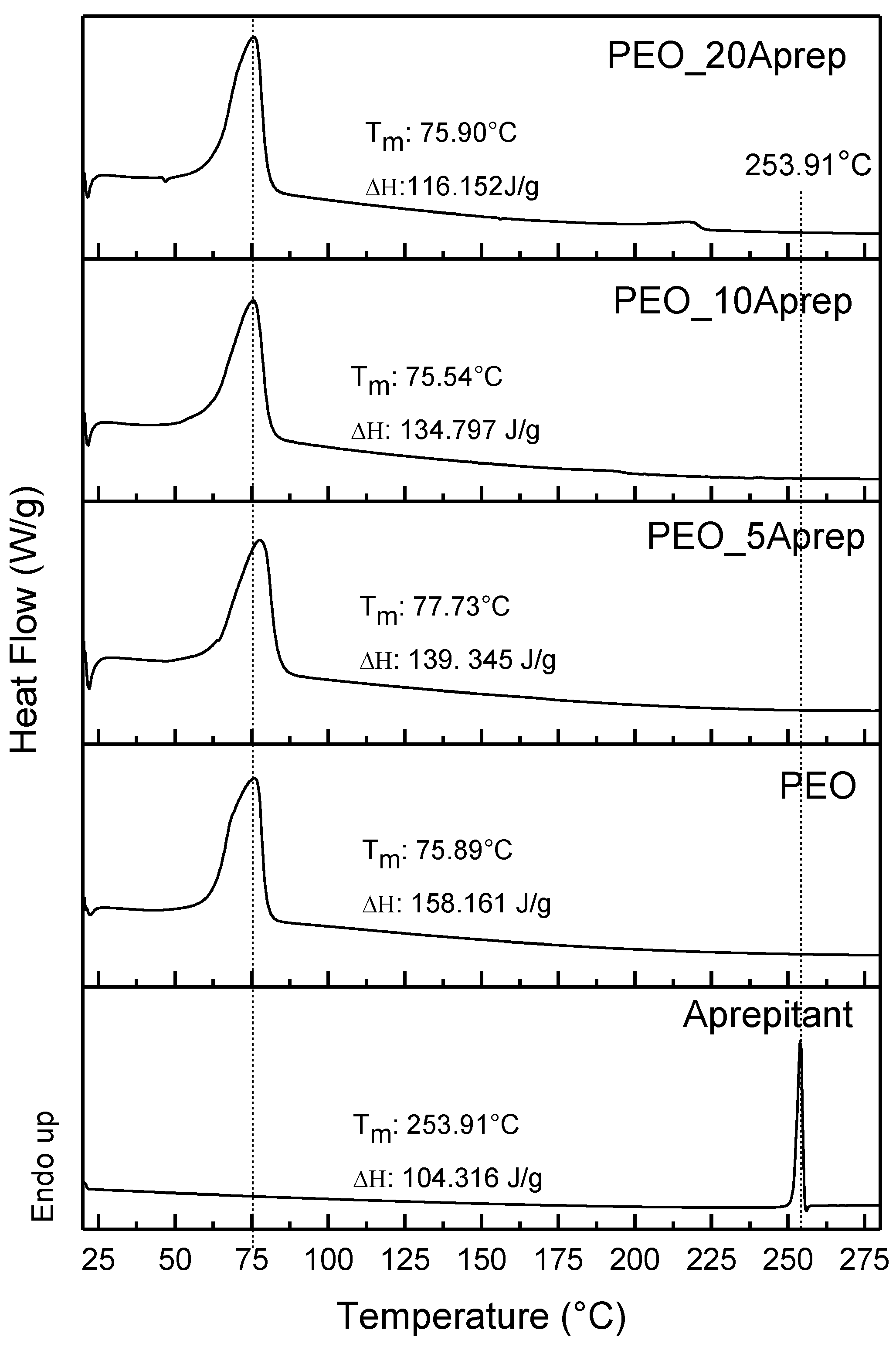
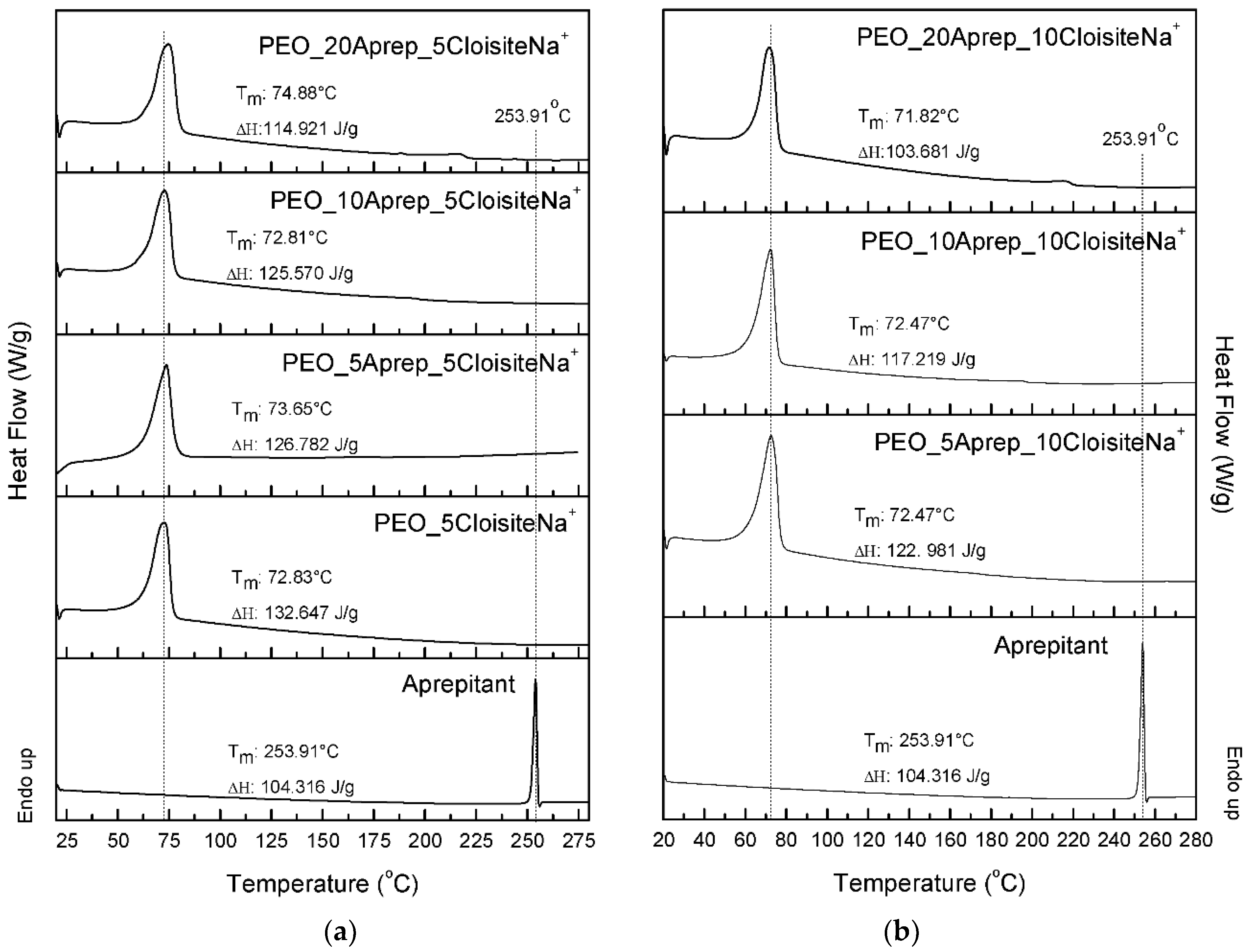
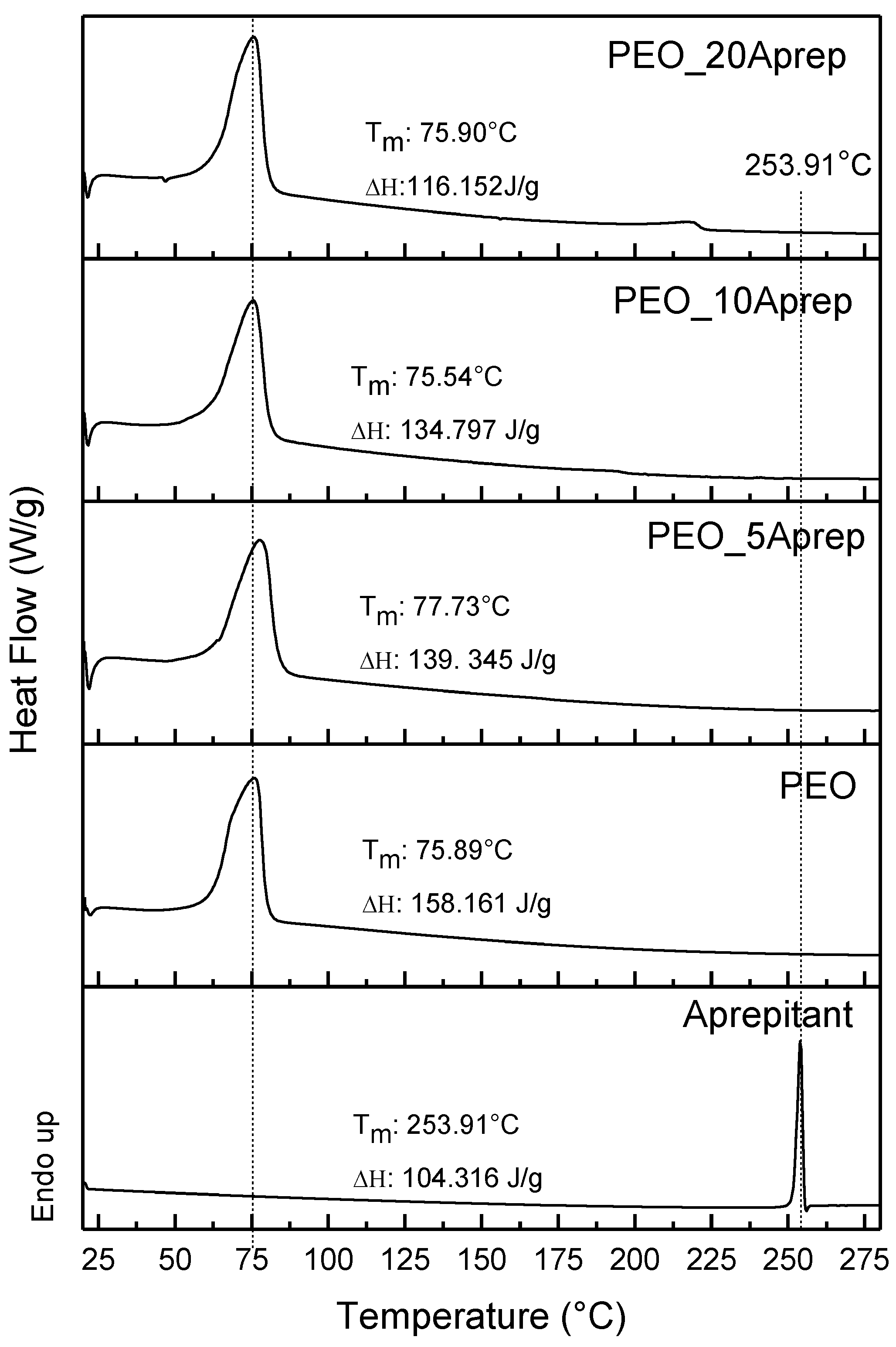
3.2. Characterization of Drug Loaded PEO
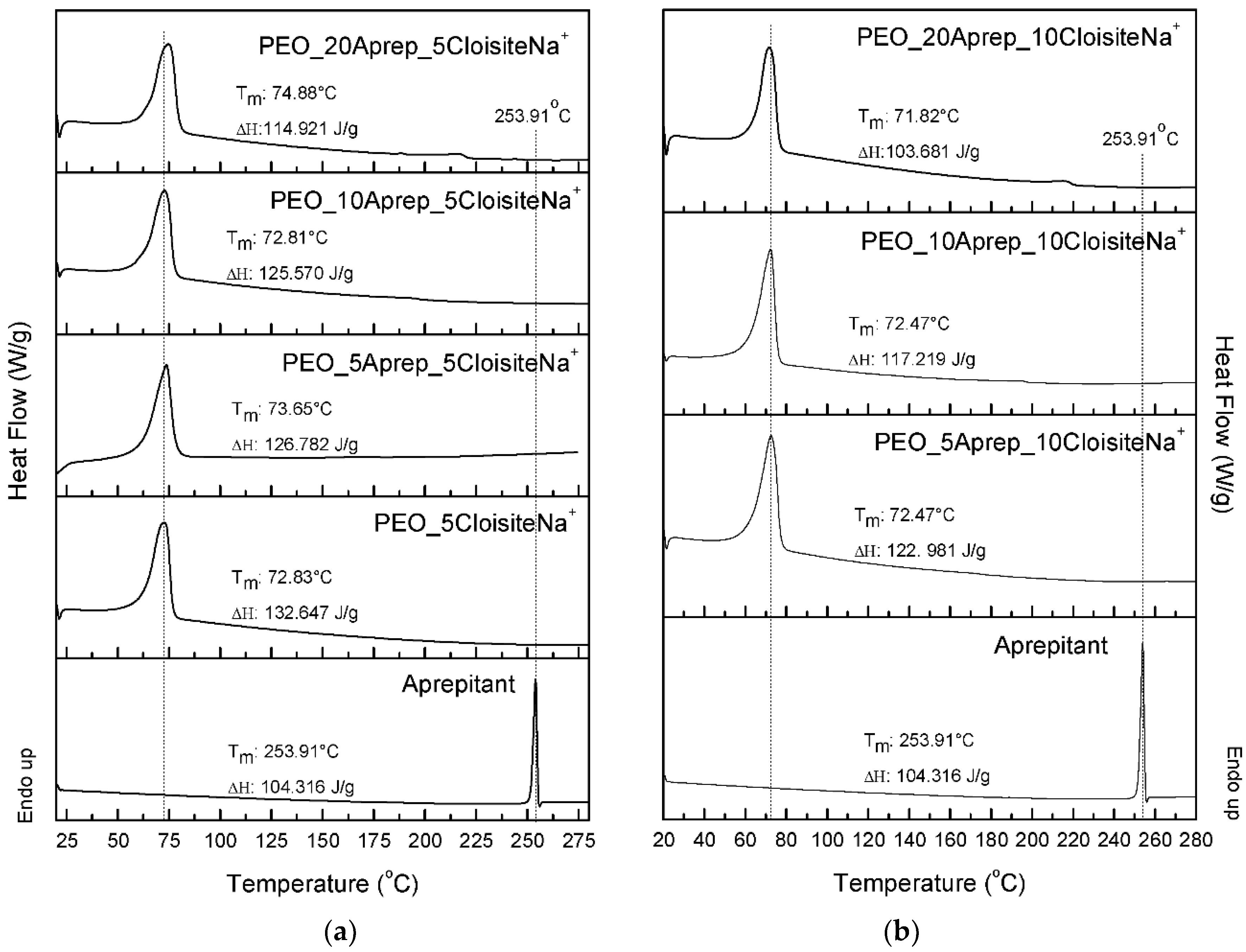
3.3. Characterization of Drug-Loaded PEO with 5 and 10 wt % Clay
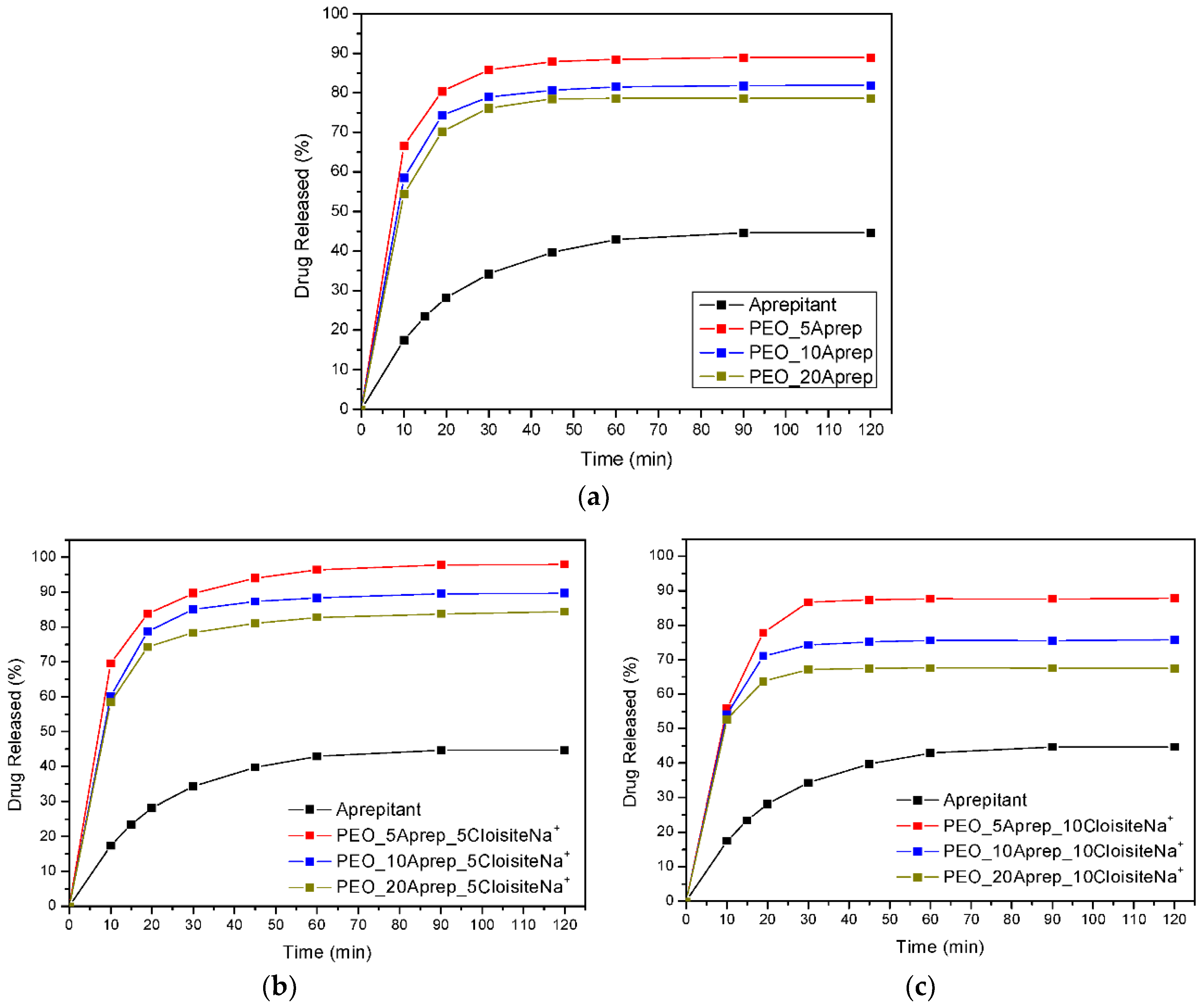
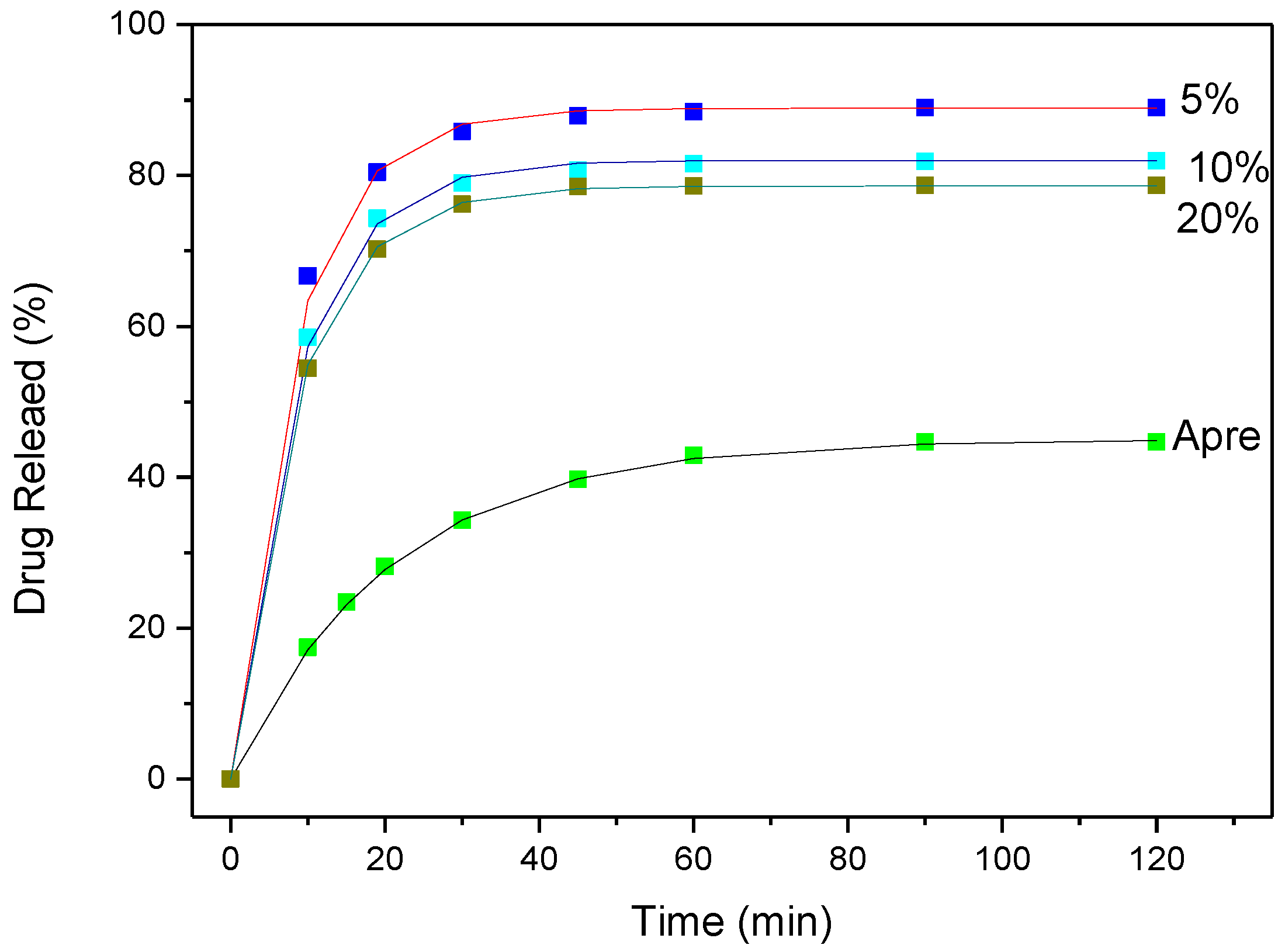
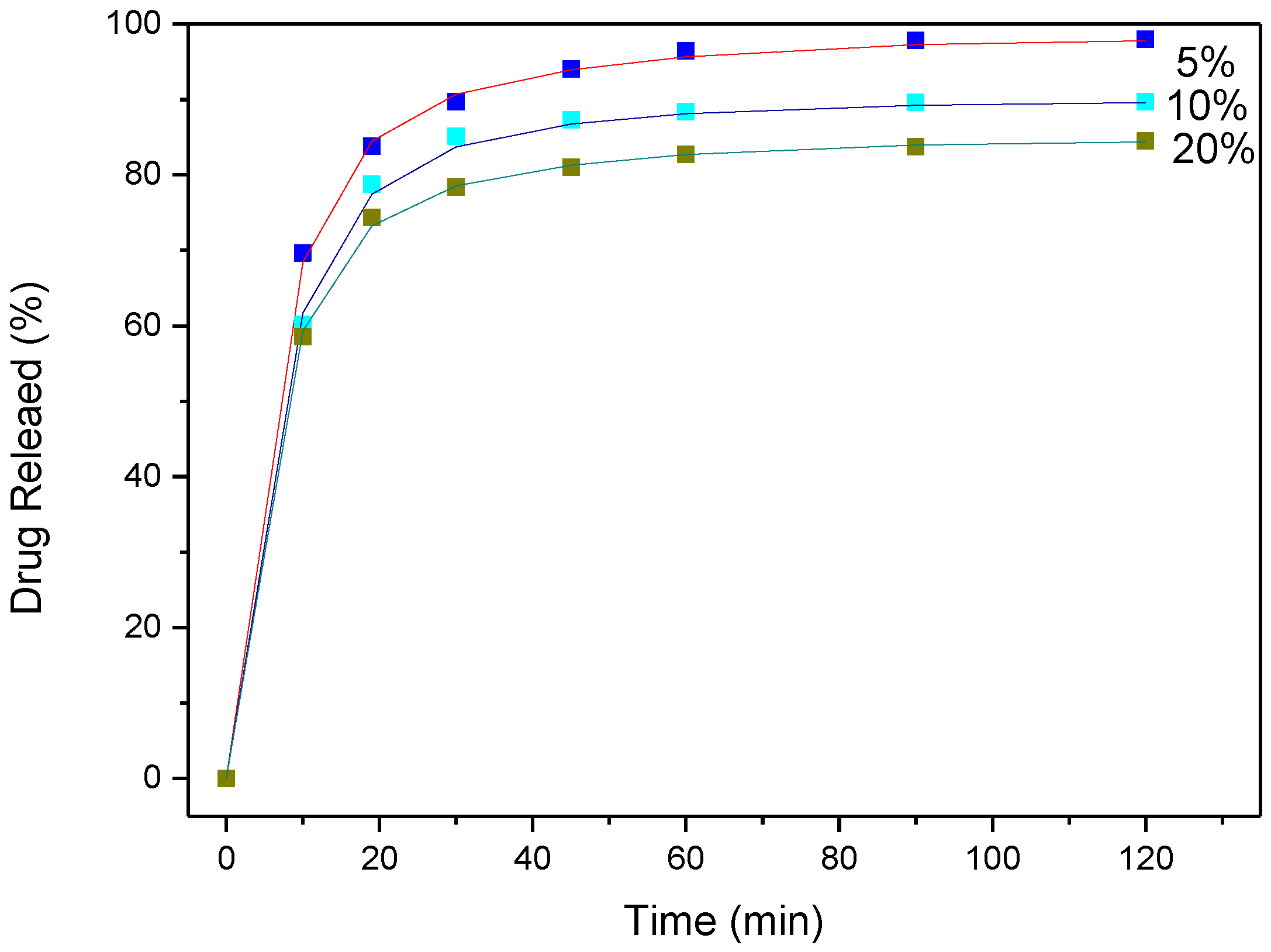
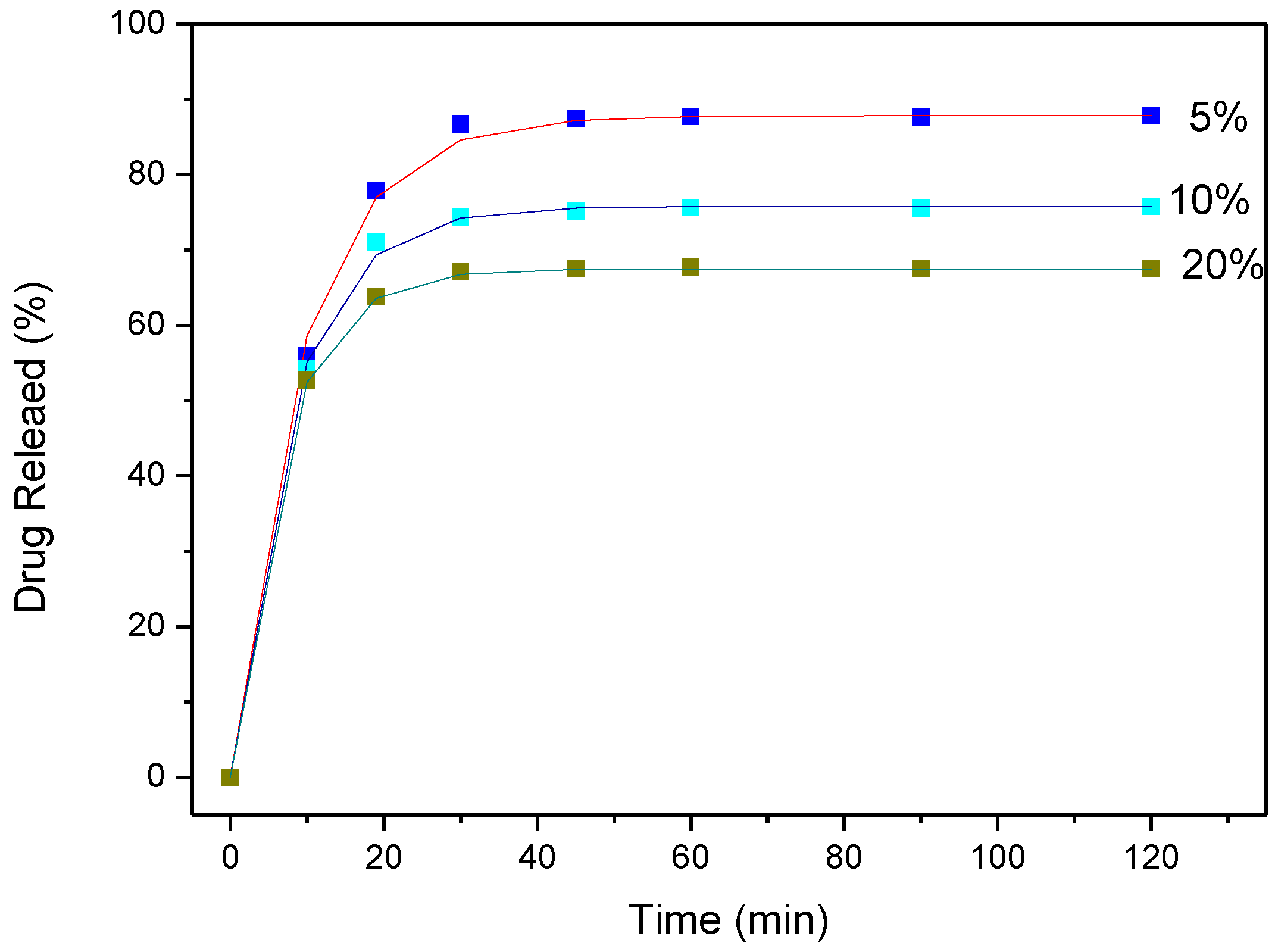
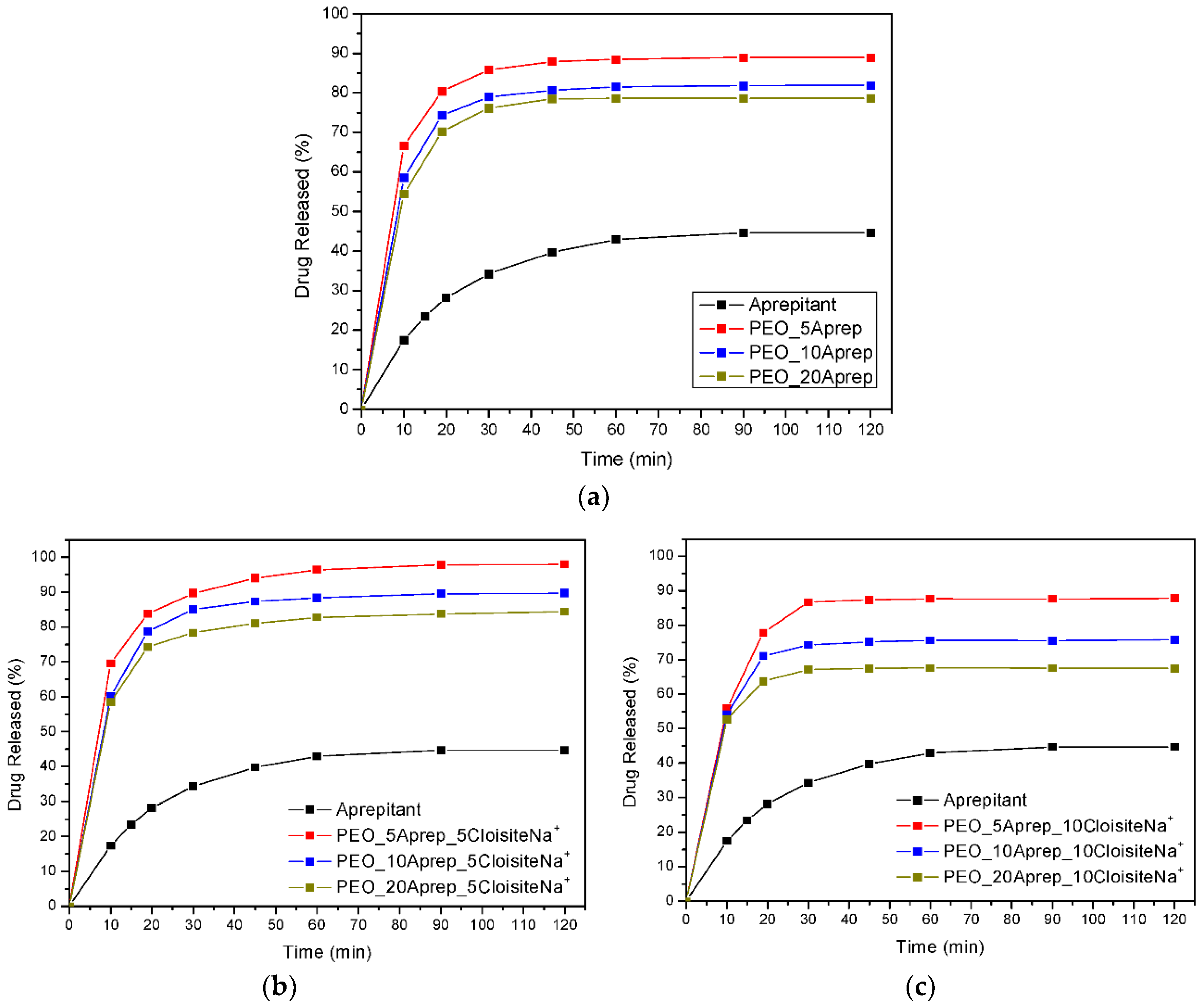
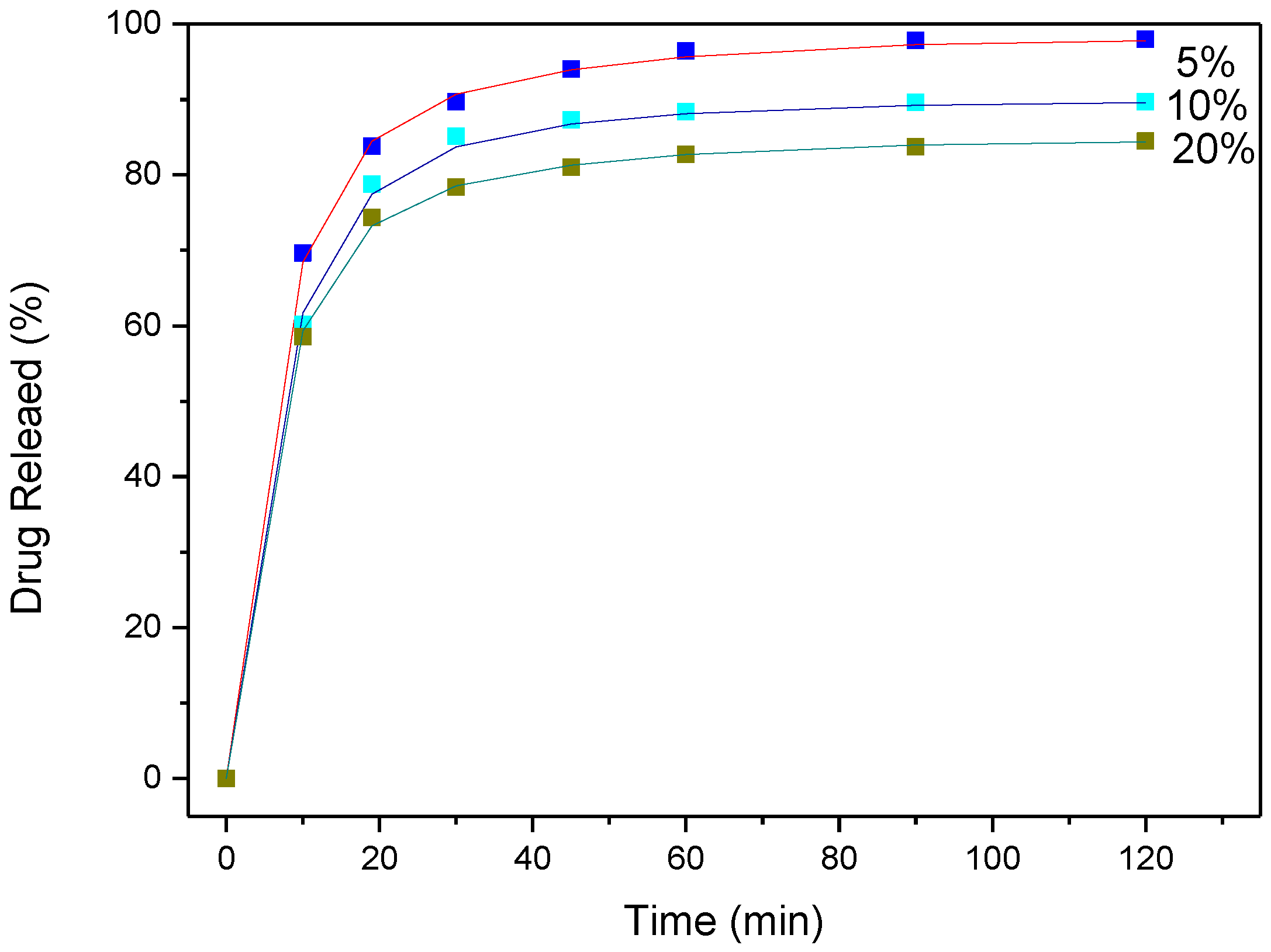
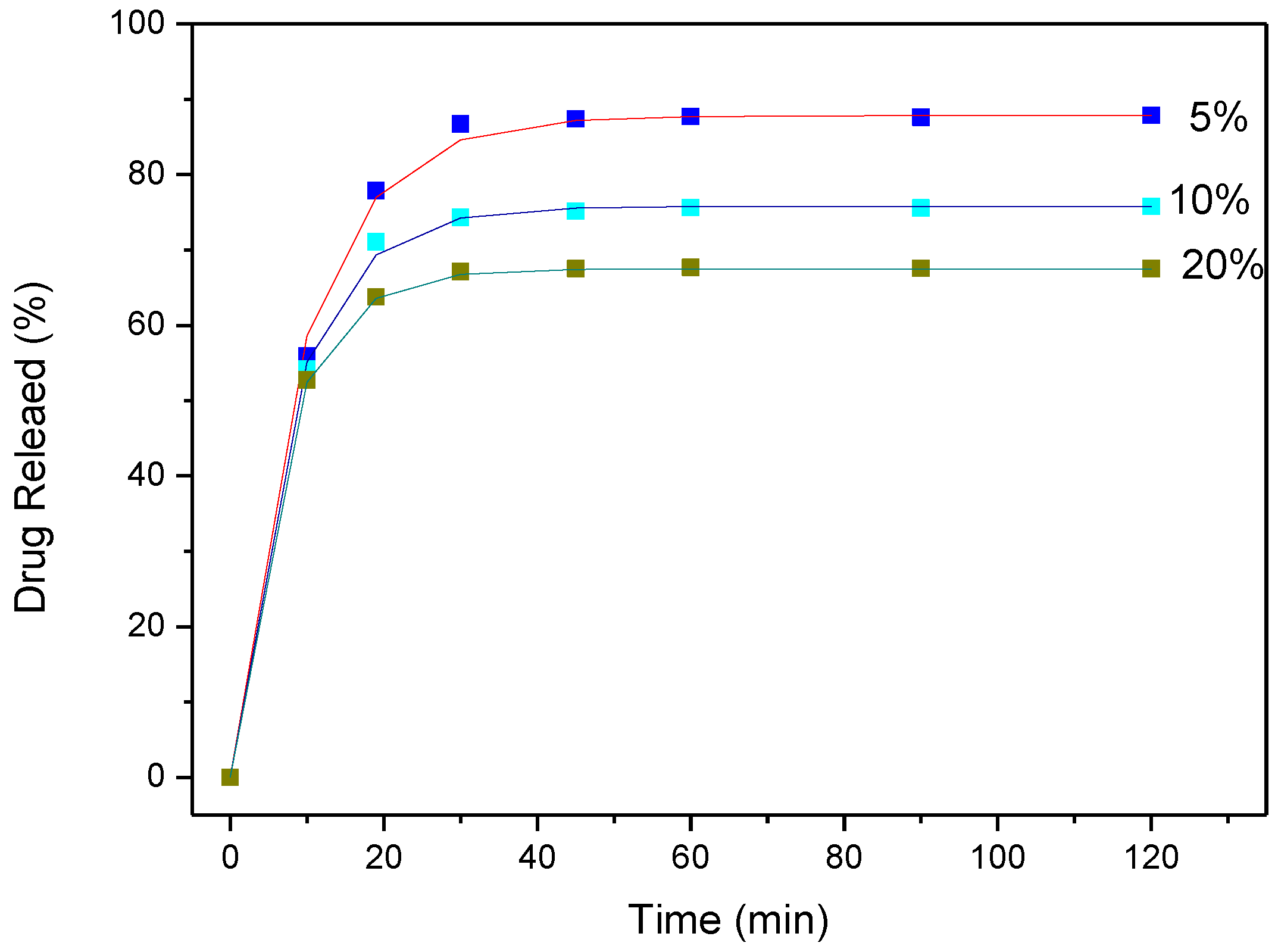
3.4. Drug (APR) Dissolution Study
3.5. Analysis and Discussion of Release Results
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Blumstein, A. Polymerization of adsorbed monolayers. Ii. Thermal degradation of the inserted polymer. J. Polym. Sci. Part A Gen. Pap. 1965, 3, 2665–2672. [Google Scholar] [CrossRef]
- Theng, B.K.G. Formation and Properties of Clay-Polymer Complexes; Elsevier: New York, NY, USA, 2012. [Google Scholar]
- Giannelis, E.P. Polymer-layered silicate nanocomposites: Synthesis, properties and applications. Appl. Organomet. Chem. 1998, 12, 675–680. [Google Scholar] [CrossRef]
- Sinha Ray, S.; Yamada, K.; Okamoto, M.; Ueda, K. Polylactide-layered silicate nanocomposite: A novel biodegradable material. Nano Lett. 2002, 2, 1093–1096. [Google Scholar] [CrossRef]
- Makoto, O.; Kazuyuki, K. Preparation of inorganic–organic nanocomposites through intercalation of organoammonium ions into layered silicates. Bull. Chem. Soc. Jpn. 1997, 70, 2593–2618. [Google Scholar]
- Theng, B.K.G. The Chemistry of Clay-Organic Reactions; Wiley: Hoboken, NJ, USA, 1974. [Google Scholar]
- Fukushima, Y.; Inagaki, S. Synthesis of an intercalated compound of montmorillonite and 6-polyamide. J. Incl. Phenom. 1987, 5, 473–482. [Google Scholar] [CrossRef]
- Burnside, S.D.; Giannelis, E.P. Synthesis and properties of new poly(dimethylsiloxane) nanocomposites. Chem. Mater. 1995, 7, 1597–1600. [Google Scholar] [CrossRef]
- Chen, B.; Evans, J.R.G. Poly(ε-caprolactone)−clay nanocomposites: Structure and mechanical properties. Macromolecules 2006, 39, 747–754. [Google Scholar] [CrossRef]
- Zhao, Q.; Samulski, E.T. Supercritical CO2-mediated intercalation of peo in clay. Macromolecules 2003, 36, 6967–6969. [Google Scholar] [CrossRef]
- Breitenbach, J. Melt extrusion: From process to drug delivery technology. Eur. J. Pharm. Biopharm. 2002, 54, 107–117. [Google Scholar] [CrossRef]
- Andrews, G.P.; Jones, D.S.; Diak, O.A.; McCoy, C.P.; Watts, A.B.; McGinity, J.W. The manufacture and characterisation of hot-melt extruded enteric tablets. Eur. J. Pharm. Biopharm. 2008, 69, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, P.; Zhang, X.; Shen, F.; Gogos, C.G. Effects of extrusion process parameters on the dissolution behavior of indomethacin in eudragit® E PO solid dispersions. Int. J. Pharm. 2010, 383, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Schilling, S.U.; Shah, N.H.; Waseem Malick, A.; McGinity, J.W. Properties of melt extruded enteric matrix pellets. Eur. J. Pharm. Biopharm. 2010, 74, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Maniruzzaman, M.; Morgan, D.J.; Mendham, A.P.; Pang, J.; Snowden, M.J.; Douroumis, D. Drug–polymer intermolecular interactions in hot-melt extruded solid dispersions. Int. J. Pharm. 2013, 443, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Maniruzzaman, M.; Rana, M.; Boateng, J.; Mitchell, J.; Douroumis, D. Dissolution enhancement of poorly water-soluble APIs processed by hot-melt extrusion using hydrophilic polymers. Drug Dev. Ind. Pharm. 2013, 39, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Maniruzzaman, M.; Boateng, J.; Snowden, M.; Douroumis, D. A review of hot-melt extrusion: Process technology to pharmaceutical products. ISRN Pharm. 2012, 2012, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Maniruzzaman, M.; Nair, A.; Scoutaris, N.; Bradley, M.S.A.; Snowden, M.J.; Douroumis, D. One-step continuous extrusion process for the manufacturing of solid dispersions. Int. J. Pharm. 2015, 496, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Vithani, K.; Maniruzzaman, M.; Slipper, I.J.; Mostafa, S.; Miolane, C.; Cuppokc, Y.; Marchaud, D.; Douroumis, D. Sustained release solid lipid matrices processed by hot-meltextrusion (HME). Colloids Surf. B Biointerfaces 2013, 110, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Alshehri, S.M.; Tiwari, R.V.; Alsulays, B.B.; Ashour, E.A.; Alshetaili, A.S.; Almutairy, B.; Park, J.-B.; Morott, J.; Sandhu, B.; Majumdar, S.; et al. Investigation of the combined effect of mgo and peg on the release profile of mefenamic acid prepared via hot-melt extrusion techniques. Pharm. Dev. Technol. 2017, 22, 740–753. [Google Scholar] [CrossRef] [PubMed]
- Patil, H.; Tiwari, R.V.; Repka, M.A. Hot-melt extrusion: From theory to application in pharmaceutical formulation. AAPS PharmSciTech 2016, 17, 20–42. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Maddineni, S.; Lu, J.; Repka, M.A. Melt extrusion with poorly soluble drugs. Int. J. Pharm. 2013, 453, 233–252. [Google Scholar] [CrossRef] [PubMed]
- Bikiaris, D.; Karavelidis, V.; Karavas, E. Effectiveness of various drug Carriers in controlled release formulations of raloxifene HCl prepared by melt mixing. Curr. Drug Deliv. 2009, 6, 425–436. [Google Scholar] [CrossRef]
- Fousteris, E.; Tarantili, P.A.; Karavas, E.; Bikiaris, D. Polyvinylpyrrolidone-poloxamer-188 solid dispersions prepared by hot melt extrusion: Thermal properties and release behavior. J. Therm. Anal. Calorim. 2013, 113, 1037–1047. [Google Scholar] [CrossRef]
- Palazi, E.; Karavas, E.; Barmpalexis, P.; Kostoglou, M.; Nanaki, S.; Christodoulou, E.; Bikiaris, D. Melt extrusion process for adjusting drug release of poorly water soluble drug felodipine using different polymer matrices. Eur. J. Pharm. Sci. 2018, 114, 332–345. [Google Scholar] [CrossRef] [PubMed]
- Prodduturi, S.; Manek, R.V.; Kolling, W.M.; Stodghill, S.P.; Repka, M.A. Solid-state stability and characterization of hot-melt extruded poly(ethylene oxide) films. J. Pharm. Sci. 2005, 94, 2232–2245. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.T.; Craig, D.Q.M.; McNally, T. Modification of ibuprofen drug release from poly(ethylene glycol) layered silicate nanocomposites prepared by hot-melt extrusion. J. Appl. Polym. Sci. 2014, 131. [Google Scholar] [CrossRef]
- Schachter, D.M.; Xiong, J.; Tirol, G.C. Solid state NMR perspective of drug–polymer solid solutions: A model system based on poly(ethylene oxide). Int. J. Pharm. 2004, 281, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Ozeki, T.; Yuasa, H.; Kanaya, Y. Application of the solid dispersion method to the controlled release of medicine. IX. Difference in the release of flurbiprofen from solid dispersions with poly(ethylene oxide) and hydroxypropylcellulose and the interaction between medicine and polymers. Int. J. Pharm. 1997, 155, 209–217. [Google Scholar] [CrossRef]
- Siepmann, J.; Siepmann, F. Mathematical modeling of drug delivery. Int. J. Pharm. 2008, 364, 328–343. [Google Scholar] [CrossRef] [PubMed]
- Siepmann, J.; Siepmann, F. Modeling of diffusion controlled drug delivery. J. Control. Release 2012, 161, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Viseras, C.; Cerezo, P.; Sanchez, R.; Salcedo, I.; Aguzzi, C. Current challenges in clay minerals for drug delivery. Appl. Clay Sci. 2010, 48, 291–295. [Google Scholar] [CrossRef]
- Mousa, M.; Evans, N.D.; Oreffo, R.O.C.; Dawson, J.I. Clay nanoparticles for regenerative medicine and biomaterial design: A review of clay bioactivity. Biomaterials 2018, 159, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Bitinis, N.; Hernandez, M.; Verdejo, R.; Kenny, J.M.; Lopez-Manchado, M.A. Recent advances in clay/polymer nanocomposites. Adv. Mater. 2011, 23, 5229–5236. [Google Scholar] [CrossRef] [PubMed]
- Mittal, G.; Rhee, K.Y.; Mišković-Stanković, V.; Hui, D. Reinforcements in multi-scale polymer composites: Processing, properties, and applications. Compos. Part B 2018, 138, 122–139. [Google Scholar] [CrossRef]
- Yang, M.; Wang, P.; Huang, C.-Y.; Ku, M.S.; Liu, H.; Gogos, C. Solid dispersion of acetaminophen and poly(ethylene oxide) prepared by hot-melt mixing. Int. J. Pharm. 2010, 395, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Poli-Bigelli, S.; Rodrigues-Pereira, J.; Carides, A.D.; Julie Ma, G.; Eldridge, K.; Hipple, A.; Evans, J.K.; Horgan, K.J.; Lawson, F.; On Behalf of the Aprepitant Protocol 054 Study Group. Addition of the neurokinin 1 receptor antagonist aprepitant to standard antiemetic therapy improves control of chemotherapy-induced nausea and vomiting. Cancer 2003, 97, 3090–3098. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Kamboj, S.; Singh, G.; Rana, V. Development of aprepitant loaded orally disintegrating films for enhanced pharmacokinetic performance. Eur. J. Pharm. Sci. 2016, 84, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Kesisoglou, F.; Mitra, A. Crystalline nanosuspensions as potential toxicology and clinical oral formulations for bcs ii/iv compounds. AAPS J. 2012, 14, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Kesisoglou, F.; Wu, Y. Understanding the effect of API properties on bioavailability through absorption modeling. AAPS J. 2008, 10, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Bikiaris, D.N. Solid dispersions, Part I: Recent evolutions and future opportunities in manufacturing methods for dissolution rate enhancement of poorly water-soluble drugs. Expert Opin. Drug Deliv. 2011, 8, 1501–1519. [Google Scholar] [CrossRef] [PubMed]
- Bikiaris, D.N. Solid dispersions, Part II: New strategies in manufacturing methods for dissolution rate enhancement of poorly water-soluble drugs. Expert Opin. Drug Deliv. 2011, 8, 1663–1680. [Google Scholar] [CrossRef] [PubMed]
- Barmpalexis, P.; Koutsidis, I.; Karavas, E.; Louka, D.; Papadimitriou, S.A.B.; Bikiaris, D.N. Development of PVP/PEG mixtures as appropriate carriers for the preparation of drug solid dispersions by melt mixing technique and optimization of dissolution using artificial neural networks. Eur. J. Pharm. Biopharm. 2013, 85, 1219–1231. [Google Scholar] [CrossRef] [PubMed]
- Papadimitriou, S.A.; Barmpalexis, P.; Karavas, E.; Bikiaris, D.N. Optimizing the ability of PVP/PEG mixtures to be used as appropriate carriers for the preparation of drug solid dispersions by melt mixing technique using artificial neural networks: I. Eur. J. Pharm. Biopharm. 2012, 82, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekhara Rao, B.; Vidyadhara, S.; Rlc, S.; Chowdary, D.Y. Dissolution enhancement of poorly soluble drug aprepitant by hot melt extrusion method using hydrophilic polymer: A solid dispersion technique. Res. J. Pharm. Biol. Chem. Sci. 2014, 5, 1469–1485. [Google Scholar]
- Prodduturi, S.; Dhawan, S.; Jatla, V.; Dintakurthi, N.; Venkata Gopal Krishna, K.; Gunturu, G.; Ansari, K.A.; Ridhurkar, D.; Rudraraju, V.; Pillai, R. Enhancement of dissolution rate of aprepitant using melt-extrusion technology. In Proceedings of the AAPS, Washington, DC, USA, 23–27 October 2011. [Google Scholar]
- Penumetcha, S.S.; Gutta, L.N.; Dhanala, H.; Yamili, S.; Challa, S.; Rudraraju, S.; Rudraraju, S.; Rudraraju, V. Hot melt extruded aprepitant–soluplus solid dispersion: Preformulation considerations, stability and in vitro study. Drug Dev. Ind. Pharm. 2016, 42, 1609–1620. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zou, M.; Piao, H.; Liu, Y.; Tang, B.; Gao, Y.; Ma, N.; Cheng, G. Characterization and pharmacokinetic study of aprepitant solid dispersions with soluplus®. Molecules 2015, 20, 11345–11356. [Google Scholar] [CrossRef] [PubMed]
- Dissolution Methods. Available online: https://www.accessdata.fda.gov/scripts/cder/dissolution (accessed on 2018).
- Hoffmann, C.L.; Rabolt, J.F. Self-assembled thin-film blends by polymer codeposition: Poly(ethylene oxide) and poly(methyl methacrylate). Macromolecules 1996, 29, 2543–2547. [Google Scholar] [CrossRef]
- Rocco, A.; Polo da Fonseca, C.; Pereira, R. A polymeric solid electrolyte based on a binary blend of poly(ethylene oxide), poly(methyl vinyl ether-maleic acid) and liclo4. Ploymer 2002, 43, 3601–3609. [Google Scholar] [CrossRef]
- Tang, Z.; Wang, J.; Chen, Q.; He, W.; Shen, C.; Mao, X.-X.; Zhang, J. A novel peo-based composite polymer electrolyte with absorptive glass mat for li-ion batteries. Electrochim. Acta 2007, 52, 6638–6643. [Google Scholar] [CrossRef]
- Sundar, M.; Selladurai, S. Effect of fillers on magnesium–poly(ethylene oxide) solid polymer electrolyte. Ionics 2006, 12, 281–286. [Google Scholar] [CrossRef]
- Yoshihara, T.; Tadokoro, H.; Murahashi, S. Normal vibrations of the polymer molecules of helical conformation. Iv. Polyethylene oxide and polyethylene-d4 oxide. J. Chem. Phys. 1964, 41, 2902–2911. [Google Scholar] [CrossRef]
- Mallakpour, S.; Barati, A. Application of modified cloisite Na+ with l-phenylalanine for the preparation of new poly(vinyl alcohol)/organoclay bionanocomposite films. Polym.-Plast. Technol. Eng. 2012, 51, 321–327. [Google Scholar] [CrossRef]
- Junior, C.R.F.; de Moura, M.R.; Aouada, F.A. Synthesis and characterization of intercalated nanocomposites based on poly(methacrylic acid) hydrogel and nanoclay cloisite-Na+ for possible application in agriculture. J. Nanosci. Nanotechnol. 2017, 17, 5878–5883. [Google Scholar] [CrossRef]
- Loyens, W.; Maurer, F.H.J.; Jannasch, P. Melt-compounded salt-containing poly(ethylene oxide)/clay nanocomposites for polymer electrolyte membranes. Ploymer 2005, 46, 7334–7345. [Google Scholar] [CrossRef]
- Loyens, W.; Jannasch, P.; Maurer, F.H.J. Effect of clay modifier and matrix molar mass on the structure and properties of poly(ethylene oxide)/cloisite nanocomposites via melt-compounding. Polymer 2005, 46, 903–914. [Google Scholar] [CrossRef]
- Ratna, D.; Divekar, S.; Samui, A.; Chakraborty, B.; Banthia, A. Poly(ethylene oxide)/clay nanocomposite: Thermomechanical properties and morphology. Polymer 2006, 47, 4068–4074. [Google Scholar] [CrossRef]
- Strawhecker, K.E.; Manias, E. Crystallization behavior of poly(ethylene oxide) in the presence of Na+ montmorillonite fillers. Chem. Mater. 2003, 15, 844–849. [Google Scholar] [CrossRef]
- Liao, B.; Song, M.; Liang, H.J.; Pang, Y.X. Polymer-layered silicate nanocomposites. 1. A study of poly(ethylene oxide)/Na+-montmorillonite nanocomposites as polyelectrolytes and polyethylene-block-poly(ethylene glycol) copolymer/Na+-montmorillonite nanocomposites as fillers for reinforcement of polyethylene. Polymer 2001, 42, 10007–10011. [Google Scholar]
- Ridhurkar, D.N.; Ansari, K.A.; Kumar, D.; Kaul, N.S.; Krishnamurthy, T.; Dhawan, S.; Pillai, R. Inclusion complex of aprepitant with cyclodextrin: Evaluation of physico-chemical and pharmacokinetic properties. Drug Dev. Ind. Pharm. 2013, 39, 1783–1792. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Zhou, Y.; Wei, P.; Li, M.; Chen, G. Preparation and pharmacokinetic study of aprepitant–sulfobutyl ether-β-cyclodextrin complex. AAPS PharmSciTech 2014, 15, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Helmy, R.; Zhou, G.X.; Chen, Y.W.; Crocker, L.; Wang, T.; Wenslow, R.M.; Vailaya, A. Characterization and quantitation of aprepitant drug substance polymorphs by attenuated total reflectance fourier transform infrared spectroscopy. Anal. Chem. 2003, 75, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Braun, D.E.; Gelbrich, T.; Kahlenberg, V.; Laus, G.; Wieser, J.; Griesser, U.J. Packing polymorphism of a conformationally flexible molecule (aprepitant). New J. Chem. 2008, 32, 1677–1685. [Google Scholar] [CrossRef]
- Van Renterghem, J.; Dhondt, H.; Verstraete, G.; De Bruyne, M.; Vervaet, C.; De Beer, T. The impact of the injection mold temperature upon polymer crystallization and resulting drug release from immediate and sustained release tablets. Int. J. Pharm. 2018, 541, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Liu, Z.; Chen, Y.; Chen, Z.; Chen, H.; Pui, Y.; Qian, F. Oral bioavailability enhancement of β-lapachone, a poorly soluble fast crystallizer, by cocrystal, amorphous solid dispersion, and crystalline solid dispersion. Eur. J. Pharm. Biopharm. 2018, 124, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Van Duong, T.; Goderis, B.; Van Humbeeck, J.; Van Den Mooter, G. Microstructure of Pharmaceutical Semicrystalline Dispersions: The Significance of Polymer Conformation. Mol. Pharm. 2018, 15, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Tien, C. Adsorption Calculations and Modeling; Butterworth-Heinemann: Boston, MA, USA, 1994. [Google Scholar]
- Kim, C.J. Effects of drug solubility, drug loading, and polymer molecular weight on drug release from Polyox tablets. Drug Dev. Ind. Pharm. 1998, 24, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Cantin, O.; Siepmann, F.; Danede, F.; Willart, J.F.; Karrout, Y.; Siepmann, J. PEO hot melt extrudates for controlled drug delivery: Importance of the molecular weight. J. Drug Deliv. Sci. Technol. 2016, 36, 130–140. [Google Scholar] [CrossRef]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pappa, C.; Nanaki, S.; Giliopoulos, D.; Triantafyllidis, K.; Kostoglou, M.; Avgeropoulos, A.; Bikiaris, D. Nanostructured Composites of Sodium Montmorillonite Clay and PEO Used in Dissolution Improvement of Aprepitant Drug by Melt Mixing. Appl. Sci. 2018, 8, 786. https://doi.org/10.3390/app8050786
Pappa C, Nanaki S, Giliopoulos D, Triantafyllidis K, Kostoglou M, Avgeropoulos A, Bikiaris D. Nanostructured Composites of Sodium Montmorillonite Clay and PEO Used in Dissolution Improvement of Aprepitant Drug by Melt Mixing. Applied Sciences. 2018; 8(5):786. https://doi.org/10.3390/app8050786
Chicago/Turabian StylePappa, Christina, Stavroula Nanaki, Dimitrios Giliopoulos, Konstantinos Triantafyllidis, Margaritis Kostoglou, Apostolos Avgeropoulos, and Dimitrios Bikiaris. 2018. "Nanostructured Composites of Sodium Montmorillonite Clay and PEO Used in Dissolution Improvement of Aprepitant Drug by Melt Mixing" Applied Sciences 8, no. 5: 786. https://doi.org/10.3390/app8050786