
Synthesis, Characterization and Shape-Dependent Catalytic CO Oxidation Performance of Ruthenium Oxide Nanomaterials: Influence of Polymer Surfactant


Abstract
:
1. Introduction
2. Experimental Section
2.1. Materials
2.2. Syntheses of Catalysts
2.3. Catalyst Characterization
3. Results
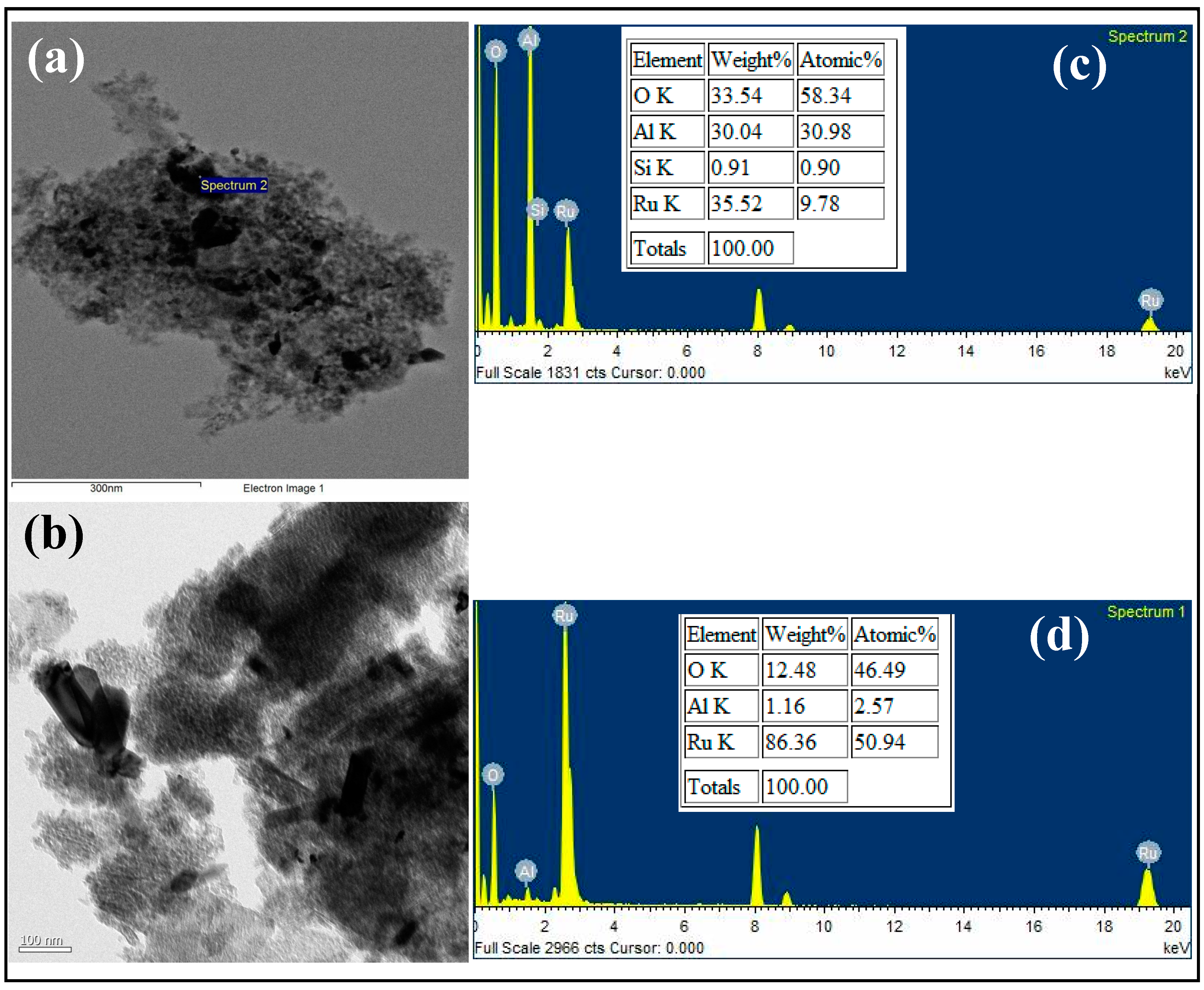
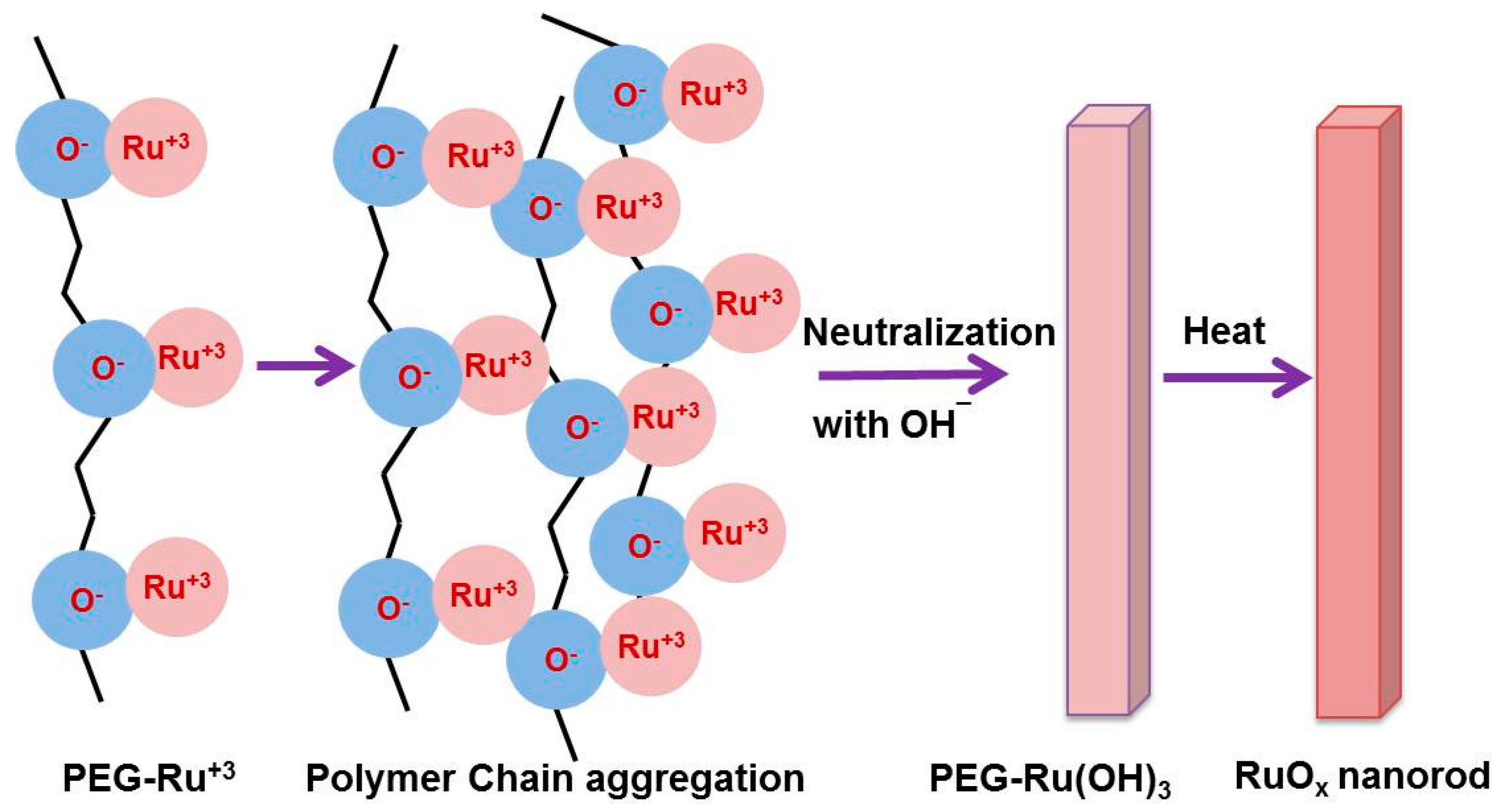
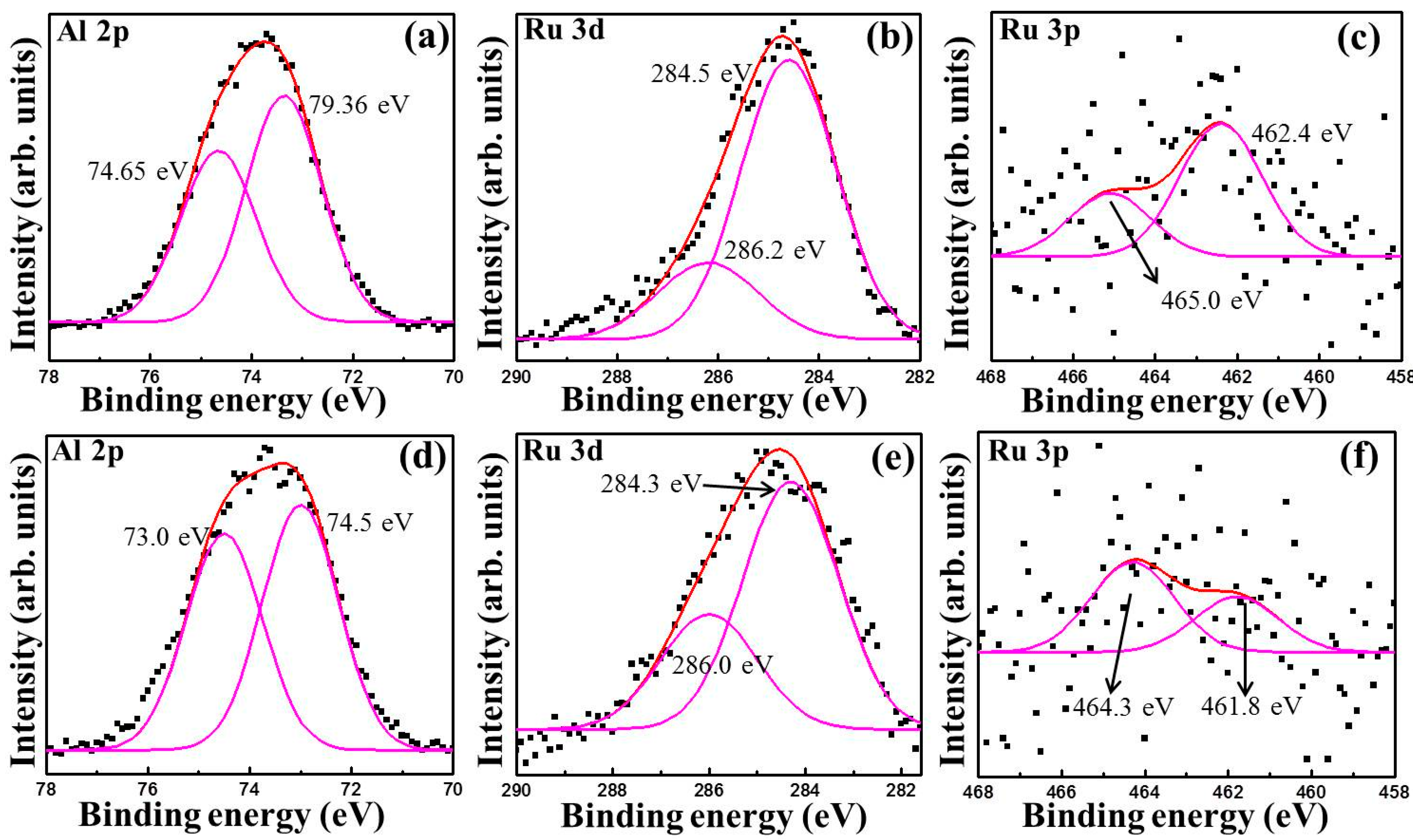
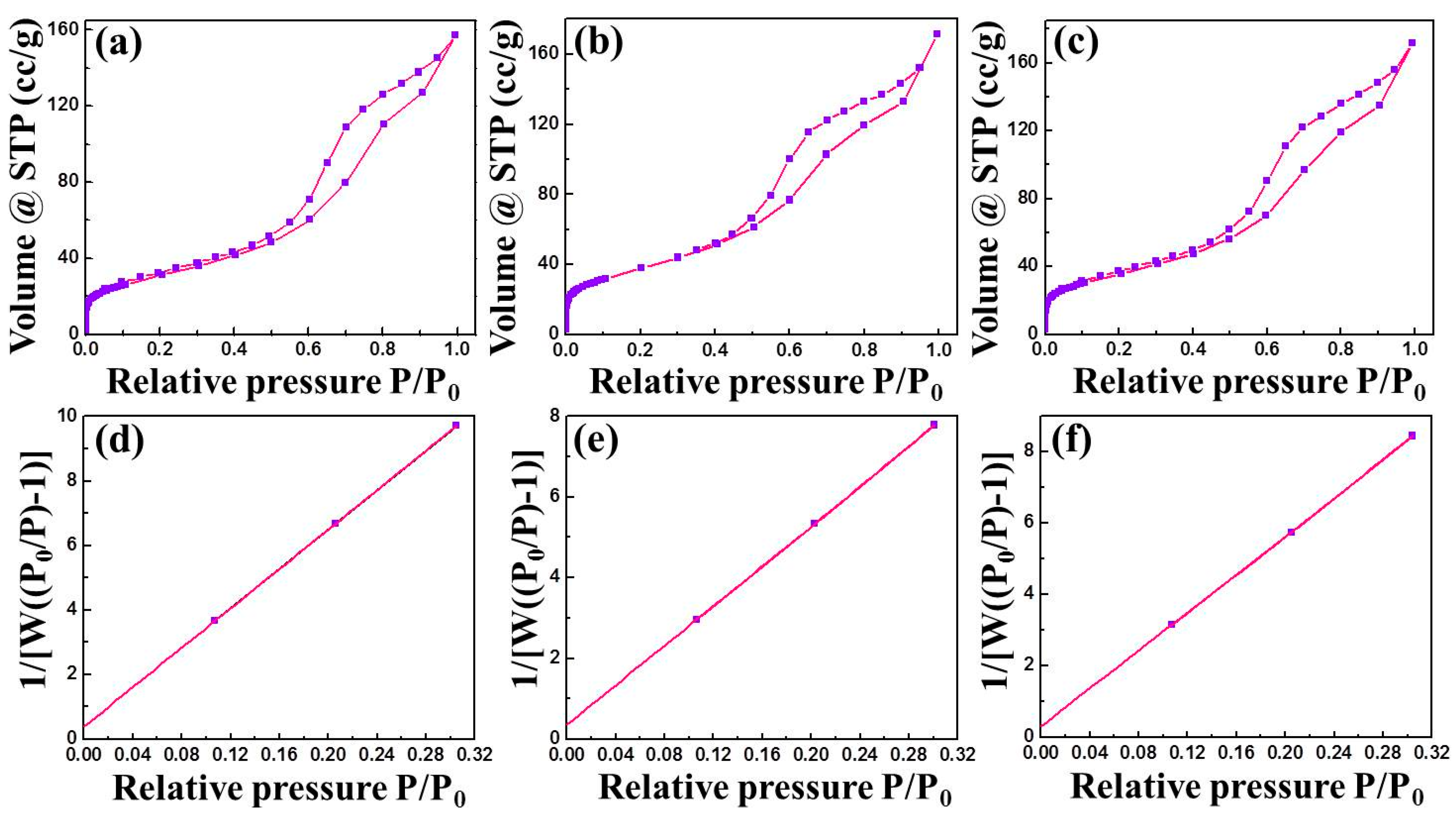
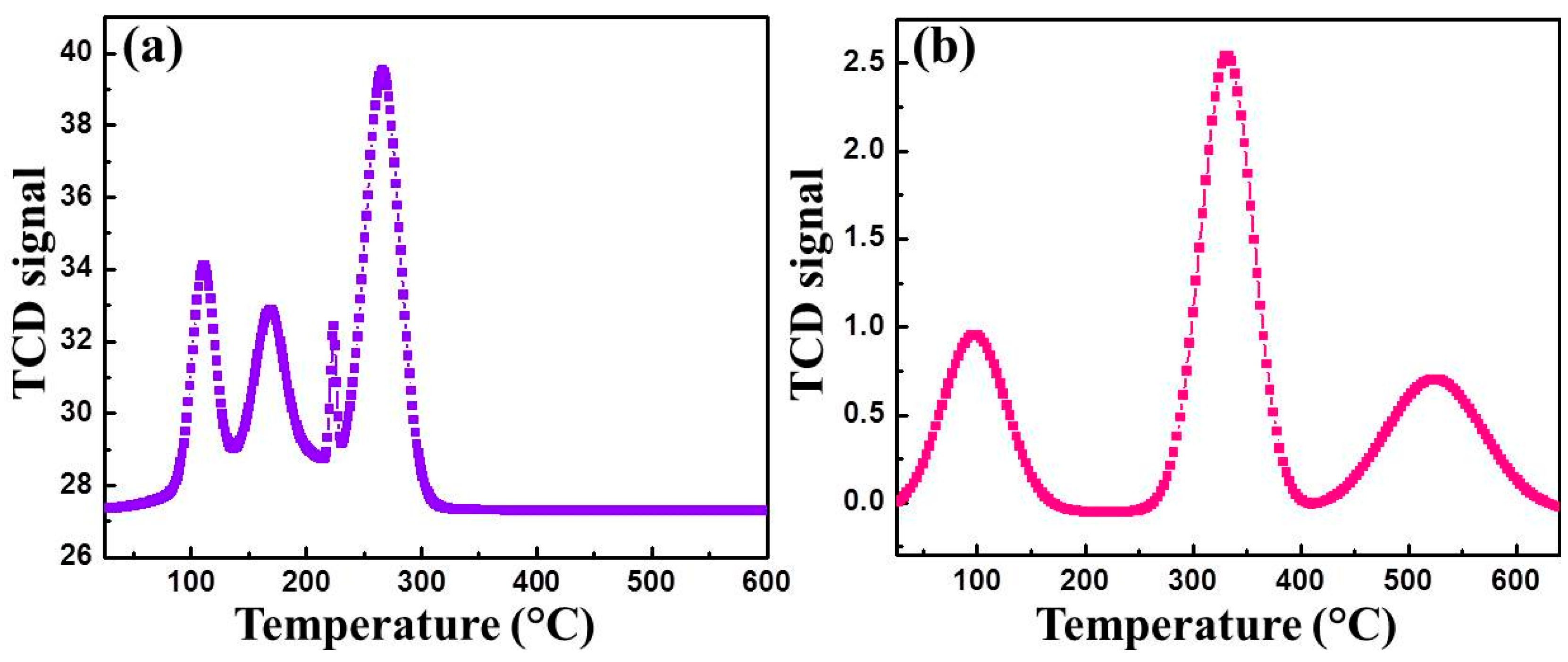
3.1. Characterization

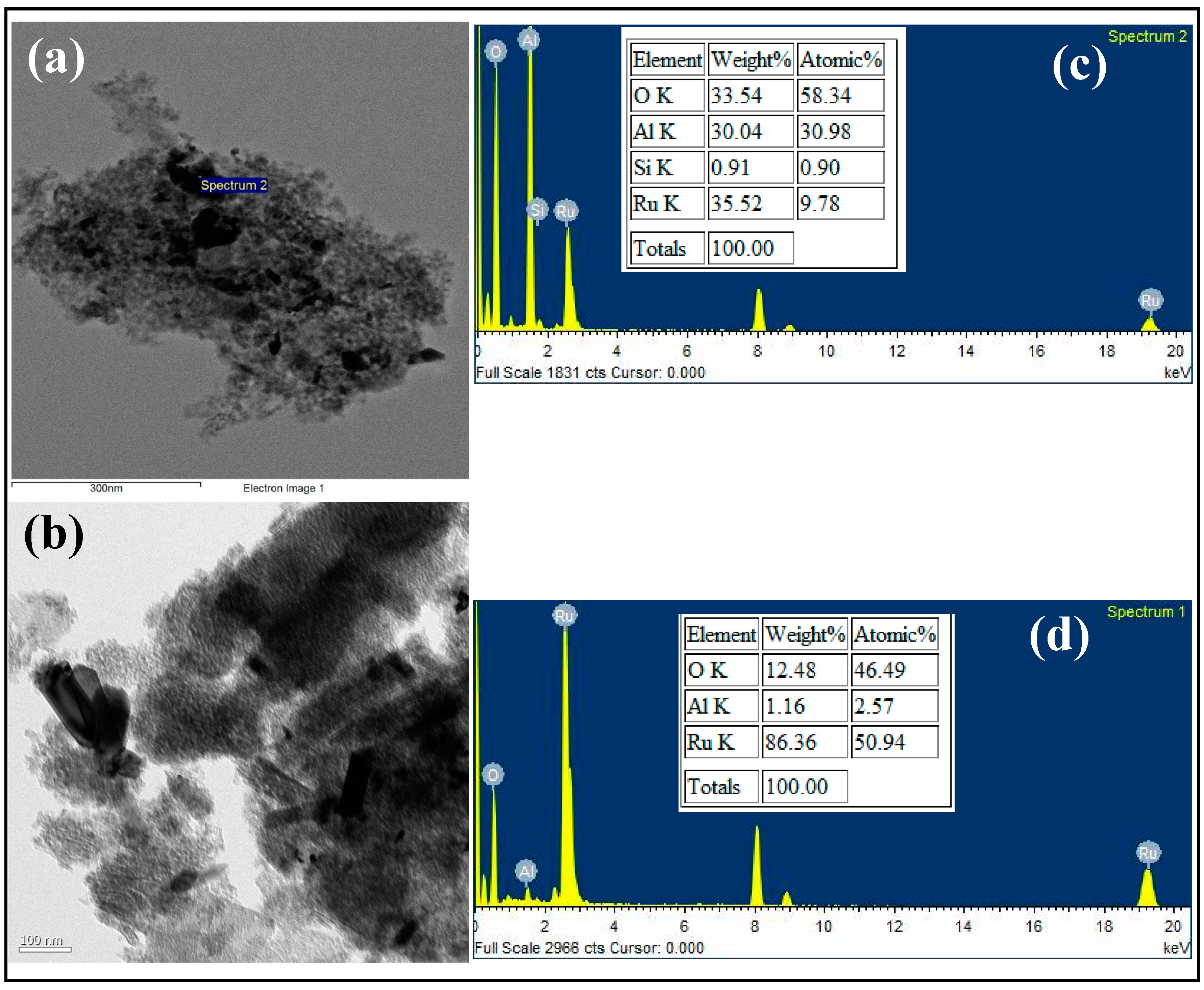

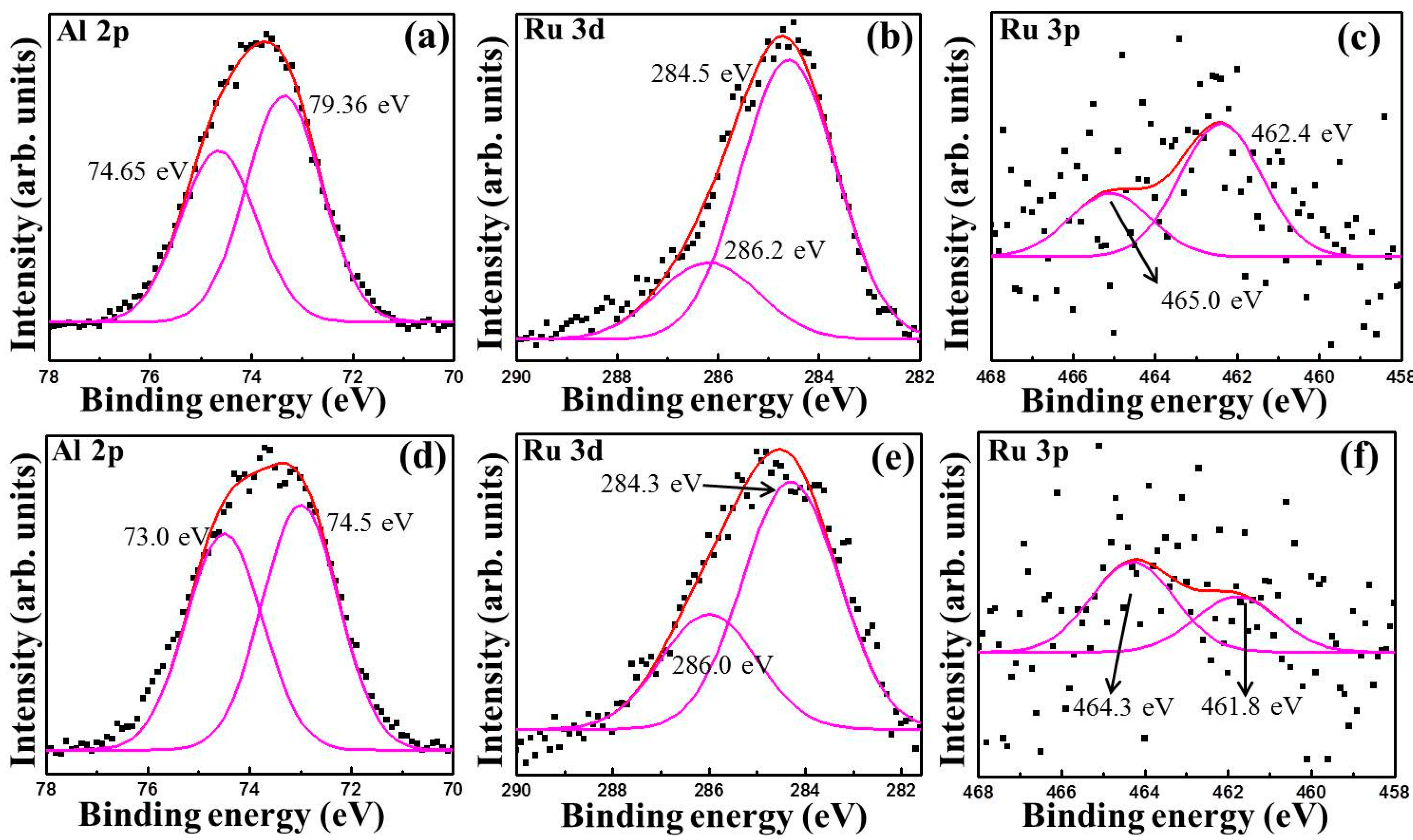


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalyst Name | Process | Surface Area (m2/g) | Pore Volume (cc/g) | Pore Radius (Mode Dv(r)) (Å) |
|---|---|---|---|---|
| ALVIR | Adsorption | 102.9 | 0.2322 | 30.72 |
| Desorption | 140.7 | 0.2480 | 28.18 | |
| RAWOS | Adsorption | 120.5 | 0.2475 | 30.58 |
| Desorption | 168.7 | 0.2694 | 24.40 | |
| RAWS | Adsorption | 119.0 | 0.2532 | 30.76 |
| Desorption | 164.8 | 0.2723 | 24.54 |
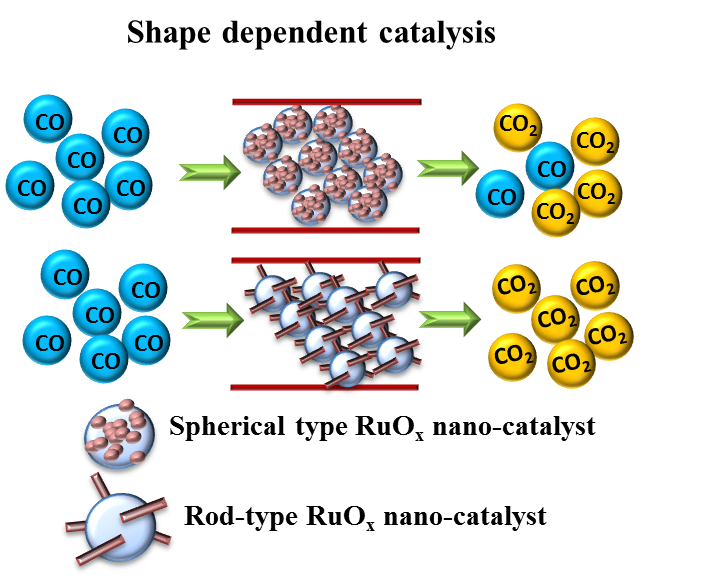
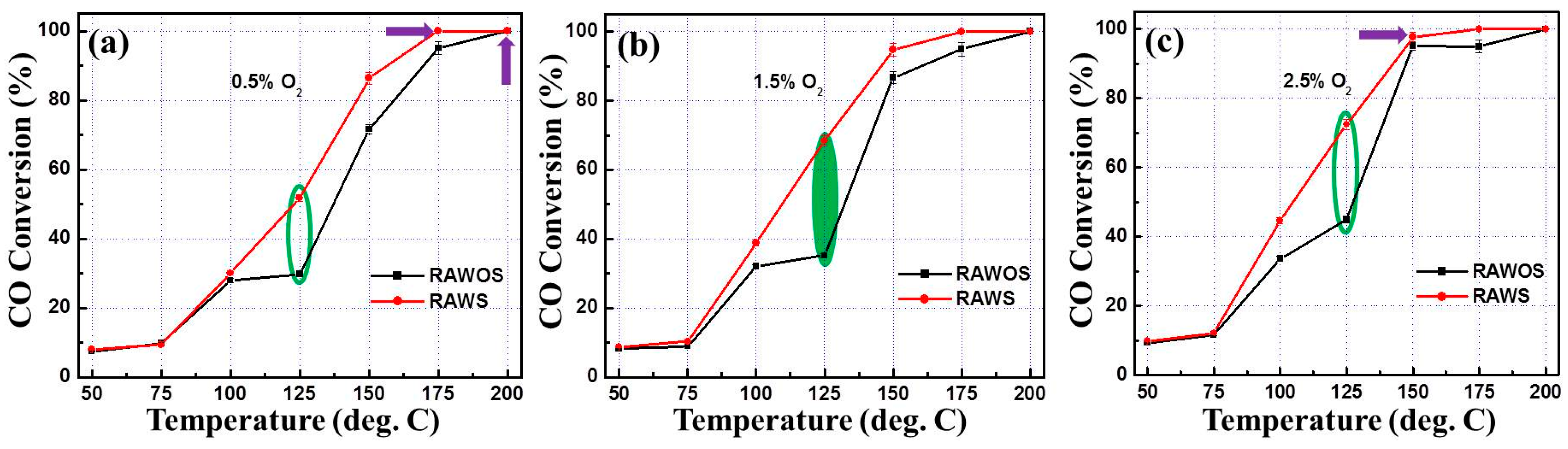
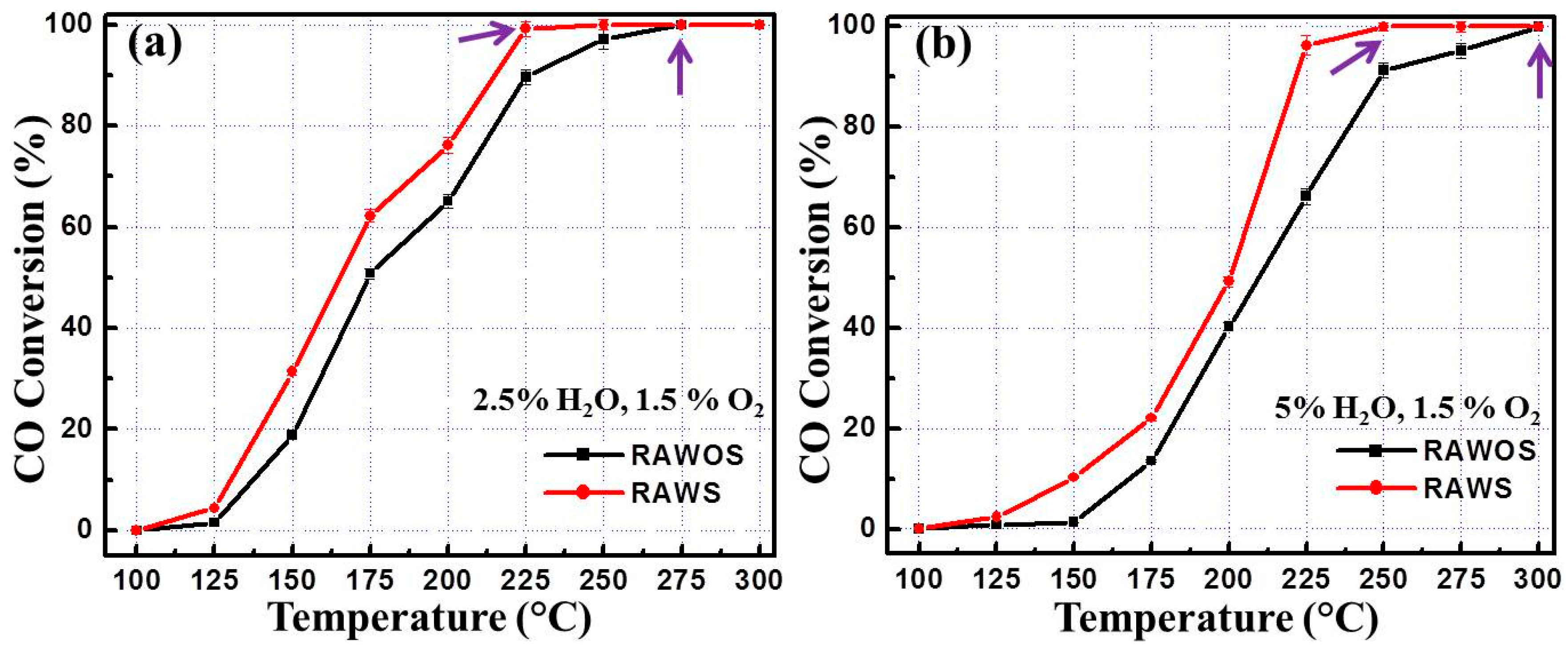
3.2. CO Oxidation Performance of the Catalysts
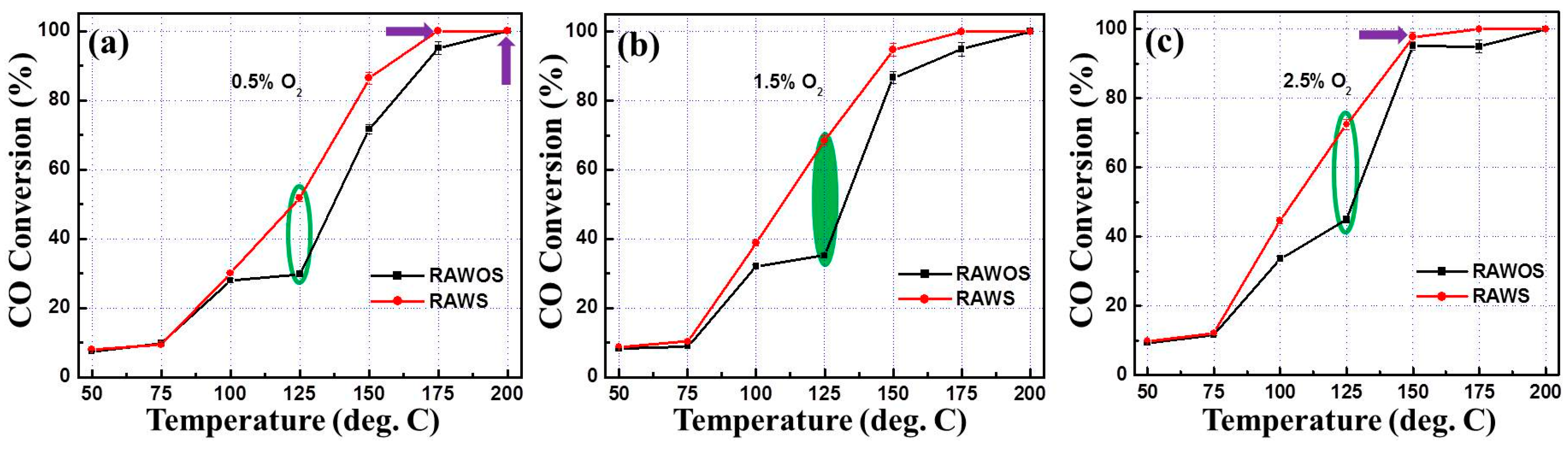
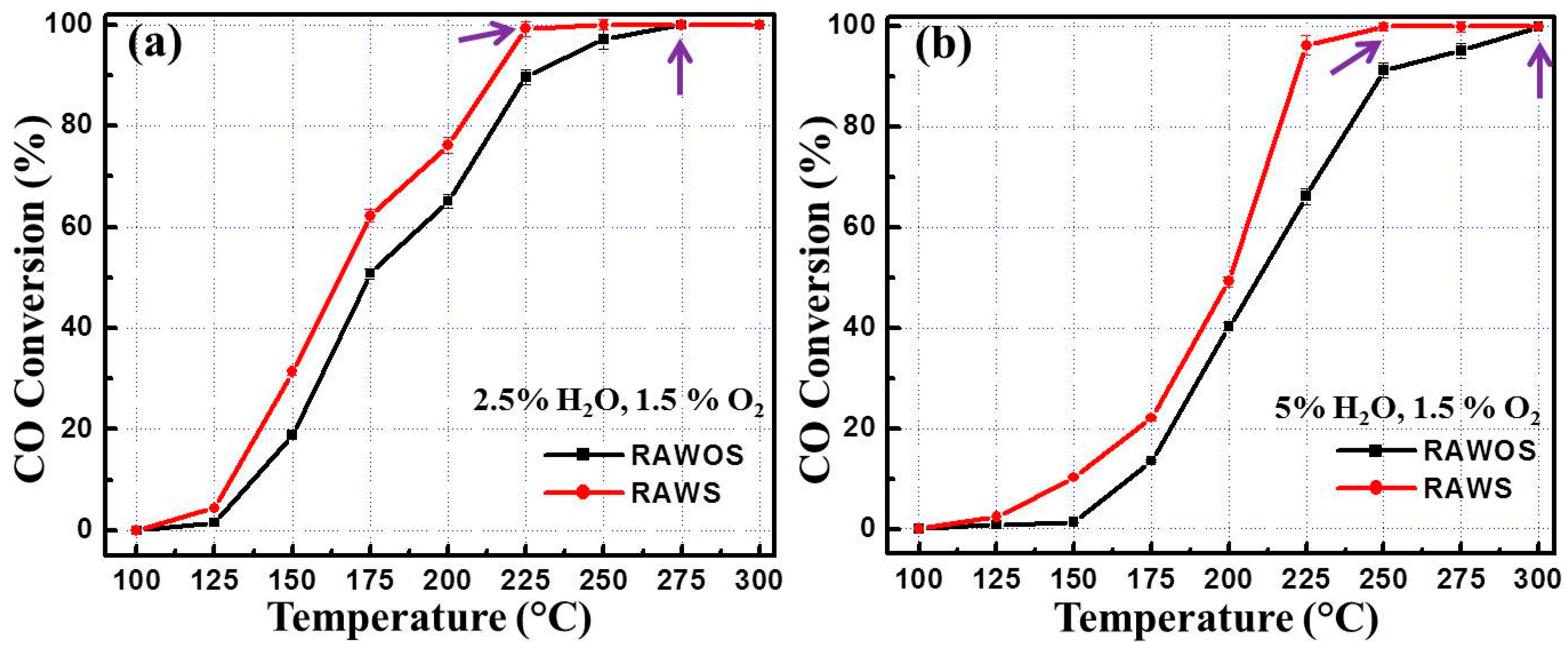
4. Discussion

5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhan, B.Z.; White, M.A.; Sham, T.K.; Pincock, J.A.; Doucet, R.J.; Rao, K.V.R.; Robertson, K.N.; Cameron, T.S. Zeolite-confined nano-RuO2: Agreen, selective, and efficient catalyst for aerobic alcohol oxidation. J. Am. Chem. Soc. 2003, 125, 2195–2199. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, W.; Iwata, H.; Yasunaga, Y.; Murakami, Y.; Takasu, Y. Preparation of ruthenic acid nanosheets and utilization of its interlayer surface for electrochemical energy storage. Angew. Chem. Int. Ed. 2003, 42, 4092–4096. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wang, C.; Wan, N.; Mao, Z. Deposited RuO2-IrO2/Pt electrocatalyst for the regenerative fuel cell. Int. J. Hydrogen Energy 2007, 32, 400–404. [Google Scholar] [CrossRef]
- Park, S.; Kim, M.; Koo, D.H.; Chang, S. Use of ruthenium/alumina as a convenient catalyst for copper-free Sonogashira coupling reactions. Adv. Syn. Catal. 2004, 346, 1638–1640. [Google Scholar] [CrossRef]
- Dutta, P.K.; Vaidyalingam, A.S. Health effects of exposure to ambient carbon monoxide. Microporous Mesoporous Mater. 2003, 62, 107–120. [Google Scholar] [CrossRef]
- Kwon, S.-H.; Kim, K.-H. Hybrid functional RuO2-Al2O3 thin films prepared by atomic layer deposition for inkjet printhead. J. Solid State Electrochem. 2010, 14, 225–229. [Google Scholar] [CrossRef]
- Li, K.; Wang, X.F.; Zeng, H.C. Kinetics of N2O decomposition on a RuO2/Al2O3 catalyst. Chem. Eng. Res. Design 1997, 75, 807–812. [Google Scholar] [CrossRef]
- Gracia-Suarez, E.J.; Tristany, M.; Gracia, A.B.; Colliere, V.; Philippot, K. Carbon-supported Ru and Pd nanoparticles: Efficient and recyclable catalysts for the aerobic oxidation of benzyl alcohol in water. Microporous Mesoporous Mater. 2012, 153, 155–162. [Google Scholar] [CrossRef]
- Zang, L.; Kisch, H. Room temperature oxidation of carbon monoxide catalyzed by hydrous ruthenium dioxide. Angew. Chem. Int. Ed. 2000, 39, 3921–3922. [Google Scholar] [CrossRef]
- Goodman, D.W.; Peden, C.H.F. CO oxidation on ruthenium: The nature of the active catalytic surface. Surf. Sci. 2007, 601, L124–L126. [Google Scholar] [CrossRef]
- Maeno, H.; Matsumoto, H. Alumina-supported ruthenium catalyst. US Patent 6429167, 6 August 2002. [Google Scholar]
- Raub, J.A. Health effects of exposure to ambient carbon monoxide. Chemosphere 1993, 1, 331–351. [Google Scholar] [CrossRef]
- Perez, Y.; Ruiz-Gonzalez, M.L.; Gonzalez-Calbet, J.M.; Concepcion, P.; Boronat, M.; Corma, A. Shape-dependent catalytic activity of palladium nanoparticles embedded in SiO2 and TiO2. Catal. Today 2012, 180, 59–67. [Google Scholar] [CrossRef]
- Martynova, Y.; Yang, B.; Yu, X.; Boscoboinik, J.A.; Shaikhutdinov, S.; Freund, H.J. Low temperature CO oxidation on ruthenium oxide thin films at near-atmospheric pressures. Catal. Lett. 2012, 142, 657–663. [Google Scholar] [CrossRef]
- Mahammadunnisa, S.K.; Reddy, P.M.K.; Lingaiah, N.; Subrahmanyam, Ch. NiO/Ce1−xNixO2-δ as an alternative to noble metal catalysts for CO oxidation. Catal. Sci. Technol. 2013, 3, 730–736. [Google Scholar] [CrossRef]
- Wang, R.; Wang, J.; Liu, L.; Dai, H. Mechanism for the high reactivity of CO oxidation on a ruthenium-oxide. Catal. Today 2013, 201, 68–78. [Google Scholar] [CrossRef]
- Liu, Z.-P.; Hu, P. Mechanism for the high reactivity of CO oxidation on a ruthenium-oxide. J. Chem. Phys. 2001, 114, 5956–5957. [Google Scholar] [CrossRef]
- Rosu, M.-C.; Suciu, R.-C.; Dreve, S.-V.; Silipas, T.-D.; Bratu, I.; Indrea, E. The influence of PEG/PPG and of the annealing temperature on TiO2-based layers properties. Rev. Roum. Chim. 2012, 57, 15–21. [Google Scholar]
- Sabori, R.; Sharifnia, S.; Aalami-Aleagha, M.E.; Panahi, M.R. Promotion of metallic catalysts by metal oxide powders in partial oxidation of methane. J. Taiwan Inst. Chem. Eng. 2012, 43, 153–158. [Google Scholar] [CrossRef]
- Vadakkekara, R.; Chakraborty, M.; Parikh, P.A. Room temperature benzaldehyde oxidation using air over gold-silver nanoalloy catalysts. J. Taiwan Inst. Chem. Eng. 2015, 50, 84–92. [Google Scholar] [CrossRef]
- Sinfelt, J.H. Structure of metal catalysts. Rev. Modern Phys. 1979, 51, 569–589. [Google Scholar] [CrossRef]
- Achouri, I.E.; Abatzoglou, N.; Fauteux-Lefebvre, C.; Braidy, N. Polymer-assisted fabrication of nanoparticles and nanocomposites. Catal. Today 2013, 207, 13–20. [Google Scholar] [CrossRef]
- Rozenberg, B.A.; Tenne, R. Polymer-assisted fabrication of nanoparticles and nanocomposites. Prog. Polym. Sci. 2008, 33, 40–112. [Google Scholar] [CrossRef]
- Zhang, H.; Feng, J.; Wang, J.; Zhang, M. Preparation of ZnOnanorods through wet chemical method. Mat. Lett. 2007, 61, 5202–5205. [Google Scholar] [CrossRef]
- Lijima, M.; Kamiya, H. Surface modification for improving the stability of nanoparticles in liquid media. KONA Powder Part. J. 2009, 27, 119–129. [Google Scholar]
- Ananth, A.; Mok, Y.S. Synthesis of RuO2 nanomaterials under dielectric barrier discharge plasma at atmospheric pressure — Influence of substrates on the morphology and application. Chem. Eng. J. 2014, 239, 290–298. [Google Scholar] [CrossRef]
- Han, S.; Kim, C.; Kwon, D. Thermal/oxidative degradation and stabilization of polyethylene glycol. Polymer 1997, 38, 317–323. [Google Scholar] [CrossRef]
- Kim, C.; Lee, H. Shape effect of Pt nanocrystals on electrocatalytic hydrogenation. Catal. Comm. 2009, 11, 7–10. [Google Scholar] [CrossRef]
- Einaga, H.; Harada, M. Photochemical preparation of poly (N-vinyl-2-pyrrolidone)-stabilized platinum colloids and their deposition on titanium dioxide. Langmuir 2005, 21, 2578–2584. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.-S.; Yoon, W.-L.; Rhee, Y.-W.; Seo, Y.-S. The surfactant-assisted Ni-Al2O3 catalyst prepared by a homogeneous precipitation method for CH4 steam reforming. Int. J. Hydrogen Energy 2012, 37, 9340–9350. [Google Scholar] [CrossRef]
- Baldwin, R.W. Preparation of catalyst pellets having sustained hardness and attrition resistance. US Patent 3146210, 25 August 1964. [Google Scholar]
- Ananth, A.; Dharaneedharan, S.; Gandhi, M.S.; Heo, M.-S.; Mok, Y.S. Novel RuO2 nanosheets—Facile synthesis, characterization and application. Chem. Eng. J. 2013, 223, 729–736. [Google Scholar] [CrossRef]
- Vidyasagar, C.C.; ArthobaNaik, Y. Surfactant (PEG 400) effects on crystallinity of ZnO nanoparticles. Arab. J. Chem. 2012. [Google Scholar] [CrossRef]
- Rozita, Y.; Brydson, R.; Scott, A.J. An investigation of commercial gamma-Al2O3 nanoparticles. J. Phys.: Conf. Ser. 2010, 241, 012096–012099. [Google Scholar]
- Xu, B.; Xiao, T.; Yan, Z.; Sun, X.; Sloan, J.; Gonazalez-Cortes, S.L.; Alshahrani, F.; Green, M.L.H. Synthesis of mesoporous alumina with highly thermal stability using glucose template in aqueous system. Microporous Mesoporous Mater. 2006, 91, 293–295. [Google Scholar] [CrossRef]
- Sifontes, A.B.; Urbina, M.; Fajardo, F.; Melo, L.; Gracia, L.; Mediavilla, M.; Carrion, N.; Brito, J.L.; Hernandez, P.; Solano, R.; et al. Preparation of γ-alumina foams of high surface area employing the polyurethane sponge replica method. Lat. Am. Appl. Res. 2010, 40, 185–191. [Google Scholar]
- Altwasser, S.; Glaser, R.; Weitkamp, J. Ruthenium-containing small-pore zeolites for shape-selective catalysis. Microporous Mesoporous Mater. 2007, 104, 281–288. [Google Scholar] [CrossRef]
- Macedo, M.I.F.; Osawa, C.C.; Bertran, C.A. Sol-gel synthesis of transparent alumina gel and pure gamma alumina by urea hydrolysis of aluminum nitrate. J. Sol-Gel Sci. Technol. 2004, 30, 135–140. [Google Scholar] [CrossRef]
- Karim, M.R.; Rahman, M.A.; Miah, M.A.J.; Ahmed, H.; Yanagisawa, M.; Ito, M. Synthesis of γ-alumina particles and surface characterization. Open Colloid Sci. J. 2011, 4, 32–36. [Google Scholar] [CrossRef]
- Hosseini, S.A.; Niaei, V.; Salari, D. Production of γ-Al2O3 from Kaolin. Open J. Phys. Chem. 2011, 1, 23–27. [Google Scholar] [CrossRef]
- Strohmeier, B.R. Characterization of an activated alumina Claus catalyst by XPS. Surf. Sci. Spectra 1994, 3, 141–146. [Google Scholar] [CrossRef]
- Shen, J.Y.; Adnot, A.; Kaliaguine, S. An ESCA study of the interaction of oxygen with the surface of ruthenium. Appl. Surf. Sci. 1991, 51, 47–60. [Google Scholar] [CrossRef]
- McEvoy, A.J.; Gissler, W. ESCA spectra and electronic properties of some ruthenium compounds. Phys. Status Solidi A 1982, 69, K91–K96. [Google Scholar] [CrossRef]
- Luo, C.; Zhang, Y.; Zeng, X.; Zeng, Y.; Wang, Y. The role of poly(ethylene glycol) in the formation of silver nanoparticles. J. Colloid Inter. Sci. 2005, 288, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Niesz, K.; Yang, P.; Somorjai, G.A. Sol-gel synthesis of ordered mesoporous alumina. Chem. Commun. 2005, 1986–1987. [Google Scholar] [CrossRef] [PubMed]
- Royer, S.; Durprez, D. Catalytic oxidation of carbon monoxide over transition metal oxides. ChemCatChem 2011, 3, 24–65. [Google Scholar] [CrossRef]
- Salek, G.; Alphonse, P.; Dufour, P.; Guillemet-Fritsch, S.; Tenailleau, C. Preparation of alumina-supported gold-ruthenium bimetallic catalysts by redox reactions and their activity in preferential CO oxidation. Appl. Catal. B 2014, 147, 1–7. [Google Scholar] [CrossRef]
- Carabineiro, S.A.C.; Bastos, S.S.T.; Orfao, J.J.M.; Pereira, M.F.R.; Delgado, J.J.; Figueiredo, J.L. Carbon monoxide oxidation catalysed by exotemplatedmanganese oxides. Catal. Lett. 2010, 134, 217–227. [Google Scholar] [CrossRef]
- Reuter, K.; Stampfl, C.; Pirovano, M.V.G.; Scheffler, M. Atomistic description of oxide formation on metal surfaces: The example of ruthenium. Chem. Phys. Lett. 2002, 352, 311–317. [Google Scholar] [CrossRef]
- Over, H.; Muhler, M.; Seitsonen, A.P. Comment on “CO oxidation on ruthenium: The nature of the active catalytic surface” by D.W. Goodman, C.H.F. Peden, M.S. Chen. Surf. Sci. 2007, 601, 5659–5662. [Google Scholar] [CrossRef]
- Goodman, D.W.; Peden, C.H.F.; Chen, M.S. Reply to comment on “CO oxidation on ruthenium: The nature of the active catalytic surface” by H. Over, M. Muhler, and A.P. Seitsonen. Surf. Sci. 2007, 601, 5663–5665. [Google Scholar] [CrossRef]
- Kirichenko, O.A.; Redina, E.A.; Davshan, N.A.; Mishin, I.V.; Kapustin, G.I.; Brueva, T.R.; Kustov, L.M.; Li, W.; Kim, C.H. Preparation of alumina-supported gold-ruthenium bimetallic catalysts by redox reactions and their activity in preferential CO oxidation. Appl. Catal. B 2013, 134–135, 123–129. [Google Scholar] [CrossRef]
- Zhou, W.-P.; Li, M.; Koenigsmann, C.; Ma, C.; Wong, S.S.; Adzic, R.R. Morphology-dependent activity of Pt nanocatalysts for ethanol oxidation in acidic media: Nanowires versus nanoparticles. Electrochim. Acta 2011, 56, 9824–9830. [Google Scholar] [CrossRef]
- Lopez, N.; Janssens, T.V.W.; Clausen, B.S.; Xu, Y.; Mavrikakis, M.; Bligaard, T.; Norskov, J.K. On the origin of the catalytic activity of gold nanoparticles for low-temperature CO oxidation. J. Catal. 2004, 223, 232–235. [Google Scholar] [CrossRef]
- Bratlie, K.M.; Lee, H.; Komvopoulos, K.; Yang, P.; Somorjai, G.A. Platinum nanoparticle shape effects on benzene hydrogenation selectivity. Nano Lett. 2007, 7, 3097–3101. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Kim, H.J.; Yoon, S.; Lee, H. Shape effect of ceria in Cu/ceria catalysts for preferential CO oxidation. J. Mol. Catal. A 2011, 335, 82–88. [Google Scholar] [CrossRef]
- Kim, Y.H.; Park, J.E.; Lee, H.C.; Choi, S.H.; Park, E.D. Active size-controlled Ru catalysts for selective CO oxidation. Appl. Catal. B 2012, 127, 129–136. [Google Scholar] [CrossRef]
- Zhang, D.; Niu, F.; Yan, T.; Shi, L.; Du, X.; Fang, J. Ceria nanospindles: Template-free solvothermal synthesis and shape-dependent catalytic activity. Appl. Surf. Sci. 2011, 257, 10161–10167. [Google Scholar] [CrossRef]
- Narayanan, R.; El-Sayed, M.A. Shape-dependent catalytic activity of platinum nanoparticles in colloidal solution. Nano Lett. 2004, 4, 1343–1348. [Google Scholar] [CrossRef]
- Mostafa, S.; Behafarid, F.; Croy, V.; Ono, L.K.; Li, L.; Yang, J.C.; Frenkel, A.I.; Cuenya, B.R. Shape-dependent catalytic properties of Pt nanoparticles. J. Am. Chem. Soc. 2010, 132, 15714–15719. [Google Scholar] [CrossRef] [PubMed]
- Lanza, R.; Jaras, S.G.; Canbu, P. Partial oxidation of methane over supported ruthenium catalysts. Appl. Catal. A 2007, 325, 57–67. [Google Scholar] [CrossRef]
- Ma, L.; He, D. Hydrogenolysis of glycerol to propanediols over highly active Ru-Re bimetallic catalysts. Top. Catal. 2009, 52, 834–844. [Google Scholar] [CrossRef]
- Betancourt, P.; Rives, A.; Hubaut, R.; Scott, C.E.; Goldwasser, J. A study of the ruthenium-alumina system. Appl. Catal. A 1998, 170, 307–314. [Google Scholar] [CrossRef]
- Bevy, L.P. Leading Edge Catalysis Research, 1st ed.; Nova Science Publishers, Inc: New York, NY, USA, 2005; pp. 116–117. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ananth, A.; Gregory, D.H.; Mok, Y.S. Synthesis, Characterization and Shape-Dependent Catalytic CO Oxidation Performance of Ruthenium Oxide Nanomaterials: Influence of Polymer Surfactant. Appl. Sci. 2015, 5, 344-358. https://doi.org/10.3390/app5030344
Ananth A, Gregory DH, Mok YS. Synthesis, Characterization and Shape-Dependent Catalytic CO Oxidation Performance of Ruthenium Oxide Nanomaterials: Influence of Polymer Surfactant. Applied Sciences. 2015; 5(3):344-358. https://doi.org/10.3390/app5030344
Chicago/Turabian StyleAnanth, Antony, Duncan H. Gregory, and Young Sun Mok. 2015. "Synthesis, Characterization and Shape-Dependent Catalytic CO Oxidation Performance of Ruthenium Oxide Nanomaterials: Influence of Polymer Surfactant" Applied Sciences 5, no. 3: 344-358. https://doi.org/10.3390/app5030344