Radiosynthesis and in Vivo Evaluation of Two PET Radioligands for Imaging α-Synuclein


Abstract
:
1. Introduction

2. Experimental Section
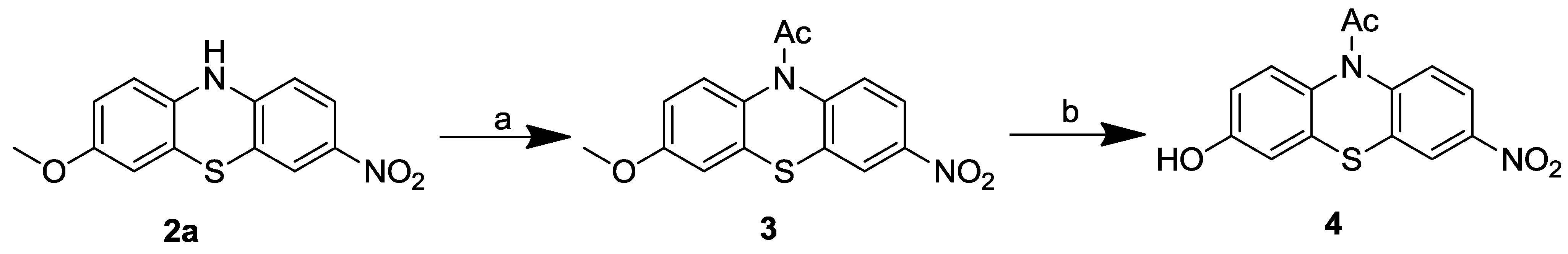
2.1. Chemistry
2.1.1. General

2.1.2. 1-(3-Methoxy-7-nitro-10H-phenothiazin-10-yl)ethanone (3)
2.1.3. 1-(3-Hydroxy-7-nitro-10H-phenothiazin-10-yl)ethanone (4)
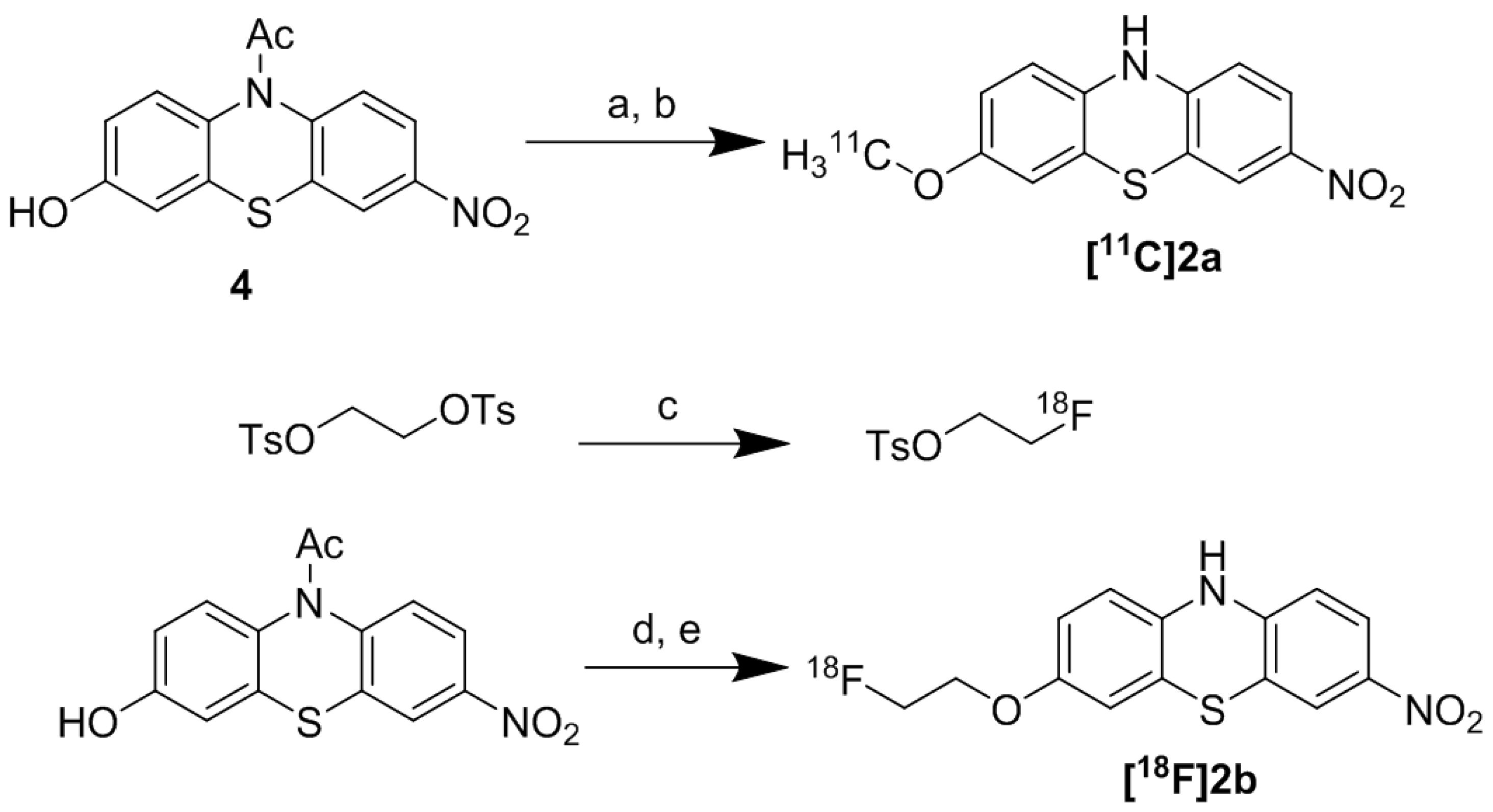
2.2. Radiochemistry
2.2.1. Radiosynthesis of [11C]2a
2.2.1.1. Production of [11C]CH3I
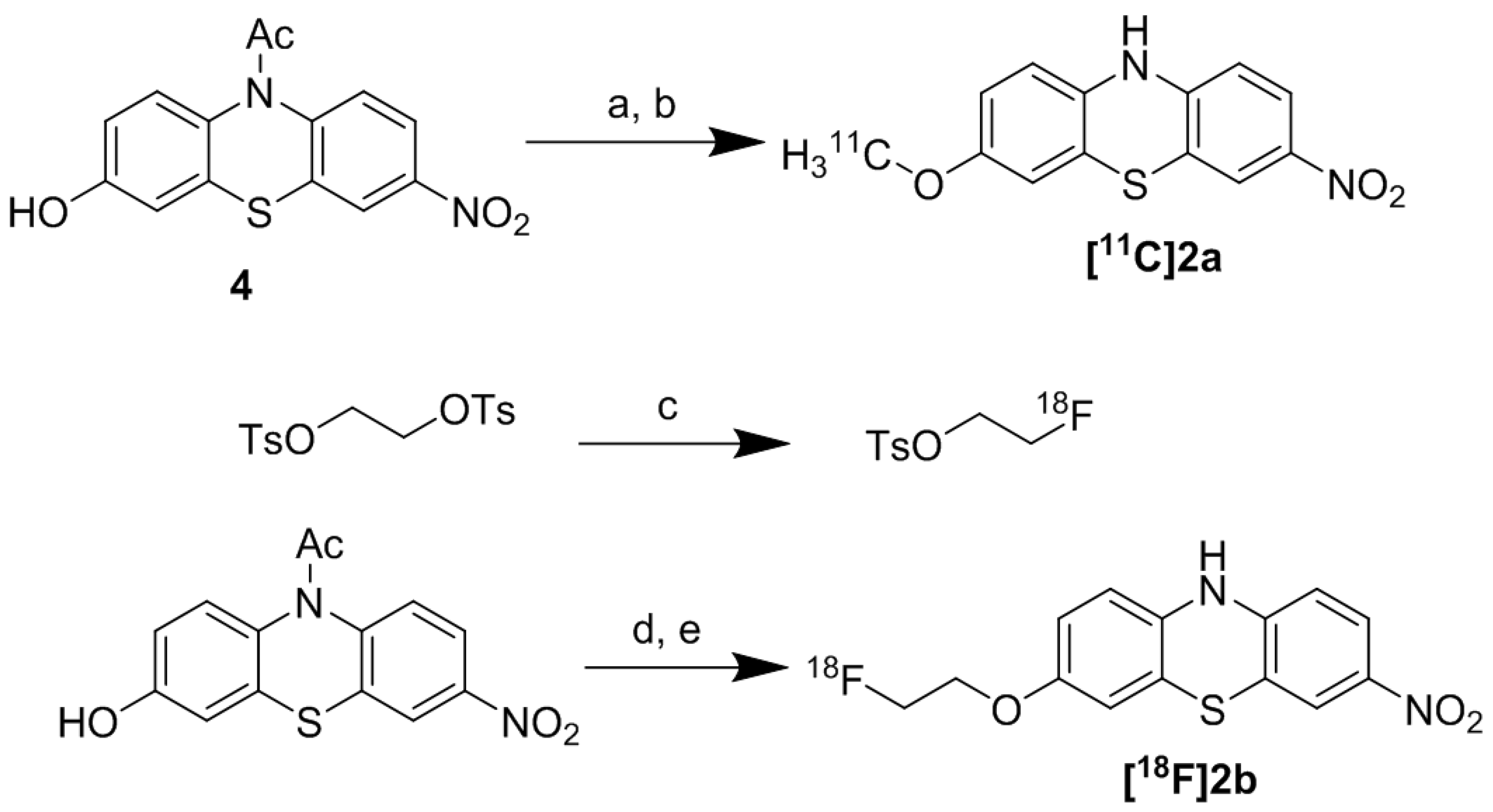
2.2.1.2. Radiosynthesis of [11C]2a
2.2.2. Radiosynthesis of [18F]2b
2.2.2.1. Production of [18F]fluoride
2.2.2.2. 2-[18F]Fluoroethyltosylate
2.2.2.3. Radiosynthesis of [18F]2b
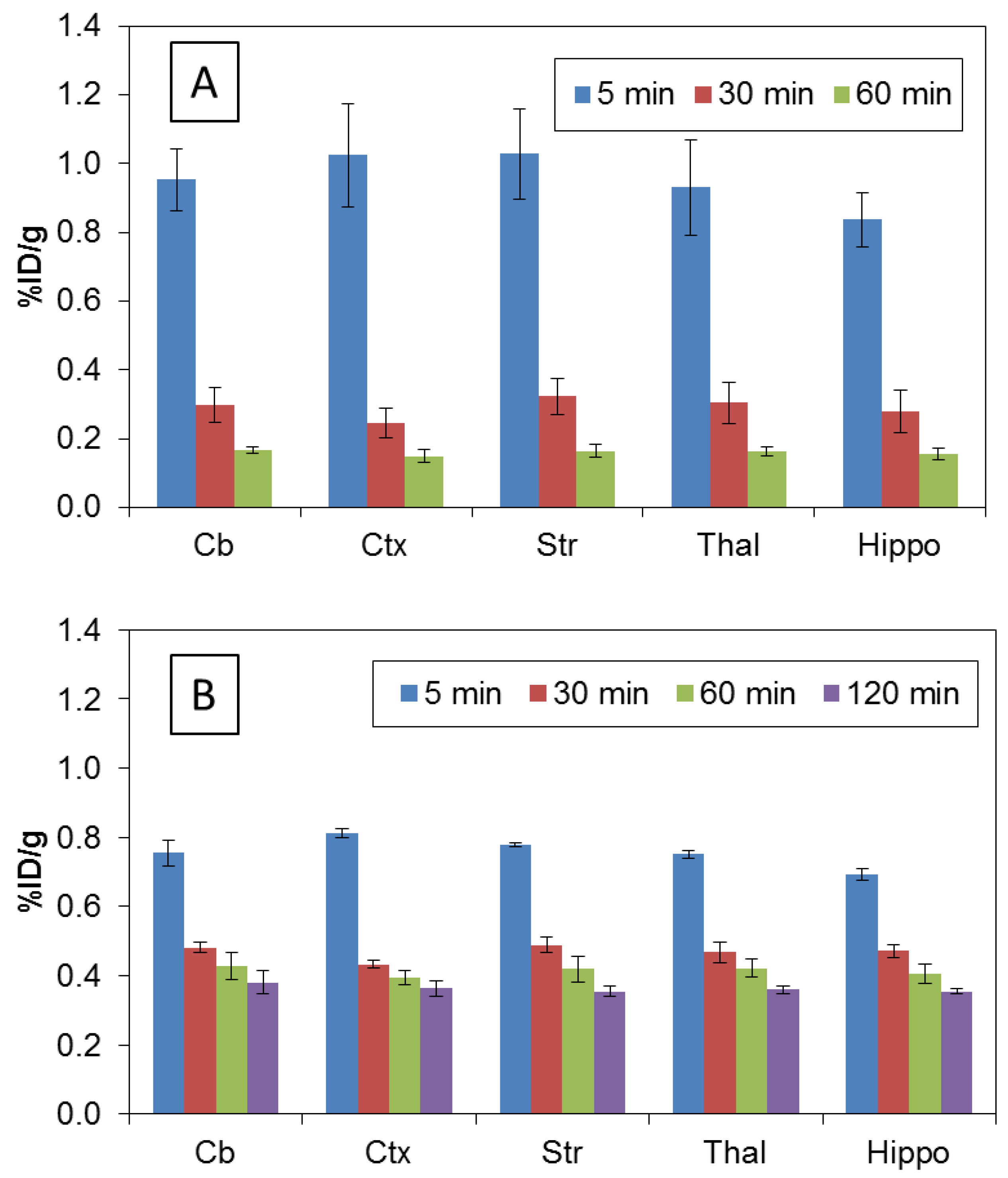
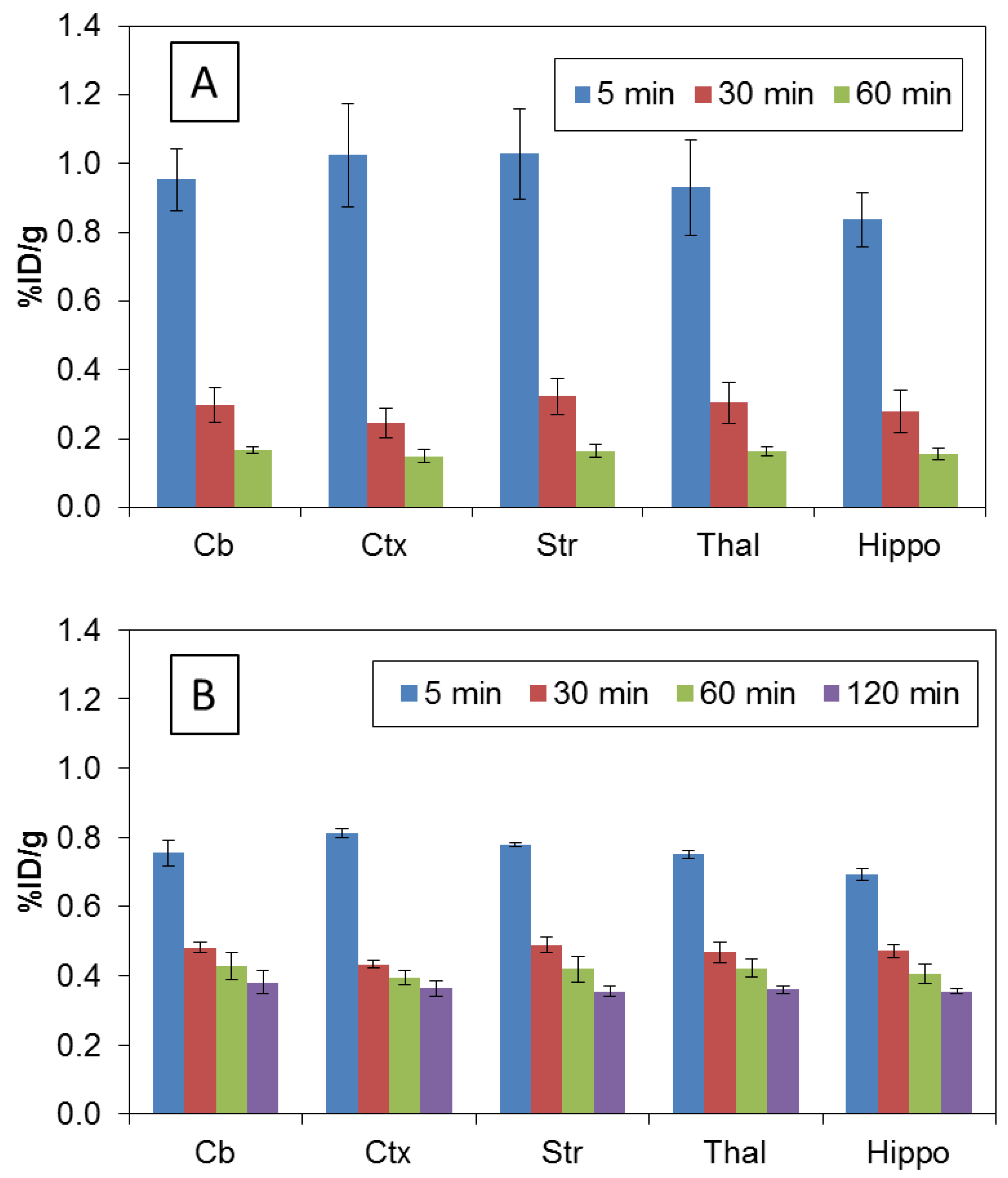
2.3. Biodistribution Studies
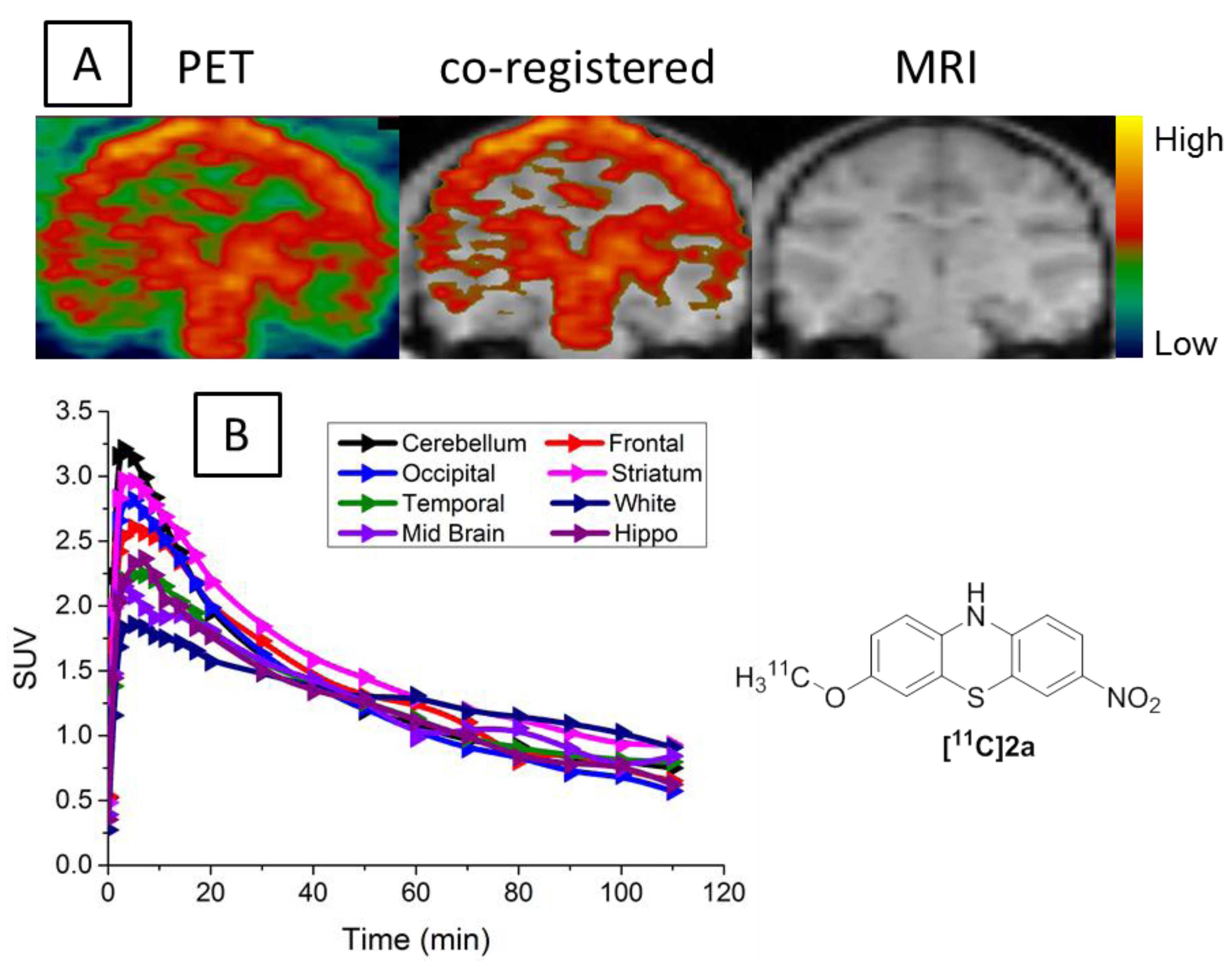
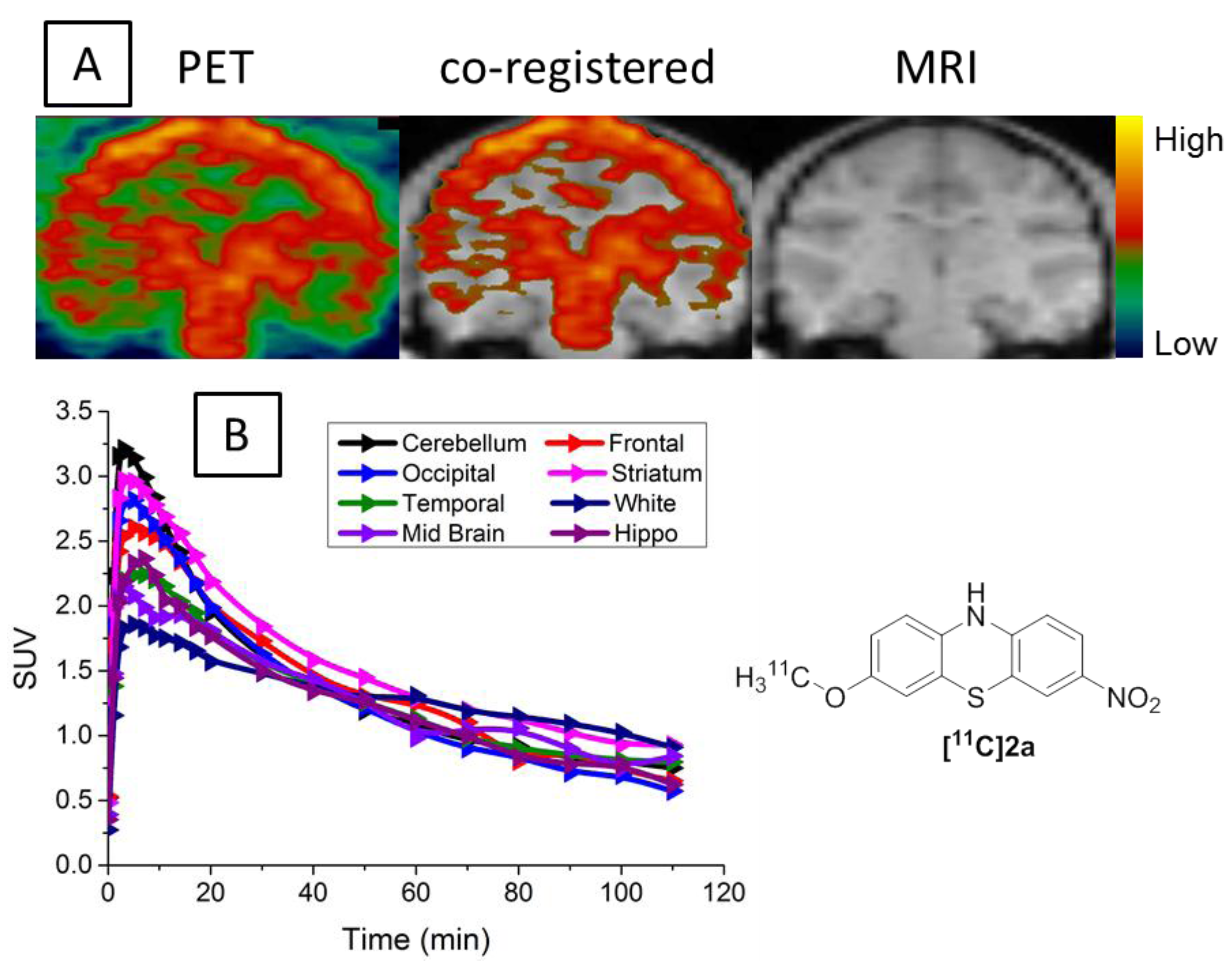
2.4. MicroPET Brain Imaging Studies of [11C]2a in Cynomolgus Macaque
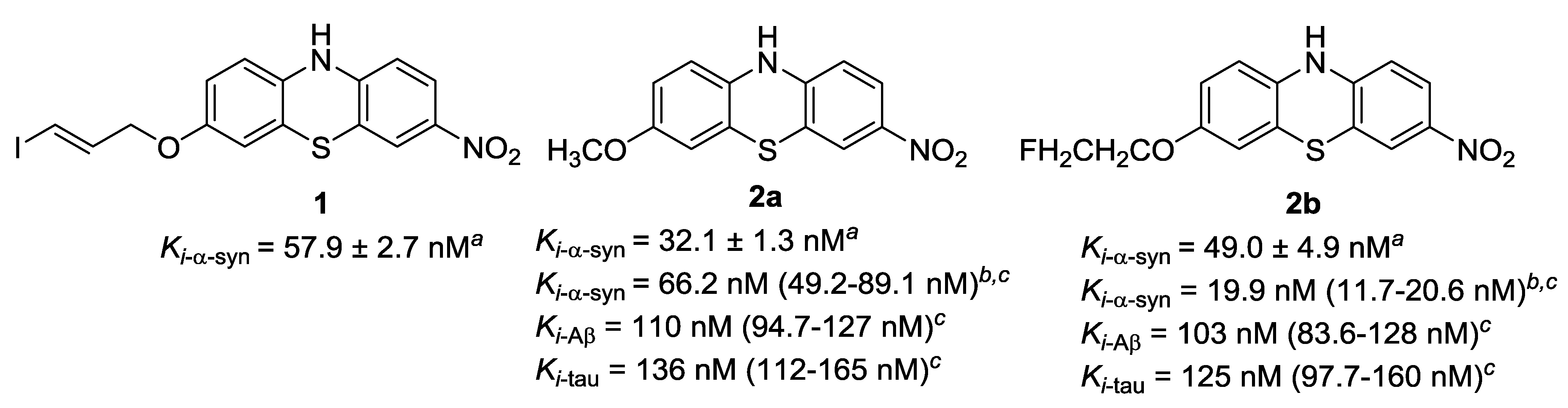
3. Results and Discussions
3.1. Chemistry
3.2. Radiochemistry

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Radioligand | Organ | 5 min | 30 min | 60 min | 120 min |
|---|---|---|---|---|---|
| [11C]2a | blood | 0.506 ± 0.040 | 0.369 ± 0.031 | 0.300 ± 0.015 | |
| heart | 0.758 ± 0.052 | 0.377 ± 0.046 | 0.245 ± 0.010 | ||
| lung | 1.149 ± 0.058 | 0.740 ± 0.038 | 0.485 ± 0.036 | ||
| muscle | 0.271 ± 0.005 | 0.325 ± 0.030 | 0.199 ± 0.016 | ||
| fat | 0.155 ± 0.023 | 0.241 ± 0.042 | 0.293 ± 0.076 | ||
| pancreas | 1.007 ± 0.262 | 0.506 ± 0.066 | 0.522 ± 0.036 | ||
| spleen | 0.659 ± 0.049 | 0.400 ± 0.052 | 0.386 ± 0.027 | ||
| kidney | 1.362 ± 0.054 | 0.807 ± 0.086 | 0.559 ± 0.053 | ||
| liver | 2.198 ± 0.111 | 1.349 ± 0.116 | 1.116 ± 0.024 | ||
| brain | 0.953 ± 0.115 | 0.287 ± 0.046 | 0.158 ± 0.013 | ||
| [18F]2b | blood | 0.553 ± 0.047 | 0.589 ± 0.016 | 0.606 ± 0.035 | 0.585 ± 0.046 |
| heart | 0.757 ± 0.033 | 0.505 ± 0.015 | 0.466 ± 0.040 | 0.410 ± 0.030 | |
| lung | 0.833 ± 0.053 | 0.561 ± 0.021 | 0.491 ± 0.030 | 0.436 ± 0.018 | |
| muscle | 0.430 ± 0.031 | 0.451 ± 0.018 | 0.376 ± 0.019 | 0.315 ± 0.015 | |
| fat | 0.255 ± 0.037 | 0.425 ± 0.023 | 0.371 ± 0.067 | 0.293 ± 0.048 | |
| pancreas | 1.004 ± 0.147 | 0.546 ± 0.068 | 0.409 ± 0.029 | 0.330 ± 0.021 | |
| spleen | 0.672 ± 0.064 | 0.509 ± 0.022 | 0.446 ± 0.038 | 0.398 ± 0.019 | |
| kidney | 1.070 ± 0.058 | 0.988 ± 0.090 | 0.678 ± 0.032 | 0.659 ± 0.027 | |
| liver | 1.626 ± 0.221 | 0.847 ± 0.027 | 0.561 ± 0.028 | 0.467 ± 0.023 | |
| bone | 0.340 ± 0.027 | 0.309 ± 0.020 | 0.407 ± 0.043 | 0.644 ± 0.071 | |
| brain | 0.758 ± 0.013 | 0.465 ± 0.018 | 0.410 ± 0.030 | 0.359 ± 0.016 |
3.3. Biodistribution in Rats
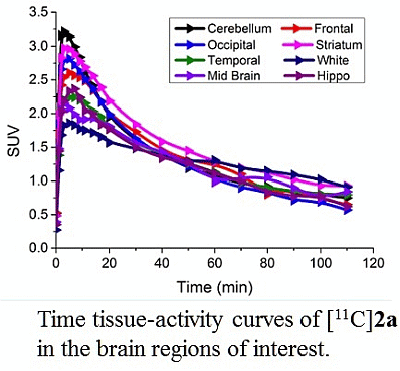
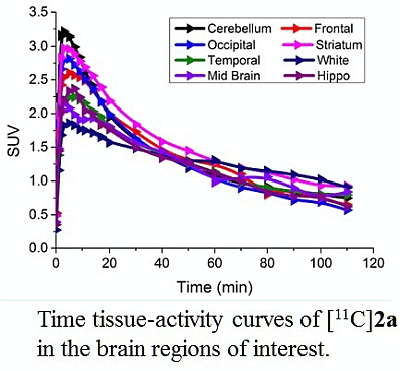
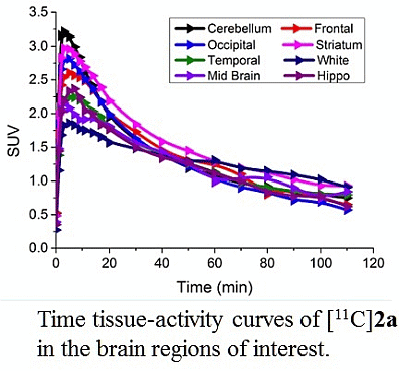
3.4. MicroPET Studies in NHP
4. Conclusions
Abbreviations
| α-syn | α-synuclein |
| BBB | blood-brain barrier |
| calcd. | calculated |
| CNS | central nervous system |
| DBU | 1,8-diazabicyclo[5.4.0]undec-7-ene |
| DCM | dichloromethane |
| DMF | N,N-dimethylformamide |
| DMSO | dimethyl sulfoxide |
| EOB | end of bombardment |
| [18F]FEOTs | [18F]fluoroethyltosylate |
| HPLC | high performance liquid chromatography |
| HRMS | high resolution mass spectrometry |
| LBs | Lewy bodies |
| LNs | Lewy neuritis |
| NHP | non-human primate |
| PD | Parkinson’s disease |
| PET | positron emission tomography |
| QC | quality control |
| RCY | radiochemical yield |
| SAR | structure-activity relationship |
| SD | Sprague Dawley |
| SUV | standardized uptake value |
| TAC | time-activity curve |
| VMAT2 | vesicular monoamine transporter type 2 |
Acknowledgments
Conflicts of Interest
References
- Hely, M.A.; Reid, W.G.J.; Adena, M.A.; Halliday, G.M.; Morris, J.G.L. The sydney multicenter study of parkinson’s disease: The inevitability of dementia at 20 years. Mov. Disord. 2008, 23, 837–844. [Google Scholar] [CrossRef]
- Hurtig, H.I.; Trojanowski, J.Q.; Galvin, J.; Ewbank, D.; Schmidt, M.L.; Lee, V.M.-Y.; Clark, C.M.; Glosser, G.; Stern, M.B.; Gollomp, S.M.; et al. Alpha-synuclein cortical lewy bodies correlate with dementia in Parkinson’s disease. Neurology 2000, 54, 1916–1921. [Google Scholar] [CrossRef]
- Jankovic, J. Parkinson’s disease: Clinical features and diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef]
- Cheng, F.; Vivacqua, G.; Yu, S. The role of alpha-synuclein in neurotransmission and synaptic plasticity. J. Chem. Neuroanat. 2011, 42, 242–248. [Google Scholar] [CrossRef]
- George, J.M.; Jin, H.; Woods, W.S.; Clayton, D.F. Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron 1995, 15, 361–372. [Google Scholar] [CrossRef]
- Clayton, D.F.; George, J.M. The synucleins: A family of proteins involved in synaptic function, plasticity, neurodegeneration and disease. Trends Neurosci. 1998, 21, 249–254. [Google Scholar] [CrossRef]
- Uéda, K.; Fukushima, H.; Masliah, E.; Xia, Y.; Iwai, A.; Yoshimoto, M.; Otero, D.A.; Kondo, J.; Ihara, Y.; Saitoh, T. Molecular cloning of cdna encoding an unrecognized component of amyloid in alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 11282–11286. [Google Scholar] [CrossRef]
- Kazantsev Ag, K.A.M. Central role of α-synuclein oligomers in neurodegeneration in Parkinson disease. Arch. Neurol. 2008, 65, 1577–1581. [Google Scholar]
- Bate, C.; Gentleman, S.; Williams, A. Alpha-synuclein induced synapse damage is enhanced by amyloid-beta1–42. Mol. Neurodegener. 2010, 5, 55. [Google Scholar] [CrossRef]
- Lee, V.M.Y.; Trojanowski, J.Q. Mechanisms of Parkinson’s disease linked to pathological α-synuclein: New targets for drug discovery. Neuron 2006, 52, 33–38. [Google Scholar] [CrossRef]
- Kramer, M.L.; Schulz-Schaeffer, W.J. Presynaptic α-synuclein aggregates, not lewy bodies, cause neurodegeneration in dementia with lewy bodies. J. Neurosci. 2007, 27, 1405–1410. [Google Scholar] [CrossRef]
- Ishiwata, K.; Kimura, Y.; Oda, K.; Ishii, K.; Sakata, M.; Kawasaki, K.; Nariai, T.; Suzuki, Y.; Ishibashi, K.; Mishina, M.; et al. Development of pet radiopharmaceuticals and their clinical applications at the positron medical center. Geriatr. Gerontol. Int. 2010, 10, S180–S196. [Google Scholar]
- Chen, M.-K.; Kuwabara, H.; Zhou, Y.; Adams, R.J.; Brašić, J.R.; McGlothan, J.L.; Verina, T.; Burton, N.C.; Alexander, M.; Kumar, A.; et al. VMAT2 and dopamine neuron loss in a primate model of Parkinson’s disease. J. Neurochem. 2008, 105, 78–90. [Google Scholar] [CrossRef]
- De la Fuente-Fernández, R. Role of datscan and clinical diagnosis in Parkinson disease. Neurology 2012, 78, 696–701. [Google Scholar] [CrossRef]
- Perlmutter, J.S.; Eidelberg, D. To scan or not to scan: Dat is the question. Neurology 2012, 78, 688–689. [Google Scholar] [CrossRef]
- Winogrodzka, A.; Wagenaar, R.C.; Booij, J.; Wolters, E.C. Rigidity and bradykinesia reduce interlimb coordination in parkinsonian gait. Arch. Phys. Med. Rehabil. 2005, 86, 183–189. [Google Scholar] [CrossRef]
- Yu, L.; Cui, J.; Padakanti, P.K.; Engel, L.; Bagchi, D.P.; Kotzbauer, P.T.; Tu, Z. Synthesis and in vitro evaluation of α-synuclein ligands. Bioorg. Med. Chem. 2012, 20, 4625–4634. [Google Scholar] [CrossRef]
- Bagchi, D.P.; Yu, L.; Perlmutter, J.S.; Xu, J.; Mach, R.H.; Tu, Z.; Kotzbauer, P.T. Binding of the radioligand SIL23 to α-synuclein fibrils in Parkinson disease brain tissue establishes feasibility and screening approaches for developing a parkinson disease imaging agent. PLoS One 2013, 8. [Google Scholar]
- Tu, Z.; Fan, J.; Li, S.; Jones, L.A.; Cui, J.; Padakanti, P.K.; Xu, J.; Zeng, D.; Shoghi, K.I.; Perlmutter, J.S.; et al. Radiosynthesis and in vivo evaluation of [11C]MP-10 as a pet probe for imaging PDE10A in rodent and non-human primate brain. Biorg. Med. Chem. 2011, 19, 1666–1673. [Google Scholar]
- Tu, Z.; Xu, J.; Jones, L.A.; Li, S.; Mach, R.H. Carbon-11 labeled papaverine as a pet tracer for imaging PDE10A: Radiosynthesis, in vitro and in vivo evaluation. Nucl. Med. Biol. 2010, 37, 509–516. [Google Scholar] [CrossRef]
- Kuhnast, B.; Bodenstein, C.; Wester, H.J.; Weber, W. Carbon-11 labelling of an n-sulfonylamino acid derivative: A potential tracer for mmp-2 and mmp-9 imaging. J. Labelled Compd. Radiopharm. 2003, 46, 539–553. [Google Scholar] [CrossRef]
- Chakrabarty, M.; Ghosh, N.; Khasnobis, S.; Chakrabarty, M. Dbu, a highly efficient reagent for the facile regeneration of (hetero)arylamines from their acetamides and benzamides: Influence of solvent, temperature, and microwave irradiation. Synth. Commun. 2002, 32, 265–272. [Google Scholar] [CrossRef]
- Prabhakaran, J.; Arango, V.; Majo, V.J.; Simpson, N.R.; Kassir, S.A.; Underwood, M.D.; Polavarapu, H.; Bruce, J.N.; Canoll, P.; John Mann, J.; et al. Synthesis and in vitro evaluation of [18F](r)-FEPAQ: A potential pet ligand for VEGFR2. Bioorg. Med. Chem. Lett. 2012, 22, 5104–5107. [Google Scholar]
- Bauman, A.; Piel, M.; Schirrmacher, R.; Rosch, F. Efficient alkali iodide promoted F-18-fluoroethylations with 2-[F-18]fluoroethyl tosylate and 1-bromo-2-[F-18]fluoroethane. Tetrahedron Lett. 2003, 44, 9165–9167. [Google Scholar] [CrossRef]
- Block, D.; Coenen, H.H.; Stöcklin, G. The n.C.A. Nucleophilic 18F-fluorination of 1,n-disubstituted alkanes as fluoroalkylation agents. J. Labelled Compd. Radiopharm. 1987, 24, 1029–1042. [Google Scholar] [CrossRef]
- Wester, H.J.; Herz, M.; Weber, W.; Heiss, P.; Senekowitsch-Schmidtke, R.; Schwaiger, M.; Stocklin, G. Synthesis and radiopharmacology of O-(2-[F-18]fluoroethyl)-l-tyrosine for tumor imaging. J. Nucl. Med. 1999, 40, 205–212. [Google Scholar]
- Block, D.; Coenen, H.H.; Stöcklin, G. N.C.A. 18F-fluoroalkylation of h-acidic compounds. J. Labelled Compd. Radiopharm. 1988, 25, 201–216. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhang, X.; Jin, H.; Padakanti, P.K.; Li, J.; Yang, H.; Fan, J.; Mach, R.H.; Kotzbauer, P.; Tu, Z. Radiosynthesis and in Vivo Evaluation of Two PET Radioligands for Imaging α-Synuclein. Appl. Sci. 2014, 4, 66-78. https://doi.org/10.3390/app4010066
Zhang X, Jin H, Padakanti PK, Li J, Yang H, Fan J, Mach RH, Kotzbauer P, Tu Z. Radiosynthesis and in Vivo Evaluation of Two PET Radioligands for Imaging α-Synuclein. Applied Sciences. 2014; 4(1):66-78. https://doi.org/10.3390/app4010066
Chicago/Turabian StyleZhang, Xiang, Hongjun Jin, Prashanth K. Padakanti, Junfeng Li, Hao Yang, Jinda Fan, Robert H. Mach, Paul Kotzbauer, and Zhude Tu. 2014. "Radiosynthesis and in Vivo Evaluation of Two PET Radioligands for Imaging α-Synuclein" Applied Sciences 4, no. 1: 66-78. https://doi.org/10.3390/app4010066