Application of Liquid-Phase Direct Fluorination: Novel Synthetic Methods for a Polyfluorinated Coating Material and a Monomer of a Perfluorinated Polymer Electrolyte Membrane

Abstract
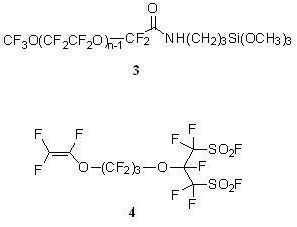
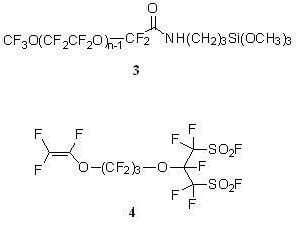
:
1. Introduction
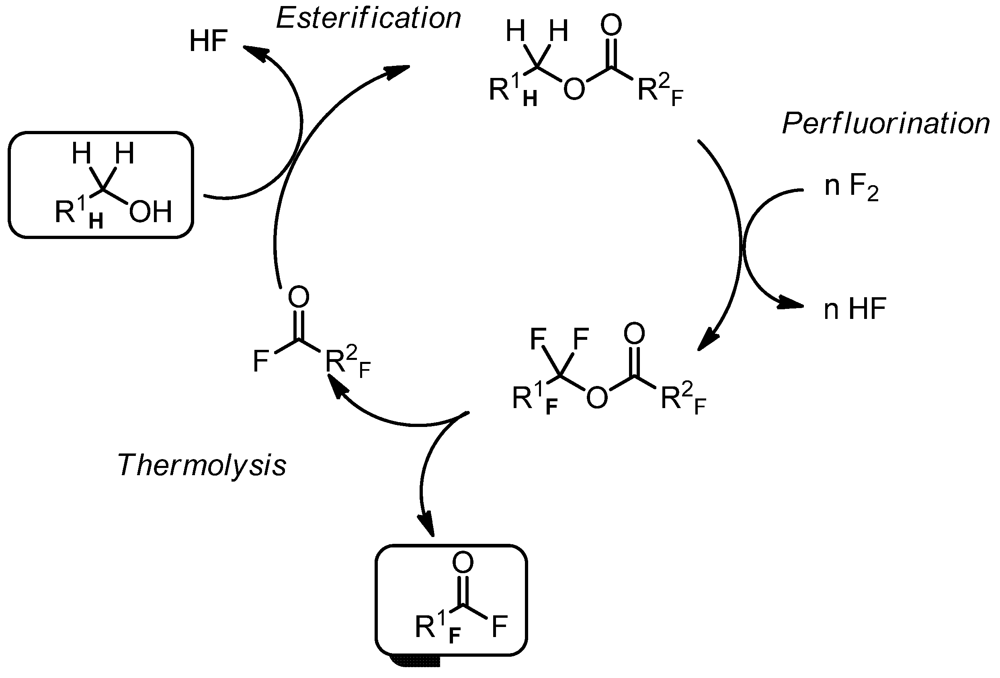
2. Experimental Section
2.1. General
2.2. Typical Procedure for the Anti-staining Coating Material
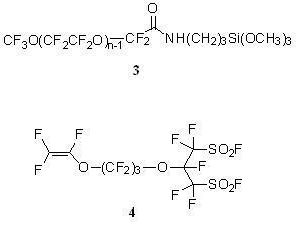
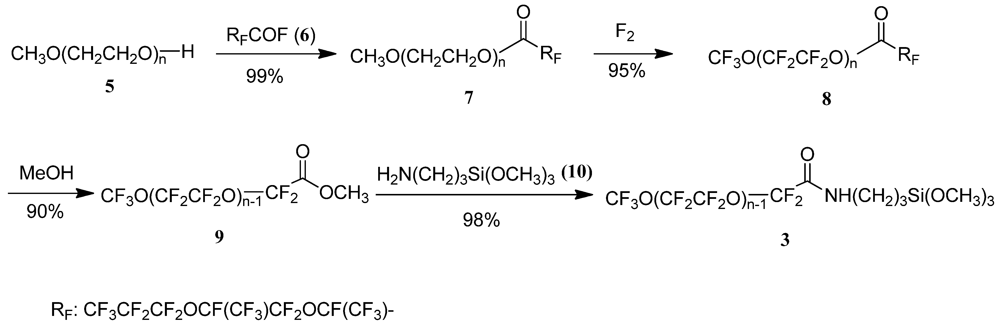
2.2.1. Synthesis of the Anti-staining Coating Material (Scheme 3)
CH3O-(CH2CH2O)nC(O)-CF(CF3)OCF2CF(CF3)OCF2CF2CF3 (7)
CF3O-(CF2CF2O)nC(O)-CF(CF3)OCF2CF(CF3)OCF2CF2CF3 (8)
CF3O-(CF2CF2O)n−1CF2COOCH3 (9)
CF3O(CF2CF2O)n−1CF2CONHCH2CH2CH2Si(OCH3)3 (3)
2.2.2. Evaluation of Anti-staining Performance
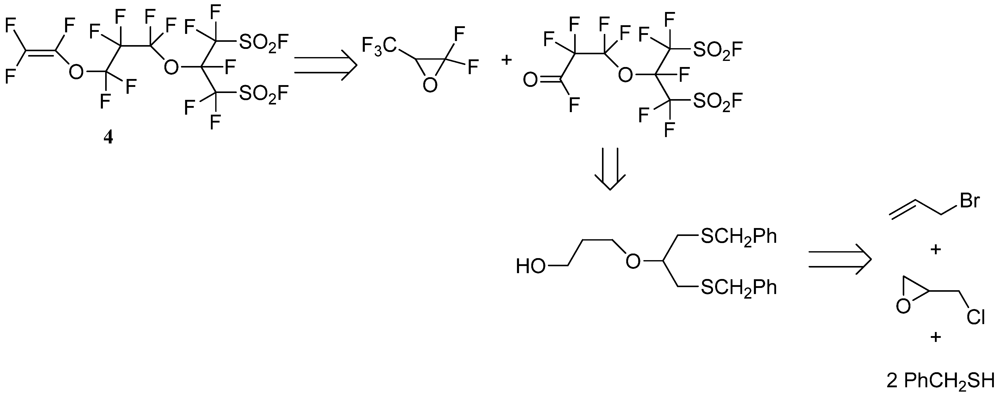
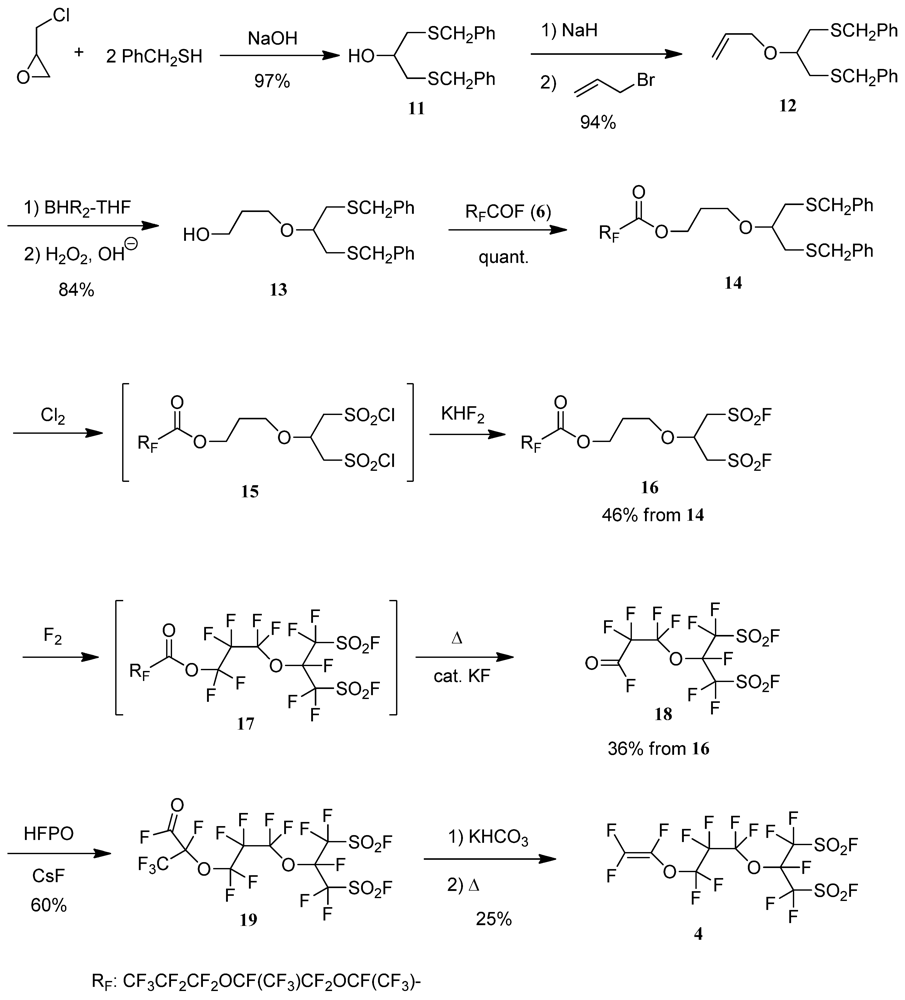
2.3. Typical Procedure for the Synthesis of the Perfluorobis(alkanesulfonyl) Monomer (Scheme 4)
2.3.1. 1,3-Bis(phenylmethylthio)propan-2-ol (11)
2.3.2. 2-Allyloxy-1,3-Bis(phenylmethylthio)propane (12)
2.3.3. 4-Oxa-8-Phenyl-5-(Phenylmethylthiomethyl)-7-Thiaoctan-1-ol (13)
2.3.4. 4-Oxa-8-Phenyl-5-(Phenylmethylthiomethyl)-7-Thiaoctyl Perfluoro(2,5-Dimethyl-3,6-Dioxanonanoate) (14)
2.3.5. 6-Fluorosulfonyl-5-(Fluorosulfonylmethyl)-4-Oxahexyl Perfluoro(2,5-Dimethyl-3,6-Dioxanonanoate) (16)
2.3.6. Perfluoro[6-Fluorosulfonyl-5-(Fluorosulfonylmethyl)-4-Oxahexanoyl] Fluoride (18)
2.3.7. Perfluoro[9-Fluorosulfonyl-8-(Fluorosulfonylmethyl)-2-Methyl-3,7-Dioxanonanoyl] Fluoride (19)
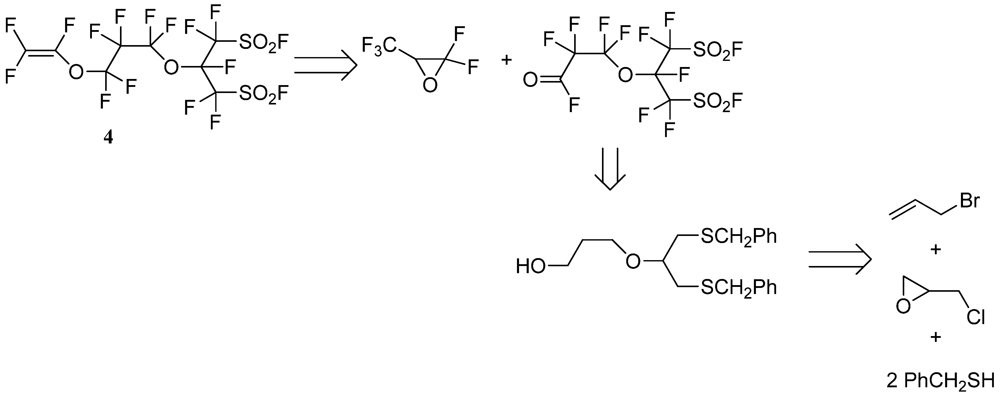
2.3.8. Perfluoro[2-(Fluorosulfonylmethyl)-3,7-Dioxa-8-Nonene]Sulfonyl Fluoride (4)
3. Results and Discussion
3.1. Synthesis and Evaluation of the Anti-staining Coating Material
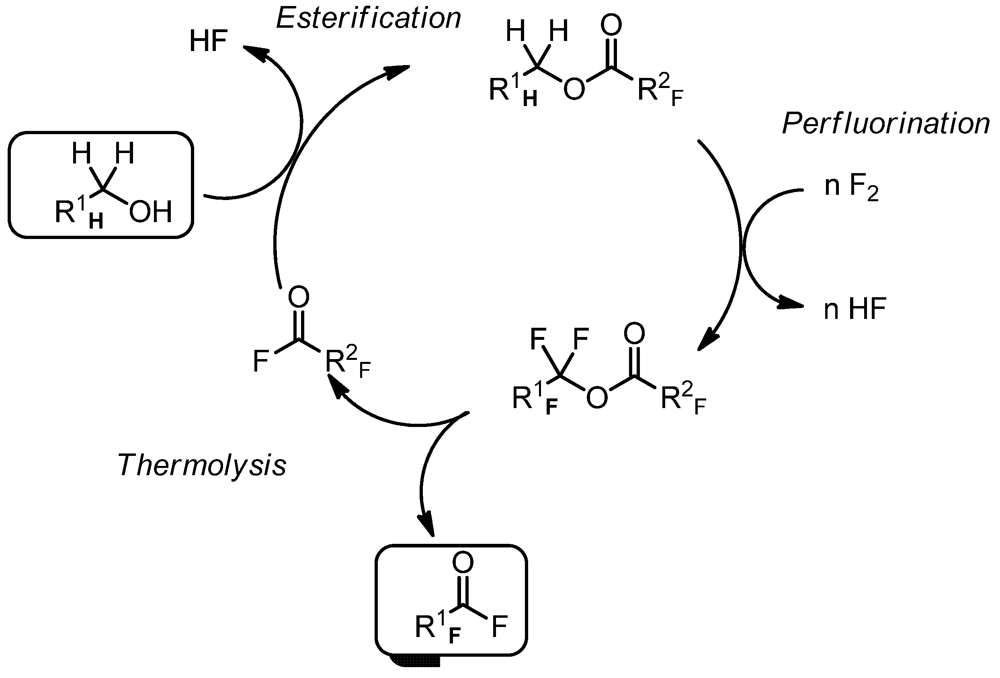
3.1.1. Synthesis of a PFPE which Possesses –(CF2CF2O)– as a Repeating Unit (Scheme 3)

3.1.2. Evaluation of Water and Oil Repellency

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | Water (deg.) | n-Hexadecane (deg.) | friction coefficient |
|---|---|---|---|
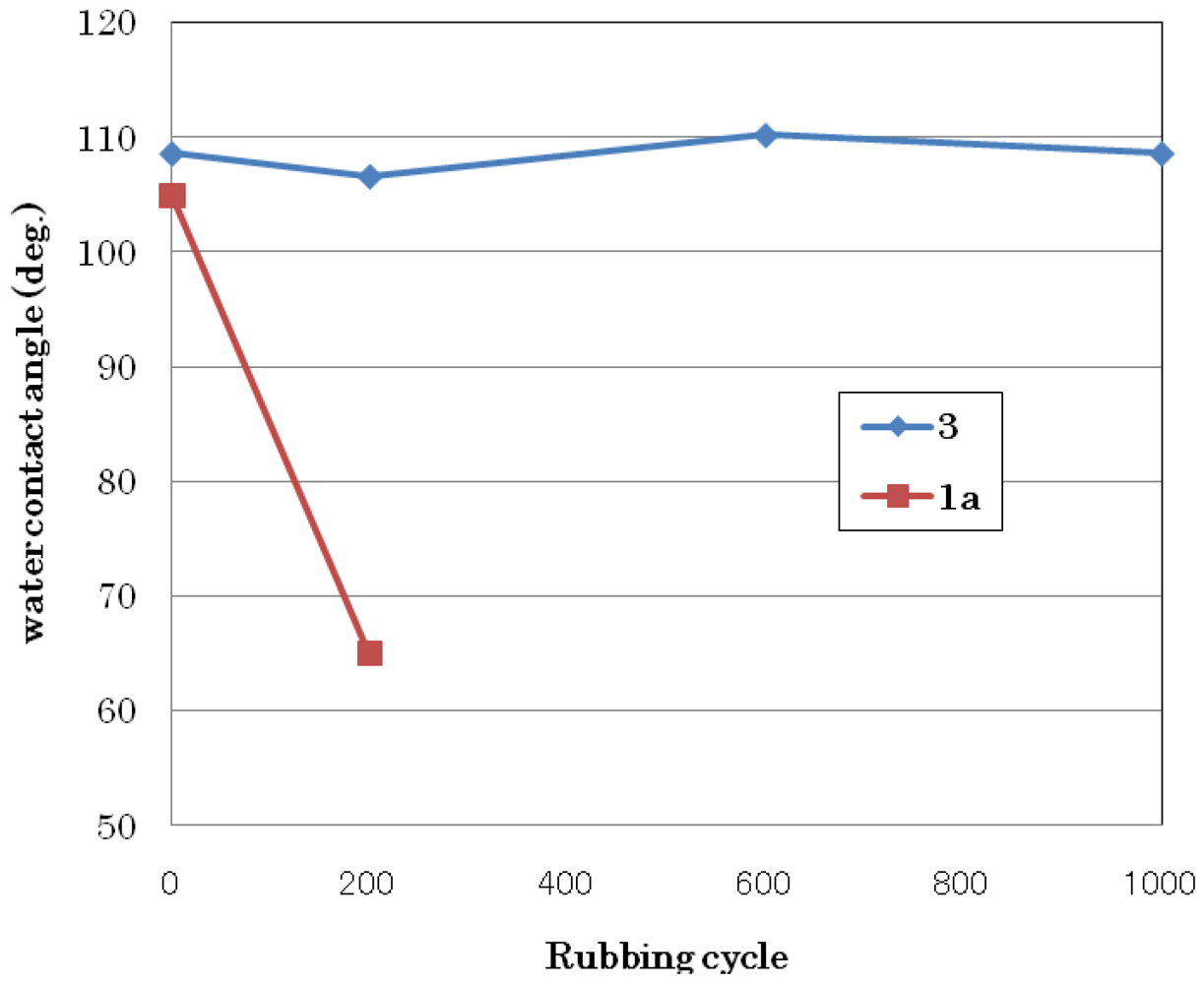
| 3 | 108 | 71 | 0.159 |
| 1a | 105 | 65 | 0.184 |
3.1.3. Abrasion Resistance Test
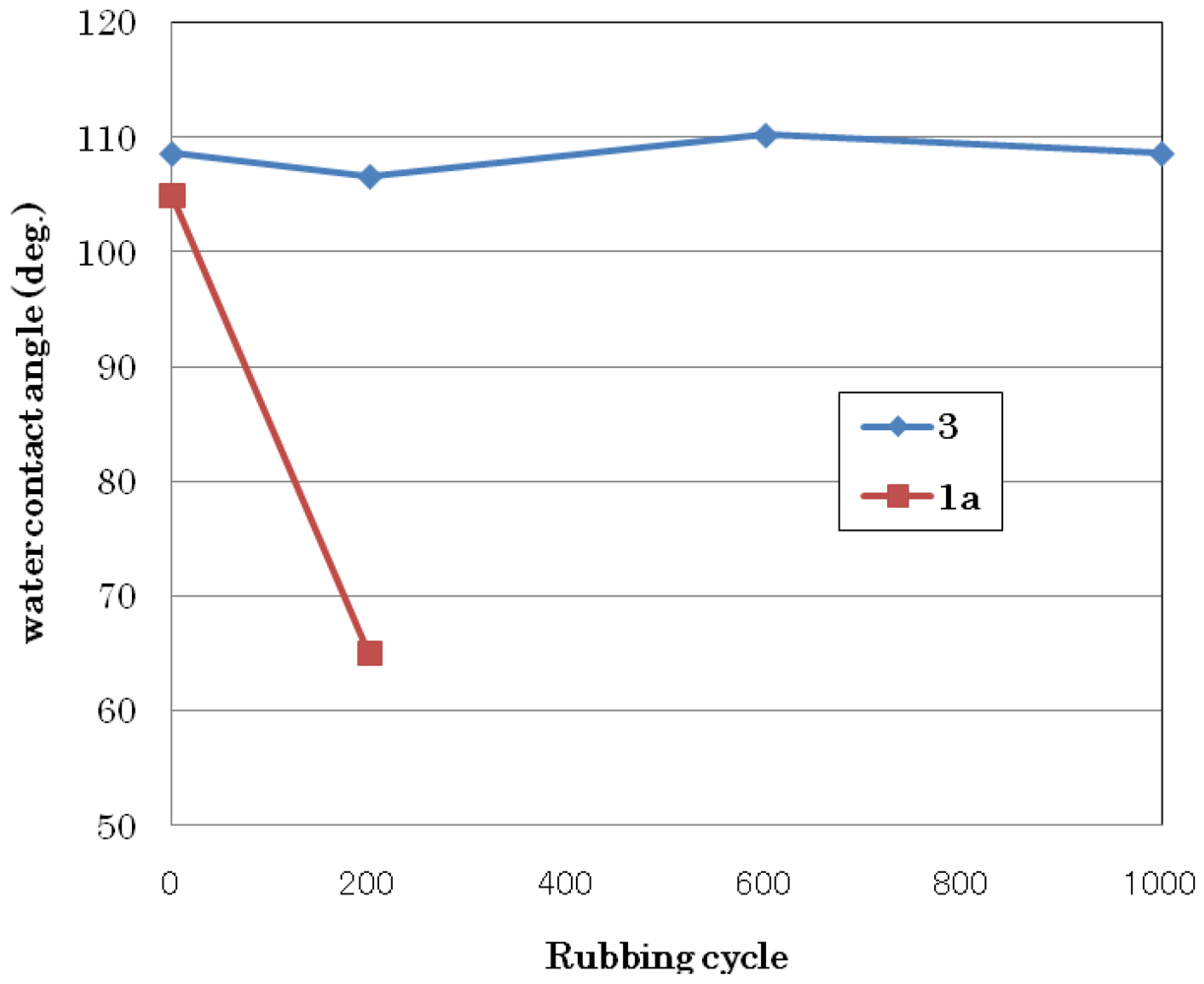
3.2. Synthesis of Perfluorobis(alkanesulfonyl) Monomer (Scheme 4)

4. Conclusions
References
- For a recent review of organofluorine compounds in materials industries, see: Okazoe, T. Overview on the history of organofluorine chemistry from the viewpoint of material industry. Proc. Jpn. Acad. Ser. B 2009, 85, 276–289. [Google Scholar] [CrossRef]
- Asai, M.; Uehara, H.; Ogawa, N.; Kamiya, K. Glass-Water Repellent Agent Composition and Method for Manufacturing Water-Repellent Glass. JP Patent 2002-145645, 22 May 2002. [Google Scholar]
- Fujita, H.; Nagai, K.; Sakagami, T.; Kai, Y.; Nakamura, I. Production of Water Repellent Glass for Automobile. JP Patent 9-286639, 4 November 1997. [Google Scholar]
- Ogawa, K.; Mino, N.; Soga, S. Stainproof Adsorbent Film and Its Production. JP Patent 2,981,043, 22 November 1999. [Google Scholar]
- Ohata, K.; Okubo, T.; Mitsuida, A.; Tomikawa, N.; Watanabe, J. Method for Forming Stainproof Layer. JP Patent 4174867, 5 November 2008. [Google Scholar]
- O’Hagan, D. Understanding organofluorine chemistry. An introduction to the C-F bond. Chem. Soc. Rev. 2008, 37, 308–319. [Google Scholar] [CrossRef]
- Kinoshita, K.; Cairns, E.J. Fuel cells. In Kirk-Othmer Encyclopedia of Chemical Technology, 4th; Howe-Grant, M., Ed.; John Wiley & Sons: New York, NY, USA, 1994; Volume 11, pp. 1098–1121. [Google Scholar]
- Okazoe, T.; Watanabe, K.; Itoh, M.; Shirakawa, D.; Tatematsu, S. A new route to perfluorinated vinyl ether monomers: Synthesis of perfluoro(alkoxyalkanoyl) fluorides from non-fluorinated compounds. J. Fluor. Chem. 2001, 112, 109–116. [Google Scholar] [CrossRef]
- Shirakawa, D.; Ohnishi, K. A study on design and analysis of new lubricant for near contact recording. IEEE Trans. Magn. 2010, 44, 3710–3713. [Google Scholar] [CrossRef]
- Takagi, H.; Matsuoka, Y.; Higuchi, S.; Funaki, H.; Seki, R.; Oharu, K.; Kamiya, H. APFO Alternative Potentiality for Perfluoropolyether Surfactants Derived from AGC’s “PERFECT” Process. In Proceedings of the 1st International Workshop “Fluorinated Surfactants: New Developments”, Idstein, Germany, 26–28 June 2008; PA3. Europa Fachhochschule Fresenius: Idstein, Germany.
- Okazoe, T.; Murotani, E.; Watanabe, K.; Itoh, M.; Shirakawa, D.; Kawahara, K.; Kaneko, I.; Tatematsu, S. An entirely new methodology for synthesizing perfluorinated compounds: Synthesis of perfluoroalkanesulfonyl fluorides from non-fluorinated compounds. J. Fluor. Chem. 2004, 125, 1695–1701. [Google Scholar] [CrossRef]
- Lagow, R.J. Reactions of fluorine in the presence of solvents. In Methoden Organic Chemistry (Houben-Weyl), 4th; Baasner, B., Hagemann, H., Tatlow, J.C., Eds.; Georg Thieme Verlag: Stuttgart, Germany, 1999; Volume E10a, pp. 194–200. [Google Scholar]
- Howell, J.L.; Shtarov, A.B.; Thrasher, J.S.; Waterfeld, A.; Murata, K.; Friesen, C.M.; Pérez, E.W. Synthesis of new linear perfluoroalkyl polyethers starting from diols and tetrafluoroethylene. Lubr. Sci. 2011, 23, 61–80. [Google Scholar]
- Forohar, F.; DesMarteau, D.D. Synthesis and reactions of the β-sultone of perfluorovinylsulfonyl fluoride. J. Fluor. Chem. 1994, 66, 101–104. [Google Scholar] [CrossRef]
- Corey, E.J.; Erickson, B.W.; Noyori, R. A new synthesis of α,β-unsaturated aldehydes using 1,3-bis(methylthio)allyllithium. J. Am. Chem. Soc. 1971, 93, 1724–1729. [Google Scholar] [CrossRef]
- Barlow, C.G.; Ollmann, R.R.; Zinn-Warner, A.S.; Nairne, R.J.D.; Beck, A.L.; Mott, A. Image Toners for Silver Halide Photographic Films. U.S. Patent 5,922,527, 13 July 1999. [Google Scholar]
- Zweifel, G.; Ayyangar, N.R.; Brown, H.C. Hydroboration. XVII. An examination of several representative dialkylboranes as selective hydroborating agents. J. Am. Chem. Soc. 1963, 85, 2072–2075. [Google Scholar] [CrossRef]
- Langler, R.F. A facile synthesis of sulfonyl chlorides. Can. J. Chem. 1976, 54, 498–499. [Google Scholar] [CrossRef]
- Gramstad, T.; Haszeldine, R.N. Perfluoroalkyl derivatives of sulphur. Part IV. Perfluoroalkanesulphonic acids. J. Chem. Soc. 1956, 173–180. [Google Scholar]
- Yamabe, M.; Matsuo, M.; Miyake, H. Fluoropolymers and fluoroelastomers. In Maclomolecular Design of Polymeric Materials; Hatada, K., Kitayama, T., Vogl, O., Eds.; Plastics Engineering Series 40; Marcel Dekker: New York, NY, USA, 1997; pp. 429–446. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Okazoe, T.; Shirakawa, D.; Murata, K. Application of Liquid-Phase Direct Fluorination: Novel Synthetic Methods for a Polyfluorinated Coating Material and a Monomer of a Perfluorinated Polymer Electrolyte Membrane. Appl. Sci. 2012, 2, 327-341. https://doi.org/10.3390/app2020327
Okazoe T, Shirakawa D, Murata K. Application of Liquid-Phase Direct Fluorination: Novel Synthetic Methods for a Polyfluorinated Coating Material and a Monomer of a Perfluorinated Polymer Electrolyte Membrane. Applied Sciences. 2012; 2(2):327-341. https://doi.org/10.3390/app2020327
Chicago/Turabian StyleOkazoe, Takashi, Daisuke Shirakawa, and Koichi Murata. 2012. "Application of Liquid-Phase Direct Fluorination: Novel Synthetic Methods for a Polyfluorinated Coating Material and a Monomer of a Perfluorinated Polymer Electrolyte Membrane" Applied Sciences 2, no. 2: 327-341. https://doi.org/10.3390/app2020327