
Extremophiles in an Antarctic Marine Ecosystem

Abstract
:1. Introduction
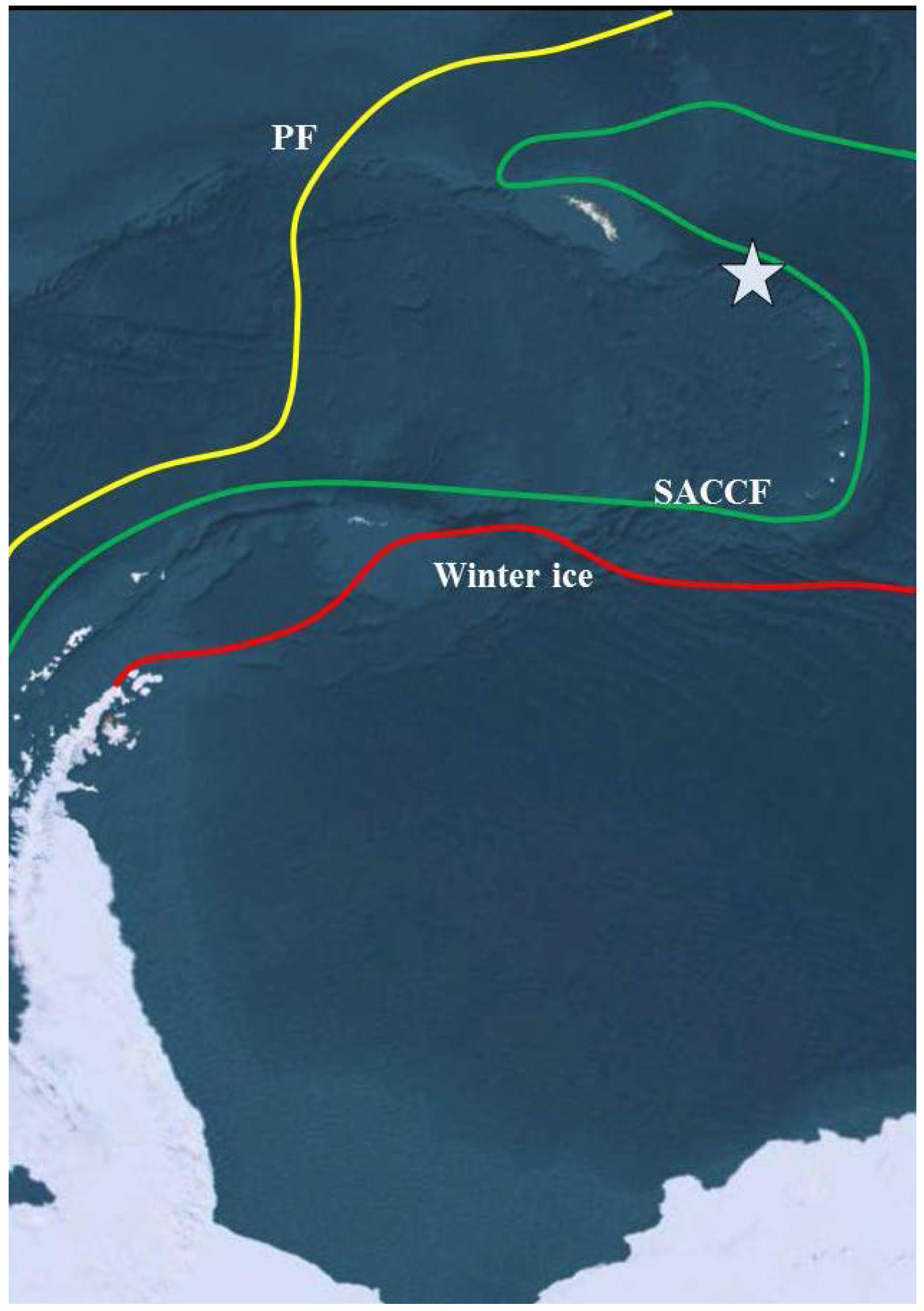
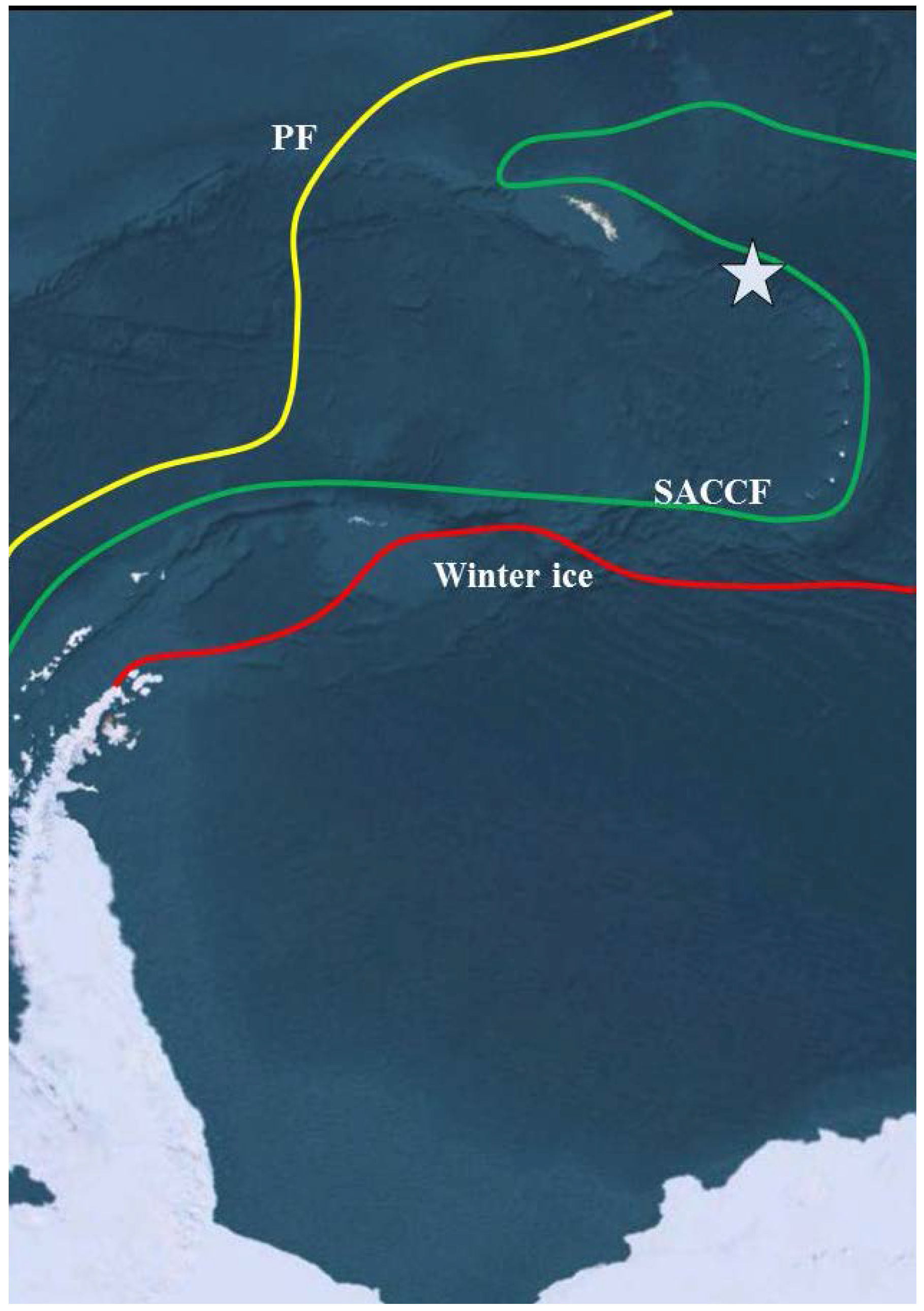
2. Experimental Section
2.1. Site Description and Sampling
2.2. Metagenomic Library Construction
2.3. 454 Pyrosequencing
2.4. MiSeq Sequencing
2.5. Data Analysis
2.6. Screening of Libraries with PCR Amplification to Identify Specific Functions
3. Results and Discussion
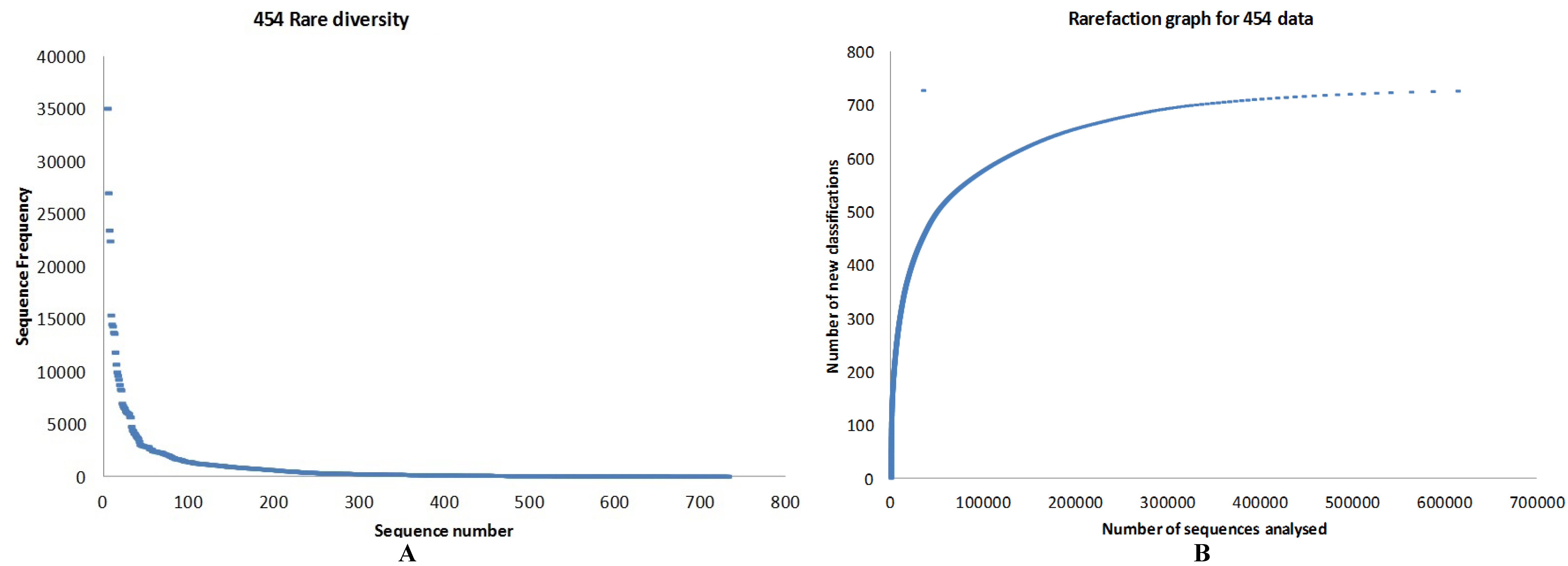
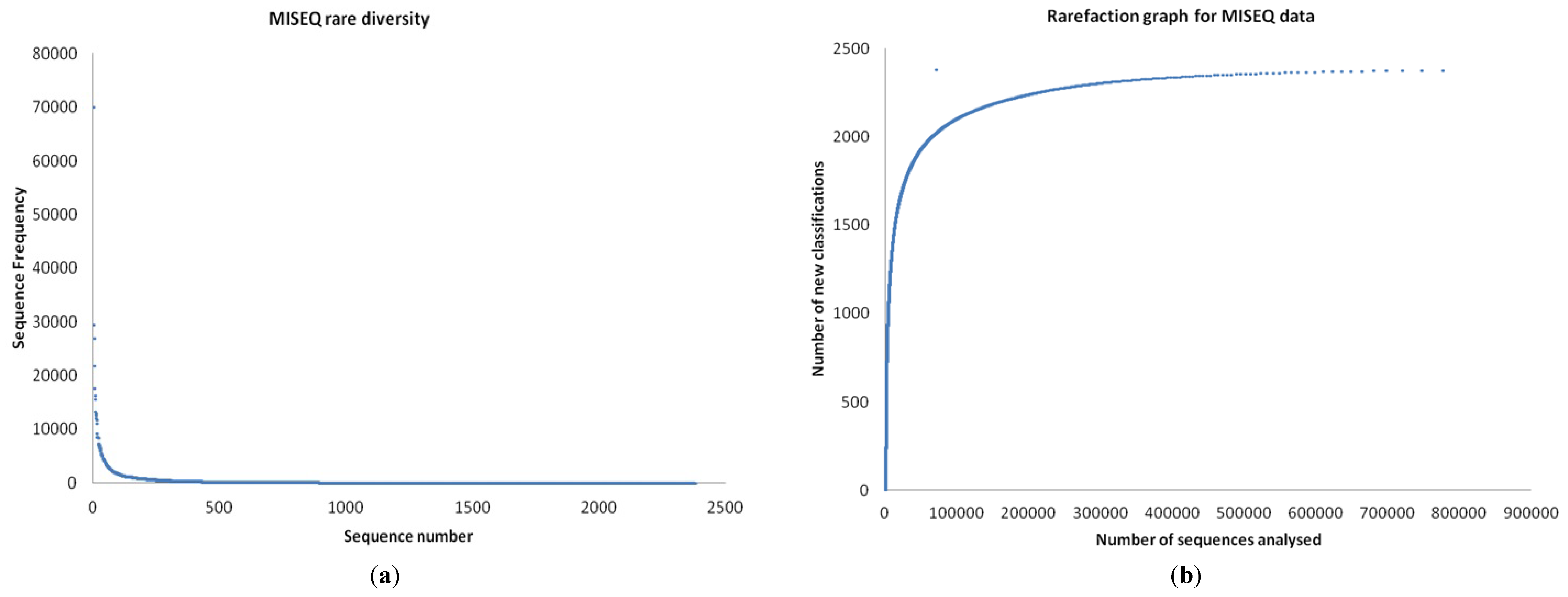
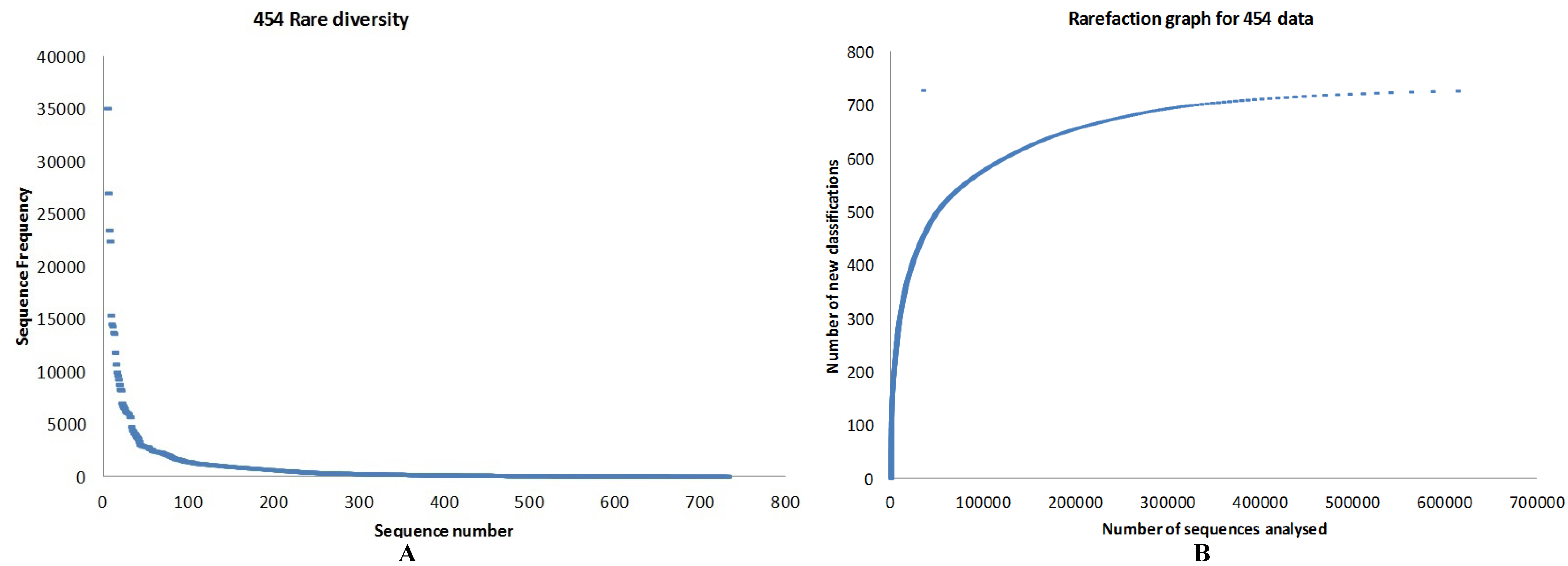
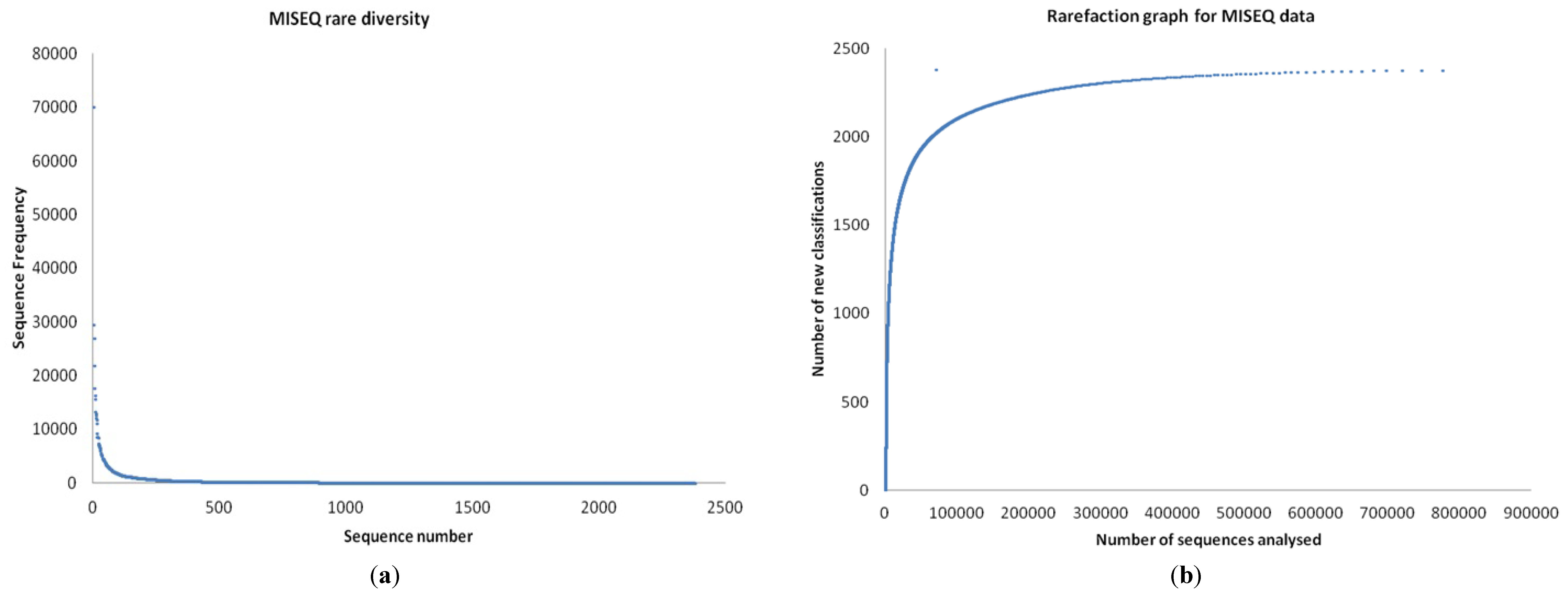
3.1. Quality Control
3.2. Preliminary Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 454 Species | MiSeq Species |
|---|---|
| Ruegeria pomeroyi (27,024) | Pseudomonas aeruginosa (21,810) |
| Roseobacter denitrificans (23,469) | Candidatus Pelagibacter ubique (17,625) |
| Candidatus Pelagibacter ubique (22,434) | Roseobacter denitrificans (16,349) |
| Rhodobacter sphaeroides (15,361) | Rhodobacterales bacterium HTCC2255 (15,625) |
| Ruegeria sp. TM1040 (14,489) | Ruegeria pomeroyi (13,319) |
| Saccharophagus degradans (14,300) | Saccharophagus degradans (11,712) |
| Pseudomonas aeruginosa (13,720) | Ruegeria sp. TM1040 (11,029) |
| Dinoroseobacter shibae (13,607) | Octadecabacter antarcticus (8624) |
| Congregibacter litoralis (11,834) | Rhodobacter sphaeroides (8519) |
| Pseudomonas fluorescens (10,692) | Cellvibrio japonicus (8484) |
| Roseovarius sp. 217 (9962) | Congregibacter litoralis (8400) |
| Roseobacter sp. MED193 (9629) | Dinoroseobacter shibae (7417) |
| Pseudomonas putida (9261) | Jannaschia sp. CCS1 (7057) |
| Roseovarius nubinhibens (8753) | Teredinibacter turnerae (6085) |
| Loktanella vestfoldensis (8319) | Roseovarius nubinhibens (5549) |
| Cellvibrio japonicus (8229) | Roseovarius sp. 217 (5291) |
| Hahella chejuensis (6977) | Roseobacter sp. MED193 (5136) |
| Sulfitobacter sp. NAS-14.1 (6741) | Loktanella vestfoldensis (4961) |
| Colwellia psychrerythraea (6581) | Marine gamma proteobacterium HTCC2143 (4509) |
| Oceanicola batsensis (6479) | Roseobacter sp. GAI101 (4494) |
| Marinobacter hydrocarbonoclasticus (6247) | Rhodobacteraceae bacterium HTCC2083 (4292) |
| Sulfitobacter sp. EE-36 (6111) | Marinobacter hydrocarbonoclasticus (4258) |
| Pseudomonas mendocina (6090) | Hahella chejuensis (4245) |
| Maritimibacter alkaliphilus (6010) | Candidatus Puniceispirillum marinum (4187) |
| Pseudoalteromonas atlantica (5972) | Marine gamma proteobacterium HTCC2148 (4100) |
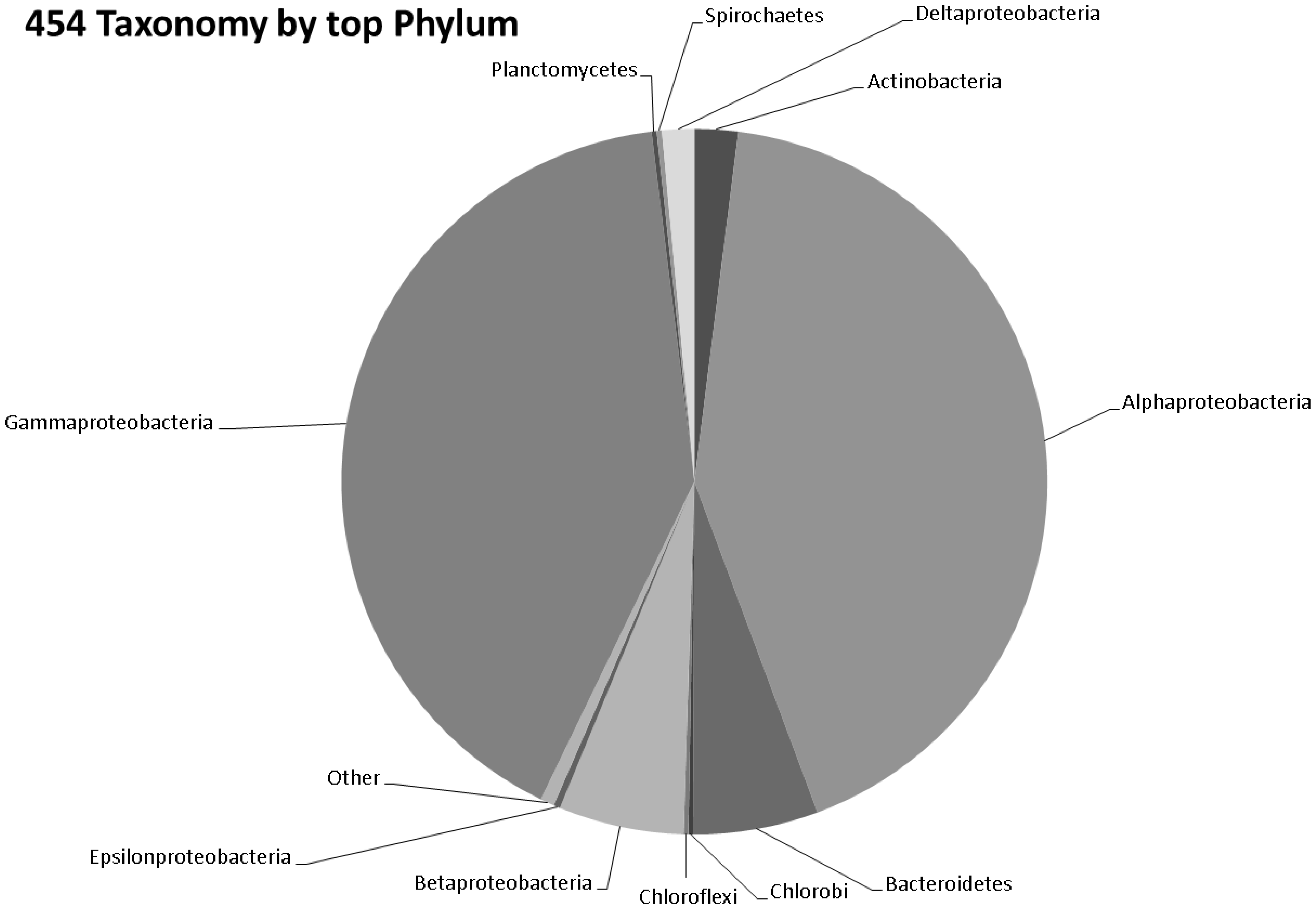
3.3. Taxonomy from 454 Pyrosequencing Data
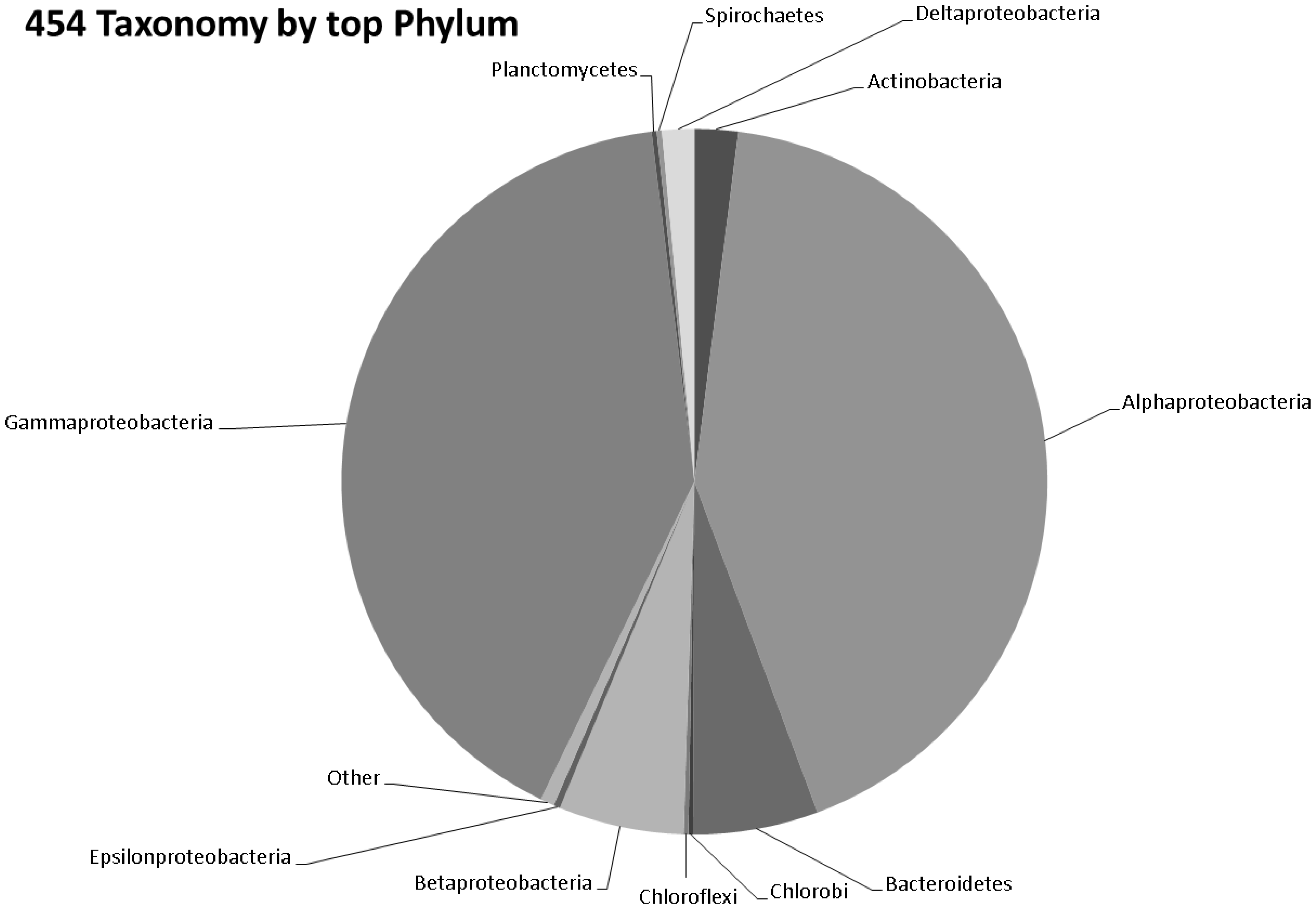
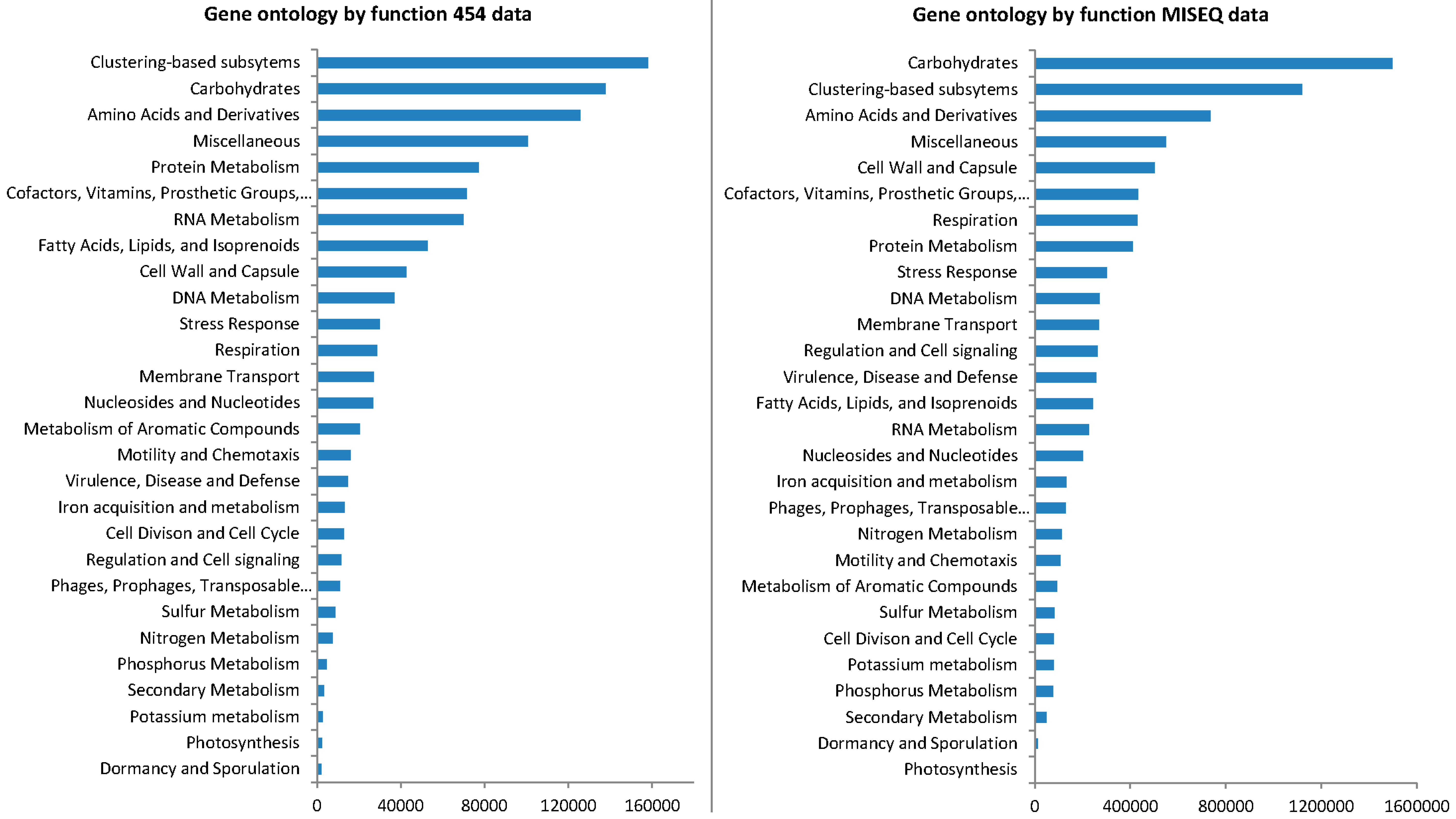
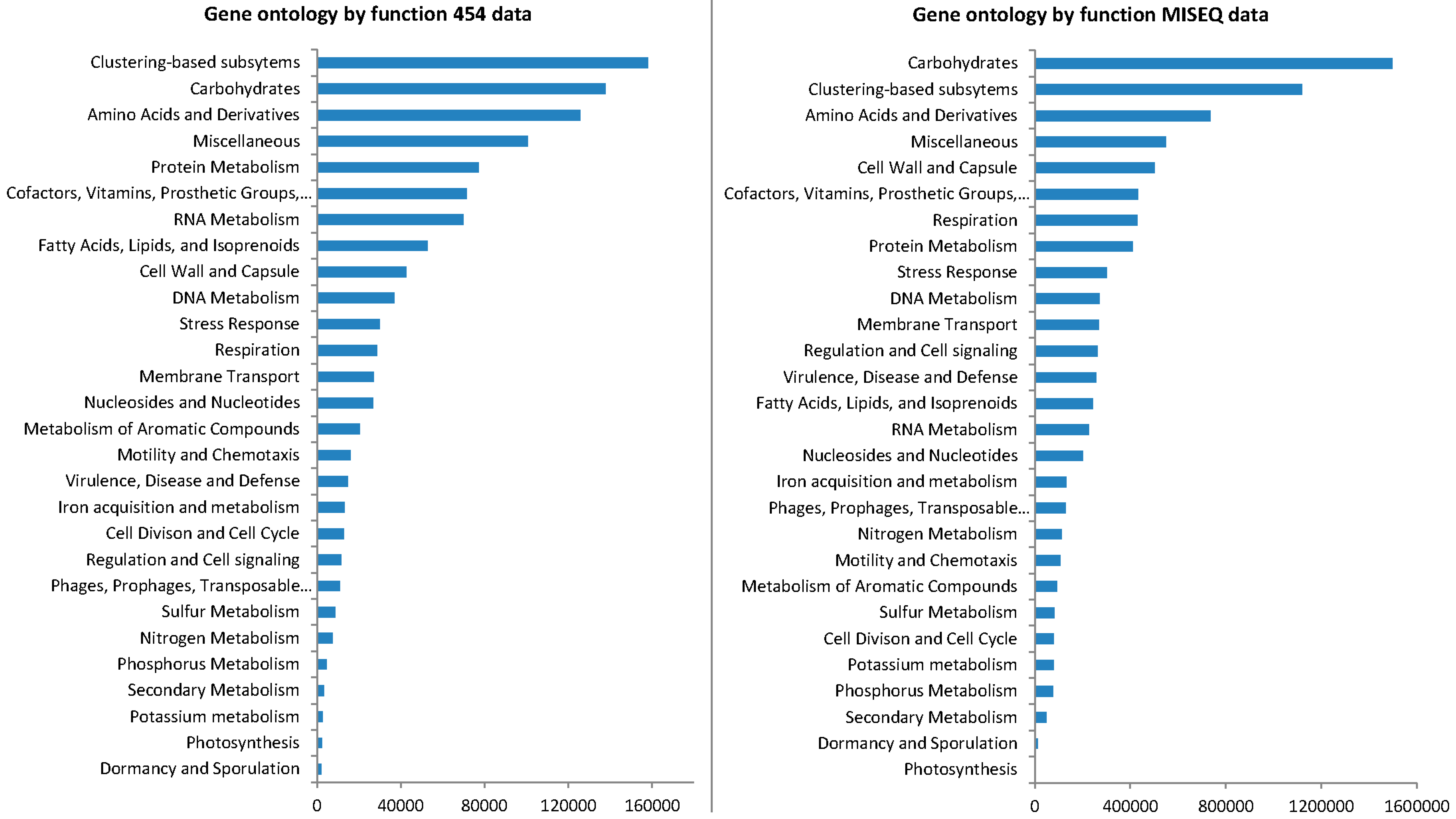
3.4. Gene Ontology from 454 Pyrosequencing Data
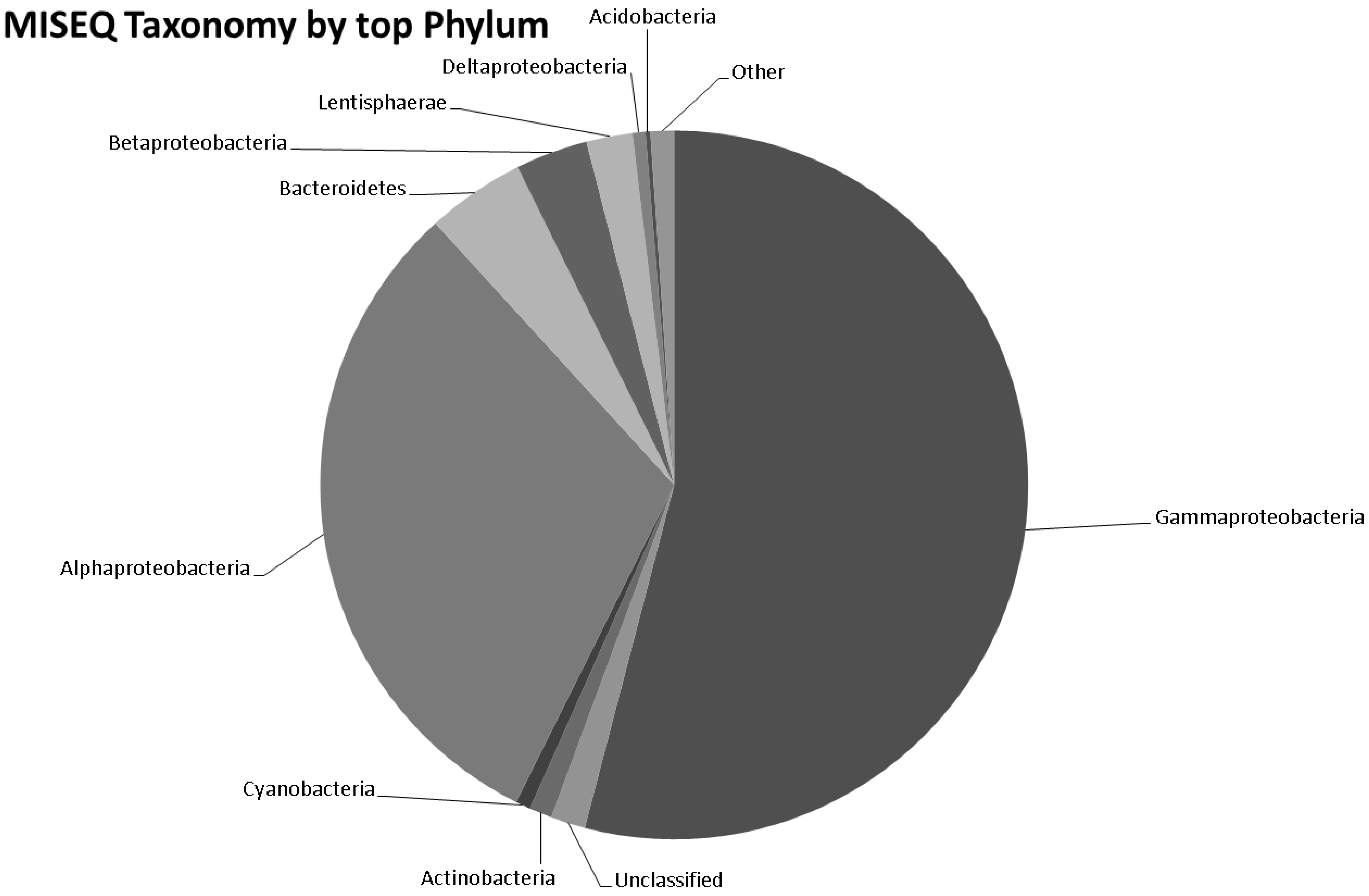
3.5. Taxonomy from MiSeq Data
3.6. Gene Ontology from MiSeq Data
3.7. Patterns of Bacterial and Functional Diversity
3.8. Viral Community
3.9. Cyanobacteria
3.10. Archaea
3.11. Actinobacteria
3.12. Methodological Potential in Bioprospecting
3.13. Ecological Characteristics of the Microbial Groups Most Frequently Identified by 454 Pyrosequencing
3.14. Further Ecological Characteristics Identified by MiSeq Sequencing
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pommier, T.; Pinhassi, J.; Hagström, Å.K. Biogeographic analysis of ribosomal RNA clusters from marine bacterioplankton. Aquat. Microb. Ecology 2005, 41, 79–89. [Google Scholar] [CrossRef]
- Hillebrand, H. On the generality of the latitudinal diversity gradient. Am. Nat. 2004, 163, 192–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giovannoni, S.J.; Stingl, U. Molecular diversity and ecology of microbial plankton. Nature 2005, 437, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Martiny, J.B.H.; Bohannan, B.J.; Brown, J.H.; Colwell, R.K.; Fuhrman, J.A.; Green, J.L.; Horner-Devine, M.C.; Kane, M.; Krumins, J.A.; Kuske, C.R.; Morin, P.J. Microbial biogeography: Putting microorganisms on the map. Nat. Rev. Microbiol. 2006, 4, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Bouvier, T.C.; del Giorgio, P.A. Compositional changes in free-living bacterial communities along a salinity gradient in two temperate estuaries. Limnol. Oceanogr. 2002, 47, 453–470. [Google Scholar] [CrossRef]
- Crump, B.C.; Hopkinson, C.S.; Sogin, M.L.; Hobbie, J.E. Microbial biogeography along an estuarine salinity gradient: combined influences of bacterial growth and residence time. Appl. Environ. Microbiol. 2004, 70, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Martinez, J.; Rodriguez-Valera, F. Microdiversity of uncultured marine prokaryotes: The SAR11 cluster and the marine Archaea of Group I. Mol. Ecol. 2000, 9, 935–948. [Google Scholar] [CrossRef] [PubMed]
- Giovannoni, S.J.; Rappé, M.S.; Vergin, K.L.; Adair, N.L. 16S rRNA genes reveal stratified open ocean bacterioplankton populations related to the Green Non-Sulfur bacteria. Proc. Natl. Acad. Sci. USA 1996, 93, 7979–7984. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.A.; Giovannoni, S.J. Detection of stratified microbial populations related to Chlorobium and Fibrobacter species in the Atlantic and Pacific oceans. Appl. Environ. Microbiol. 1996, 62, 1171–1177. [Google Scholar] [PubMed]
- Riemann, L.; Steward, G.F.; Fandino, L.B.; Campbell, L.; Landry, M.R.; Azam, F. Bacterial community composition during two consecutive NE Monsoon periods in the Arabian Sea studied by denaturing gradient gel electrophoresis (DGGE) of rRNA genes. Deep Sea Res. Part II: Top. Stud. Oceanogr. 1999, 46, 1791–1811. [Google Scholar] [CrossRef]
- Pinhassi, J.; Berman, T. Differential growth response of colony-forming alpha- and gamma-proteobacteria in dilution culture and nutrient addition experiments from Lake Kinneret (Israel), the eastern Mediterranean Sea, and the Gulf of Eilat. Appl. Environ. Microbiol. 2003, 69, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Venter, J.C.; Remington, K.; Heidelberg, J.F.; Halpern, A.L.; Rusch, D.; Eisen, J.A.; Wu, D.; Paulsen, I.; Nelson, K.E.; Nelson, W.; Fouts, D.E. Environmental genome shotgun sequencing of the Sargasso Sea. Science 2004, 304, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Sunagawa, S.; Coelho, L.P.; Chaffron, S.; Kultima, J.R.; Labadie, K.; Salazar, G.; Djahanschiri, B.; Zeller, G.; Mende, D.R.; Alberti, A.; Cornejo-Castillo, F.M. Structure and function of the global ocean microbiome. Science 2015, 348, 1261359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, J.P.; McCammon, S.A.; Brown, M.V.; Nichols, D.S.; McMeekin, T.A. Diversity and association of psychrophilic bacteria in Antarctic sea ice. Appl. Environ. Microbiol. 1997, 63, 3068–3078. [Google Scholar] [PubMed]
- Kottmeier, S.; Sullivan, C. Sea ice microbial communities (SIMCO). Polar Biol. 1988, 8, 293–304. [Google Scholar] [CrossRef]
- Delille, D. Marine bacterioplankton at the Weddell Sea ice edge, distribution of psychrophilic and psychrotrophic populations. Polar Biol. 1992, 12, 205–210. [Google Scholar] [CrossRef]
- Reichardt, W. Impact of the Antarctic benthic fauna on the enrichment of biopolymer degrading psychrophilic bacteria. Microb. Ecol. 1988, 15, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Ruger, H.J. Benthic studies of the northwest African upwelling region: Psychrophilic and psychrotrophic bacterial communities from areas with different upwelling intensities. Mar. Ecol. Prog. Ser. 1989, 57, 45–52. [Google Scholar] [CrossRef]
- Straka, R.; Stokes, J.L. Psychrophilic Bacteria from Antarctica. J. Bacteriol. 1960, 80, 622–625. [Google Scholar] [PubMed]
- Herbert, R.A.; Tanner, A.C. The isolation and some characteristics of photosynthetic bacteria (Chromatiaceae and Chlorobiaceae) from Antarctic marine sediments. J. Appl. Bacteriol. 1977, 43, 437–445. [Google Scholar] [CrossRef]
- McMeekin, T.A.; Franzmann, P.D. Effect of temperature on the growth rates of halotolerant and halophilic bacteria isolated from Antarctic saline lakes. Polar Biol. 1988, 8, 281–285. [Google Scholar] [CrossRef]
- Delille, D.; Perret, E. Influence of temperature on the growth potential of Southern polar marine bacteria. Microb. Ecol. 1989, 18, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Nedwell, D.B.; Rutter, M. Influence of temperature on growth rate and competition between two psychrotolerant Antarctic bacteria: Low temperature diminishes affinity for substrate uptake. Appl. Environ. Microbiol. 1994, 60, 1984–1992. [Google Scholar] [PubMed]
- Selje, N.; Simon, M.; Brinkhoff, T. A newly discovered Roseobacter cluster in temperate and polar oceans. Nature 2004, 427, 445–448. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, R.E.; Rogers, A.D.; Billett, D.S.; Smale, D.A.; Pearce, D.A. Patterns of marine bacterioplankton biodiversity in the surface waters of the Scotia Arc, Southern Ocean. FEMS Microbiol. Ecol. 2012, 80, 452–468. [Google Scholar] [CrossRef] [PubMed]
- Kazuoka, T.; Oikawa, T.; Muraoka, I.; Kuroda, S.I.; Soda, K. A cold-active and thermostable alcohol dehydrogenase of a psychrotorelant from Antarctic seawater, Flavobacterium frigidimaris KUC-1. Extremophiles 2007, 11, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Acevedo, J.P.; Reyes, F.; Parra, L.P.; Salazar, O.; Andrews, B.A.; Asenjo, J.A. Cloning of complete genes for novel hydrolytic enzymes from Antarctic sea water bacteria by use of an improved genome walking technique. J. Biotechnol. 2008, 133, 277–286. [Google Scholar] [CrossRef] [PubMed]
- D’Auria, S.; Aurilia, V.; Marabotti, A.; Gonnelli, M.; Strambini, G. Structure and dynamics of cold-adapted enzymes as investigated by phosphorescence spectroscopy and molecular dynamics studies. 2. The case of an esterase from Pseudoalteromonas haloplanktis. J. Phys. Chem. B 2009, 113, 13171–13178. [Google Scholar] [CrossRef] [PubMed]
- De Pascale, D.; Cusano, A.M.; Autore, F.; Parrilli, E.; di Prisco, G.; Marino, G.; Tutino, M.L. The cold-active Lip1 lipase from the Antarctic bacterium Pseudoalteromonas haloplanktis TAC125 is a member of a new bacterial lipolytic enzyme family. Extremophiles 2008, 12, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Ghiglione, J.F.; Murray, A.E. Pronounced summer to winter differences and higher wintertime richness in coastal Antarctic marine bacterioplankton. Environ. Microbiol. 2012, 14, 617–629. [Google Scholar] [CrossRef] [PubMed]
- Grzymski, J.J.; Riesenfeld, C.S.; Williams, T.J.; Dussaq, A.M.; Ducklow, H.; Erickson, M.; Cavicchioli, R.; Murray, A.E. A metagenomic assessment of winter and summer bacterioplankton from Antarctica Peninsula coastal surface waters. ISME J. 2012, 6, 1901–1915. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, D.; Lauro, F.M.; Williams, T.J.; Demaere, M.Z.; Brown, M.V.; Hoffman, J.M.; Andrews-Pfannkoch, C.; Mcquaid, J.B.; Riddle, M.J.; Rintoul, S.R.; Cavicchioli, R. Biogeographic partitioning of Southern Ocean microorganisms revealed by metagenomics. Environ. Microbiol. 2013, 15, 1318–1333. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.J.; Long, E.; Evans, F.; DeMaere, M.Z.; Lauro, F.M.; Raftery, M.J.; Ducklow, H.; Grzymski, J.J.; Murray, A.E.; Cavicchioli, R. A metaproteomic assessment of winter and summer bacterioplankton from Antarctic Peninsula coastal surface waters. ISME J. 2012, 6, 1883–1900. [Google Scholar] [CrossRef] [PubMed]
- Grzymski, J.J.; Carter, B.J.; DeLong, E.F.; Feldman, R.A.; Ghadiri, A.; Murray, A.E. Comparative genomics of DNA fragments from six antarctic marine planktonic bacteria. Appl. Environ. Microbiol. 2006, 72, 1532–1541. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Sáez, L.; Waller, A.S.; Mende, D.R.; Bakker, K.; Farnelid, H.; Yager, P.L.; Lovejoy, C.; Tremblay, J.É.; Potvin, M.; Heinrich, F.; Estrada, M. Role for urea in nitrification by polar marine Archaea. Proc. Natl. Acad. Sci. USA 2012, 109, 17989–17994. [Google Scholar] [CrossRef] [PubMed]
- Moore, J.K.; Abbott, M.R. Surface chlorophyll concentrations in relation to the Antarctic Polar Front: Seasonal and spatial patterns from satellite observations. J. Mar. Syst. 2002, 37, 69–86. [Google Scholar] [CrossRef]
- Talley, L. Descriptive Physical Oceanography: An Introduction; Academic Press: San Diego, CA, USA, 2011. [Google Scholar]
- Geneious Website. Available online: http://www.geneious.com/ (accessed on 28 June 2015).
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; Thierer, T. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; Sahl, J.W. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Alvarez, V.; Teal, T.K.; Schmidt, T.M. Systematic artifacts in metagenomes from complex microbial communities. ISME J. 2009, 3, 1314–1317. [Google Scholar] [CrossRef] [PubMed]
- Meyer, F.; Paarmann, D.; D’Souza, M.; Olson, R.; Glass, E.M.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A.; Wilkening, J. The metagenomics RAST server—A public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinf. 2008, 9, 386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.M.; Ludwig, W.; Peplies, J.; Glöckner, F.O. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maidak, B.L.; Larsen, N.; McCaughey, M.J.; Overbeek, R.; Olsen, G.J.; Fogel, K.; Blandy, J.; Woese, C.R. The Ribosomal Database Project. Nucleic Acids Res 1994, 22, 3485–3487. [Google Scholar] [CrossRef] [PubMed]
- Andersson, A.F.; Lindberg, M.; Jakobsson, H.; Backhed, F.; Nyrén, P.; Engstrand, L. Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS ONE 2008, 3, e2836. [Google Scholar] [CrossRef] [PubMed]
- Fuller, N.J.; Wilson, W.H.; Joint, I.R.; Mann, N.H. Occurrence of a sequence in marine cyanophages similar to that of T4 g20 and its application to PCR-based detection and quantification techniques. Appl. Environ. Microbiol. 1998, 64, 2051–2060. [Google Scholar] [PubMed]
- Wilson, W.H.; Fuller, N.J.; Joint, I.R.; Mann, N.H. Analysis of Cyanophage Diversity and Population Structure in a South-North Transect of the Atlantic Ocean; Oceanographic Museum: Monaco-Ville, Monaco, 1999. [Google Scholar]
- Chen, F.; Suttle, C.A. Amplification of DNA polymerase gene fragments from viruses infecting microalgae. Appl. Environ. Microbiol. 1995, 61, 1274–1278. [Google Scholar] [PubMed]
- White, T.J.; Bruns, T.; Lee, S.J.W.T.; Taylor, J.W. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innis, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Academic Press: San Diego, CA, USA, 1990; pp. 315–322. [Google Scholar]
- Gardes, M.; Bruns, T.D. ITS primers with enhanced specificity for basidiomycetes—Application to the identification of mycorrhizae and rusts. Mol. Ecol. 1993, 2, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, J.A.; Thomas, S.; Cooley, N.A.; Kulakova, A.; Field, D.; Booth, T.; McGrath, J.W.; Quinn, J.P.; Joint, I. Potential for phosphonoacetate utilization by marine bacteria in temperate coastal waters. Environ. Microbiol. 2009, 11, 111–125. [Google Scholar] [CrossRef] [PubMed]
- Throbäck, I.N.; Enwall, K.; Jarvis, Å.; Hallin, S. Reassessing PCR primers targeting nirS, nirK and nosZ genes for community surveys of denitrifying bacteria with DGGE. FEMS Microbiol. Ecol. 2004, 49, 401–417. [Google Scholar] [CrossRef] [PubMed]
- Rösch, C.; Bothe, H. Improved assessment of denitrifying, N2-fixing, and total-community bacteria by terminal restriction fragment length polymorphism analysis using multiple restriction enzymes. Appl. Environ. Microbiol. 2005, 71, 2026–2035. [Google Scholar] [CrossRef] [PubMed]
- Pedros-Alio, C. Marine microbial diversity: Can it be determined? Trends Microbiol. 2006, 14, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Andersson, A.F.; Riemann, L.; Bertilsson, S. Pyrosequencing reveals contrasting seasonal dynamics of taxa within Baltic Sea bacterioplankton communities. ISME J. 2009, 4, 171–181. [Google Scholar] [CrossRef] [PubMed]
- Church, M.J.; Hutchins, D.A.; Ducklow, H.W. Limitation of bacterial growth by dissolved organic matter and iron in the Southern ocean. Appl. Environ. Microbiol. 2000, 66, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Giudice, A.L.; Casella, P.; Bruni, V.; Michaud, L. Response of bacterial isolates from Antarctic shallow sediments towards heavy metals, antibiotics and polychlorinated biphenyls. Ecotoxicology 2013, 22, 240–250. [Google Scholar] [CrossRef] [PubMed]
- Azam, F. Microbial control of oceanic carbon flux: The plot thickens. Science 1998, 280, 694–696. [Google Scholar] [CrossRef]
- Bidle, K.D.; Brzezinski, M.A.; Long, R.A.; Jones, J.L.; Azam, F. Diminished efficiency in the oceanic silica pump caused by bacteria-mediated silica dissolution. Limnol. Oceanogr. 2003, 48, 1855–1868. [Google Scholar] [CrossRef]
- Brown, G.E.; Parks, G.A. Sorption of trace elements on mineral surfaces: modern perspectives from spectroscopic studies, and comments on sorption in the marine environment. Int. Geol. Rev. 2001, 43, 963–1073. [Google Scholar] [CrossRef]
- DeLong, E.F.; Wu, K.Y.; Prézelin, B.B.; Jovine, R.V. High abundance of Archaea in Antarctic marine picoplankton. Nature 1994, 371, 695–697. [Google Scholar] [CrossRef] [PubMed]
- Staley, J.T.; Gosink, J.J. Poles apart: Biodiversity and biogeography of sea ice bacteria. Annu. Rev. Microbiol. 1999, 53, 189–215. [Google Scholar] [CrossRef] [PubMed]
- Massana, R.; DeLong, E.F.; Pedros-Alio, C. A few cosmopolitan phylotypes dominate planktonic archaeal assemblages in widely different oceanic provinces. Appl. Environ. Microbiol. 2000, 66, 1777–1787. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.G.; Park, S.J.; Quan, Z.X.; Jung, M.Y.; Cha, I.T.; Kim, S.J.; Kim, K.H.; Yang, E.J.; Kim, Y.N.; Lee, S.H.; Rhee, S.K. Unveiling abundance and distribution of planktonic Bacteria and Archaea in a polynya in Amundsen Sea, Antarctica. Environ. Microbiol. 2014, 16, 1566–1578. [Google Scholar] [CrossRef] [PubMed]
- Luria, C.M.; Ducklow, H.W.; Amaral-Zettler, L.A. Marine bacterial, archaeal and eukaryotic diversity and community structure on the continental shelf of the western Antarctic Peninsula. Aquat. Microb. Ecol. 2014, 73, 107–121. [Google Scholar] [CrossRef]
- Humphry, D.R.; Black, G.W.; Cummings, S.P. Reclassification of “Pseudomonas fluorescens subsp. cellulosa” NCIMB 10462 (Ueda et al., 1952) as Cellvibrio japonicus sp. nov. and revival of Cellvibrio vulgaris sp. nov., nom. rev. and Cellvibrio fulvus sp. nov., nom. rev. Int. J. Syst. Evol. Microbiol. 2003, 53 Pt 2, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Moran, M.A.; Belas, R.; Schell, M.A.; González, J.M.; Sun, F.; Sun, S.; Binder, B.J.; Edmonds, J.; Ye, W.; Orcutt, B.; Howard, E.C. Ecological genomics of marine Roseobacters. Appl. Environ. Microbiol. 2007, 73, 4559–4569. [Google Scholar] [CrossRef] [PubMed]
- Costerton, W.; Anwar, H. Pseudomonas aeruginosa: The microbe and pathogen. In Pseudomonas Aeruginosa Infections and Treatment; Baltch, A., Smith, R., Eds.; CRC Press: Boca Raton, FL, USA, 1994; pp. 1–17. [Google Scholar]
- Fuchs, B.M.; Spring, S.; Teeling, H.; Quast, C.; Wulf, J.; Schattenhofer, M.; Yan, S.; Ferriera, S.; Johnson, J.; Glöckner, F.O.; Amann, R. Characterization of a marine gammaproteobacterium capable of aerobic anoxygenic photosynthesis. Proc. Natl. Acad. Sci. USA 2007, 104, 2891–2896. [Google Scholar] [CrossRef] [PubMed]
- Pseudoaltermonas Atlantica T6c Genome Home Page. Available online: http://genome.jgi-psf.org/finished_microbes/pseat/pseat.home.html (accessed on 28 June 2015).
- Roseobase: Genomic resource for marine Roseobacters HTCC2597. Available online: http://www.roseobase.org/Species/htcc2597.html (accessed on 28 June 2015).
- Gauthier, M.J.; Lafay, B.; Christen, R.; Fernandez, L.; Acquaviva, M.; Bonin, P.; Bertrand, J.C. Marinobacter hydrocarbonoclasticus gen. nov., sp. nov., a new, extremely halotolerant, hydrocarbon-degrading marine bacterium. Int. J. Syst. Bacteriol. 1992, 42, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Ekborg, N.A.; Gonzalez, J.M.; Howard, M.B.; Taylor, L.E.; Hutcheson, S.W.; Weiner, R.M. Saccharophagus degradans gen. nov., sp. nov., a versatile marine degrader of complex polysaccharides. Int. J. Syst. Evol. Microbiol. 2005, 55, 1545–1549. [Google Scholar] [CrossRef] [PubMed]
- Ratkowsky, D.A.; Lowry, R.K.; McMeekin, T.A.; Stokes, A.N.; Chandler, R.E. Model for bacterial culture growth rate throughout the entire biokinetic temperature range. J. Bacteriol. 1983, 154, 1222–1226. [Google Scholar] [PubMed]
- Palleroni, N.J.; Doudoroff, M.; Stanier, R.Y.; Solanes, R.E.; Mandel, M. Taxonomy of the Aerobic Pseudomonads: The properties of the Pseudomonas stutzeri group. Microbiology 1970, 60, 215–231. [Google Scholar] [CrossRef] [PubMed]
- Methé, B.A.; Nelson, K.E.; Deming, J.W.; Momen, B.; Melamud, E.; Zhang, X.; Moult, J.; Madupu, R.; Nelson, W.C.; Dodson, R.J.; Brinkac, L.M. The psychrophilic lifestyle as revealed by the genome sequence of Colwellia psychrerythraea 34H through genomic and proteomic analyses. Proc. Natl. Acad. Sci. USA 2005, 102, 10913–10918. [Google Scholar] [CrossRef] [PubMed]
- Swingley, W.D.; Sadekar, S.; Mastrian, S.D.; Matthies, H.J.; Hao, J.; Ramos, H.; Acharya, C.R.; Conrad, A.L.; Taylor, H.L.; Dejesa, L.C.; Shah, M.K. The complete genome sequence of Roseobacter denitrificans reveals a mixotrophic rather than photosynthetic metabolism. J. Bacteriol. 2007, 189, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Roseobase: Genomic resource for marine Roseobacters Sulfitobacter sp. NAS-14.1. Available online: http://www.roseobase.org/Species/sulf_nas14_1.html (accessed on 28 June 2015).
- González, J.M.; Covert, J.S.; Whitman, W.B.; Henriksen, J.R.; Mayer, F.; Scharf, B.; Schmitt, R.; Buchan, A.; Fuhrman, J.A.; Kiene, R.P.; Moran, M.A. Silicibacter pomeroyi sp. nov. and Roseovarius nubinhibens sp. nov., dimethylsulfoniopropionate-demethylating bacteria from marine environments. Int. J. Syst. Evol. Microbiol. 2003, 53, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Roseobase: Genomic Resource for Marine Roseobacters Roseovarius sp. 217. Available online: http://www.roseobase.org/Species/roseovarius217.html (accessed on 28 June 2015).
- Lee, H.K.; Chun, J.; Moon, E.Y.; Ko, S.H.; Lee, D.S.; Lee, H.S.; Bae, K.S. Hahella chejuensis gen. nov., sp. nov., an extracellular-polysaccharide-producing marine bacterium. Int. J. Syst. Evol. Microbiol. 2001, 51, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Van Trappen, S.; Mergaert, J.; Swings, J. Loktanella salsilacus gen. nov., sp. nov., Loktanella fryxellensis sp. nov. and Loktanella vestfoldensis sp. nov., new members of the Rhodobacter group, isolated from microbial mats in Antarctic lakes. Int. J. Syst. Evol. Microbiol. 2004, 54 Pt 4, 1263–1269. [Google Scholar] [CrossRef] [PubMed]
- Kang, I.; Vergin, K.L.; Oh, H.M.; Choi, A.; Giovannoni, S.J.; Cho, J.C. Genome sequence of strain HTCC2083, a novel member of the marine clade Roseobacter. J. Bacteriol. 2011, 193, 319–320. [Google Scholar] [CrossRef] [PubMed]
- Vollmers, J.; Voget, S.; Dietrich, S.; Gollnow, K.; Smits, M.; Meyer, K.; Brinkhoff, T.; Simon, M.; Daniel, R. Poles Apart: Arctic and Antarctic Octadecabacter strains share high genome plasticity and a new type of xanthorhodopsin. PLoS ONE 2013, 8, e63422. [Google Scholar] [CrossRef] [PubMed]
- Jannaschia sp. Strain CCS1 Genome Page. Available online: http://genome.jgi-psf.org/jan_c/jan_c.home.html (accessed on 28 June 2015).
- Oh, H.M.; Kwon, K.K.; Kang, I.; Kang, S.G.; Lee, J.H.; Kim, S.J.; Cho, J.C. Complete genome sequence of “Candidatus Puniceispirillum marinum” IMCC1322, a representative of the SAR116 clade in the Alphaproteobacteria. J. Bacteriol. 2010, 192, 3240–3241. [Google Scholar] [CrossRef] [PubMed]
- Distel, D.L.; Morrill, W.; MacLaren-Toussaint, N.; Franks, D.; Waterbury, J. Teredinibacter turnerae gen. nov., sp. nov., a dinitrogen-fixing, cellulolytic, endosymbiotic gamma-proteobacterium isolated from the gills of wood-boring molluscs (Bivalvia: Teredinidae). Int. J. Syst. Evol. Microbiol. 2002, 52, 2261–2269. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.C.; Madupu, R.; Durkin, A.S.; Ekborg, N.A.; Pedamallu, C.S.; Hostetler, J.B.; Radune, D.; Toms, B.S.; Henrissat, B.; Coutinho, P.M.; Schwarz, S. The complete genome of Teredinibacter turnerae T7901: An intracellular endosymbiont of marine wood-boring bivalves (Shipworms). PLoS ONE 2009, 4, e6085. [Google Scholar] [CrossRef] [PubMed]
- Pucciarelli, S.; Devaraj, R.R.; Mancini, A.; Ballarini, P.; Castelli, M.; Schrallhammer, M.; Petroni, G.; Miceli, C. Microbial consortium associated with the antarctic marine ciliate Euplotes focardii: An investigation from genomic sequences. Microb. Ecol. 2015, 70, 484–497. [Google Scholar] [CrossRef] [PubMed]
- Béja, O.; Aravind, L.; Koonin, E.V.; Suzuki, M.T.; Hadd, A.; Nguyen, L.P.; Jovanovich, S.B.; Gates, C.M.; Feldman, R.A.; Spudich, J.L.; Spudich, E.N. Bacterial rhodopsin: Evidence for a new type of phototrophy in the sea. Science 2000, 289, 1902–1906. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dickinson, I.; Goodall-Copestake, W.; Thorne, M.A.S.; Schlitt, T.; Ávila-Jiménez, M.L.; Pearce, D.A. Extremophiles in an Antarctic Marine Ecosystem. Microorganisms 2016, 4, 8. https://doi.org/10.3390/microorganisms4010008
Dickinson I, Goodall-Copestake W, Thorne MAS, Schlitt T, Ávila-Jiménez ML, Pearce DA. Extremophiles in an Antarctic Marine Ecosystem. Microorganisms. 2016; 4(1):8. https://doi.org/10.3390/microorganisms4010008
Chicago/Turabian StyleDickinson, Iain, William Goodall-Copestake, Michael A.S. Thorne, Thomas Schlitt, Maria L. Ávila-Jiménez, and David A. Pearce. 2016. "Extremophiles in an Antarctic Marine Ecosystem" Microorganisms 4, no. 1: 8. https://doi.org/10.3390/microorganisms4010008