
Genomics of Methylotrophy in Gram-Positive Methylamine-Utilizing Bacteria

Abstract
:1. Introduction
2. Materials and Methods
2.1. Permissions
2.2. Strain Isolation and Cultivation
2.3. DNA Isolation, Whole Genome Sequencing, Assembly and Genome Annotation

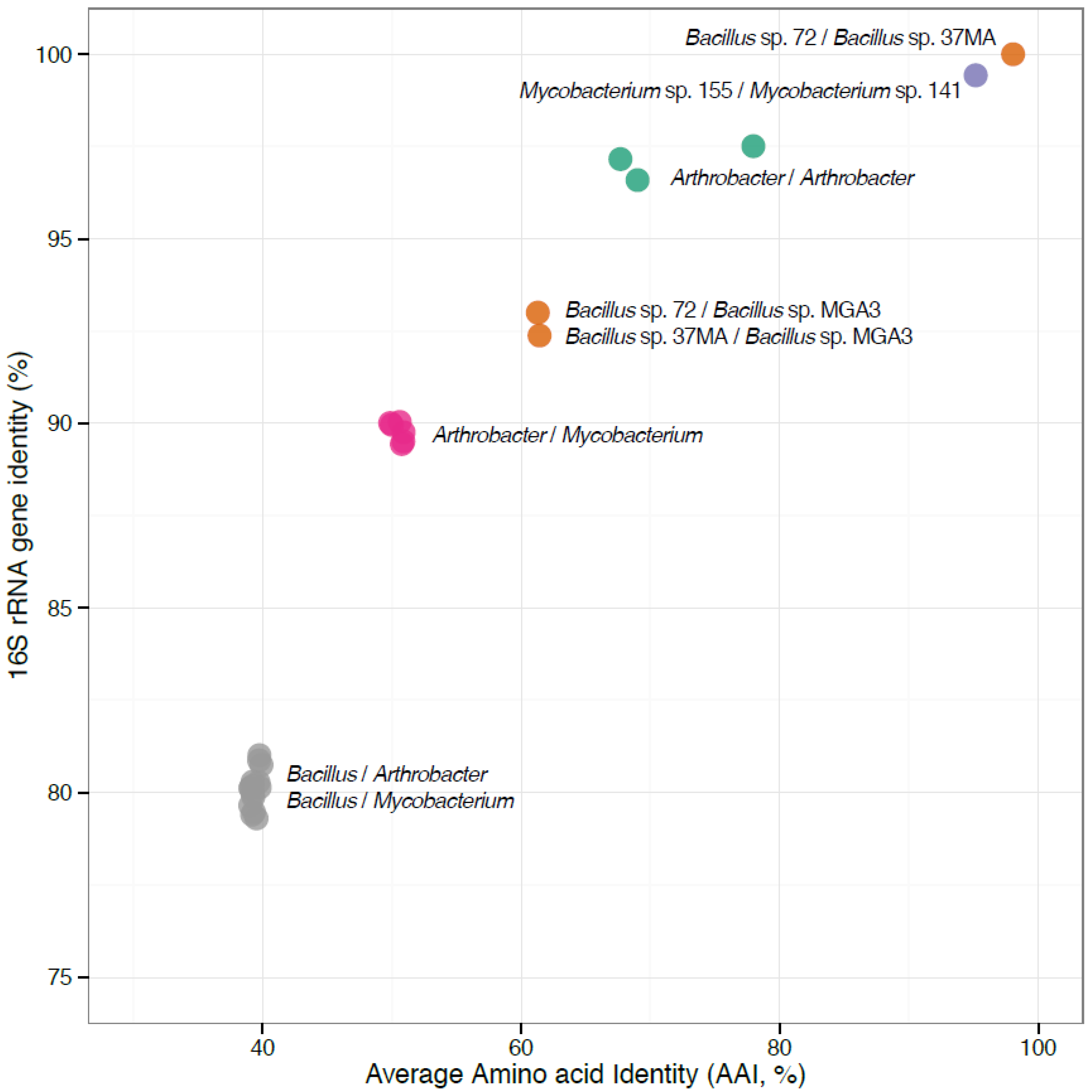
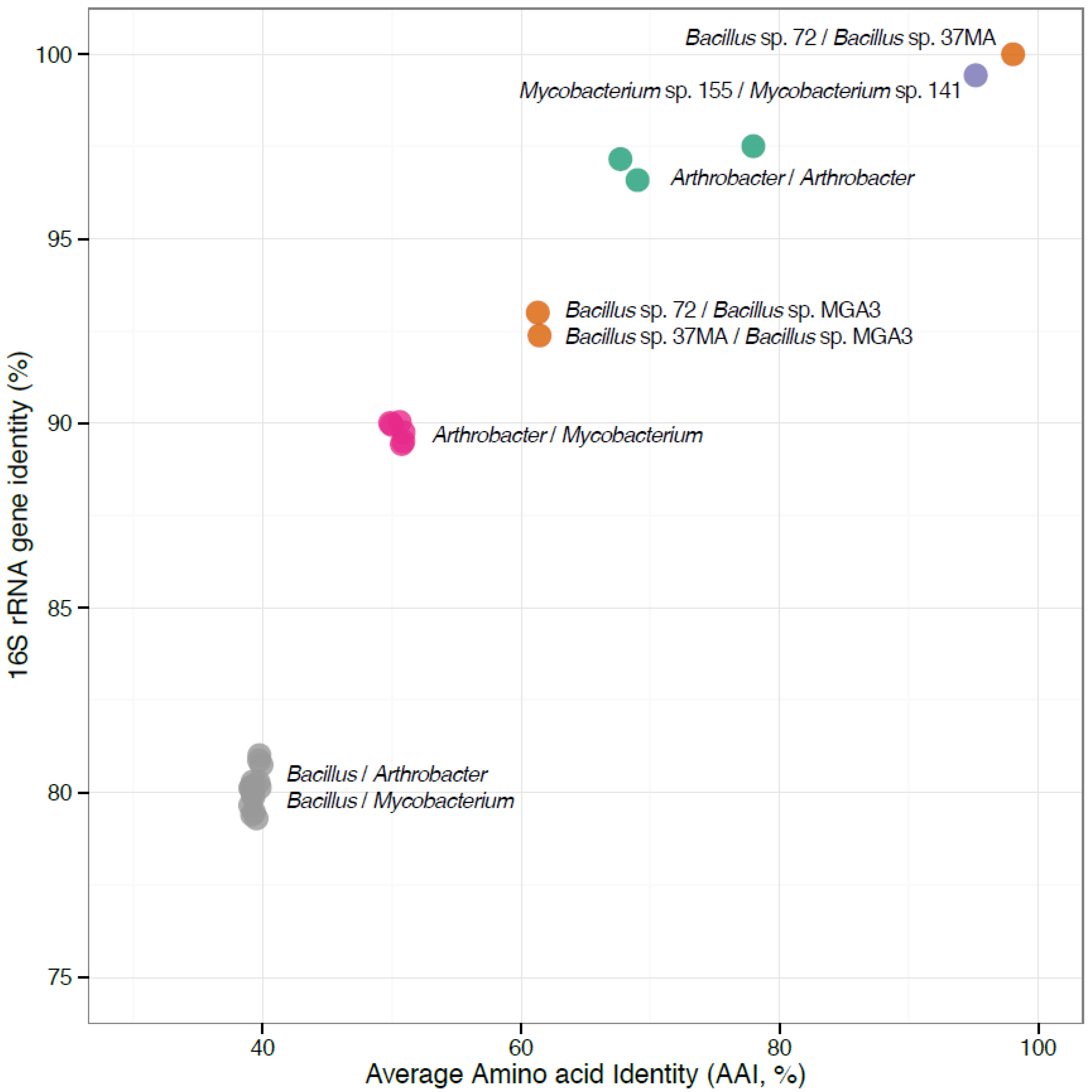{kind=link}
{kind=link}
| Strain | Year Isolated | Enrichment Temperature °C | Total Nucleotides | GC% | Sequencing Technology | Scaffolds | Coverage (X) | NCBI Accession Number |
|---|---|---|---|---|---|---|---|---|
| Arthrobacter sp. 31Y | 2011 | 10 | 5,079,550 | 61.94 | PacBio | 2 | 140 | JAFW00000000.1 |
| Arthrobacter sp. 35W | 2011 | 10 | 4,660,196 | 66.71 | Illumina/PacBio | 4 | 1647/130 | AXVQ00000000.1 |
| Arthrobacter sp. MA-N2 | 2004 | Room | 4,833,792 | 62.96 | Illumina | 5 | 793 | AQRI01000000.1 |
| Bacillus sp. 37MA | 2011 | 10 | 3,981,584 | 40.52 | Illumina | 5 | 1091 | ARCN01000000.1 |
| Bacillus sp. 72 | 2011 | 10 | 3,908,751 | 40.85 | PacBio | 26 | 180 | JQMI01000000.1 |
| Mycobacterium sp. 141 | 2011 | 30 | 4,544,736 | 65.57 | Illumina | 3 | 1171 | ARNS01000000.1 |
| Mycobacterium sp. 155 | 2011 | 30 | 4,609,894 | 65.62 | Illumina | 2 | 1451 | AREU01000000.1 |
2.4. Phylogenetic Analysis
2.5. Reconstruction of Methylotrophy Pathways
3. Results and Discussion
3.1. Gram-Positive Methylotrophs Isolated from Lake Washington Are All Facultative Methylated Amine Utilizers
3.2. The Newly-Sequenced Genomes Represent a Diversity of Gram-Positive Methylotrophs
3.3. Methylotrophy Pathways Deduced from the Novel Genomes Agree with Prior Knowledge, but Suggest Novel Primary Oxidation Modules
3.3.1. Primary C1 Oxidation Modules
| Arthrobacter sp. 31Y | Arthrobacter sp. 35W | Arthrobacter sp. M-N2 | Bacillus sp. 37MA | Bacillus sp. 72 | Mycobacterium sp. 141 | Mycobacterium sp. 155 | |
|---|---|---|---|---|---|---|---|
| Tmo | 1611 | 3598 | 1313, 1316 | - | - | 2303 | 3391 |
| Tmd | 2725 | 3579 | 1325 | - | - | 2887 | 3922 |
| Mao | 2723, 2743 | 3577, 3611 | 1074, 1322/1323, 3581, 4240, 4249 | 1646, 4043, 4264/4265 | 0038, 0528, 3848 | 2885 (partial) | 3920 (partial) |
| EutQ * | 1610, 1786, 2727, 2952 | 1944, 3581, 3597 | 1314, 1315, 1327 | 2690, 2768, 4276, 4302 | 0506, 0538 | 2304, 2889 | 1719, 3392, 3929 |
| Gma | - | - | - | - | - | 2874 | 3910 |
| MgsA | - | - | - | - | - | 2875 | 3911 |
| MgsB | - | - | - | - | - | 2876 | 3912 |
| MgsC | - | - | - | - | - | 2877 | 3913 |
| MgdA | 2010 | - | 1962 | - | - | 1509 | 2645 |
| MgdB | 2011 | - | 1963 | - | - | 1510 | 2646 |
| MgdC | 2012 | - | 1964 | - | - | 1511 | 2647 |
| MgdD | 2013 | - | 1965 | - | - | 1512 | 2648 |
| FolD1 | 3659 | 0111 | 2114 | 2264 | 1211 | 1451 | 2593 |
| FolD2 | 2859 | - | - | - | - | - | - |
| Mch | 2860 | - | - | - | - | - | - |
| PurU1 | 3656 | 0108 | 2111 | 3324 | 2413 | 1098, 2609 | 2235, 3667 |
| PurU2 | 2015 | - | 1967 | - | - | - | - |
| PurU3 | 2862 | - | - | - | - | - | - |
| Fhs | 0972 | - | 0487 | - | - | - | - |
| FaDH | 2857 | - | - | - | - | 2016 | - |
| Fdh1A | - | - | 3202 | - | - | 1890 | 3030/3031 |
| Fdh1B | - | - | 3201 | - | - | 1889 | 3029 |
| dh1C | - | - | 3200 | - | - | 1888 | 3028 |
| Fdh2 | - | - | - | 3013 | 1944 | - | - |
| Fdh3 | 3415 | - | 1898 | - | - | - | - |
| Fdh4 | 2858 | 0024 | - | - | - | - | - |
| Fdh5 | - | - | 3418 | - | - | - | - |
| Pgi1 | 4707 | 2121 | 1329 | - | - | - | - |
| Pgi2 | - | - | - | 2633 | 1591 | - | - |
| Pgi3 | - | - | - | - | - | 1659, 1660 | 2797, 2798 |
| Zwf1 | 2734, 4706 | 1501, 3590, -4201 | 1079, 1690 | - | - | 0485 | 1591 |
| Zwf2 | 0544 | 2030 | 0009 | - | - | - | - |
| Zwf3 | - | - | - | - | - | 0102, 3386 | 0013 |
| Zwf4 | - | - | - | 2231 | 1177 | - | - |
| Zwf5 | - | - | - | 4315 | 0509 | - | - |
| OpcA * | 2735, 4705 | 1502, 3591, 4202 | 1078, 1330/1331, 1691, 3346, 3572/3573 | - | - | 0486 | 1592 |
| Pgl | 4704 | 1503 | 3347, 3572 | 0487 | 1593 | 3368, 3220 | 2456,2308 |
| Gnd1 | 0504 | 1989, 3592 | 1692, 3535 | 2234, 3220 | 1184, 2308 | ||
| Gnd2 | 2377 | 3418 | 1072, 4056 | - | - | 0096, 2519 | 3586 |
| Gnd3 | - | - | - | 4314 | 0510 | - | - |
| Hps | 2720, 2732 | 2137, 3588 | 1082, 1667, 1687 | 2007, 2010, 4272/4273, 4303, 4317 | 0507, 0521, 0535, 2219, 2222 | 2900 | 3939 |
| Hpi | 2731 | 2136, 3587 | 1083, 1668, 1686 | 4274 | 0522, 0536 | 3940, 3946 | 2901, 2907 |
| Pfk1 | 1057, 2718 | 2691, 3605 | 0573, 1061, 4242, 4265 | 4309 | 0515 | 2904 | 3943 |
| Pfk2 | - | - | - | 2525 | 1481 | 0976 | 2045 |
| Pfk3 | 4492 | - | - | - | - | - | - |
| Fba | 2746, 2929 | 3604, 3822 | 1066, 1250 | 3273, 4307 | 0517, 2360 | 1980, 2894 | 3125, 3933 |
| Tkt | 2737, 4709 | 1498, 3600 | 1070, 1091, 3342 | 1128, 4308 | 0372, 0576 | 0483, 2898 | 1589, 3937 |
| Tal | 2738, 4708 | 1499, 3601 | 1069, 1090, 3343 | 3279 | 2359 | 0484, 2897 | 1590, 3936 |
| GlpX1 | 3729 | 0180 | 2184 | 3270 | 2355 | 3603 | 0231 |
| GlpX2 | - | - | - | 3354, 4306 | 0578, 2442 | - | - |
| Rpe | 2739, 4222 | 0804, 3602 | 1068, 1089, 2663 | 4305 | 0519 | 2896 | 3935 |
| Rpi1 | 0371, 2740 | 1824, 3603 | 1067, 1088, 3504 | 4310 | 0514 | 2895 | 3934 |
| Rpi2 | 2802, 3510 | 1140 | 0961 | 3357 | 2445 | - | - |
| Tpi1 | 4701 | 1505 | 3370 | 2917 | 1868 | 0491 | 1597 |
| Tpi2 | 2801 | 1141 | - | - | - | - | - |
| Tpi3 | 3515 | - | - | - | - | - | - |
| Gap1 | 4699 | 1507 | 3352 | 2512, 2919 | 1468, 1870 | 0493 | 1599 |
| Gap2 | 0418, 2719 | 1975, 3607 | 1060, 3526, 4241 | - | - | - | - |
| Pgk | 4700 | 1506 | 3351 | 2918 | 1869 | 0492 | 1598 |
| Eda | 2236 | - | 4368, 4405 | - | - | - | - |
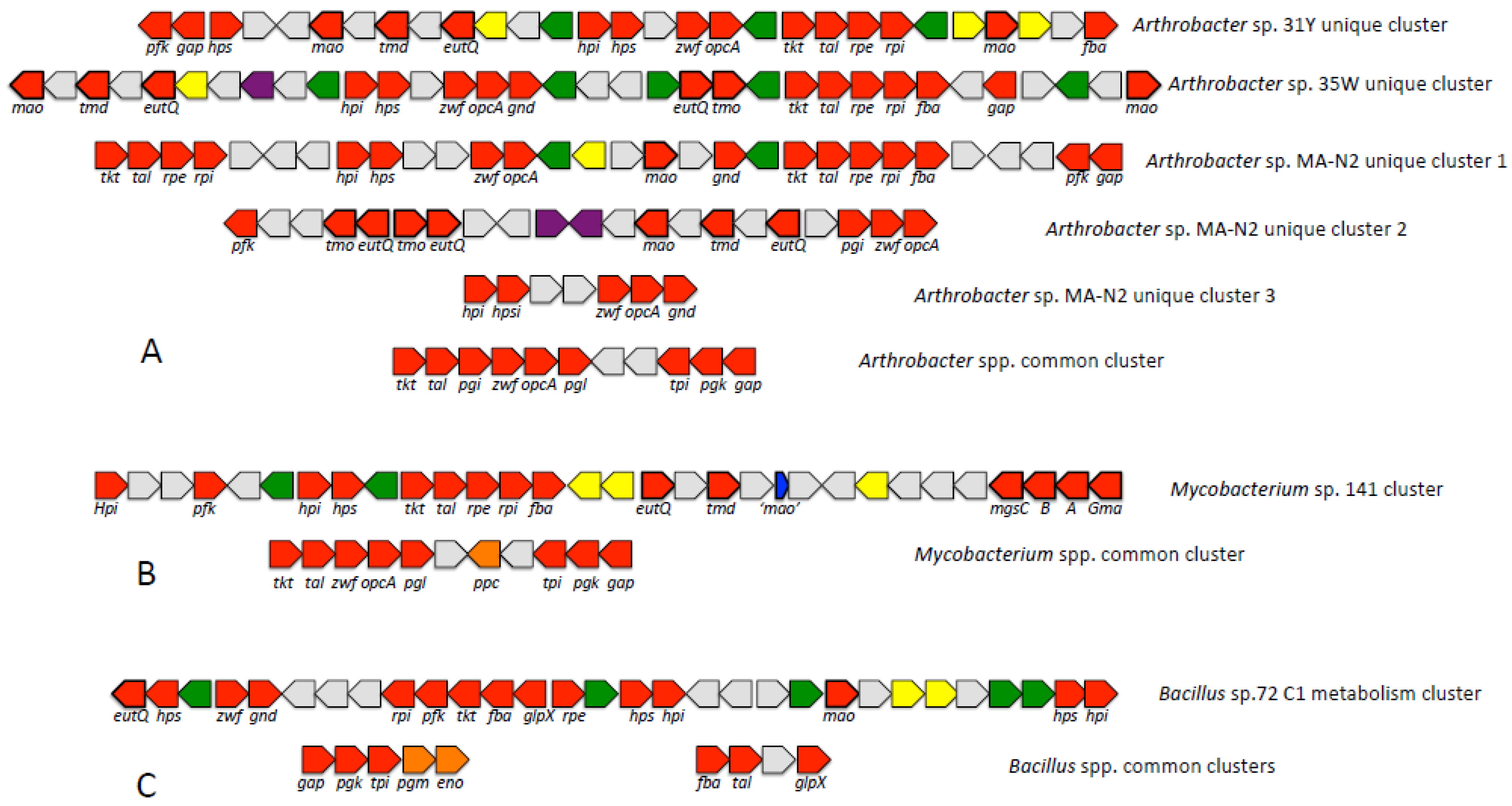
3.3.2. Potential for Oxidation of Formaldehyde to CO2 via Linear Pathways
3.3.3. Oxidation of Formaldehyde to CO2 by the Cyclic Pathway
3.3.4. The Assimilatory RuMP Cycle
3.4. Analysis of Distinct C1 Gene Clusters Suggests a Means for the Evolution of Methylotrophy in Gram-Positive Methylotrophs
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Anthony, C. The Biochemistry of Methylotrophs; Academic Press: London, UK, 1982. [Google Scholar]
- Chistoserdova, L.; Lidstrom, M.E. Aerobic methylotrophic prokaryotes. In The Prokaryotes, 4th ed.; Rosenberg, E., DeLong, E.F., Thompson, F., Lory, S., Stackebrandt, E., Eds.; Springer: Berlin Heidelberg, Germany, 2013; pp. 267–285. [Google Scholar]
- Ward, N.; Larsen, Ø.; Sakwa, J.; Bruseth, L.; Khouri, H.; Durkin, A.S.; Dimitrov, G.; Jiang, L.; Scanlan, D.; Kang, K.H.; et al. Genomic insights into methanotrophy: The complete genome sequence of Methylococcus capsulatus (Bath). PLoS Biol. 2004, 2, e303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, S.; Makarova, K.S.; Saw, J.H.; Senin, P.; Ly, B.V.; Zhou, Z.; Ren, Y.; Wang, J.; Galperin, M.Y.; Omelchenko, M.V.; et al. Complete genome sequence of the extremely acidophilic methanotroph isolate V4, Methylacidiphilum infernorum, a representative of the bacterial phylum Verrucomicrobia. Biol. Direct 2008, 3, 26. [Google Scholar] [CrossRef] [PubMed]
- Khadem, A.F.; Wieczorek, A.S.; Pol, A.; Vuilleumier, S.; Harhangi, H.R.; Dunfield, P.F.; Kalyuzhnaya, M.G.; Murrell, J.C.; Francoijs, K.J.; Stunnenberg, H.G.; et al. Draft genome sequence of the volcano-inhabiting thermoacidophilic methanotroph Methylacidiphilum fumariolicum strain SolV. J. Bacteriol. 2012, 194, 3729–3730. [Google Scholar] [CrossRef] [PubMed]
- Ettwig, K.F.; Butler, M.K.; le Paslier, D.; Pelletier, E.; Mangenot, S.; Kuypers, M.M.; Schreiber, F.; Dutilh, B.E.; Zedelius, J.; de Beer, D.; et al. Nitrite-driven anaerobic methane oxidation by oxygenic bacteria. Nature 2010, 464, 543–548. [Google Scholar] [CrossRef] [PubMed]
- De Boer, L.; Brouwer, J.W.; van Hassel, C.W.; Levering, P.R.; Dijkhuizen, L. Nitrogen metabolism in the facultative methylotroph Arthrobacter P1 grown with various amines or ammonia as nitrogen sources. Antonie Van Leeuwenhoek 1989, 56, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Vonck, J.; Arfman, N.; de Vries, G.E.; van Beeumen, J.; van Bruggen, E.F.; Dijkhuizen, L. Electron microscopic analysis and biochemical characterization of a novel methanol dehydrogenase from the thermotolerant Bacillus sp. C1. J. Biol. Chem. 1991, 266, 3949–3954. [Google Scholar] [PubMed]
- De Vries, G.E.; Arfman, N.; Terpstra, P.; Dijkhuizen, L. Cloning, expression, and sequence analysis of the Bacillus methanolicus C1 methanol dehydrogenase gene. J. Bacteriol. 1992, 174, 5346–5353. [Google Scholar]
- Zhang, X.; Fuller, J.H.; McIntire, W.S. Cloning, sequencing, expression, and regulation of the structural gene for the copper/topa quinone-containing methylamine oxidase from Arthrobacter strain P1, a Gram-positive facultative methylotroph. J. Bacteriol. 1993, 175, 5617–5627. [Google Scholar] [PubMed]
- Bystrykh, L.V.; Vonck, J.; van Bruggen, E.F.; van Beeumen, J.; Samyn, B.; Govorukhina, N.I.; Arfman, N.; Duine, J.A.; Dijkhuizen, L. Electron microscopic analysis and structural characterization of novel NADP(H)-containing methanol: N,N′-dimethyl-4-nitrosoaniline oxidoreductases from the Gram-positive methylotrophic bacteria Amycolatopsis methanolica and Mycobacterium gastri MB19. J. Bacteriol. 1993, 175, 1814–1822. [Google Scholar] [PubMed]
- Park, H.; Lee, H.; Ro, Y.T.; Kim, Y.M. Identification and functional characterization of a gene for the methanol : N,N′-dimethyl-4-nitrosoaniline oxidoreductase from Mycobacterium sp. strain JC1 (DSM 3803). Microbiology 2010, 156, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Heggeset, T.M.; Krog, A.; Balzer, S.; Wentzel, A.; Ellingsen, T.E.; Brautaset, T. Genome sequence of thermotolerant Bacillus methanolicus: features and regulation related to methylotrophy and production of l-lysine and l-glutamate from methanol. Appl. Environ. Microbiol. 2012, 78, 5170–5181. [Google Scholar] [CrossRef] [PubMed]
- Irla, M.; Neshat, A.; Winkler, A.; Albersmeier, A.; Heggeset, T.M.; Brautaset, T.; Kalinowski, J.; Wendisch, V.F.; Rückert, C. Complete genome sequence of Bacillus methanolicus MGA3, a thermotolerant amino acid producing methylotroph. J. Biotechnol. 2014, 188C, 110–111. [Google Scholar] [CrossRef]
- Müller, J.E.; Heggeset, T.M.; Wendisch, V.F.; Vorholt, J.A.; Brautaset, T. Methylotrophy in the thermophilic Bacillus methanolicus, basic insights and application for commodity production from methanol. Appl. Microbiol. Biotechnol. 2015, 99, 535–551. [Google Scholar] [CrossRef] [PubMed]
- Colby, J.; Zatman, L.J. Enzymological aspects of the pathways for trimethylamine oxidation and C1 assimilation of obligate methylotrophs and restricted facultative methylotrophs. Biochem. J. 1975, 148, 513–520. [Google Scholar] [PubMed]
- Boulton, C.A.; Large, P.J. Inactivation of trimethylamine N-oxide aldolase (demethylase) during preparation of bacterial extracts. FEMS Microbiol. Lett. 1979, 5, 159–162. [Google Scholar] [CrossRef]
- Chen, Y.; Patel, N.A.; Crombie, A.; Scrivens, J.H.; Murrell, J.C. Bacterial flavin-containing monooxygenase is trimethylamine monooxygenase. Proc. Natl. Acad. Sci. USA 2011, 108, 17791–17796. [Google Scholar] [CrossRef] [PubMed]
- Kalyuzhnaya, M.G.; Lidstrom, M.E.; Chistoserdova, L. Utility of environmental probes targeting ancient enzymes: Methylotroph detection in Lake Washington. Microb. Ecol. 2004, 48, 436–472. [Google Scholar] [CrossRef]
- Beck, D.A.C.; McTaggart, T.L.; Setboonsarng, U.; Vorobev, A.; Kalyuzhnaya, M.G.; Ivanova, N.; Goodwin, L.; Woyke, T.; Lidstrom, M.E.; Chistoserdova, L. The expanded diversity of Methylophilaceae from Lake Washington through cultivation and genomic sequencing of novel ecotypes. PLoS One 2014, 9, e102458. [Google Scholar] [CrossRef] [PubMed]
- Beck, D.A.C.; McTaggart, T.L.; Setboonsarng, U.; Vorobev, A.; Kalyuzhnaya, M.G.; Goodwin, L.; Shapiro, N.; Woyke, T.; Lidstrom, M.E.; Chistoserdova, L. Multiphyletic origins of methylotrophy in Alphaproteobacteria, exemplified by comparative genomics of Lake Washington isolates. Environ. Microbiol. 2015, 17, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Kalyuzhnaya, M.G.; Bowerman, S.; Lara, J.C.; Lidstrom, M.E.; Chistoserdova, L. Methylotenera mobilis gen. nov., sp. nov., an obligately methylamine-utilizing bacterium within the family Methylophilaceae. Int. J. Syst. Evol. Microbiol. 2006, 56, 2819–2823. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.; Alexander, D.; Marks, P.; Klammer, A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.; et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Gnerre, S.; MacCallum, I.; Przybylski, D.; Ribeiro, F.J.; Burton, J.N.; Walker, B.J.; Sharpe, T.; Hall, G.; Shea, T.P.; Sykes, S.; et al. High-quality draft assemblies of mammalian genomes from massively parallel sequence data. Proc. Natl. Acad. Sci. USA 2011, 108, 1513–1518. [Google Scholar] [CrossRef] [PubMed]
- Zerbino, D.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.L.; Lacascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Pati, A.; Ivanova, N.N.; Mikhailova, N.; Ovchinnikova, G.; Hooper, S.D.; Lykidis, A.; Kyrpides, N.C. GenePRIMP: A gene prediction improvement pipeline for prokaryotic genomes. Nat. Methods 2010, 7, 455–457. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucl. Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.M.; Ludwig, W.; Peplies, J.; Glckner, F.O. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucl. Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef]
- INFERNAL. Inference of RNA Alignments. Available online: http://infernal.janelia.org (accessed on 1 April 2014).
- Markowitz, V.M.; Chen, I.M.; Palaniappan, K.; Chu, K.; Szeto, E.; Pillay, M.; Ratner, A.; Huang, J.; Woyke, T.; Huntemann, M.; et al. IMG 4 version of the integrated microbial genomes comparative analysis system. Nucl. Acids Res. 2014, 42, D560–D567. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidis, K.T.; Tiedje, J.M. Towards a genome-based taxonomy for prokaryotes. J. Bacteriol. 2005, 187, 6258–6264. [Google Scholar] [CrossRef] [PubMed]
- Summers, M.L.; Wallis, J.G.; Campbell, E.L.; Meeks, J.C. Genetic evidence of a major role for glucose-6-phosphate dehydrogenase in nitrogen fixation and dark growth of the cyanobacterium Nostoc sp. strain ATCC 29133. J. Bacteriol. 1995, 177, 6184–6194. [Google Scholar] [PubMed]
- Latypova, E.; Yang, S.; Wang, Y.S.; Wang, T.; Chavkin, T.A.; Hackett, M.; Schäfer, H.; Kalyuzhnaya, M.G. Genetics of the glutamate-mediated methylamine utilization pathway in the facultative methylotrophic beta-proteobacterium Methyloversatilis universalis FAM5. Mol. Microbiol. 2010, 75, 426–439. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Scanlan, J.; Song, L.; Crombie, A.; Rahman, M.T.; Schäfer, H.; Murrell, J.C. γ-glutamylmethylamide is an essential intermediate in the metabolism of methylamine by Methylocella silvestris. Appl. Environ. Microbiol. 2010, 76, 4530–4537. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; McAleer, K.L.; Murrell, J.C. Monomethylamine as a nitrogen source for a nonmethylotrophic bacterium, Agrobacterium tumefaciens. Appl. Environ. Microbiol. 2010, 76, 4102–4104. [Google Scholar] [CrossRef] [PubMed]
- Parkin, K.L.; Hultinj, H.O. Characterization of trimethylamine-N-oxide (TMAO) demethylase activity from fish muscle microsomes. Biochemistry 1986, 100, 77–86. [Google Scholar]
- Lidbury, I.; Murrell, J.C.; Chen, Y. Trimethylamine N-oxide metabolism by abundant marine heterotrophic bacteria. Proc. Natl. Acad. Sci. USA 2014, 111, 2710–2740. [Google Scholar] [CrossRef] [PubMed]
- Chistoserdova, L. Modularity of methylotrophy, revisited. Environ. Microbiol. 2011, 13, 2603–2622. [Google Scholar] [CrossRef] [PubMed]
- Peyraud, R.; Schneider, K.; Kiefer, P.; Massou, S.; Vorholt, J.A.; Portais, J.C. Genome-scale reconstruction and system level investigation of the metabolic network of Methylobacterium extorquens AM1. BMC Syst. Biol. 2011, 5, 189. [Google Scholar] [CrossRef] [PubMed]
- Kalyuzhnaya, M.G.; Lidstrom, M.E. QscR-mediated transcriptional activation of serine cycle genes in Methylobacterium extorquens AM1. J. Bacteriol. 2005, 187, 7511–7517. [Google Scholar] [CrossRef] [PubMed]
- Kato, N.; Yurimoto, H.; Thauer, R.K. The physiological role of the ribulose monophosphate pathway in bacteria and archaea. Biosci. Biotechnol. Biochem. 2006, 70, 10–21. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McTaggart, T.L.; Beck, D.A.C.; Setboonsarng, U.; Shapiro, N.; Woyke, T.; Lidstrom, M.E.; Kalyuzhnaya, M.G.; Chistoserdova, L. Genomics of Methylotrophy in Gram-Positive Methylamine-Utilizing Bacteria. Microorganisms 2015, 3, 94-112. https://doi.org/10.3390/microorganisms3010094
McTaggart TL, Beck DAC, Setboonsarng U, Shapiro N, Woyke T, Lidstrom ME, Kalyuzhnaya MG, Chistoserdova L. Genomics of Methylotrophy in Gram-Positive Methylamine-Utilizing Bacteria. Microorganisms. 2015; 3(1):94-112. https://doi.org/10.3390/microorganisms3010094
Chicago/Turabian StyleMcTaggart, Tami L., David A. C. Beck, Usanisa Setboonsarng, Nicole Shapiro, Tanja Woyke, Mary E. Lidstrom, Marina G. Kalyuzhnaya, and Ludmila Chistoserdova. 2015. "Genomics of Methylotrophy in Gram-Positive Methylamine-Utilizing Bacteria" Microorganisms 3, no. 1: 94-112. https://doi.org/10.3390/microorganisms3010094