
The Role of Autophagy-Related Proteins in Candida albicans Infections

 ,
,
Abstract
:1. Introduction
2. Fungal Recognition by Dectin-1
3. Autophagy
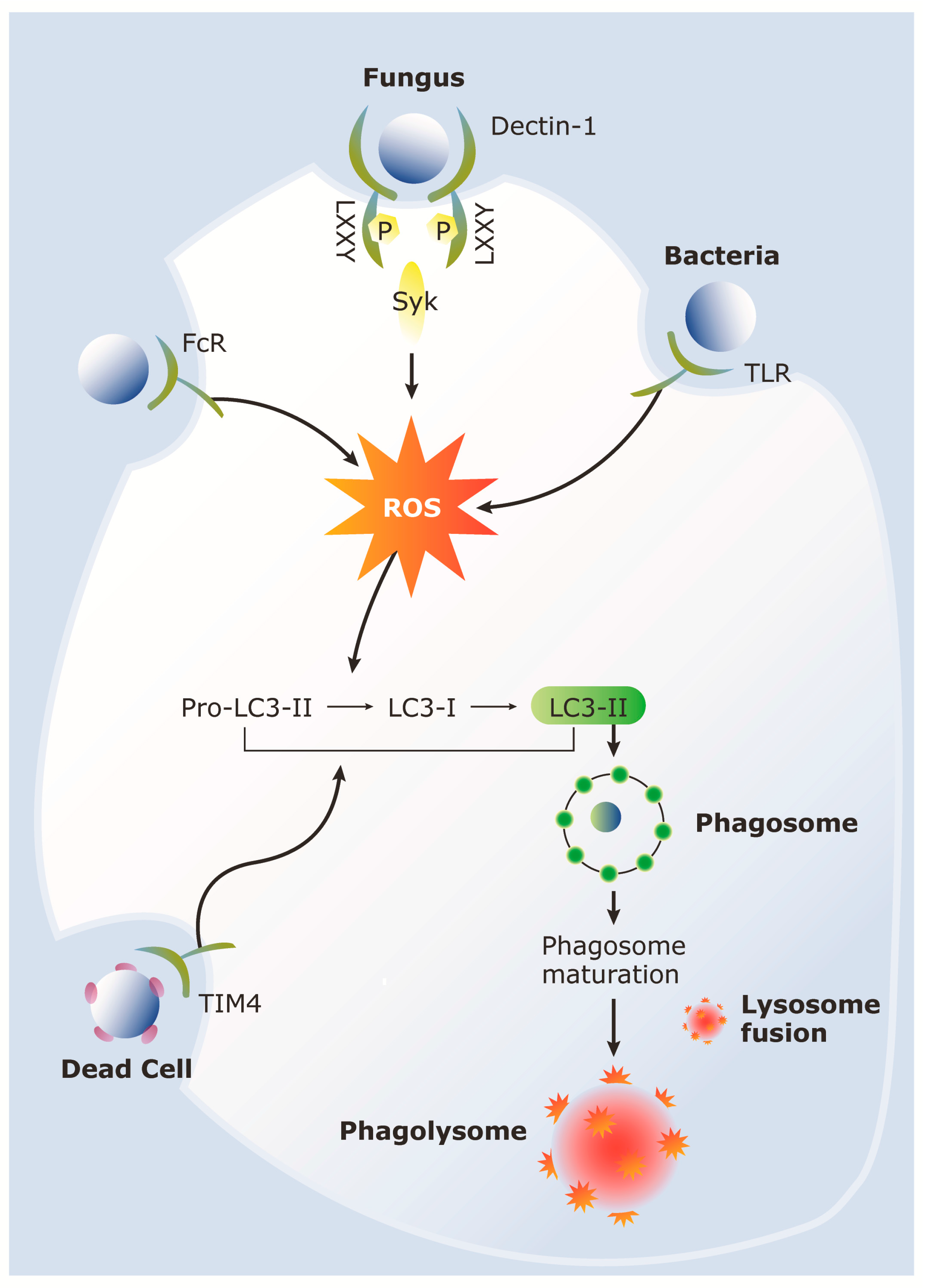
4. LC3-Associated Phagocytosis (LAP)
5. Role of LAP during Host Response to C. albicans
6. Role of Autophagy Proteins in Aspergillus Infections
7. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| LC3 | Microtubule-associated light chain 3 |
| LAP | LC3-associated phagocytosis |
| Syk | Spleen tyrosine kinase |
| Atg | Autophagy-related gene |
| APC | Antigen presenting cell |
| DC | dendritic cell |
| PAMP | Pathogen associated molecular pattern |
| CLR | C-type lectin receptor |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| NFAT | Nuclear factor of activated T cells |
| NLR | Nod-like receptor |
| TLR | Toll-like receptor |
| PRR | Pattern recognition receptor |
| ITAM | Immunoreceptor tyrosine-activation motif |
| ULK1 | Unc-51-like kinase1 |
| ER | Endoplasmic reticulum |
| ROS | Reactive oxygen species |
| PI3K | Phosphatidylinositol-3-OH kinase |
| Rubicon | RUN domain protein as Beclin-1 interacting and cysteine-rich containing |
| mTOR | Mammalian target of rapamycin |
| MHC | Major histocompatibility complex |
| SNP | Single nucleotide polymorphism |
| CGD | Chronic granulomatous disease |
References
- Pfaller, M.A.; Diekema, D.J. Epidemiology of invasive candidiasis: A persistent public health problem. Clin. Microbiol. Rev. 2007, 20, 133–163. [Google Scholar] [CrossRef] [PubMed]
- Gudlaugsson, O.; Gillespie, S.; Lee, K.; Vande Berg, J.; Hu, J.; Messer, S.; Herwaldt, L.; Pfaller, M.; Diekema, D. Attributable mortality of nosocomial candidemia, revisited. Clin. Infect. Dis. 2003, 37, 1172–1177. [Google Scholar] [CrossRef] [PubMed]
- Stuart, L.M.; Ezekowitz, R.A. Phagocytosis and comparative innate immunity: Learning on the fly. Nat. Rev. Immunol. 2008, 8, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Vyas, J.; Van Der Veen, A.; Ploegh, H. The known unknowns of antigen processing and presentation. Nat. Rev. Immunol. 2008, 8, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Kanayama, M.; Inoue, M.; Danzaki, K.; Hammer, G.; He, Y.-W.; Shinohara, M.L. Autophagy enhances NFκB activity in specific tissue macrophages by sequestering A20 to boost antifungal immunity. Nat. Commun. 2015, 6, 5779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Becker, C.; Lowell, C.A.; Underhill, D.M. Dectin-1 triggered recruitment of LC3 to phagosomes facilitates MHC class II presentation of fungal-derived antigens. J. Biol. Chem. 2012, 287, 34149–34156. [Google Scholar] [CrossRef] [PubMed]
- Rosentul, D.C.; Plantinga, T.S.; Farcas, M.; Oosting, M.; Hamza, O.J.M.; Scott, W.K.; Alexander, B.D.; Yang, J.C.; Laird, G.M.; Joosten, L.A.B.; et al. Role of autophagy genetic variants for the risk of Candida infections. Med. Mycol. 2014, 52, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Smeekens, S.P.; Malireddi, R.K.; Plantinga, T.S.; Buffen, K.; Oosting, M.; Joosten, L.A.B.; Kullberg, B.J.; Perfect, J.R.; Scott, W.K.; Van De Veerdonk, F.L.; et al. Autophagy is redundant for the host defense against systemic Candida albicans infections. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Tam, J.M.; Mansour, M.K.; Khan, N.S.; Seward, M.; Puranam, S.; Tanne, A.; Sokolovska, A.; Becker, C.E.; Acharya, M.; Baird, M.A.; et al. Dectin-1-Dependent LC3 Recruitment to Phagosomes Enhances Fungicidal Activity in Macrophages. J. Infect. Dis. 2014, 210, 1844–1854. [Google Scholar] [CrossRef] [PubMed]
- Kyrmizi, I.; Gresnigt, M.S.; Akoumianaki, T.; Samonis, G.; Sidiropoulos, P.; Boumpas, D.; Netea, M.G.; Van De Veerdonk, F.L.; Kontoyiannis, D.P.; Chamilos, G. Corticosteroids block autophagy protein recruitment in Aspergillus fumigatus phagosomes via targeting dectin-1/Syk kinase signaling. J. Immunol. 2013, 191, 1287–1299. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Smeekens, S.P.; Casagrande, A.; Iannitti, R.; Conway, K.L.; Gresnigt, M.S.; Begun, J.; Plantinga, T.S.; Joosten, L.A.B.; Van Der Meer, J.W.M.; et al. IL-1 receptor blockade restores autophagy and reduces inflammation in chronic granulomatous disease in mice and in humans. Proc. Natl. Acad. Sci. USA 2014, 111, 3526–3531. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.; Malireddi, R.K.S.; Lu, Q.; Cunha, L.D.; Pelletier, S.; Gingras, S.; Orchard, R.; Guan, J.-L.; Tan, H.; Peng, J.; et al. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat. Cell Biol. 2015, 17, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Van De Veerdonk, F.L.; Dinarello, C.A. Deficient autophagy unravels the ROS paradox in chronic granulomatous disease. Autophagy 2014, 10, 1141–1142. [Google Scholar] [CrossRef] [PubMed]
- Nicola, A.M.; Albuquerque, P.; Martinez, L.R.; Dal-Rosso, R.A.; Saylor, C.; De Jesus, M.; Nosanchuk, J.D.; Casadevall, A. Macrophage Autophagy in Immunity to Cryptococcus neoformans and Candida albicans. Infect. Immun. 2012, 80, 3065–3076. [Google Scholar] [CrossRef] [PubMed]
- Lai, S.-C.; Devenish, R.J. LC3-Associated Phagocytosis (LAP): Connections with Host Autophagy. Cells 2012, 1, 396–408. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Underhill, D.M. β-Glucan signaling connects phagocytosis to autophagy. Glycobiology 2013, 23, 1047–1051. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.D. Dectin-1: A signalling non-TLR pattern-recognition receptor. Nat. Rev. Immunol. 2006, 6, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Tsoni, S.V.; Brown, G.D. beta-Glucans and dectin-1. Ann. N. Y. Acad. Sci. 2008, 1143, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Riggi, S.J.; Di Luzio, N.R. Identification of a reticuloendothelial stimulating agent in zymosan. Am. J. Physiol. 1961, 200, 297–300. [Google Scholar] [PubMed]
- Cunha, C.; Di Ianni, M.; Bozza, S.; Giovannini, G.; Zagarella, S.; Zelante, T.; D’angelo, C.; Pierini, A.; Pitzurra, L.; Falzetti, F.; et al. Dectin-1 Y238X polymorphism associates with susceptibility to invasive aspergillosis in hematopoietic transplantation through impairment of both recipient- and donor-dependent mechanisms of antifungal immunity. Blood 2010, 116, 5394–5402. [Google Scholar] [CrossRef] [PubMed]
- Ferwerda, B.; Ferwerda, G.; Plantinga, T.S.; Willment, J.A.; Van Spriel, A.B.; Venselaar, H.; Elbers, C.C.; Johnson, M.D.; Cambi, A.; Huysamen, C.; et al. Human dectin-1 deficiency and mucocutaneous fungal infections. N. Engl. J. Med. 2009, 361, 1760–1767. [Google Scholar] [CrossRef] [PubMed]
- Iliev, I.D.; Funari, V.A.; Taylor, K.D.; Nguyen, Q.; Reyes, C.N.; Strom, S.P.; Brown, J.; Becker, C.A.; Fleshner, P.R.; Dubinsky, M.; et al. Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science 2012, 336, 1314–1317. [Google Scholar] [CrossRef] [PubMed]
- Ariizumi, K.; Shen, G.L.; Shikano, S.; Xu, S.; Ritter, R.; Kumamoto, T.; Edelbaum, D.; Morita, A.; Bergstresser, P.R.; Takashima, A. Identification of a novel, dendritic cell-associated molecule, dectin-1, by subtractive cDNA cloning. J. Biol. Chem. 2000, 275, 20157–20167. [Google Scholar] [CrossRef] [PubMed]
- Rogers, N.C.; Slack, E.C.; Edwards, A.D.; Nolte, M.A.; Schulz, O.; Schweighoffer, E.; Williams, D.L.; Gordon, S.; Tybulewicz, V.L.; Brown, G.D.; et al. Syk-dependent cytokine induction by Dectin-1 reveals a novel pattern recognition pathway for C type lectins. Immunity 2005, 22, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Underhill, D.M.; Rossnagle, E.; Lowell, C.A.; Simmons, R.M. Dectin-1 activates Syk tyrosine kinase in a dynamic subset of macrophages for reactive oxygen production. Blood 2005, 106, 2543–2550. [Google Scholar] [CrossRef] [PubMed]
- Herre, J.; Marshall, A.S.J.; Caron, E.; Edwards, A.D.; Williams, D.L.; Schweighoffer, E.; Tybulewicz, V.; Reis E Sousa, C.; Gordon, S.; et al. Dectin-1 uses novel mechanisms for yeast phagocytosis in macrophages. Blood 2004, 104, 4038–4045. [Google Scholar] [CrossRef] [PubMed]
- Hara, H.; Ishihara, C.; Takeuchi, A.; Imanishi, T.; Xue, L.; Morris, S.W.; Inui, M.; Takai, T.; Shibuya, A.; Saijo, S.; et al. The adaptor protein CARD9 is essential for the activation of myeloid cells through ITAM-associated and Toll-like receptors. Nat. Immunol. 2007, 8, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Gantner, B.N.; Simmons, R.M.; Canavera, S.J.; Akira, S.; Underhill, D.M. Collaborative induction of inflammatory responses by dectin-1 and Toll-like receptor 2. J. Exp. Med. 2003, 197, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Hernanz-Falcón, P.; Joffre, O.; Williams, D.L.; Reis E Sousa, C. Internalization of Dectin-1 terminates induction of inflammatory responses. Eur. J. Immunol. 2009, 39, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Mansour, M.K.; Tam, J.M.; Khan, N.S.; Seward, M.; Davids, P.J.; Puranam, S.; Sokolovska, A.; Sykes, D.B.; Dagher, Z.; Becker, C.; et al. Dectin-1 activation controls maturation of β-1,3-glucan-containing phagosomes. J. Biol. Chem. 2013, 288, 16043–16054. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Shinohara, M.L. Clustering of pattern recognition receptors for fungal detection. PLoS Pathog. 2014, 10, e1003873. [Google Scholar] [CrossRef] [PubMed]
- Miyazato, A.; Nakamura, K.; Yamamoto, N.; Mora-Montes, H.M.; Tanaka, M.; Abe, Y.; Tanno, D.; Inden, K.; Gang, X.; Ishii, K.; et al. Toll-like receptor 9-dependent activation of myeloid dendritic cells by Deoxynucleic acids from Candida albicans. Infect. Immun. 2009, 77, 3056–3064. [Google Scholar] [CrossRef] [PubMed]
- Kasperkovitz, P.V.; Cardenas, M.L.; Vyas, J.M. TLR9 is actively recruited to Aspergillus fumigatus phagosomes and requires the N-terminal proteolytic cleavage domain for proper intracellular trafficking. J. Immunol. 2010, 185, 7614–7622. [Google Scholar] [CrossRef] [PubMed]
- Kasperkovitz, P.V.; Khan, N.S.; Tam, J.M.; Mansour, M.K.; Davids, P.J.; Vyas, J.M. Toll-like receptor 9 modulates macrophage antifungal effector function during innate recognition of Candida albicans and Saccharomyces cerevisiae. Infect. Immun. 2011, 79, 4858–4867. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.S.; Kasperkovitz, P.V.; Timmons, A.K.; Mansour, M.K.; Tam, J.M.; Seward, M.W.; Reedy, J.L.; Puranam, S.; Feliu, M.; Vyas, J.M. Dectin-1 Controls TLR9 Trafficking to Phagosomes Containing β-1,3 Glucan. J. Immunol. 2016, 196, 2249–2261. [Google Scholar] [CrossRef] [PubMed]
- Nedjic, J.; Aichinger, M.; Emmerich, J.; Mizushima, N.; Klein, L. Autophagy in thymic epithelium shapes the T-cell repertoire and is essential for tolerance. Nature 2008, 455, 396–400. [Google Scholar] [CrossRef] [PubMed]
- Paludan, C.; Schmid, D.; Landthaler, M.; Vockerodt, M.; Kube, D.; Tuschl, T.; Münz, C. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 2005, 307, 593–596. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, T.; Akira, S. Regulation of innate immune responses by autophagy-related proteins. J. Cell Biol. 2010, 189, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.-S.; Kehrl, J.H. TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci. Signal. 2010, 3, ra42–ra42. [Google Scholar] [CrossRef] [PubMed]
- Delgado, M.; Elmaoued, R.; Davis, A.; Kyei, G.; Deretic, V. Toll-like receptors control autophagy. EMBO J. 2008, 27, 1110–1121. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.-S.; Kehrl, J.H. MyD88 and Trif target Beclin 1 to trigger autophagy in macrophages. J. Biol. Chem. 2008, 283, 33175–33182. [Google Scholar] [CrossRef] [PubMed]
- Travassos, L.H.; Carneiro, L.A.M.; Ramjeet, M.; Hussey, S.; Kim, Y.-G.; Magalhães, J.G.; Yuan, L.; Soares, F.; Chea, E.; Le Bourhis, L.; et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 2010, 11, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.B.; Ornatowski, W.; Vergne, I.; Naylor, J.; Delgado, M.; Roberts, E.; Ponpuak, M.; Master, S.; Pilli, M.; White, E.; et al. Human IRGM regulates autophagy and cell-autonomous immunity functions through mitochondria. Nat. Cell Biol. 2010, 12, 1154–1165. [Google Scholar] [CrossRef] [PubMed]
- Hartman, M.L.; Kornfeld, H. Interactions between naïve and infected macrophages reduce Mycobacterium tuberculosis viability. PLoS ONE 2011, 6, e27972. [Google Scholar] [CrossRef] [PubMed]
- Mostowy, S.; Sancho-Shimizu, V.; Hamon, M.A.; Simeone, R.; Brosch, R.; Johansen, T.; Cossart, P. p62 and NDP52 proteins target intracytosolic Shigella and Listeria to different autophagy pathways. J. Biol. Chem. 2011, 286, 26987–26995. [Google Scholar] [CrossRef] [PubMed]
- Orvedahl, A.; Levine, B. Eating the enemy within: Autophagy in infectious diseases. Cell Death Differ. 2009, 16, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.G.; Master, S.S.; Singh, S.B.; Taylor, G.A.; Colombo, M.I.; Deretic, V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 2004, 119, 753–766. [Google Scholar] [CrossRef] [PubMed]
- Castillo, E.F.; Dekonenko, A.; Arko-Mensah, J.; Mandell, M.A.; Dupont, N.; Jiang, S.; Delgado-Vargas, M.; Timmins, G.S.; Bhattacharya, D.; Yang, H.; et al. Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc. Natl. Acad. Sci. USA 2012, 109, E3168–E3176. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Fux, B.; Goodwin, M.; Dunay, I.R.; Strong, D.; Miller, B.C.; Cadwell, K.; Delgado, M.A.; Ponpuak, M.; Green, K.G.; et al. Autophagosome-independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell Host Microbe 2008, 4, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Turner, M.J.; Delay, M.L.; Klenk, E.I.; Sowders, D.P.; Colbert, R.A. Endoplasmic reticulum stress and the unfolded protein response are linked to synergistic IFN-beta induction via X-box binding protein 1. Eur. J. Immunol. 2008, 38, 1194–1203. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Elazar, Z. ROS, mitochondria and the regulation of autophagy. Trends Cell Biol. 2007, 17, 422–427. [Google Scholar] [CrossRef] [PubMed]
- Acharya, M.; Sokolovska, A.; Tam, J.M.; Conway, K.L.; Stefani, C.; Raso, F.; Mukhopadhyay, S.; Paul, E.; Savill, J.; Hynes, R.O.; et al. αv Integrin-Triggered Association of Autophagy Components With Toll-like Receptors Regulates B cell Responses. Nat. Commun. 2016, 7, 10917. [Google Scholar] [CrossRef] [PubMed]
- Deretic, V.; Levine, B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe 2009, 5, 527–549. [Google Scholar] [CrossRef] [PubMed]
- Orvedahl, A.; Levine, B. Autophagy and viral neurovirulence. Cell. Microbiol. 2008, 10, 1747–1756. [Google Scholar] [CrossRef] [PubMed]
- Orvedahl, A.; Macpherson, S.; Sumpter, R.; Tallóczy, Z.; Zou, Z.; Levine, B. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe 2010, 7, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Lund, J.; Ramanathan, B.; Mizushima, N.; Iwasaki, A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 2007, 315, 1398–1401. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Kang, K.H.; Spector, S.A. Production of interferon α by human immunodeficiency virus type 1 in human plasmacytoid dendritic cells is dependent on induction of autophagy. J. Infect. Dis. 2012, 205, 1258–1267. [Google Scholar] [CrossRef] [PubMed]
- Schmid, D.; Münz, C. Innate and adaptive immunity through autophagy. Immunity 2007, 27, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Uhl, M.; Kepp, O.; Jusforgues-Saklani, H.; Vicencio, J.-M.; Kroemer, G.; Albert, M.L. Autophagy within the antigen donor cell facilitates efficient antigen cross-priming of virus-specific CD8+ T cells. Cell Death Differ. 2009, 16, 991–1005. [Google Scholar] [CrossRef] [PubMed]
- Yordy, B.; Tal, M.C.; Hayashi, K.; Arojo, O.; Iwasaki, A. Autophagy and selective deployment of Atg proteins in antiviral defense. Int. Immunol. 2013, 25, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Henault, J.; Martinez, J.; Riggs, J.M.; Tian, J.; Mehta, P.; Clarke, L.; Sasai, M.; Latz, E.; Brinkmann, M.M.; Iwasaki, A.; et al. Noncanonical Autophagy Is Required for Type I Interferon Secretion in Response to DNA-Immune Complexes. Immunity 2012, 37, 986–997. [Google Scholar] [CrossRef] [PubMed]
- Florey, O.; Kim, S.E.; Sandoval, C.P.; Haynes, C.M.; Overholtzer, M. Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nat. Cell Biol. 2011, 13, 1335–1343. [Google Scholar] [CrossRef] [PubMed]
- Sanjuan, M.A.; Dillon, C.P.; Tait, S.W.G.; Moshiach, S.; Dorsey, F.; Connell, S.; Komatsu, M.; Tanaka, K.; Cleveland, J.L.; Withoff, S.; et al. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 2007, 450, 1253–1257. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.; Almendinger, J.; Oberst, A.; Ness, R.; Dillon, C.P.; Fitzgerald, P.; Hengartner, M.O.; Green, D.R. Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc. Natl. Acad. Sci. USA 2011, 108, 17396–17401. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-Y.; Zhao, H.; Martinez, J.; Doggett, T.A.; Kolesnikov, A.V.; Tang, P.H.; Ablonczy, Z.; Chan, C.-C.; Zhou, Z.; Green, D.R.; et al. Noncanonical autophagy promotes the visual cycle. Cell 2013, 154, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Canadien, V.; Lam, G.Y.; Steinberg, B.E.; Dinauer, M.C.; Magalhaes, M.A.O.; Glogauer, M.; Grinstein, S.; Brumell, J.H. Activation of antibacterial autophagy by NADPH oxidases. Proc. Natl. Acad. Sci. USA 2009, 106, 6226–6231. [Google Scholar] [CrossRef] [PubMed]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Becker, C.; Reyes, C.; Underhill, D.M. Cutting edge: FYCO1 recruitment to dectin-1 phagosomes is accelerated by light chain 3 protein and regulates phagosome maturation and reactive oxygen production. J. Immunol. 2014, 192, 1356–1360. [Google Scholar] [CrossRef] [PubMed]
- Pagano, L.; Caira, M.; Candoni, A.; Offidani, M.; Martino, B.; Specchia, G.; Pastore, D.; Stanzani, M.; Cattaneo, C.; Fanci, R.; et al. Invasive aspergillosis in patients with acute myeloid leukemia: A SEIFEM-2008 registry study. Haematologica 2010, 95, 644–650. [Google Scholar] [CrossRef] [PubMed]
- Perkhofer, S.; Lass-Flörl, C.; Hell, M.; Russ, G.; Krause, R.; Hönigl, M.; Geltner, C.; Auberger, J.; Gastl, G.; Mitterbauer, M.; et al. The Nationwide Austrian Aspergillus Registry: A prospective data collection on epidemiology, therapy and outcome of invasive mould infections in immunocompromised and/or immunosuppressed patients. Int. J. Antimicrob. Agents 2010, 36, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Marciano, B.E.; Spalding, C.; Fitzgerald, A.; Mann, D.; Brown, T.; Osgood, S.; Yockey, L.; Darnell, D.N.; Barnhart, L.; Daub, J.; et al. Common severe infections in chronic granulomatous disease. Clin. Infect. Dis. 2015, 60, 1176–1183. [Google Scholar] [CrossRef] [PubMed]
- Bortoletto, P.; Lyman, K.; Camacho, A.; Fricchione, M.; Khanolkar, A.; Katz, B.Z. Chronic Granulomatous Disease: A Large, Single-center US Experience. Pediatr. Infect. Dis. J. 2015, 34, 1110–1114. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Zhang, J.; Fan, W.; Wong, K.N.; Ding, X.; Chen, S.; Zhong, Q. The RUN domain of rubicon is important for hVps34 binding, lipid kinase inhibition, and autophagy suppression. J. Biol. Chem. 2011, 286, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Smeekens, S.P.; Ng, A.; Kumar, V.; Johnson, M.D.; Plantinga, T.S.; van Diemen, C.; Arts, P.; Verwiel, E.T.; Gresnigt, M.S.; Fransen, K.; et al. Functional genomics identifies type I interferon pathway as central for host defense against Candida albicans. Nat. Commun. 2012, 4, 1342. [Google Scholar] [CrossRef] [PubMed]
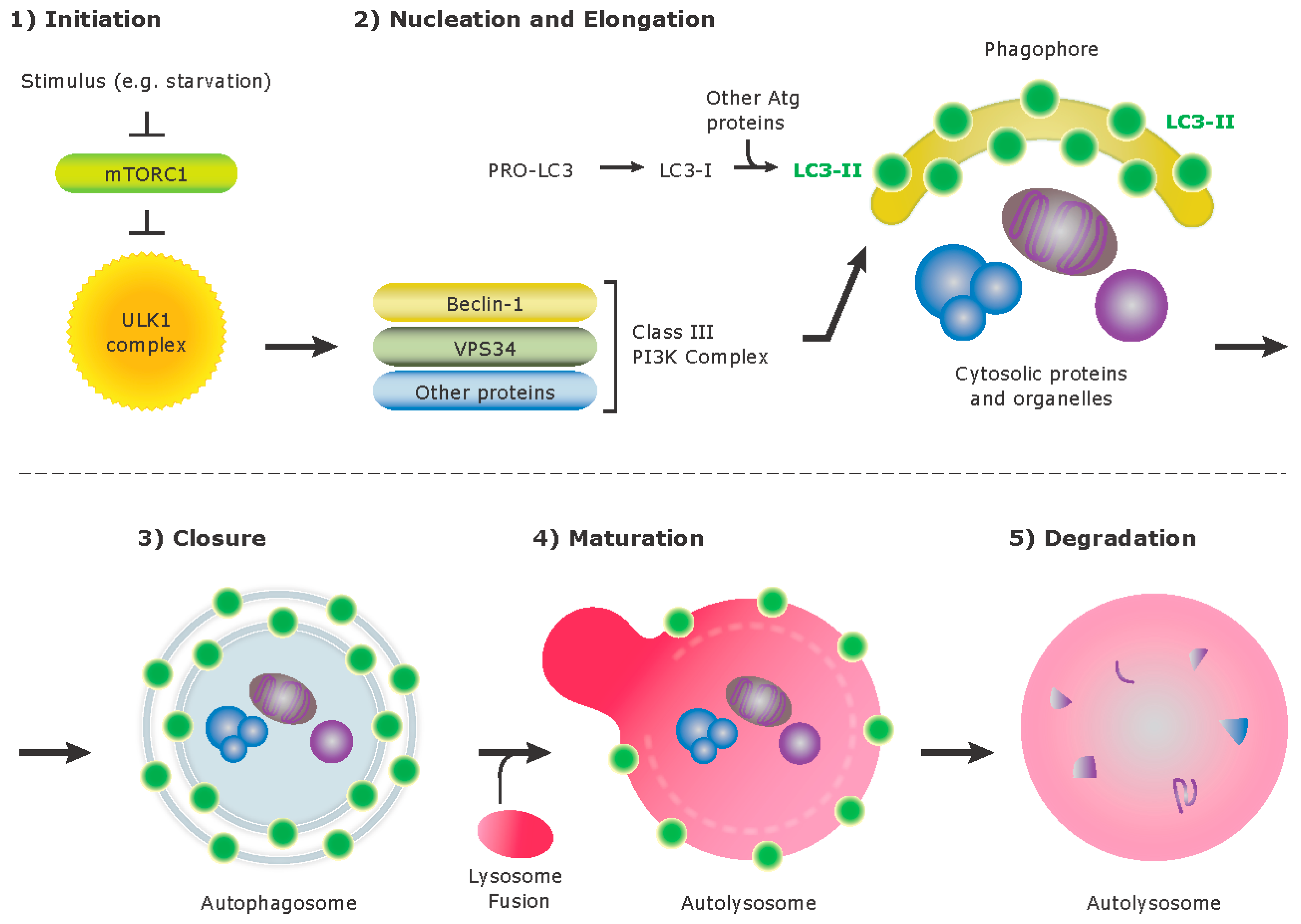

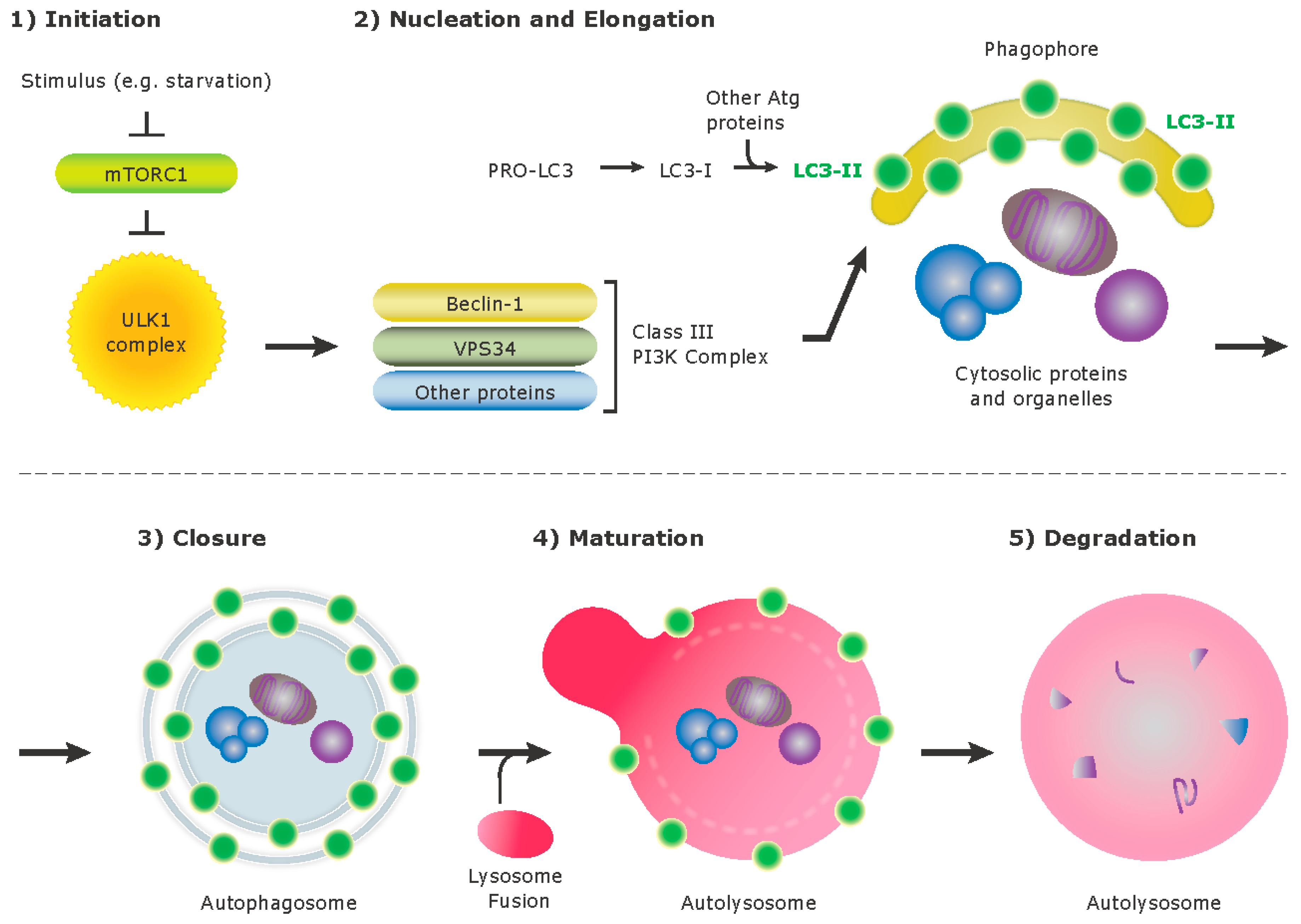{kind=link}
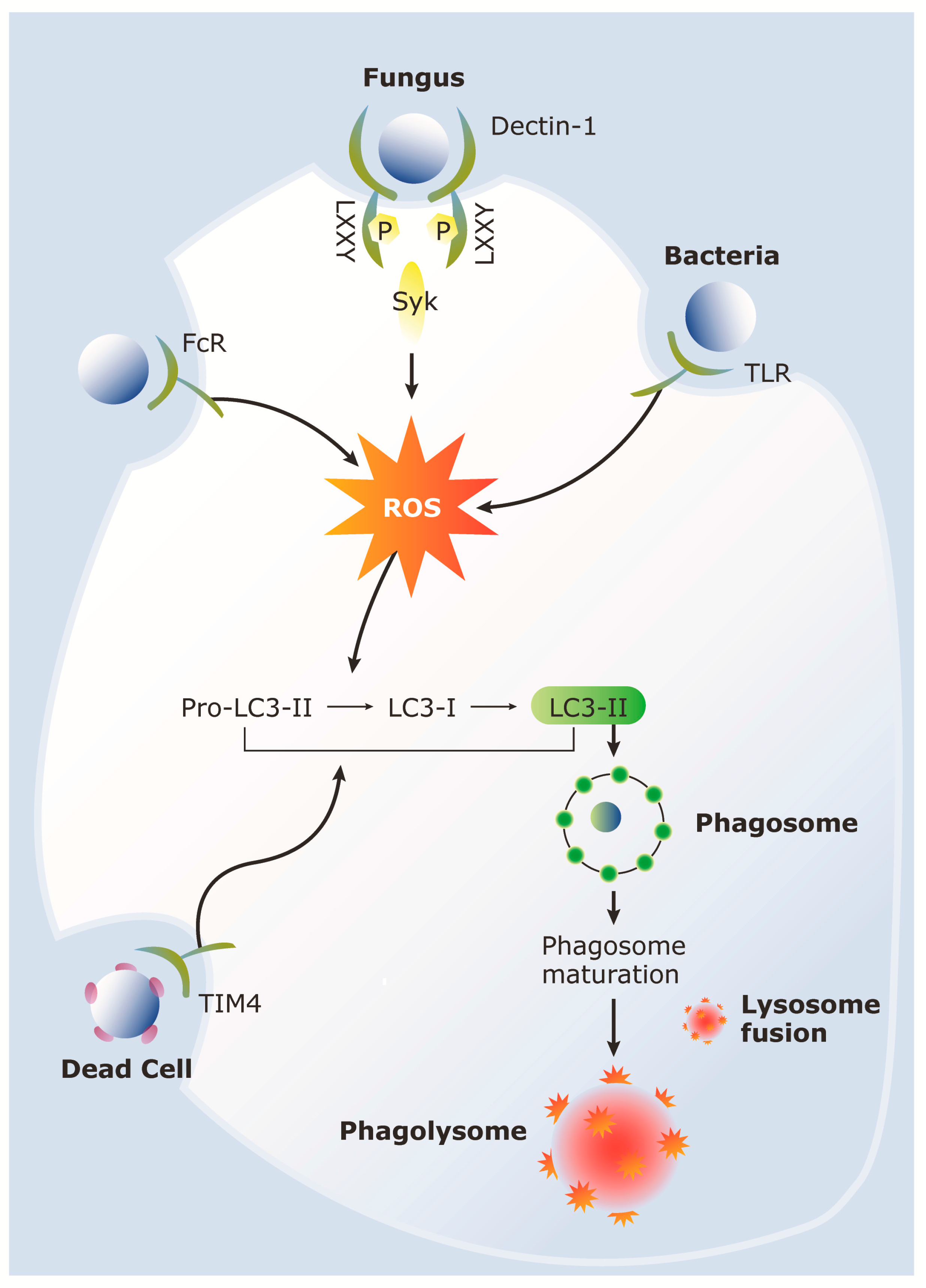{kind=link}
| Fungi | Effects of Autophagy Proteins | Reference |
|---|---|---|
| Candida albicans | Candida albicans induces a shift of LC3-I to LC3-II in macrophages. | [7,10,75] |
| LC3 is recruited to phagosomes in macrophages infected with Candida albicans. | [7,10,15] | |
| LC3-II recruitment to phagosomes requires Dectin-1 –dependent Syk phosphorylation. | [7,10] | |
| LC3-II recruitment to phagosomes is required for killing of Candida albicans by macrophages. | [10] | |
| Knockdown of Atg5 in macrophages in vitro results in reduced phagocytosis and killing of Candida albicans. The conditional knockout of Atg5 in murine myeloid cells increases mortality in response to Candida albicans in vivo. | [15] | |
| Atg7 is essential in mouse myeloid cells for host resistance to Candida albicans by enhancing neutrophil recruitment in vivo. | [6] | |
| Conditional knockout of Atg7 in mouse myeloid-cells did not lead to increased mortality due to Candida albicans. | [9] | |
| Aspergillus fumigatus | LC3-II is recruited to phagosomes containing Aspergillus fumigatus. | [11,13] |
| LC3-II recruitment is Dectin-1 dependent and regulated by Syk kinase dependent ROS production. | [11] | |
| Knockdown of Atg5 in human macrophages results in a reduction in the number of fungal spores contained within acidified lysosomes and reduced fungal killing. | [11] | |
| Macrophages deficient in autophagy proteins (Beclin-1, Rubicon, Atg7, and NOX2) fail to recruit LC3-II to pathogen containing phagosomes and display defects in pathogen clearance. | [13] | |
| Human monocytes from CGD patients (defective ROS production) and CGD murine macrophages have minimal LC3 recruitment and increased release of IL-1β in response to Aspergillus fumigatus. Inhibition of IL-1β in CGD mice increases LC3 recruitment and autophagy gene expression, and protects CGD mice from invasive aspergillosis. | [12] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tam, J.M.; Mansour, M.K.; Acharya, M.; Sokolovska, A.; Timmons, A.K.; Lacy-Hulbert, A.; Vyas, J.M. The Role of Autophagy-Related Proteins in Candida albicans Infections. Pathogens 2016, 5, 34. https://doi.org/10.3390/pathogens5020034
Tam JM, Mansour MK, Acharya M, Sokolovska A, Timmons AK, Lacy-Hulbert A, Vyas JM. The Role of Autophagy-Related Proteins in Candida albicans Infections. Pathogens. 2016; 5(2):34. https://doi.org/10.3390/pathogens5020034
Chicago/Turabian StyleTam, Jenny M., Michael K. Mansour, Mridu Acharya, Anna Sokolovska, Allison K. Timmons, Adam Lacy-Hulbert, and Jatin M. Vyas. 2016. "The Role of Autophagy-Related Proteins in Candida albicans Infections" Pathogens 5, no. 2: 34. https://doi.org/10.3390/pathogens5020034