
Evasion of Neutrophil Killing by Staphylococcus aureus

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
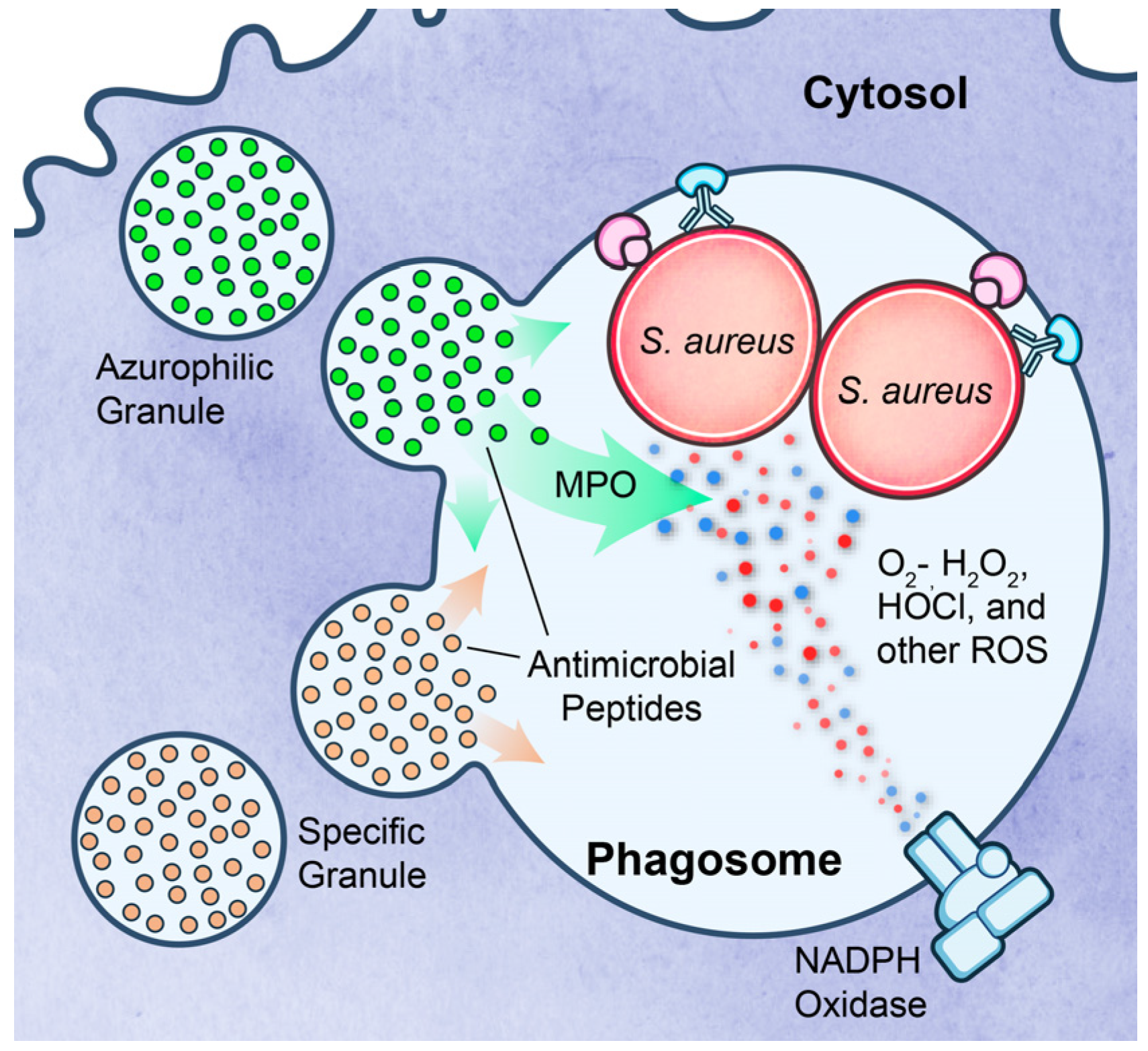
2. Neutrophils in Host Defense
3. Evasion of S. aureus Killing by Neutrophils
3.1. Inhibition of Neutrophil Recruitment
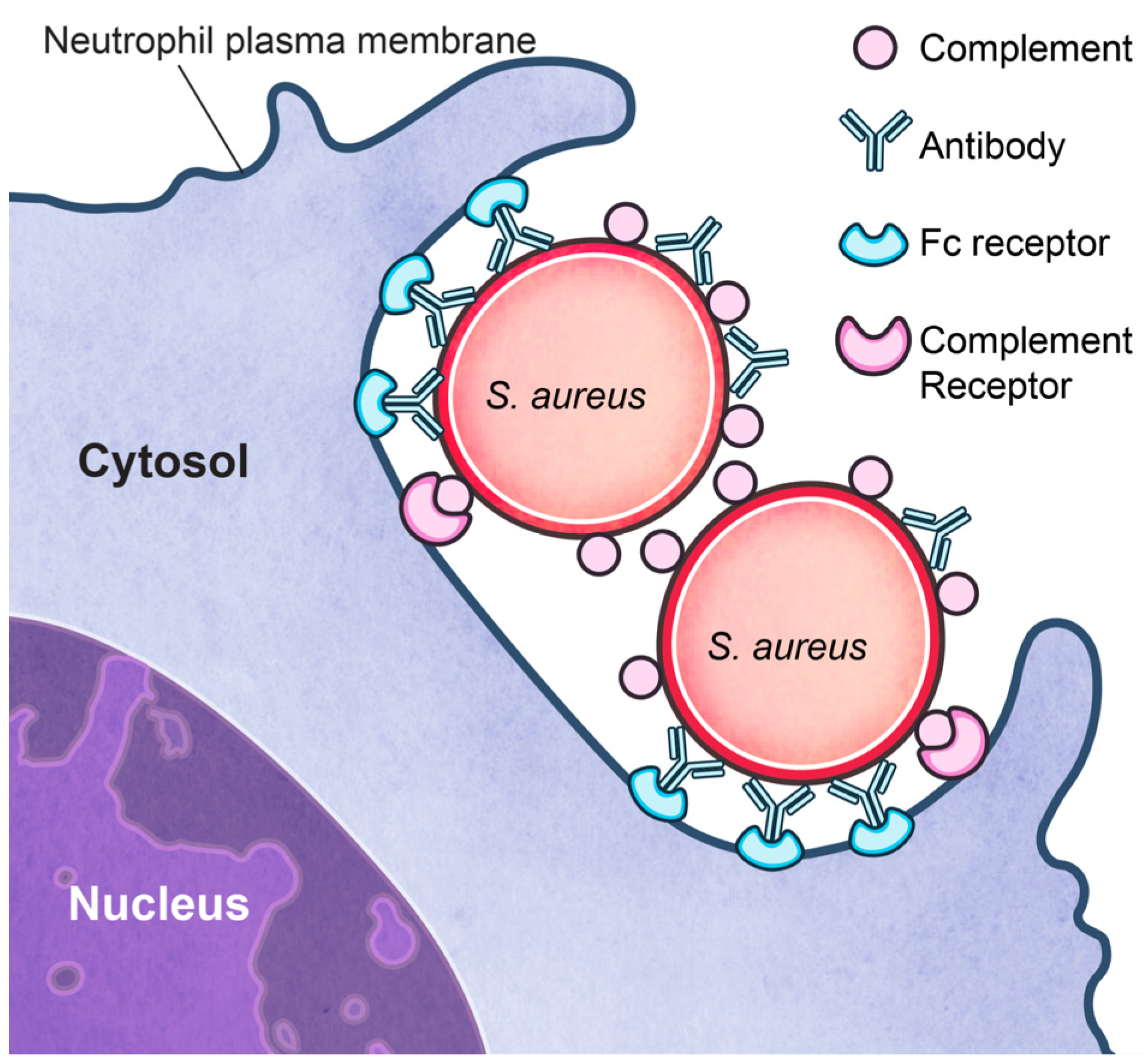
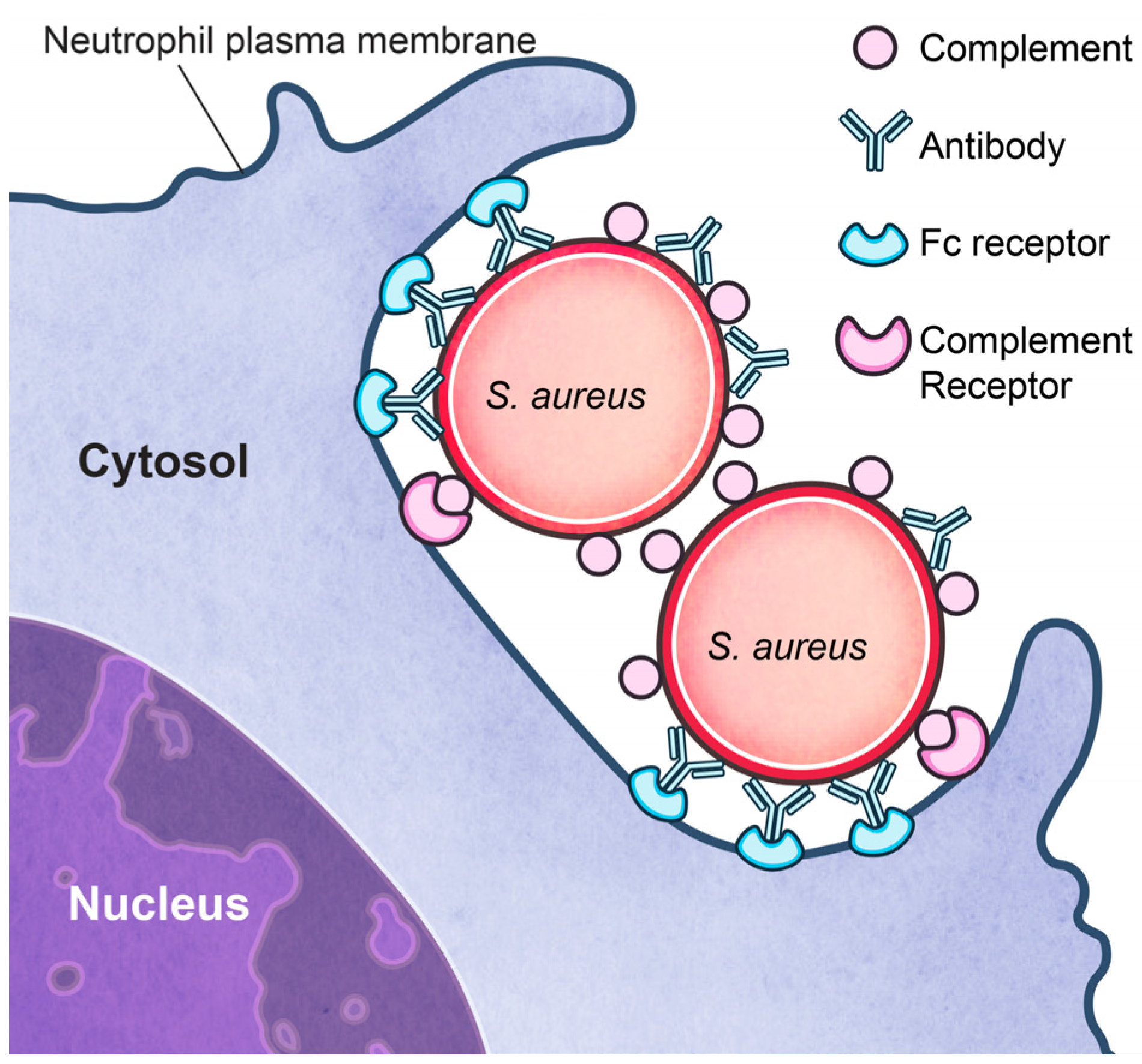
3.2. Inhibition of Phagocytosis?
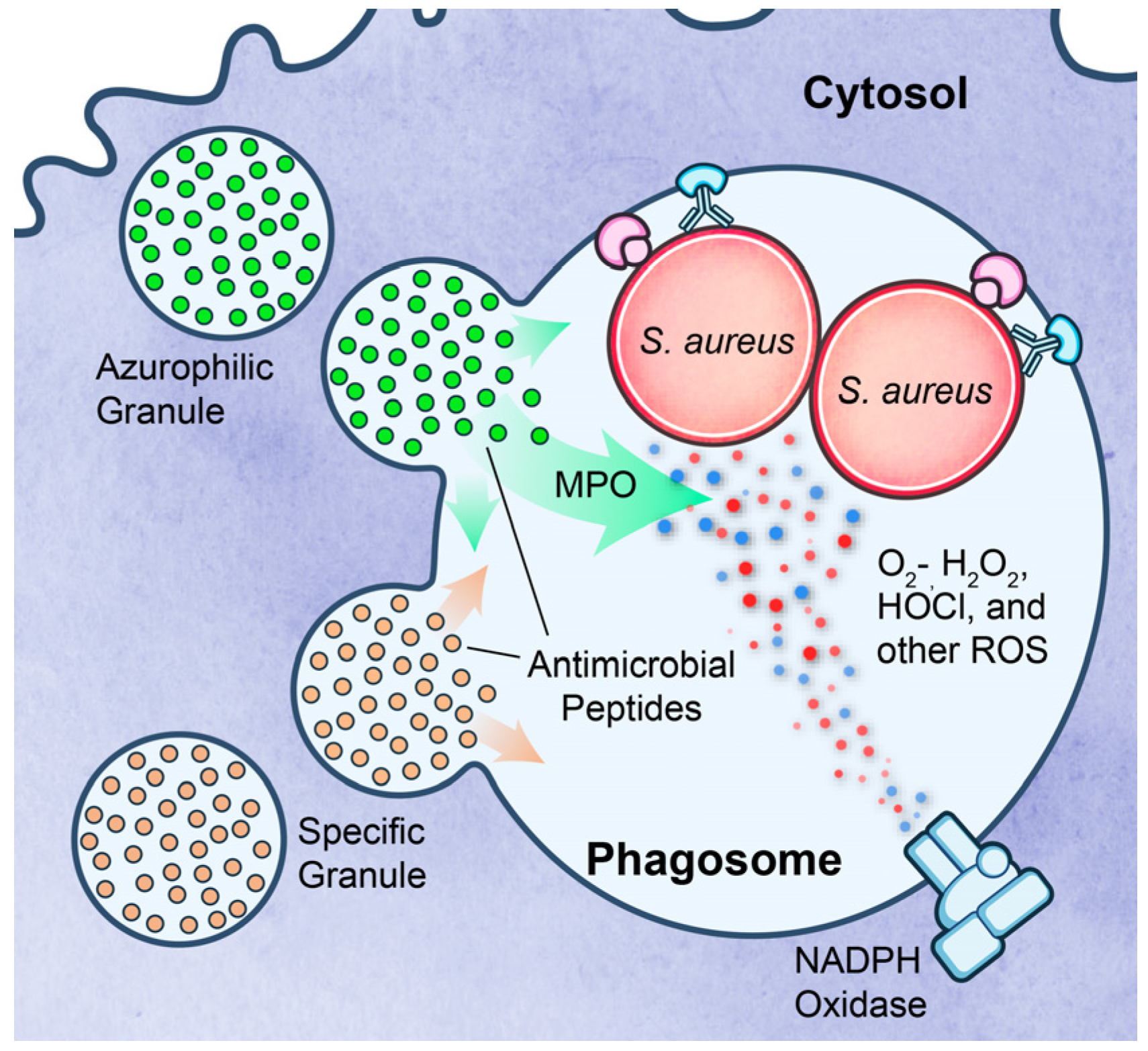
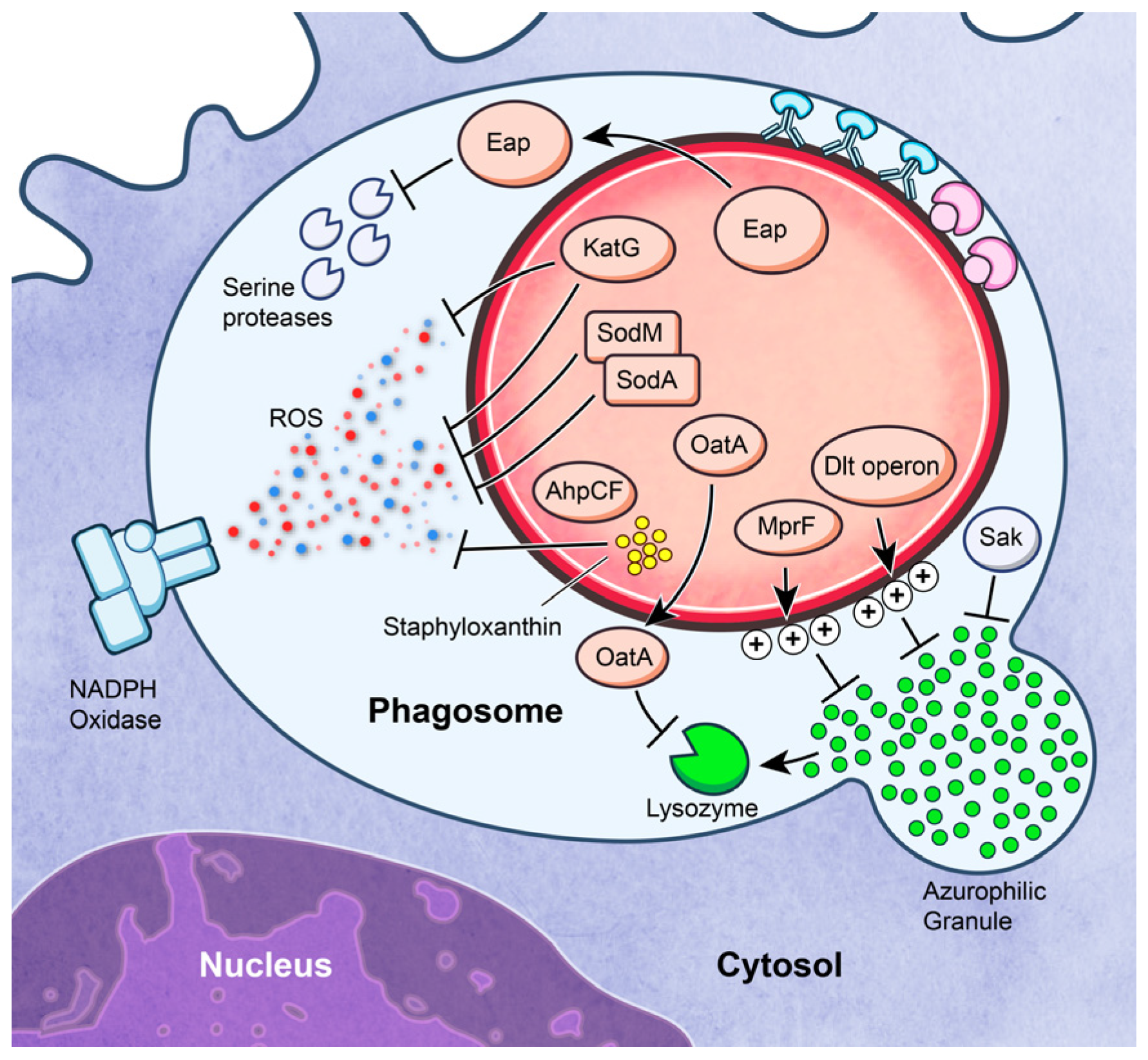
3.3. Survival after Phagocytosis
3.4. Lysis after Phagocytosis
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| PMN | Polymorphonuclear leukocyte |
| LFA-1 | Lymphocyte function-associated antigen-1 |
| PRR | Pattern recognition receptor |
| PAMP | Pathogen-associated molecular pattern |
| TLR | Toll-like receptor |
| ROS | Reactive oxygen species |
| AMP | Antimicrobial peptide |
| CHIPS | Chemotaxis inhibiting protein of S. aureus |
| FPR | Formyl peptide receptor |
| SCIN | Staphylococcal complement inhibitor |
| SAK | Staphylokinase |
| SEA | Staphylococcal enterotoxin A |
| IEC | Immune evasion cluster |
| Eap | Extracellular adherence protein |
| Ssls | Staphylococcal-like protein |
| PSGL-1 | P-selectin glycoprotein ligand-1 |
| Efb | Extracellular fibrinogen-binding protein |
| Ecb | Extracellular complement-binding protein |
| FLIPr | FPR-like 1 inhibitory protein |
| Hla | alpha-hemolysin |
| SpA | S. aureus protein A |
| ScpA | Staphopain A |
| Sbi | Second binding protein of immunoglobulin |
| IsdH | Iron-regulated surface determinant protein H |
| SOD | Superoxide dismutase |
| MPO | Myeloperoxidase |
| PSM | Phenol soluble modulin |
| PVL | Panton-Valentine leukocidin |
| Hlg | Gamma-hemolysin |
References
- Diekema, D.J.; Pfaller, M.A.; Schmitz, F.J.; Smayevsky, J.; Bell, J.; Jones, R.N.; Beach, M.; Group, S.P. Survey of infections due to staphylococcus species: Frequency of occurrence and antimicrobial susceptibility of isolates collected in the united states, canada, latin america, europe, and the western pacific region for the sentry antimicrobial surveillance program, 1997–1999. Clin. Infect. Dis. 2001, 32 (Suppl. 2), S114–S132. [Google Scholar] [PubMed]
- Gorwitz, R.J.; Kruszon-Moran, D.; McAllister, S.K.; McQuillan, G.; McDougal, L.K.; Fosheim, G.E.; Jensen, B.J.; Killgore, G.; Tenover, F.C.; Kuehnert, M.J. Changes in the prevalence of nasal colonization with Staphylococcus aureus in the united states, 2001–2004. J. Infect. Dis. 2008, 197, 1226–1234. [Google Scholar] [CrossRef] [PubMed]
- DeLeo, F.R.; Otto, M.; Kreiswirth, B.N.; Chambers, H.F. Community-associated meticillin-resistant Staphylococcus aureus. Lancet 2010, 375, 1557–1568. [Google Scholar] [CrossRef]
- Talan, D.A.; Krishnadasan, A.; Gorwitz, R.J.; Fosheim, G.E.; Limbago, B.; Albrecht, V.; Moran, G.J.; Group, E.M.I.N.S. Comparison of Staphylococcus aureus from skin and soft-tissue infections in us emergency department patients, 2004 and 2008. Clin. Infect. Dis. 2011, 53, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Klevens, R.M.; Morrison, M.A.; Nadle, J.; Petit, S.; Gershman, K.; Ray, S.; Harrison, L.H.; Lynfield, R.; Dumyati, G.; Townes, J.M.; et al. Invasive methicillin-resistant Staphylococcus aureus infections in the united states. JAMA 2007, 298, 1763–1771. [Google Scholar] [CrossRef] [PubMed]
- Fridkin, S.K.; Hageman, J.C.; Morrison, M.; Sanza, L.T.; Como-Sabetti, K.; Jernigan, J.A.; Harriman, K.; Harrison, L.H.; Lynfield, R.; Farley, M.M.; et al. Methicillin-resistant Staphylococcus aureus disease in three communities. N Engl. J. Med. 2005, 352, 1436–1444. [Google Scholar] [CrossRef] [PubMed]
- Chambers, H.F.; Deleo, F.R. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat. Rev. Microbiol. 2009, 7, 629–641. [Google Scholar] [CrossRef] [PubMed]
- DeLeo, F.R.; Chambers, H.F. Reemergence of antibiotic-resistant Staphylococcus aureus in the genomics era. J. Clin. Investig. 2009, 119, 2464–2474. [Google Scholar] [CrossRef] [PubMed]
- Lekstrom-Himes, J.A.; Gallin, J.I. Immunodeficiency diseases caused by defects in phagocytes. N. Engl. J. Med. 2000, 343, 1703–1714. [Google Scholar] [PubMed]
- Rankin, S.M. The bone marrow: A site of neutrophil clearance. J. Leukoc. Biol. 2010, 88, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Summers, C.; Rankin, S.M.; Condliffe, A.M.; Singh, N.; Peters, A.M.; Chilvers, E.R. Neutrophil kinetics in health and disease. Trends. Immunol. 2010, 31, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Phillipson, M.; Kubes, P. The neutrophil in vascular inflammation. Nat. Med. 2011, 17, 1381–1390. [Google Scholar] [CrossRef] [PubMed]
- Ley, K.; Laudanna, C.; Cybulsky, M.I.; Nourshargh, S. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007, 7, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Sadik, C.D.; Kim, N.D.; Luster, A.D. Neutrophils cascading their way to inflammation. Trends Immunol. 2011, 32, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Gillrie, M.R.; Zbytnuik, L.; McAvoy, E.; Kapadia, R.; Lee, K.; Waterhouse, C.C.; Davis, S.P.; Muruve, D.A.; Kubes, P.; Ho, M. Divergent roles of toll-like receptor 2 in response to lipoteichoic acid and Staphylococcus aureus in vivo. Eur. J. Immunol. 2010, 40, 1639–1650. [Google Scholar] [CrossRef] [PubMed]
- Von Aulock, S.; Morath, S.; Hareng, L.; Knapp, S.; Van Kessel, K.P.; Van Strijp, J.A.; Hartung, T. Lipoteichoic acid from Staphylococcus aureus is a potent stimulus for neutrophil recruitment. Immunobiology 2003, 208, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Leemans, J.C.; Heikens, M.; van Kessel, K.P.; Florquin, S.; van der Poll, T. Lipoteichoic acid and peptidoglycan from Staphylococcus aureus synergistically induce neutrophil influx into the lungs of mice. Clin. Diagn. Lab. Immunol. 2003, 10, 950–953. [Google Scholar] [CrossRef] [PubMed]
- Lotz, S.; Aga, E.; Wilde, I.; van Zandbergen, G.; Hartung, T.; Solbach, W.; Laskay, T. Highly purified lipoteichoic acid activates neutrophil granulocytes and delays their spontaneous apoptosis via cd14 and tlr2. J. Leukoc. Biol. 2004, 75, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Mullaly, S.C.; Kubes, P. The role of tlr2 in vivo following challenge with Staphylococcus aureus and prototypic ligands. J. Immunol. 2006, 177, 8154–8163. [Google Scholar] [CrossRef] [PubMed]
- Hoogerwerf, J.J.; de Vos, A.F.; Bresser, P.; van der Zee, J.S.; Pater, J.M.; de Boer, A.; Tanck, M.; Lundell, D.L.; Her-Jenh, C.; Draing, C.; et al. Lung inflammation induced by lipoteichoic acid or lipopolysaccharide in humans. Am. J. Respir. Crit. Care Med. 2008, 178, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Standiford, T.J.; Arenberg, D.A.; Danforth, J.M.; Kunkel, S.L.; VanOtteren, G.M.; Strieter, R.M. Lipoteichoic acid induces secretion of interleukin-8 from human blood monocytes: A cellular and molecular analysis. Infect. Immun. 1994, 62, 119–125. [Google Scholar] [PubMed]
- Soell, M.; Diab, M.; Haan-Archipoff, G.; Beretz, A.; Herbelin, C.; Poutrel, B.; Klein, J.P. Capsular polysaccharide types 5 and 8 of Staphylococcus aureus bind specifically to human epithelial (kb) cells, endothelial cells, and monocytes and induce release of cytokines. Infect. Immun. 1995, 63, 1380–1386. [Google Scholar] [PubMed]
- Yao, L.; Lowy, F.D.; Berman, J.W. Interleukin-8 gene expression in Staphylococcus aureus-infected endothelial cells. Infect. Immun. 1996, 64, 3407–3409. [Google Scholar] [PubMed]
- Abram, C.L.; Lowell, C.A. The ins and outs of leukocyte integrin signaling. Annu. Rev. Immunol. 2009, 27, 339–362. [Google Scholar] [CrossRef] [PubMed]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Kanneganti, T.D.; Lamkanfi, M.; Nunez, G. Intracellular nod-like receptors in host defense and disease. Immunity 2007, 27, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Xu, Z.; Gupta, D.; Dziarski, R. Peptidoglycan recognition proteins: A novel family of four human innate immunity pattern recognition molecules. J. Biol. Chem. 2001, 276, 34686–34694. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, S.E.; Schreiber, A.D. Fc gamma receptors in phagocytes. Curr. Opin. Hematol. 1998, 5, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Sengelov, H. Complement receptors in neutrophils. Crit. Rev. Immunol. 1995, 15, 107–131. [Google Scholar] [CrossRef] [PubMed]
- Nunes, P.; Demaurex, N.; Dinauer, M.C. Regulation of the nadph oxidase and associated ion fluxes during phagocytosis. Traffic 2013, 14, 1118–1131. [Google Scholar] [CrossRef] [PubMed]
- DeLeo, F.R.; Allen, L.A.; Apicella, M.; Nauseef, W.M. Nadph oxidase activation and assembly during phagocytosis. J. Immunol. 1999, 163, 6732–6740. [Google Scholar] [PubMed]
- Nauseef, W.M.; Borregaard, N. Neutrophils at work. Nat. Immunol. 2014, 15, 602–611. [Google Scholar] [CrossRef] [PubMed]
- Nauseef, W.M. Detection of superoxide anion and hydrogen peroxide production by cellular nadph oxidases. Biochim. Biophys. Acta 2014, 1840, 757–767. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C.; Kettle, A.J. Redox reactions and microbial killing in the neutrophil phagosome. Antioxid. Redox Signal. 2013, 18, 642–660. [Google Scholar] [CrossRef] [PubMed]
- Faurschou, M.; Borregaard, N. Neutrophil granules and secretory vesicles in inflammation. Microbes Infect. 2003, 5, 1317–1327. [Google Scholar] [CrossRef] [PubMed]
- Borregaard, N.; Sorensen, O.E.; Theilgaard-Monch, K. Neutrophil granules: A library of innate immunity proteins. Trends Immunol. 2007, 28, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, J.G.; Cohn, Z.A. Degranulation of polymorphonuclear leucocytes following phagocytosis of microorganisms. J. Exp. Med. 1960, 112, 1005–1014. [Google Scholar] [CrossRef] [PubMed]
- Lominadze, G.; Powell, D.W.; Luerman, G.C.; Link, A.J.; Ward, R.A.; McLeish, K.R. Proteomic analysis of human neutrophil granules. Mol. Cell. Proteom. 2005, 4, 1503–1521. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.D.; DeLeo, F.R. Role of neutrophils in innate immunity: A systems biology-level approach. Wiley Interdiscip. Rev. Syst. Biol. Med. 2009, 1, 309–333. [Google Scholar] [CrossRef] [PubMed]
- Buckley, C.D.; Gilroy, D.W.; Serhan, C.N. Proresolving lipid mediators and mechanisms in the resolution of acute inflammation. Immunity 2014, 40, 315–327. [Google Scholar] [CrossRef] [PubMed]
- DeLeo, F.R.; Diep, B.A.; Otto, M. Host defense and pathogenesis in Staphylococcus aureus infections. Infect. Dis. Clin. N. Am. 2009, 23, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Rigby, K.M.; DeLeo, F.R. Neutrophils in innate host defense against Staphylococcus aureus infections. Semin. Immunopathol. 2012, 34, 237–259. [Google Scholar] [CrossRef] [PubMed]
- De Haas, C.J.; Veldkamp, K.E.; Peschel, A.; Weerkamp, F.; Van Wamel, W.J.; Heezius, E.C.; Poppelier, M.J.; Van Kessel, K.P.; van Strijp, J.A. Chemotaxis inhibitory protein of Staphylococcus aureus, a bacterial antiinflammatory agent. J. Exp. Med. 2004, 199, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Postma, B.; Poppelier, M.J.; van Galen, J.C.; Prossnitz, E.R.; van Strijp, J.A.; de Haas, C.J.; van Kessel, K.P. Chemotaxis inhibitory protein of Staphylococcus aureus binds specifically to the c5a and formylated peptide receptor. J. Immunol. 2004, 172, 6994–7001. [Google Scholar] [CrossRef] [PubMed]
- Van Wamel, W.J.; Rooijakkers, S.H.; Ruyken, M.; van Kessel, K.P.; van Strijp, J.A. The innate immune modulators staphylococcal complement inhibitor and chemotaxis inhibitory protein of Staphylococcus aureus are located on beta-hemolysin-converting bacteriophages. J. Bacteriol. 2006, 188, 1310–1315. [Google Scholar] [CrossRef] [PubMed]
- Jin, T.; Bokarewa, M.; Foster, T.; Mitchell, J.; Higgins, J.; Tarkowski, A. Staphylococcus aureus resists human defensins by production of staphylokinase, a novel bacterial evasion mechanism. J. Immunol. 2004, 172, 1169–1176. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.X.; Gilmore, K.J.; Szabo, P.A.; Zeppa, J.J.; Baroja, M.L.; Haeryfar, S.M.; McCormick, J.K. Superantigens subvert the neutrophil response to promote abscess formation and enhance Staphylococcus aureus survival in vivo. Infect. Immun. 2014, 82, 3588–3598. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Altstaedt, J.; von der Ohe, M.; Proft, T.; Gross, U.; Rink, L. Induction of interleukin-8 in human neutrophils after mhc class ii cross-linking with superantigens. J. Leukoc. Biol. 2001, 70, 80–86. [Google Scholar] [PubMed]
- Hopkins, P.A.; Fraser, J.D.; Pridmore, A.C.; Russell, H.H.; Read, R.C.; Sriskandan, S. Superantigen recognition by hla class ii on monocytes up-regulates toll-like receptor 4 and enhances proinflammatory responses to endotoxin. Blood 2005, 105, 3655–3662. [Google Scholar] [CrossRef] [PubMed]
- Stapels, D.A.; Ramyar, K.X.; Bischoff, M.; von Kockritz-Blickwede, M.; Milder, F.J.; Ruyken, M.; Eisenbeis, J.; McWhorter, W.J.; Herrmann, M.; van Kessel, K.P.; et al. Staphylococcus aureus secretes a unique class of neutrophil serine protease inhibitors. Proc. Natl. Acad. Sci. USA 2014, 111, 13187–13192. [Google Scholar] [CrossRef] [PubMed]
- Stapels, D.A.; Kuipers, A.; von Kockritz-Blickwede, M.; Ruyken, M.; Tromp, A.T.; Horsburgh, M.J.; de Haas, C.J.; van Strijp, J.A.; van Kessel, K.P.; Rooijakkers, S.H. Staphylococcus aureus protects its immune-evasion proteins against degradation by neutrophil serine proteases. Cell. Microbiol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Bestebroer, J.; Aerts, P.C.; Rooijakkers, S.H.; Pandey, M.K.; Kohl, J.; van Strijp, J.A.; de Haas, C.J. Functional basis for complement evasion by staphylococcal superantigen-like 7. Cell. Microbiol. 2010, 12, 1506–1516. [Google Scholar] [CrossRef] [PubMed]
- Langley, R.; Wines, B.; Willoughby, N.; Basu, I.; Proft, T.; Fraser, J.D. The staphylococcal superantigen-like protein 7 binds iga and complement c5 and inhibits iga-fc alpha ri binding and serum killing of bacteria. J. Immunol. 2005, 174, 2926–2933. [Google Scholar] [CrossRef] [PubMed]
- Bestebroer, J.; Poppelier, M.J.; Ulfman, L.H.; Lenting, P.J.; Denis, C.V.; van Kessel, K.P.; van Strijp, J.A.; de Haas, C.J. Staphylococcal superantigen-like 5 binds psgl-1 and inhibits p-selectin-mediated neutrophil rolling. Blood 2007, 109, 2936–2943. [Google Scholar] [CrossRef] [PubMed]
- Bestebroer, J.; van Kessel, K.P.; Azouagh, H.; Walenkamp, A.M.; Boer, I.G.; Romijn, R.A.; van Strijp, J.A.; de Haas, C.J. Staphylococcal ssl5 inhibits leukocyte activation by chemokines and anaphylatoxins. Blood 2009, 113, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Rooijakkers, S.H.; Ruyken, M.; Roos, A.; Daha, M.R.; Presanis, J.S.; Sim, R.B.; van Wamel, W.J.; van Kessel, K.P.; van Strijp, J.A. Immune evasion by a staphylococcal complement inhibitor that acts on c3 convertases. Nat. Immunol. 2005, 6, 920–927. [Google Scholar] [CrossRef] [PubMed]
- Jongerius, I.; Kohl, J.; Pandey, M.K.; Ruyken, M.; van Kessel, K.P.; van Strijp, J.A.; Rooijakkers, S.H. Staphylococcal complement evasion by various convertase-blocking molecules. J. Exp. Med. 2007, 204, 2461–2471. [Google Scholar] [CrossRef] [PubMed]
- Rooijakkers, S.H.; Ruyken, M.; van Roon, J.; van Kessel, K.P.; van Strijp, J.A.; van Wamel, W.J. Early expression of scin and chips drives instant immune evasion by staphylococcus aureus. Cell. Microbiol. 2006, 8, 1282–1293. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.P.; Kuipers, A.; Freitag, C.M.; Jongerius, I.; Medina, E.; van Rooijen, W.J.; Spaan, A.N.; van Kessel, K.P.; Hook, M.; Rooijakkers, S.H. Phagocytosis escape by a Staphylococcus aureus protein that connects complement and coagulation proteins at the bacterial surface. PLoS Pathog. 2013, 9, e1003816. [Google Scholar] [CrossRef] [PubMed]
- Laarman, A.J.; Mijnheer, G.; Mootz, J.M.; van Rooijen, W.J.; Ruyken, M.; Malone, C.L.; Heezius, E.C.; Ward, R.; Milligan, G.; van Strijp, J.A.; et al. Staphylococcus aureus staphopain a inhibits cxcr2-dependent neutrophil activation and chemotaxis. EMBO J. 2012, 31, 3607–3619. [Google Scholar] [CrossRef] [PubMed]
- Dossett, J.H.; Kronvall, G.; Williams, R.C., Jr.; Quie, P.G. Antiphagocytic effects of staphylococcal protein a. J. Immunol. 1969, 103, 1405–1410. [Google Scholar] [PubMed]
- Peterson, P.K.; Verhoef, J.; Sabath, L.D.; Quie, P.G. Effect of protein a on staphylococcal opsonization. Infect. Immun. 1977, 15, 760–764. [Google Scholar] [PubMed]
- Zhang, L.; Jacobsson, K.; Vasi, J.; Lindberg, M.; Frykberg, L. A second igg-binding protein in Staphylococcus aureus. Microbiology 1998, 144, 985–991. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.J.; Visai, L.; Kerrigan, S.W.; Speziale, P.; Foster, T.J. The sbi protein is a multifunctional immune evasion factor of Staphylococcus aureus. Infect. Immun. 2011, 79, 3801–3809. [Google Scholar] [CrossRef] [PubMed]
- Thakker, M.; Park, J.S.; Carey, V.; Lee, J.C. Staphylococcus aureus serotype 5 capsular polysaccharide is antiphagocytic and enhances bacterial virulence in a murine bacteremia model. Infect. Immun. 1998, 66, 5183–5189. [Google Scholar] [PubMed]
- Nanra, J.S.; Buitrago, S.M.; Crawford, S.; Ng, J.; Fink, P.S.; Hawkins, J.; Scully, I.L.; McNeil, L.K.; Aste-Amezaga, J.M.; Cooper, D.; et al. Capsular polysaccharides are an important immune evasion mechanism for Staphylococcus aureus. Hum. Vaccines Immunother. 2013, 9, 480–487. [Google Scholar] [CrossRef]
- Higgins, J.; Loughman, A.; van Kessel, K.P.; van Strijp, J.A.; Foster, T.J. Clumping factor a of Staphylococcus aureus inhibits phagocytosis by human polymorphonuclear leucocytes. FEMS Microbiol. Lett. 2006, 258, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Visai, L.; Yanagisawa, N.; Josefsson, E.; Tarkowski, A.; Pezzali, I.; Rooijakkers, S.H.; Foster, T.J.; Speziale, P. Immune evasion by Staphylococcus aureus conferred by iron-regulated surface determinant protein isdh. Microbiology 2009, 155, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Rogers, D.E. Studies on bacteriemia. I. Mechanisms relating to the persistence of bacteriemia in rabbits following the intravenous injection of staphylococci. J. Exp. Med. 1956, 103, 713–742. [Google Scholar] [CrossRef] [PubMed]
- Gregory, S.H.; Sagnimeni, A.J.; Wing, E.J. Bacteria in the bloodstream are trapped in the liver and killed by immigrating neutrophils. J. Immunol. 1996, 157, 2514–2520. [Google Scholar] [PubMed]
- Voyich, J.M.; Braughton, K.R.; Sturdevant, D.E.; Whitney, A.R.; Said-Salim, B.; Porcella, S.F.; Long, R.D.; Dorward, D.W.; Gardner, D.J.; Kreiswirth, B.N.; et al. Insights into mechanisms used by Staphylococcus aureus to avoid destruction by human neutrophils. J. Immunol. 2005, 175, 3907–3919. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Porter, A.R.; Kennedy, A.D.; Kobayashi, S.D.; DeLeo, F.R. Phagocytosis and killing of Staphylococcus aureus by human neutrophils. J. Innate. Immun. 2014, 6, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Rogers, D.E.; Tompsett, R. The survival of staphylococci within human leukocytes. J. Exp. Med. 1952, 95, 209–230. [Google Scholar] [CrossRef] [PubMed]
- Rogers, D.E. Experimental observations on staphylococcal disease. Postep. Mirobiologii. 1966, 5, 279–296. [Google Scholar]
- Gresham, H.D.; Lowrance, J.H.; Caver, T.E.; Wilson, B.S.; Cheung, A.L.; Lindberg, F.P. Survival of Staphylococcus aureus inside neutrophils contributes to infection. J. Immunol. 2000, 164, 3713–3722. [Google Scholar] [CrossRef] [PubMed]
- Kapral, F.A.; Shayegani, M.G. Intracellular survival of staphylococci. J. Exp. Med. 1959, 110, 123–138. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.D.; Braughton, K.R.; Palazzolo-Ballance, A.M.; Kennedy, A.D.; Sampaio, E.; Kristosturyan, E.; Whitney, A.R.; Sturdevant, D.E.; Dorward, D.W.; Holland, S.M.; et al. Rapid neutrophil destruction following phagocytosis of Staphylococcus aureus. J. Innate. Immun. 2010, 2, 560–575. [Google Scholar] [CrossRef] [PubMed]
- Karavolos, M.H.; Horsburgh, M.J.; Ingham, E.; Foster, S.J. Role and regulation of the superoxide dismutases of Staphylococcus aureus. Microbiology 2003, 149, 2749–2758. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, K.; Coutts, G.; Jonsson, I.M.; Tarkowski, A.; Kokai-Kun, J.F.; Mond, J.J.; Foster, S.J. Catalase (kata) and alkyl hydroperoxide reductase (ahpc) have compensatory roles in peroxide stress resistance and are required for survival, persistence, and nasal colonization in Staphylococcus aureus. J. Bacteriol. 2007, 189, 1025–1035. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.Y.; Essex, A.; Buchanan, J.T.; Datta, V.; Hoffman, H.M.; Bastian, J.F.; Fierer, J.; Nizet, V. Staphylococcus aureus golden pigment impairs neutrophil killing and promotes virulence through its antioxidant activity. J. Exp. Med. 2005, 202, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Cassat, J.E.; Skaar, E.P. Iron in infection and immunity. Cell Host Microbe 2013, 13, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Holland, S.M. Chronic granulomatous disease. Hematol. Oncol. Clin. N. Am. 2013, 27, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Pincus, S.H.; Klebanoff, S.J. Quantitative leukocyte iodination. N. Engl. J. Med. 1971, 284, 744–750. [Google Scholar] [CrossRef] [PubMed]
- Klebanoff, S.J.; Kettle, A.J.; Rosen, H.; Winterbourn, C.C.; Nauseef, W.M. Myeloperoxidase: A front-line defender against phagocytosed microorganisms. J. Leukoc. Biol. 2013, 93, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Chapman, A.L.; Hampton, M.B.; Senthilmohan, R.; Winterbourn, C.C.; Kettle, A.J. Chlorination of bacterial and neutrophil proteins during phagocytosis and killing of Staphylococcus aureus. J. Biol. Chem. 2002, 277, 9757–9762. [Google Scholar] [CrossRef] [PubMed]
- Peschel, A.; Otto, M.; Jack, R.W.; Kalbacher, H.; Jung, G.; Gotz, F. Inactivation of the dlt operon in Staphylococcus aureus confers sensitivity to defensins, protegrins, and other antimicrobial peptides. J. Biol. Chem. 1999, 274, 8405–8410. [Google Scholar] [CrossRef] [PubMed]
- Collins, L.V.; Kristian, S.A.; Weidenmaier, C.; Faigle, M.; Van Kessel, K.P.; Van Strijp, J.A.; Gotz, F.; Neumeister, B.; Peschel, A. Staphylococcus aureus strains lacking d-alanine modifications of teichoic acids are highly susceptible to human neutrophil killing and are virulence attenuated in mice. J. Infect. Dis. 2002, 186, 214–219. [Google Scholar] [CrossRef] [PubMed]
- Peschel, A.; Jack, R.W.; Otto, M.; Collins, L.V.; Staubitz, P.; Nicholson, G.; Kalbacher, H.; Nieuwenhuizen, W.F.; Jung, G.; Tarkowski, A.; et al. Staphylococcus aureus resistance to human defensins and evasion of neutrophil killing via the novel virulence factor mprf is based on modification of membrane lipids with l-lysine. J. Exp. Med. 2001, 193, 1067–1076. [Google Scholar] [CrossRef] [PubMed]
- Bera, A.; Herbert, S.; Jakob, A.; Vollmer, W.; Gotz, F. Why are pathogenic staphylococci so lysozyme resistant? The peptidoglycan o-acetyltransferase oata is the major determinant for lysozyme resistance of Staphylococcus aureus. Mol. Microbiol. 2005, 55, 778–787. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Braughton, K.R.; Kretschmer, D.; Bach, T.H.; Queck, S.Y.; Li, M.; Kennedy, A.D.; Dorward, D.W.; Klebanoff, S.J.; Peschel, A.; et al. Identification of novel cytolytic peptides as key virulence determinants for community-associated mrsa. Nat. Med. 2007, 13, 1510–1514. [Google Scholar] [CrossRef] [PubMed]
- Gladstone, G.P.; Van Heyningen, W.E. Staphylococcal leucocidins. Br. J. Exp. Pathol. 1957, 38, 123–137. [Google Scholar] [PubMed]
- Woodin, A.M. Staphylococcal leukocidin. Ann. N Y Acad. Sci. 1965, 128, 152–164. [Google Scholar] [CrossRef] [PubMed]
- Panton, P.N.; Valentine, F.C.O. Staphylococcal toxin. Lancet 1932, 219, 506–508. [Google Scholar] [CrossRef]
- Ventura, C.L.; Malachowa, N.; Hammer, C.H.; Nardone, G.A.; Robinson, M.A.; Kobayashi, S.D.; DeLeo, F.R. Identification of a novel Staphylococcus aureus two-component leukotoxin using cell surface proteomics. PLoS ONE 2010, 5, e11634. [Google Scholar] [CrossRef] [PubMed]
- Dumont, A.L.; Nygaard, T.K.; Watkins, R.L.; Smith, A.; Kozhaya, L.; Kreiswirth, B.N.; Shopsin, B.; Unutmaz, D.; Voyich, J.M.; Torres, V.J. Characterization of a new cytotoxin that contributes to Staphylococcus aureus pathogenesis. Mol. Microbiol. 2011, 79, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Malachowa, N.; Whitney, A.R.; Kobayashi, S.D.; Sturdevant, D.E.; Kennedy, A.D.; Braughton, K.R.; Shabb, D.W.; Diep, B.A.; Chambers, H.F.; Otto, M.; et al. Global changes in Staphylococcus aureus gene expression in human blood. PLoS ONE 2011, 6, e18617. [Google Scholar] [CrossRef] [PubMed]
- Surewaard, B.G.; de Haas, C.J.; Vervoort, F.; Rigby, K.M.; DeLeo, F.R.; Otto, M.; van Strijp, J.A.; Nijland, R. Staphylococcal alpha-phenol soluble modulins contribute to neutrophil lysis after phagocytosis. Cell. Microbiol. 2013, 15, 1427–1437. [Google Scholar] [CrossRef] [PubMed]
- Greenlee-Wacker, M.C.; Rigby, K.M.; Kobayashi, S.D.; Porter, A.R.; DeLeo, F.R.; Nauseef, W.M. Phagocytosis of Staphylococcus aureus by human neutrophils prevents macrophage efferocytosis and induces programmed necrosis. J. Immunol. 2014, 192, 4709–4717. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McGuinness, W.A.; Kobayashi, S.D.; DeLeo, F.R. Evasion of Neutrophil Killing by Staphylococcus aureus. Pathogens 2016, 5, 32. https://doi.org/10.3390/pathogens5010032
McGuinness WA, Kobayashi SD, DeLeo FR. Evasion of Neutrophil Killing by Staphylococcus aureus. Pathogens. 2016; 5(1):32. https://doi.org/10.3390/pathogens5010032
Chicago/Turabian StyleMcGuinness, Will A., Scott D. Kobayashi, and Frank R. DeLeo. 2016. "Evasion of Neutrophil Killing by Staphylococcus aureus" Pathogens 5, no. 1: 32. https://doi.org/10.3390/pathogens5010032