
Importance of B Lymphocytes and the IgG-Binding Protein Sbi in Staphylococcus aureus Skin Infection


Abstract
:1. Introduction
2. Results
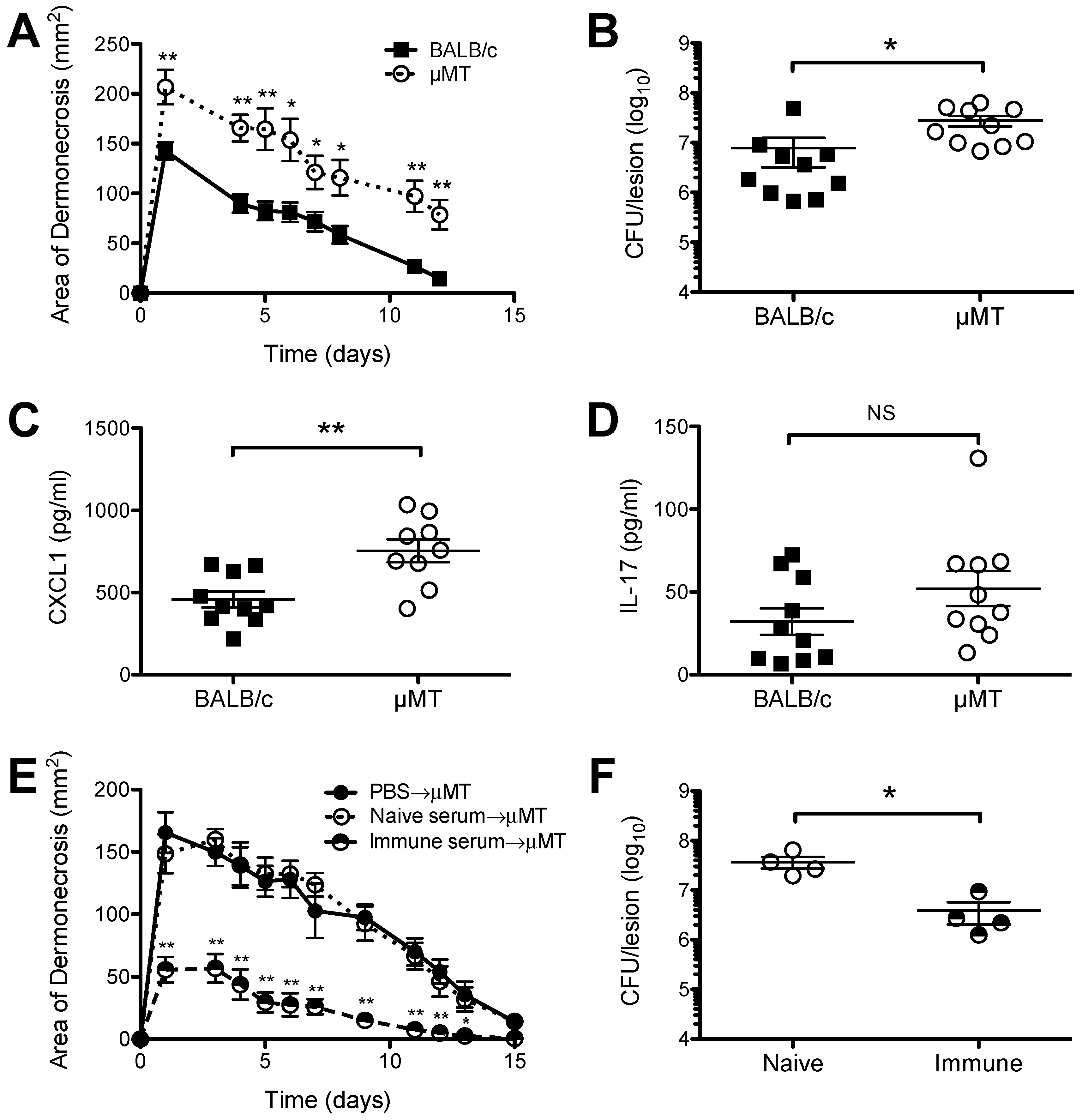
2.1. Increased Susceptibility of B Cell Deficient Mice to S. aureus SSTI
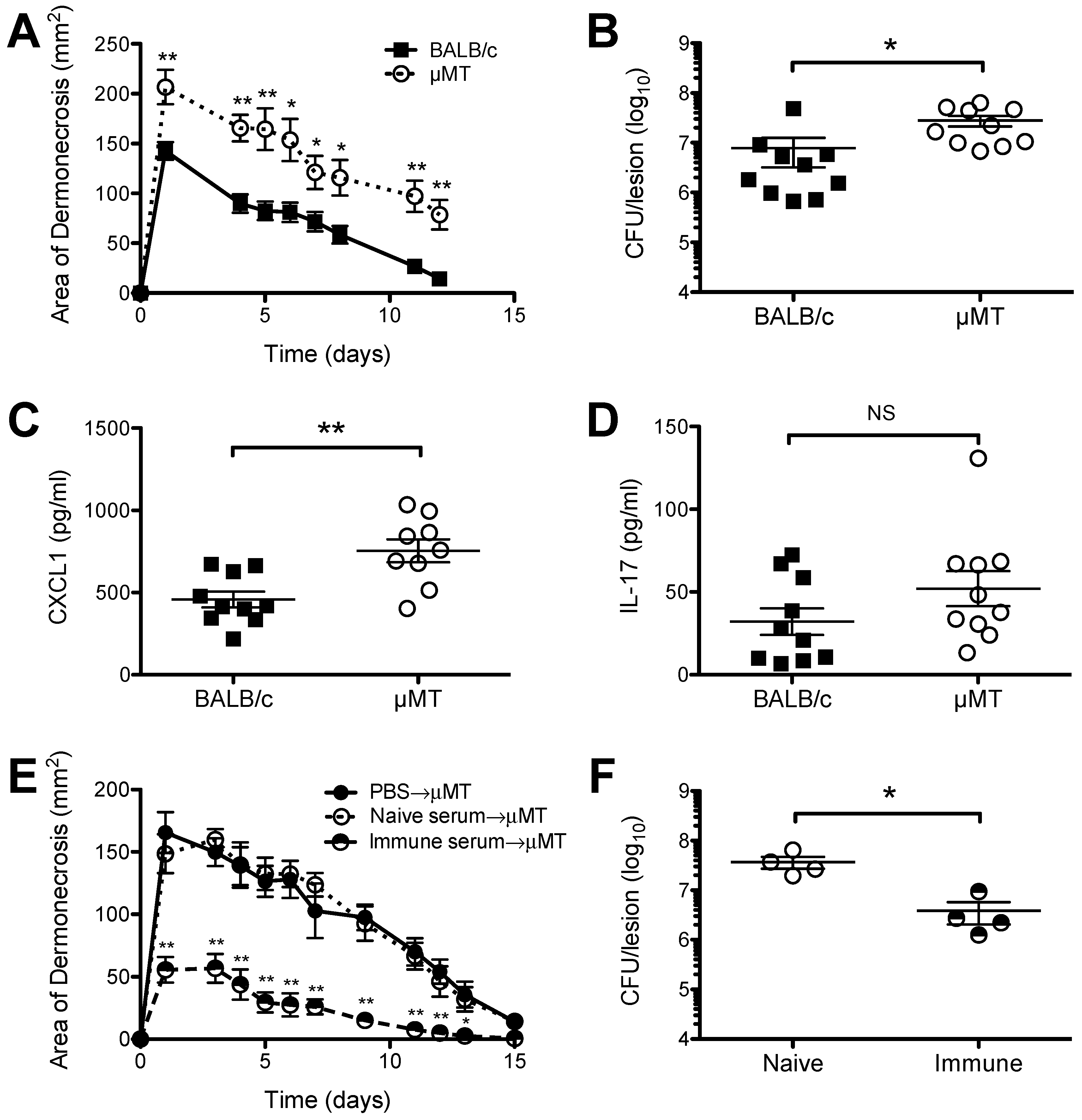
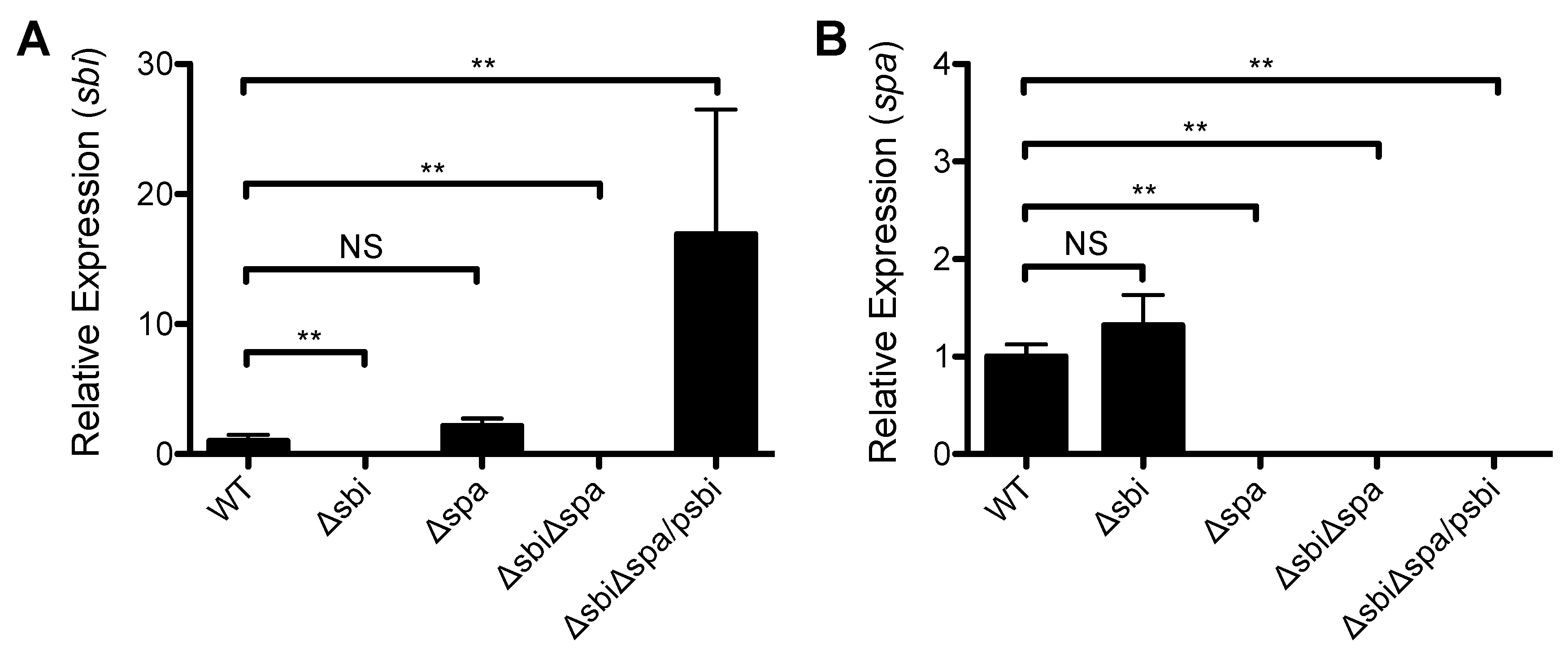
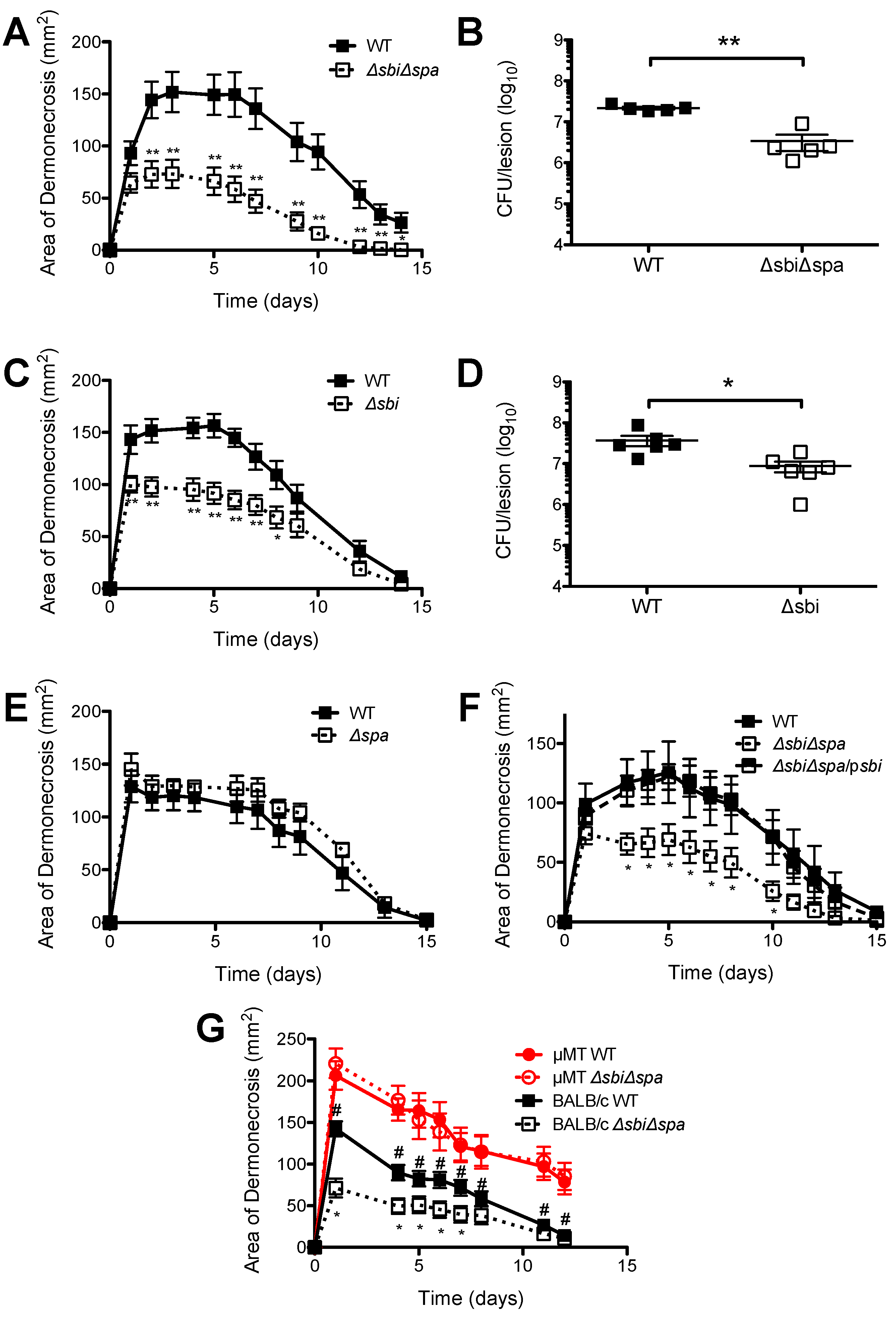
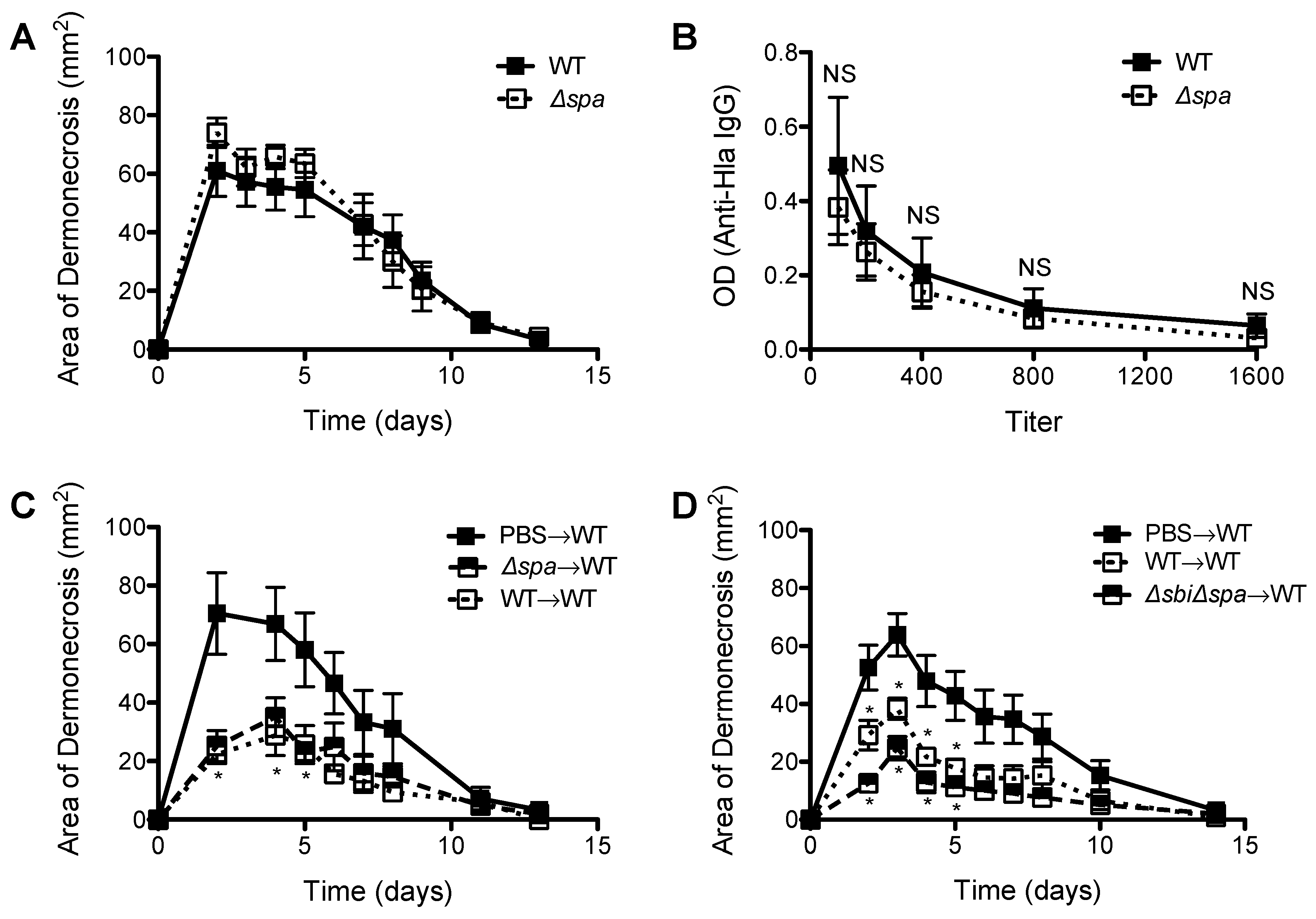
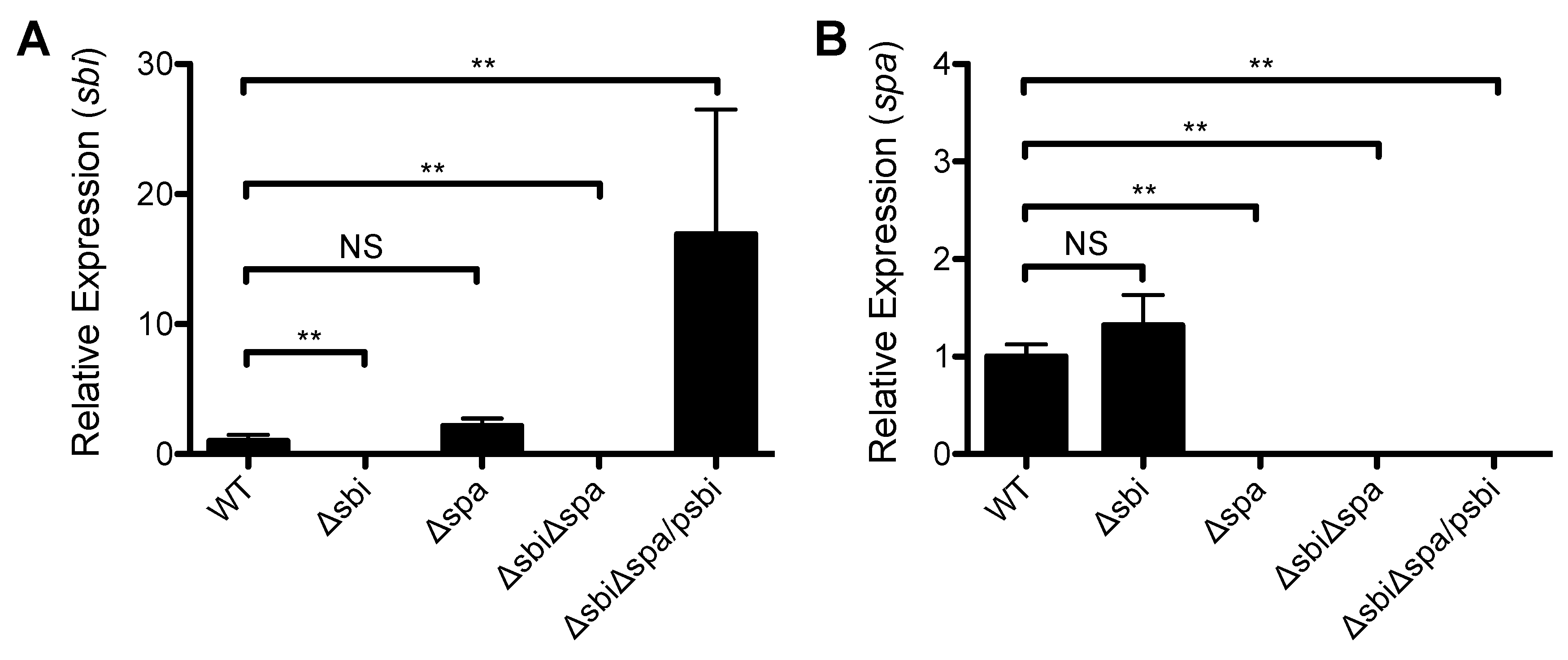
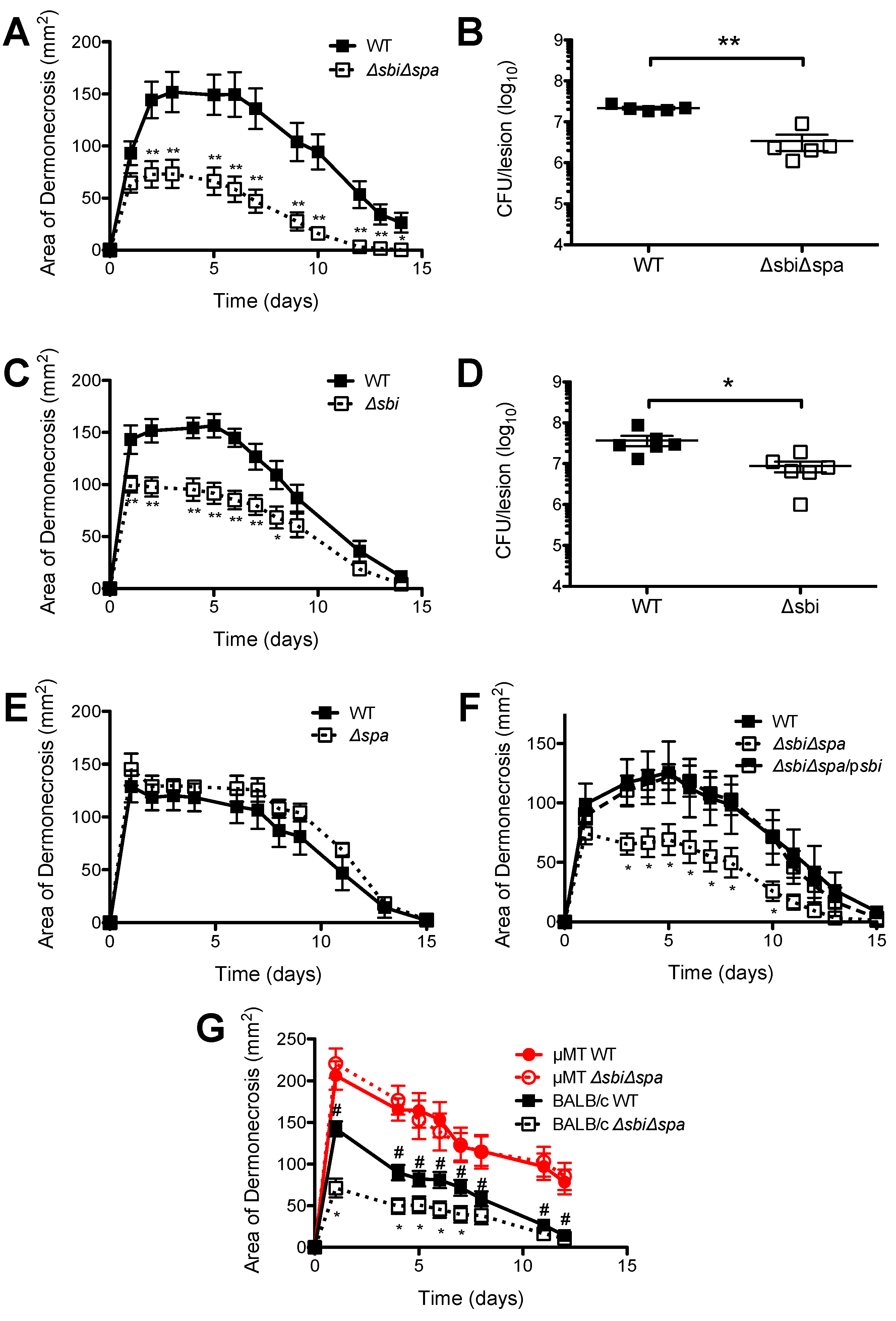
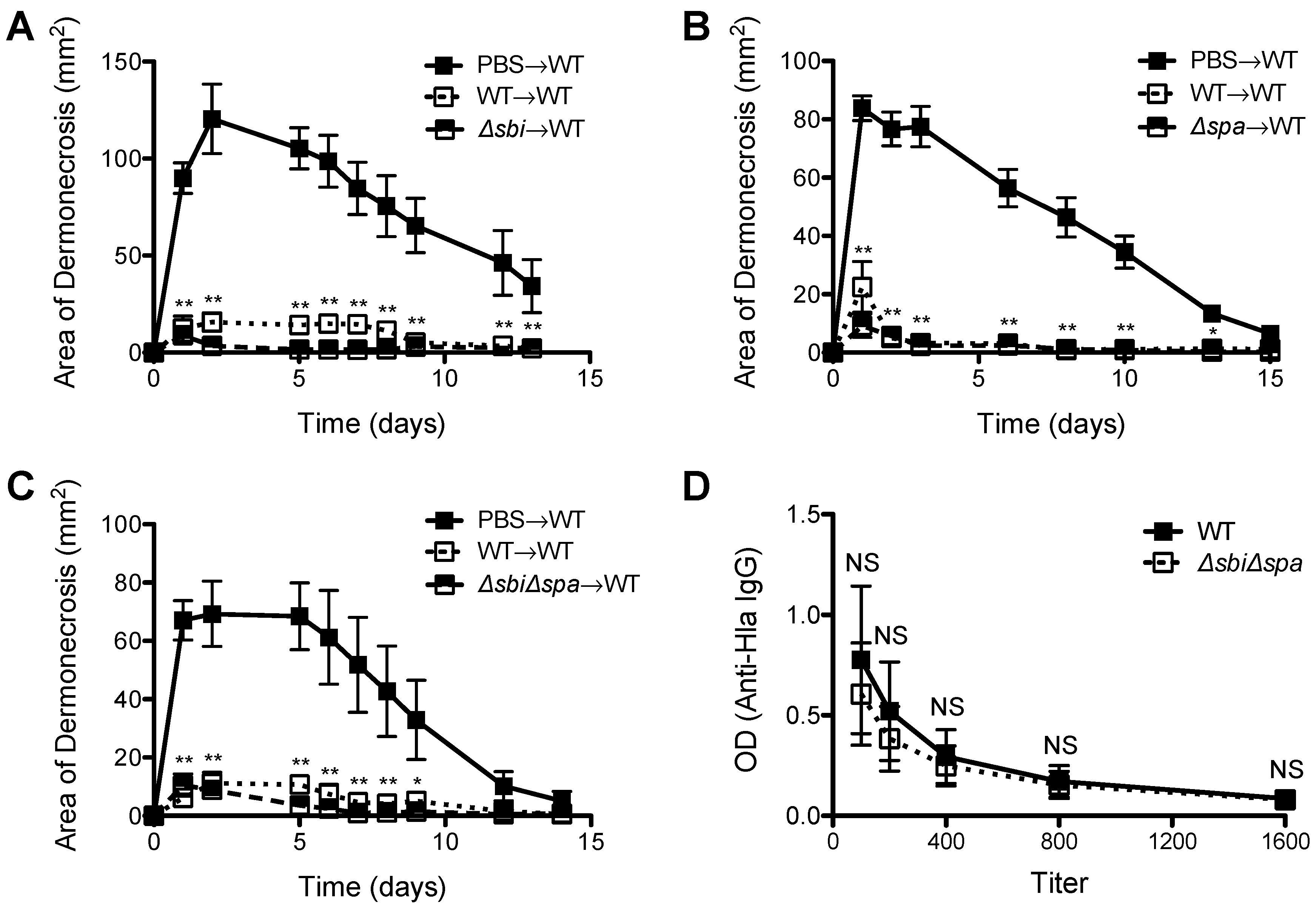
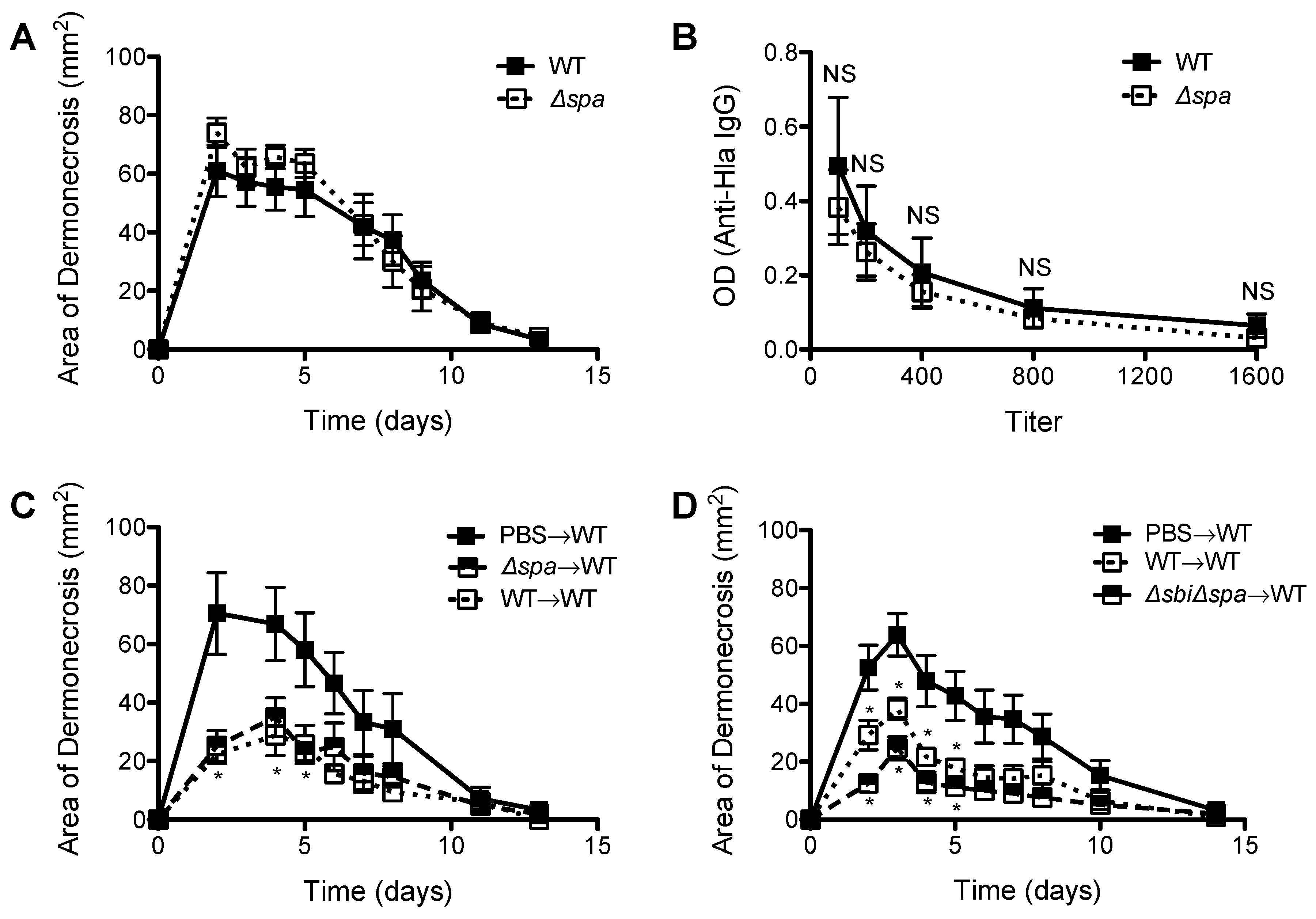
2.2. A Role for Sbi, but not SpA, in the Pathogenesis of SSTI Caused by USA300
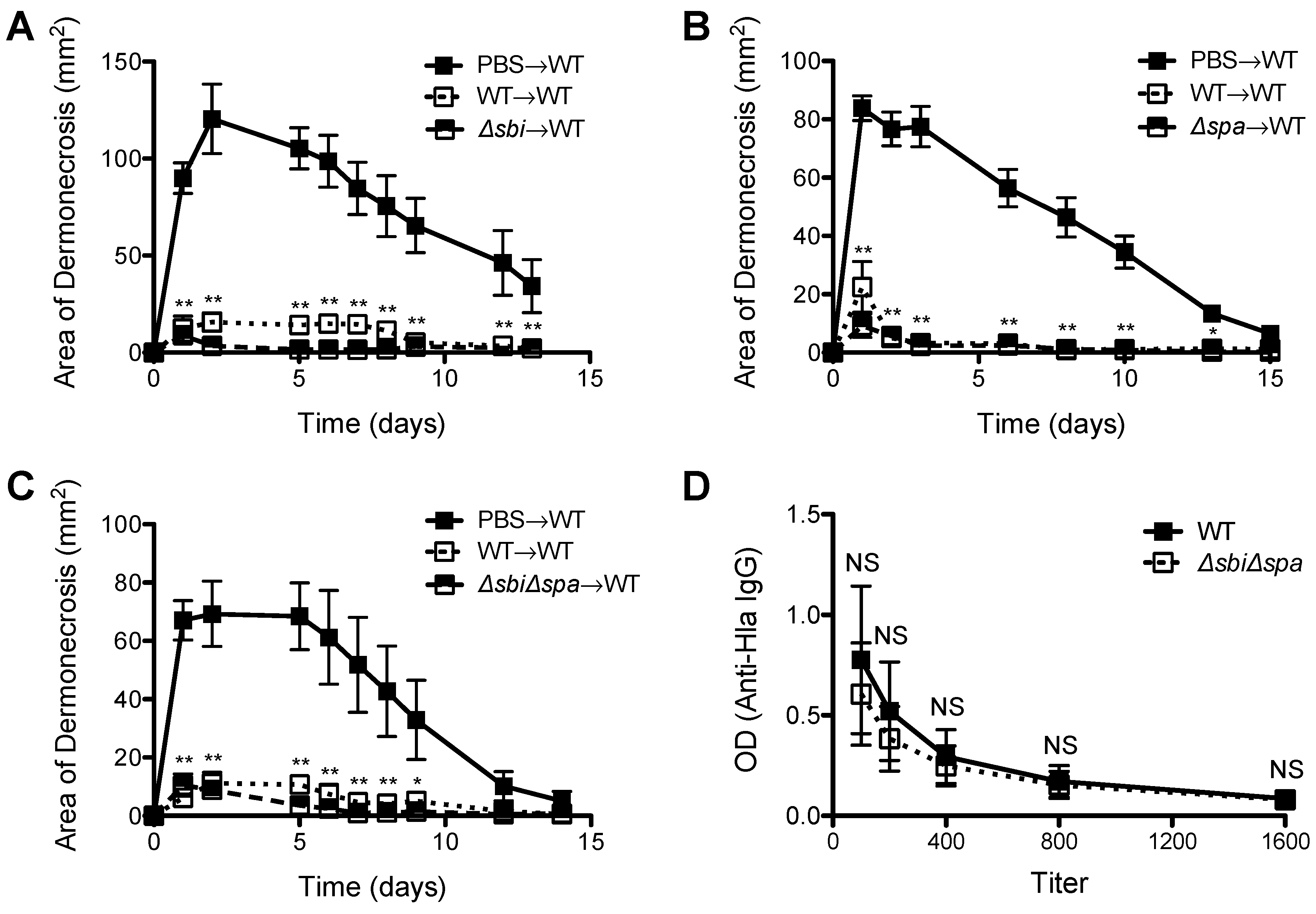
2.3. Neither Sbi nor SpA Impacted Protective Immunity against Recurrent S. aureus SSTI
3. Discussion
4. Materials and Methods
4.1. Bacterial Isolates

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sequence (5′-3′) | Application | Reference |
|---|---|---|---|
| sbi (downstream flanking region) | F—AAAGAATTCTCAATCAAAAATATCTTCTCT | deletion | This work |
| R—AAACCATGGATATATAATAATCCATTT | |||
| sbi (upstream flanking region) | F—AAAGGATCCATTAGTATAGTAACAATATTATG | deletion | This work |
| R—AAAGAATTCGTGTATTCCCTTTCTTTT | |||
| aad9 | F—AAAGAATTCATCGAATCCCTTCGTGAGCG | deletion | [33] |
| R—AAAGAATTCTAATAAACTATCGAAGGAAC | |||
| sbi | F—AAAGGATCCATGAAAAATAAATATATC | complementation | This work |
| R—AAAGAATTCTTATTTCCAGAATGATAA | |||
| sbi | F—GGGCAGCAACAATTACGTTAG | qRT-PCR | This work |
| R—TGTTTGGTGCTTAGTTGAAGTTTG | |||
| Probe—TGGGGAAGCAAAAGCGAGTGAAAAC | |||
| spa | F—TTTGTCAGCAGTAGTGCCGTTTGC | qRT-PCR | [33] |
| R—GGCAACAAGCCTGGCAAAGAAGAT | |||
| Probe—AAATGGGACGTCCAGCTGTCGAAGTT |
4.2. Expression Analysis by Semiquantitative Reverse-Transcription PCR (qRT-PCR)
4.3. Mouse Models of Primary and Secondary S. aureus SSTI
4.4. Quantification of Bacteria and Cytokines in the Skin Lesions
4.5. Serum Transfer
4.6. Assessment of Antibody Levels by ELISA
4.7. Data Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- David, M.Z.; Daum, R.S. Community-associated methicillin-resistant staphylococcus aureus: Epidemiology and clinical consequences of an emerging epidemic. Clin. Microbiol. Rev. 2010, 23, 616–687. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.G.; Eells, S.J.; David, M.Z.; Ortiz, N.; Taylor, A.R.; Kumar, N.; Cruz, D.; Boyle-Vavra, S.; Daum, R.S. Staphylococcus aureus skin infection recurrences among household members: An examination of host, behavioral, and pathogen-level predictors. Clin. Infect. Dis. 2015, 60, 753–763. [Google Scholar] [CrossRef] [PubMed]
- Milner, J.D.; Brenchley, J.M.; Laurence, A.; Freeman, A.F.; Hill, B.J.; Elias, K.M.; Kanno, Y.; Spalding, C.; Elloumi, H.Z.; Paulson, M.L.; et al. Impaired t(h)17 cell differentiation in subjects with autosomal dominant hyper-ige syndrome. Nature 2008, 452, 773–776. [Google Scholar] [CrossRef] [PubMed]
- Vyas, K.J.; Shadyab, A.H.; Lin, C.D.; Crum-Cianflone, N.F. Trends and factors associated with initial and recurrent methicillin-resistant staphylococcus aureus (mrsa) skin and soft-tissue infections among hiv-infected persons: An 18-year study. J. Int. Assoc. Prov. AIDS Care 2014, 13, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Hemmige, V.; McNulty, M.; Silverman, E.; David, M.Z. Predictors of skin and soft tissue infections in hiv-infected outpatients in the community-associated methicillin-resistant staphylococcus aureus era. Eur. J. Clin. Microbiol. Infect. Dis. 2015, 34, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Hausser, C.; Virelizier, J.L.; Buriot, D.; Griscelli, C. Common variable hypogammaglobulinemia in children. Clinical and immunologic observations in 30 patients. Am. J. Dis. Child 1983, 137, 833–837. [Google Scholar] [CrossRef] [PubMed]
- Hermaszewski, R.A.; Webster, A.D. Primary hypogammaglobulinaemia: A survey of clinical manifestations and complications. Q. J. Med. 1993, 86, 31–42. [Google Scholar] [PubMed]
- Verkaik, N.J.; Boelens, H.A.; de Vogel, C.P.; Tavakol, M.; Bode, L.G.; Verbrugh, H.A.; van Belkum, A.; van Wamel, W.J. Heterogeneity of the humoral immune response following staphylococcus aureus bacteremia. Eur. J. Clin. Microbiol. Infect. Dis. 2010, 29, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, I.P.; Dumont, A.L.; James, D.B.; Yoong, P.; Saville, B.R.; Soper, N.; Torres, V.J.; Creech, C.B. Children with invasive staphylococcus aureus disease exhibit a potently neutralizing antibody response to the cytotoxin lukab. Infect. Immun. 2014, 82, 1234–1242. [Google Scholar] [CrossRef] [PubMed]
- Fritz, S.A.; Tiemann, K.M.; Hogan, P.G.; Epplin, E.K.; Rodriguez, M.; Al-Zubeidi, D.N.; Bubeck Wardenburg, J.; Hunstad, D.A. A serologic correlate of protective immunity against community-onset staphylococcus aureus infection. Clin. Infect. Dis. 2013, 56, 1554–1561. [Google Scholar] [CrossRef] [PubMed]
- Goodyear, C.S.; Narita, M.; Silverman, G.J. In vivo vl-targeted activation-induced apoptotic supraclonal deletion by a microbial b cell toxin. J. Immunol. 2004, 172, 2870–2877. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Cheng, A.G.; Kim, H.Y.; Missiakas, D.M.; Schneewind, O. Nontoxigenic protein a vaccine for methicillin-resistant staphylococcus aureus infections in mice. J. Exp. Med. 2010, 207, 1863–1870. [Google Scholar] [CrossRef] [PubMed]
- Falugi, F.; Kim, H.K.; Missiakas, D.M.; Schneewind, O. Role of protein a in the evasion of host adaptive immune responses by staphylococcus aureus. MBio 2013, 4, e00575-13. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K.; Kim, H.Y.; Schneewind, O.; Missiakas, D. Identifying protective antigens of staphylococcus aureus, a pathogen that suppresses host immune responses. FASEB J. 2011, 25, 3605–3612. [Google Scholar] [CrossRef] [PubMed]
- Pauli, N.T.; Kim, H.K.; Falugi, F.; Huang, M.; Dulac, J.; Dunand, C.H.; Zheng, N.Y.; Kaur, K.; Andrews, S.F.; Huang, Y.; et al. Staphylococcus aureus infection induces protein a-mediated immune evasion in humans. J. Exp. Med. 2014, 211, 2331–2339. [Google Scholar] [CrossRef] [PubMed]
- Gomez, M.I.; Lee, A.; Reddy, B.; Muir, A.; Soong, G.; Pitt, A.; Cheung, A.; Prince, A. Staphylococcus aureus protein a induces airway epithelial inflammatory responses by activating tnfr1. Nat. Med. 2004, 10, 842–848. [Google Scholar] [CrossRef] [PubMed]
- Bubeck Wardenburg, J.; Patel, R.J.; Schneewind, O. Surface proteins and exotoxins are required for the pathogenesis of staphylococcus aureus pneumonia. Infect. Immun. 2007, 75, 1040–1044. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.G.; Kim, H.K.; Burts, M.L.; Krausz, T.; Schneewind, O.; Missiakas, D.M. Genetic requirements for staphylococcus aureus abscess formation and persistence in host tissues. FASEB J. 2009, 23, 3393–3404. [Google Scholar] [CrossRef] [PubMed]
- Atkins, K.L.; Burman, J.D.; Chamberlain, E.S.; Cooper, J.E.; Poutrel, B.; Bagby, S.; Jenkins, A.T.; Feil, E.J.; van den Elsen, J.M. S. Aureus igg-binding proteins spa and sbi: Host specificity and mechanisms of immune complex formation. Mol. Immunol. 2008, 45, 1600–1611. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.J.; Visai, L.; Kerrigan, S.W.; Speziale, P.; Foster, T.J. The sbi protein is a multifunctional immune evasion factor of staphylococcus aureus. Infect. Immun. 2011, 79, 3801–3809. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.J. Immune evasion by staphylococci. Nat. Rev. Microbiol. 2005, 3, 948–958. [Google Scholar] [CrossRef] [PubMed]
- Burman, J.D.; Leung, E.; Atkins, K.L.; O'Seaghdha, M.N.; Lango, L.; Bernado, P.; Bagby, S.; Svergun, D.I.; Foster, T.J.; Isenman, D.E.; et al. Interaction of human complement with sbi, a staphylococcal immunoglobulin-binding protein: Indications of a novel mechanism of complement evasion by staphylococcus aureus. J. Biol. Chem. 2008, 283, 17579–17593. [Google Scholar] [CrossRef] [PubMed]
- Clark, E.A.; Crennell, S.; Upadhyay, A.; Zozulya, A.V.; Mackay, J.D.; Svergun, D.I.; Bagby, S.; van den Elsen, J.M. A structural basis for staphylococcal complement subversion: X-ray structure of the complement-binding domain of staphylococcus aureus protein sbi in complex with ligand c3d. Mol. Immunol. 2011, 48, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Koch, T.K.; Reuter, M.; Barthel, D.; Bohm, S.; van den Elsen, J.; Kraiczy, P.; Zipfel, P.F.; Skerka, C. Staphylococcus aureus proteins sbi and efb recruit human plasmin to degrade complement c3 and c3b. PLoS ONE 2012, 7, e47638. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, C.P.; Daniels, M.; Zhao, F.; Alegre, M.L.; Chong, A.S.; Daum, R.S. Protective immunity against recurrent staphylococcus aureus skin infection requires antibody and interleukin-17a. Infect. Immun. 2014, 82, 2125–2134. [Google Scholar] [CrossRef] [PubMed]
- Bubeck Wardenburg, J.; Schneewind, O. Vaccine protection against staphylococcus aureus pneumonia. J. Exp. Med. 2008, 205, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, A.D.; Bubeck Wardenburg, J.; Gardner, D.J.; Long, D.; Whitney, A.R.; Braughton, K.R.; Schneewind, O.; DeLeo, F.R. Targeting of alpha-hemolysin by active or passive immunization decreases severity of usa300 skin infection in a mouse model. J. Infect. Dis. 2010, 202, 1050–1058. [Google Scholar] [CrossRef] [PubMed]
- Spellberg, B.; Ibrahim, A.S.; Yeaman, M.R.; Lin, L.; Fu, Y.; Avanesian, V.; Bayer, A.S.; Filler, S.G.; Lipke, P.; Otoo, H.; et al. The antifungal vaccine derived from the recombinant n terminus of als3p protects mice against the bacterium staphylococcus aureus. Infect. Immun. 2008, 76, 4574–4580. [Google Scholar] [CrossRef] [PubMed]
- Schmaler, M.; Jann, N.J.; Ferracin, F.; Landmann, R. T and b cells are not required for clearing staphylococcus aureus in systemic infection despite a strong tlr2-myd88-dependent t cell activation. J. Immunol. 2011, 186, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Archer, N.K.; Harro, J.M.; Shirtliff, M.E. Clearance of staphylococcus aureus nasal carriage is t cell dependent and mediated through interleukin-17a expression and neutrophil influx. Infect. Immun. 2013, 81, 2070–2075. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, C.P.; Daniels, M.D.; Zhao, F.; Spellberg, B.; Chong, A.S.; Daum, R.S. Local inflammation exacerbates the severity of staphylococcus aureus skin infection. PLoS ONE 2013, 8, e69508. [Google Scholar] [CrossRef] [PubMed]
- Sampedro, G.R.; DeDent, A.C.; Becker, R.E.; Berube, B.J.; Gebhardt, M.J.; Cao, H.; Bubeck Wardenburg, J. Targeting staphylococcus aureus alpha-toxin as a novel approach to reduce severity of recurrent skin and soft-tissue infections. J. Infect. Dis. 2014, 210, 1012–1018. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, C.P.; Boyle-Vavra, S.; Daum, R.S. Importance of the global regulators agr and saers in the pathogenesis of ca-mrsa usa300 infection. PLoS ONE 2010, 5, e15177. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.S.; Pietras, E.M.; Garcia, N.C.; Ramos, R.I.; Farzam, D.M.; Monroe, H.R.; Magorien, J.E.; Blauvelt, A.; Kolls, J.K.; Cheung, A.L.; et al. Il-17 is essential for host defense against cutaneous staphylococcus aureus infection in mice. J. Clin. Investig. 2010, 120, 1762–1773. [Google Scholar] [CrossRef] [PubMed]
- Miller, L.S.; O'Connell, R.M.; Gutierrez, M.A.; Pietras, E.M.; Shahangian, A.; Gross, C.E.; Thirumala, A.; Cheung, A.L.; Cheng, G.; Modlin, R.L. Myd88 mediates neutrophil recruitment initiated by il-1r but not tlr2 activation in immunity against staphylococcus aureus. Immunity 2006, 24, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Kelly-Scumpia, K.M.; Scumpia, P.O.; Weinstein, J.S.; Delano, M.J.; Cuenca, A.G.; Nacionales, D.C.; Wynn, J.L.; Lee, P.Y.; Kumagai, Y.; Efron, P.A.; et al. B cells enhance early innate immune responses during bacterial sepsis. J. Exp. Med. 2011, 208, 1673–1682. [Google Scholar] [CrossRef] [PubMed]
- Kwiecinski, J.; Jin, T.; Josefsson, E. Surface proteins of staphylococcus aureus play an important role in experimental skin infection. Apmis 2014, 122, 1240–1250. [Google Scholar] [CrossRef] [PubMed]
- Soong, G.; Chun, J.; Parker, D.; Prince, A. Staphylococcus aureus activation of caspase 1/calpain signaling mediates invasion through human keratinocytes. J. Infect. Dis. 2012, 205, 1571–1579. [Google Scholar] [CrossRef] [PubMed]
- Prabhakara, R.; Foreman, O.; De Pascalis, R.; Lee, G.M.; Plaut, R.D.; Kim, S.Y.; Stibitz, S.; Elkins, K.L.; Merkel, T.J. Epicutaneous model of community-acquired staphylococcus aureus skin infections. Infect. Immun. 2013, 81, 1306–1315. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Cheng, B.L.; Boyle-Vavra, S.; Alegre, M.L.; Daum, R.S.; Chong, A.S.; Montgomery, C.P. Proteomic identification of saers-dependent targets critical for protective humoral immunity against staphylococcus aureus skin infection. Infect. Immun. 2015, 83, 3712–3721. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, C.P.; Boyle-Vavra, S.; Adem, P.V.; Lee, J.C.; Husain, A.N.; Clasen, J.; Daum, R.S. Comparison of virulence in community-associated methicillin-resistant staphylococcus aureus pulsotypes usa300 and usa400 in a rat model of pneumonia. J. Infect. Dis. 2008, 198, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Arnaud, M.; Chastanet, A.; Debarbouille, M. New vector for efficient allelic replacement in naturally nontransformable, low-gc-content, gram-positive bacteria. Appl. Environ. Microbiol. 2004, 70, 6887–6891. [Google Scholar] [CrossRef] [PubMed]
- Hartleib, J.; Kohler, N.; Dickinson, R.B.; Chhatwal, G.S.; Sixma, J.J.; Hartford, O.M.; Foster, T.J.; Peters, G.; Kehrel, B.E.; Herrmann, M. Protein a is the von willebrand factor binding protein on staphylococcus aureus. Blood 2000, 96, 2149–2156. [Google Scholar] [PubMed]
- Charpentier, E.; Anton, A.I.; Barry, P.; Alfonso, B.; Fang, Y.; Novick, R.P. Novel cassette-based shuttle vector system for gram-positive bacteria. Appl. Environ. Microbiol. 2004, 70, 6076–6085. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, F.; Chong, A.S.; Montgomery, C.P. Importance of B Lymphocytes and the IgG-Binding Protein Sbi in Staphylococcus aureus Skin Infection. Pathogens 2016, 5, 12. https://doi.org/10.3390/pathogens5010012
Zhao F, Chong AS, Montgomery CP. Importance of B Lymphocytes and the IgG-Binding Protein Sbi in Staphylococcus aureus Skin Infection. Pathogens. 2016; 5(1):12. https://doi.org/10.3390/pathogens5010012
Chicago/Turabian StyleZhao, Fan, Anita S. Chong, and Christopher P. Montgomery. 2016. "Importance of B Lymphocytes and the IgG-Binding Protein Sbi in Staphylococcus aureus Skin Infection" Pathogens 5, no. 1: 12. https://doi.org/10.3390/pathogens5010012