
Human Hemorrhagic Fever Causing Arenaviruses: Molecular Mechanisms Contributing to Virus Virulence and Disease Pathogenesis

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
3. Basic Biology of Arenaviruses
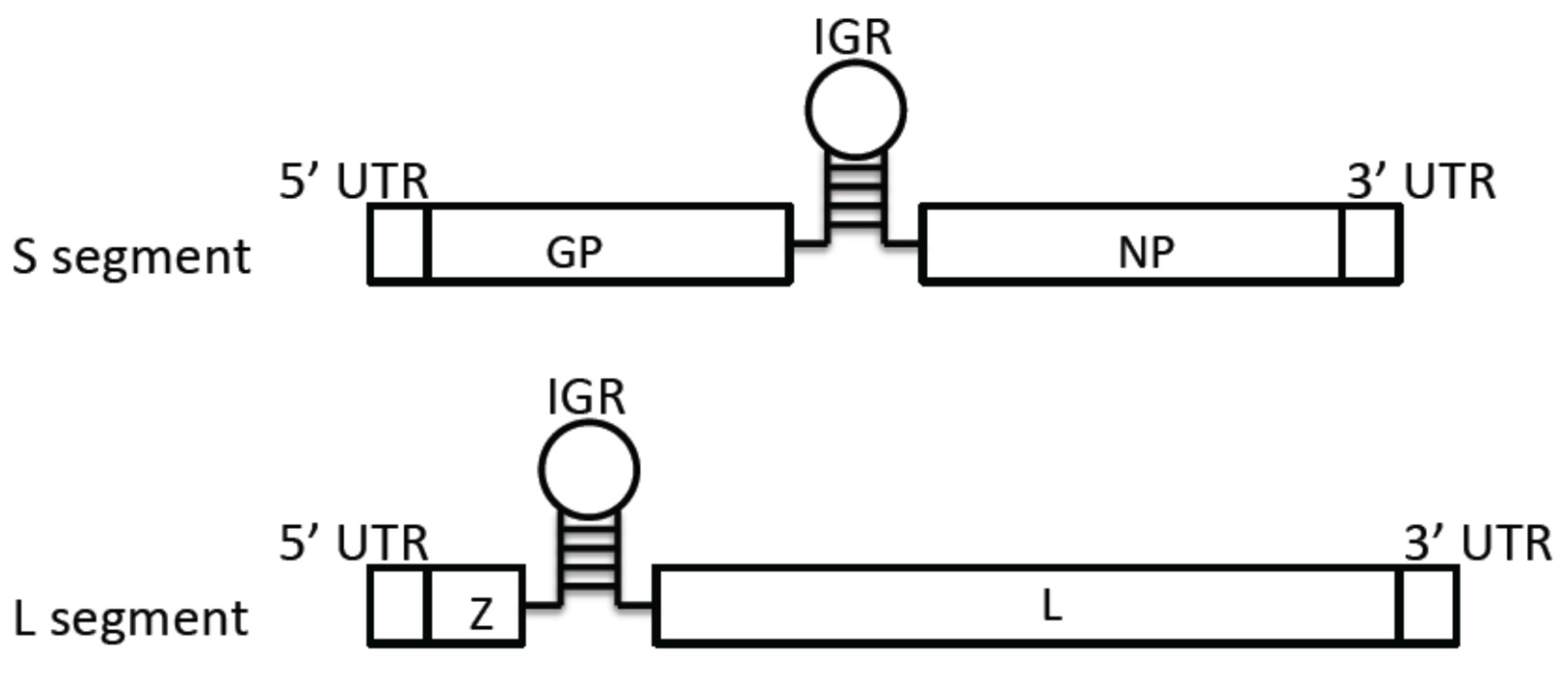
3.1. Genome Structure
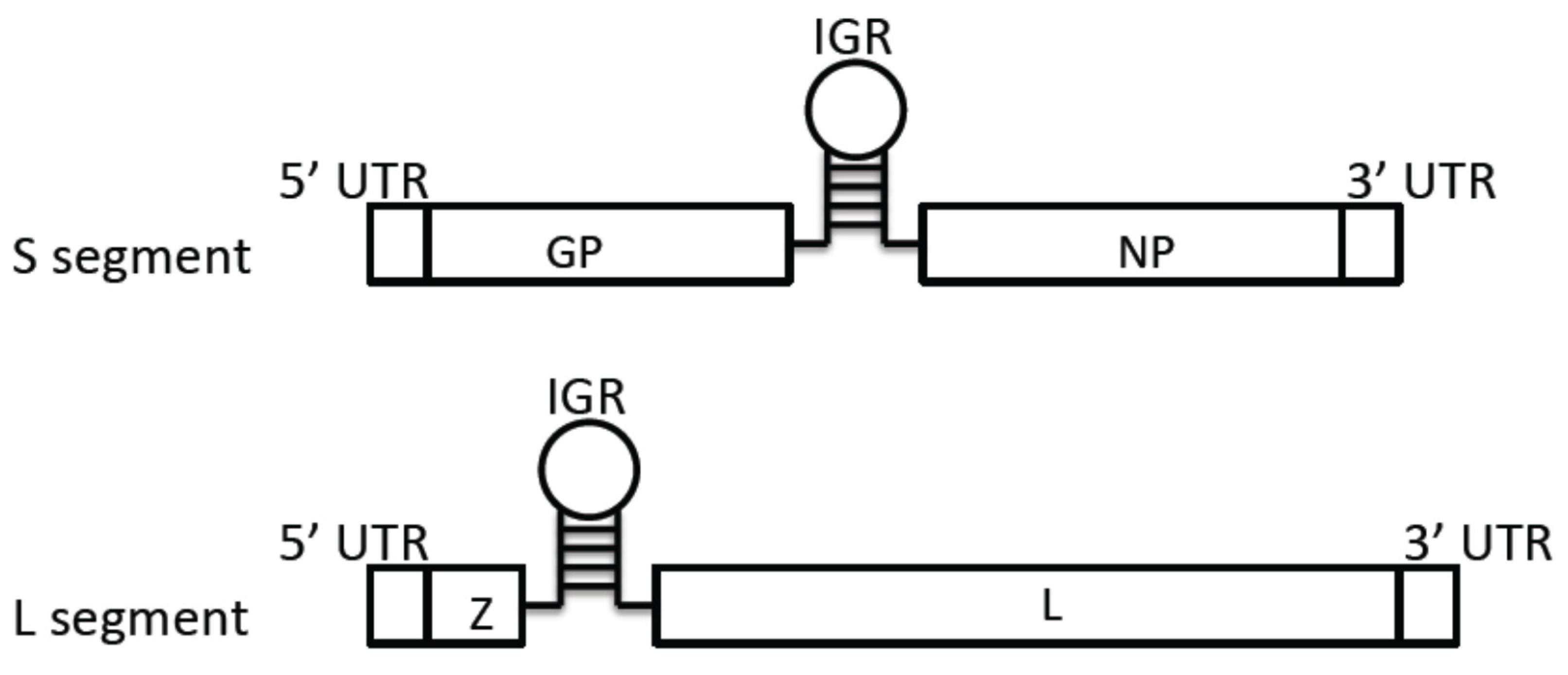
3.2. Virus Entry Mechanisms

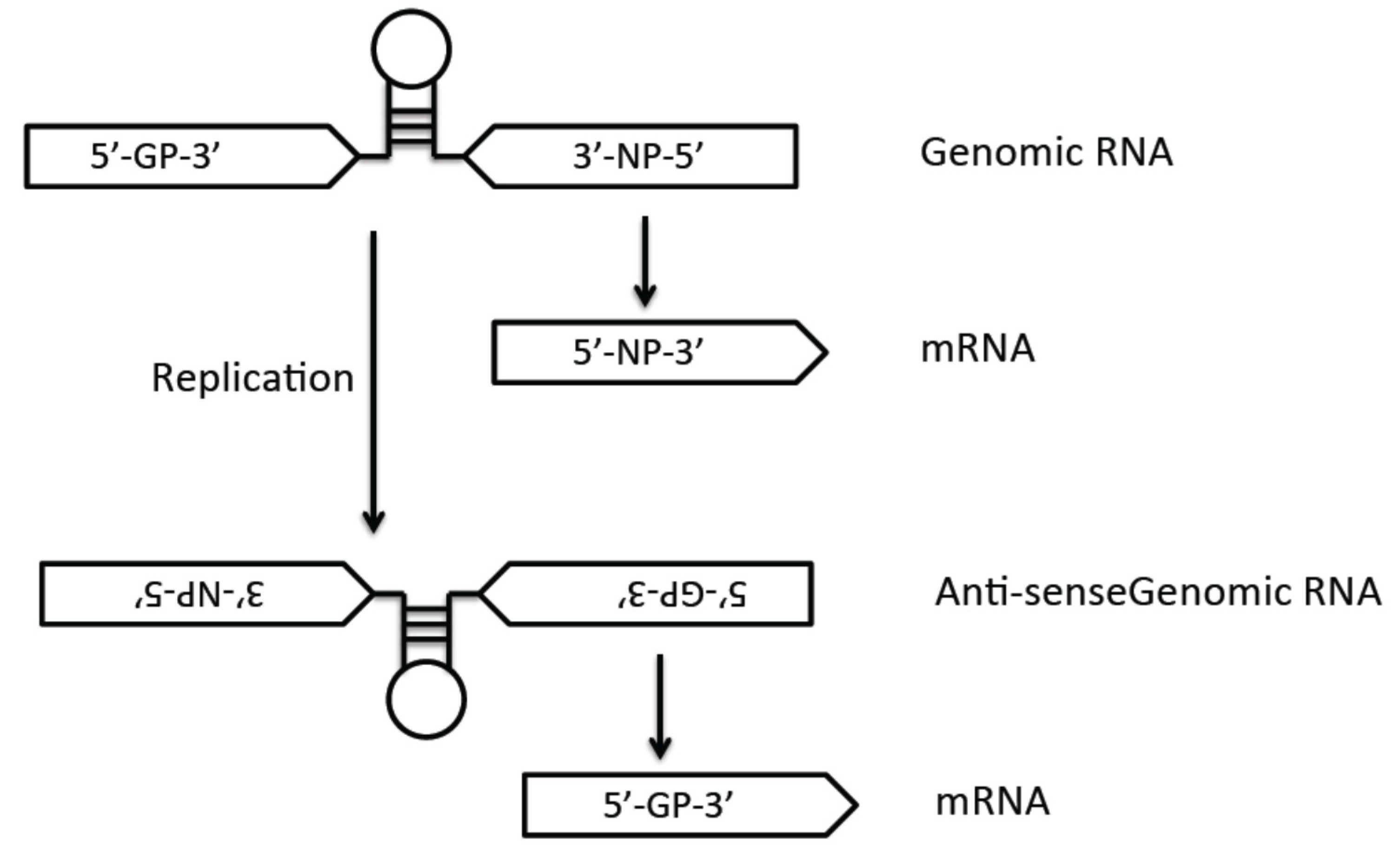
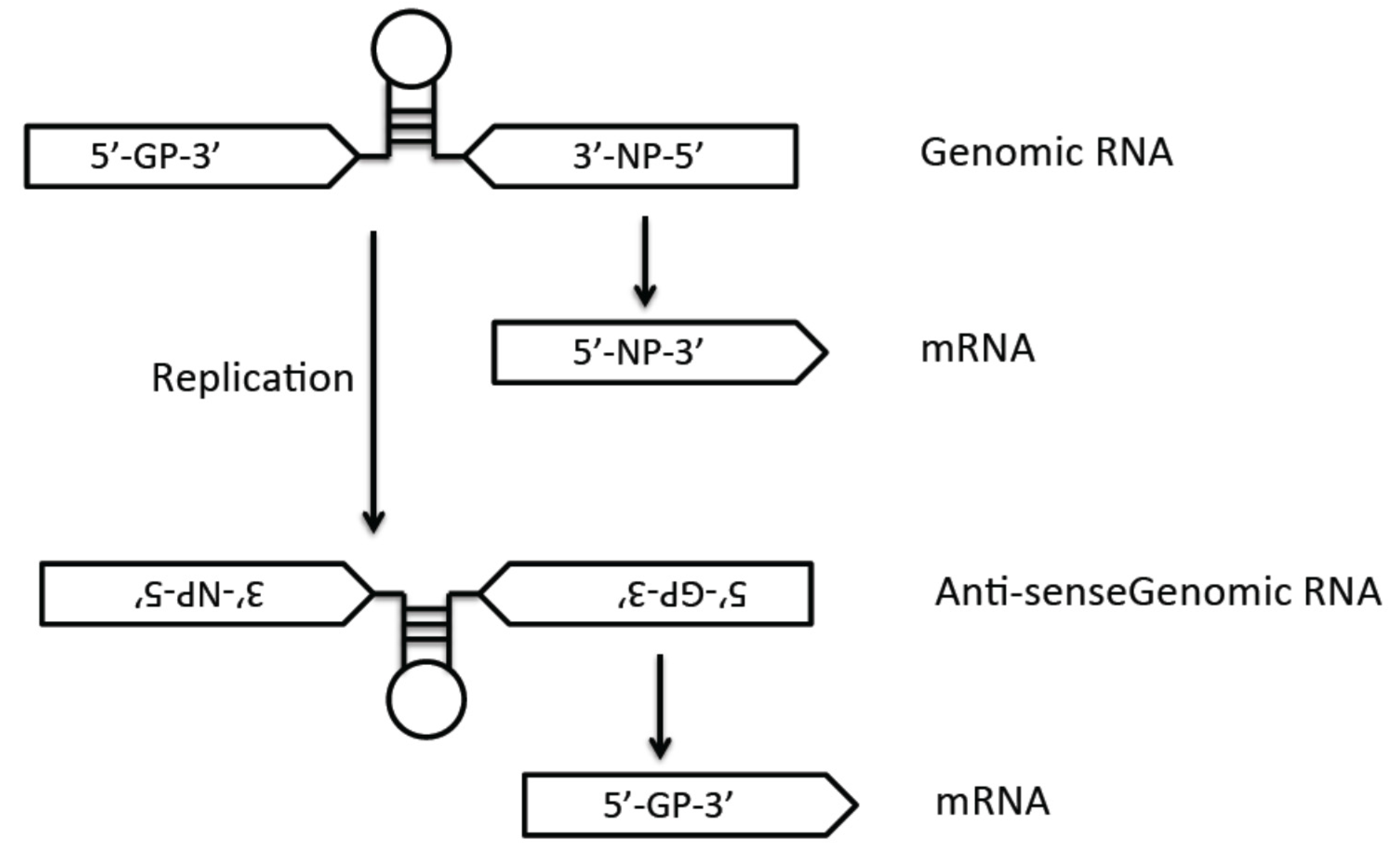
3.3. Viral Genome Replication and Transcription
3.4. Post-Translational Protein Processing
3.5. Virion Assembly
4. Molecular Mechanisms Contributing to Virus Virulence and HF Disease Pathogenesis
4.1. Roles of the Glycoprotein (GP) and of the Host Cell Receptors in HF Disease Susceptibility and Pathogenesis
4.2. Role of the Polymerase (L) Protein in Viral Virulence
4.3. Role of the Nucleoprotein (NP) in Innate Immune Suppression
4.4. Roles of the Z Protein in Innate Immune Suppression and Viral Pathogenicity
6. Conclusions
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Maiztegui, J.I.; McKee, K.T.; Oro, J.G.B.; Harrison, L.H.; Gibbs, P.H.; Feuillade, M.R.; Enria, D.A.; Briggiler, A.M.; Levis, S.C.; Ambrosio, A.M.; et al. Protective efficacy of a live attenuated vaccine against Argentine hemorrhagic fever. J. Infect. Dis. 1998, 177, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Günther, S.; Lenz, O. Lassa virus. Crit. Rev. Clin. Lab. Sci. 2004, 41, 339–390. [Google Scholar] [CrossRef] [PubMed]
- Zong, M.; Fofana, I.; Choe, H. Human and host species transferrin receptor 1 use by North American arenaviruses. J. Virol. 2014, 88, 9418–9428. [Google Scholar] [CrossRef] [PubMed]
- McLay, L.; Liang, Y.; Ly, H. Comparative analysis of disease pathogenesis and molecular mechanisms of New World and Old World arenavirus infections. J. Gen. Virol. 2014, 95, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Radoshitzky, S.R.; Bao, Y.; Buchmeier, M.J.; Charrel, R.N.; Clawson, A.N.; Clegg, C.S.; DeRisi, J.L.; Emonet, S.; Gonzalez, J.P.; Kuhn, J.H.; et al. Past, present, and future of arenavirus taxonomy. Arch. Virol. 2015. [Google Scholar]
- Moraz, M.-L.; Kunz, S. Pathogenesis of arenavirus hemorrhagic fevers. Expert Rev. Anti-infective Ther. 2010, 9, 49–59. [Google Scholar] [CrossRef]
- Cummins, D.; McCormick, J.B.; Bennett, D.; Samba, J.A.; Farrar, B.; Machin, S.J.; Fisher-Hoch, S.P. Acute sensorineural deafness in Lassa fever. JAMA: J. Am. Med. Assoc. 1990, 264, 2093–2096. [Google Scholar] [CrossRef]
- Briese, T.; Paweska, J.T.; McMullan, L.K.; Hutchison, S.K.; Street, C.; Palacios, G.; Khristova, M.L.; Weyer, J.; Swanepoel, R.; Egholm, M.; et al. Genetic detection and characterization of Lujo virus, a new hemorrhagic fever–associated arenavirus from Southern Africa. PLoS Pathogens 2009, 5, e1000455. [Google Scholar] [CrossRef] [PubMed]
- Paweska, J.T.; Sewlall, N.H.; Ksiazek, T.G.; Blumberg, L.H.; Hale, M.J.; Lipkin, W.I.; Weyer, J.; Nichol, S.T.; Rollin, P.E.; McMullan, L.K.; et al. Nosocomial outbreak of novel arenavirus infection, Southern Africa. Emerg. Infect. Dis. 2009, 15, 1598–1602. [Google Scholar] [CrossRef]
- Peters, C.J. Lymphocytic choriomeningitis virus — an old enemy up to new tricks. N. Engl. J. Med. 2006, 354, 2208–2211. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, M.C.; Saron, M.F.; Brouqui, P.; Bourgeade, A. Lymphocytic choriomeningitis virus in Southern France: Four case reports and a review of the literature. Eur. J. Epidemiol. 1997, 13, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Wright, R.; Johnson, D.; Neumann, M.; Ksiazek, T.G.; Rollin, P.; Keech, R.V.; Bonthius, D.J.; Hitchon, P.; Grose, C.F.; Bell, W.E.; et al. Congenital lymphocytic choriomeningitis virus syndrome: A disease that mimics congenital toxoplasmosis or cytomegalovirus infection. Pediatrics 1997, 100, e9. [Google Scholar] [CrossRef] [PubMed]
- Bonthius, D.J. Lymphocytic choriomeningitis virus: An underrecognized cause of neurologic disease in the fetus, child, and adult. Semin. Pediatr. Neurol. 2012, 19, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Fischer, S.A.; Graham, M.B.; Kuehnert, M.J.; Kotton, C.N.; Srinivasan, A.; Marty, F.M.; Comer, J.A.; Guarner, J.; Paddock, C.D.; DeMeo, D.L.; et al. Transmission of lymphocytic choriomeningitis virus by organ transplantation. N. Engl. J. Med. 2006, 354, 2235–2249. [Google Scholar] [CrossRef] [PubMed]
- MacNeil, A.; Stroher, U.; Farnon, E.; Campbell, S.; Cannon, D.; Paddock, C.D.; Drew, C.P.; Kuehnert, M.; Knust, B.; Gruenenfelder, R.; et al. Solid organ transplant-associated lymphocytic choriomeningitis, United States, 2011. Emerg. Infect. Dis. 2012, 18, 1256–1262. [Google Scholar] [CrossRef] [PubMed]
- Prevention, C.F.D.C.A. Brief report: Lymphocytic choriomeningitis virus transmitted through solid organ transplantation - Massachussetts, 2008. MMWR Morb Mortal Wkly Rep. 2008, 57, 799–801. [Google Scholar] [PubMed]
- Delgado, S.; Erickson, B.R.; Agudo, R.; Blair, P.J.; Vallejo, E.; Albariño, C.G.; Vargas, J.; Comer, J.A.; Rollin, P.E.; Ksiazek, T.G.; et al. Chapare virus, a newly discovered arenavirus isolated from a fatal hemorrhagic fever case in Bolivia. PLoS Pathogens 2008, 4, e1000047. [Google Scholar] [CrossRef] [PubMed]
- Lisieux, T.; Coimbra, M.; Nassar, E.S.; Burattini, M.N.; Souza, L.T.M.D.; Ferreira, I.B.; Rocco, I.M.; Rosa, A.P.A.T.D.; Vasconcelos, P.F.C.; Pinheiro, F.P.; et al. New arenavirus isolated in Brazil. Lancet 1994, 343, 391–392. [Google Scholar]
- Aguilar, P.V.; Camargo, W.; Vargas, J.; Guevara, C.; Roca, Y.; Felices, V.; Laguna-Torres, V.A.; Tesh, R.; Ksiazek, T.G.; Kochel, T.J. Reemergence of Bolivian hemorrhagic fever, 2007–2008. Emerg. Infect. Dis. 2009, 15, 1526–1528. [Google Scholar] [CrossRef] [PubMed]
- Ambrosio, A.; Saavedra, M.; Mariani, M.; Gamboa, G.; Maiza, A. Argentine hemorrhagic fever vaccines. Hum. Vaccines 2011, 7, 694–700. [Google Scholar] [CrossRef]
- Charrel, R.N.; Lamballerie, X.D. Arenaviruses other than Lassa virus. Antivir. Res. 2003, 57, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Enria, D.A.; Briggiler, A.M.; Sánchez, Z. Treatment of Argentine hemorrhagic fever. Antivir. Res. 2008, 78, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Fulhorst, C.F.; Cajimat, M.N.B.; Milazzo, M.L.; Paredes, H.; de Manzione, N.M.C.; Salas, R.A.; Rollin, P.E.; Ksiazek, T.G. Genetic diversity between and within the arenavirus species indigenous to Western Venezuela. Virology 2008, 378, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Harrison, L.H.; Halsey, N.A.; McKee, K.T.; Peters, C.J.; Barrera Oro, J.G.; Briggiler, A.M.; Feuillade, M.R.; Maiztegui, J.I. Clinical case definitions for Argentine hemorrhagic fever. Clin. Infect. Dis. 1999, 28, 1091–1094. [Google Scholar] [CrossRef] [PubMed]
- Manzione, N.D.; Salas, R.A.; Paredes, H.; Godoy, O.; Rojas, L.; Araoz, F.; Fulhorst, C.F.; Ksiazek, T.G.; Mills, J.N.; Ellis, B.A.; et al. Venezuelan hemorrhagic fever: Clinical and epidemiological studies of 165 cases. Clin. Infect. Dis. 1998, 26, 308–313. [Google Scholar] [CrossRef] [PubMed]
- Pfau, C.J. Arenaviruses. In Medical Microbiology, 4th ed.; Baron, S., Ed.; The University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996. [Google Scholar]
- Meyer, B.J.; de la Torre, J.C.; Southern, P.J. Arenaviruses: Genomic RNAs, transcription, and replication. Curr. Top. Microbiol. Immunol. 2002, 262, 139–157. [Google Scholar] [PubMed]
- Knipe, D.M.H.; Peter, M. Fields Virology, 5th ed.; Lippincott Williams & Wilkins: Philadelphia, USA, 2007. [Google Scholar]
- Cao, W.; Henry, M.D.; Borrow, P.; Yamada, H.; Elder, J.H.; Ravkov, E.V.; Nichol, S.T.; Compans, R.W.; Campbell, K.P.; Oldstone, M.B. Identification of alpha-dystroglycan as a receptor for lymphocytic choriomeningitis virus and Lassa fever virus. Science 1998, 282, 2079–2081. [Google Scholar] [CrossRef] [PubMed]
- Spiropoulou, C.F.; Kunz, S.; Rollin, P.E.; Campbell, K.P.; Oldstone, M.B. New world arenavirus Clade C, but not Clade A and B viruses, utilizes alpha-dystroglycan as its major receptor. J. Virol. 2002, 76, 5140–5146. [Google Scholar] [CrossRef] [PubMed]
- Kunz, S.; Campbell, K.P.; Oldstone, M.B. Alpha-dystroglycan can mediate arenavirus infection in the absence of beta-dystroglycan. Virology 2003, 316, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Rojek, J.M.; Spiropoulou, C.F.; Campbell, K.P.; Kunz, S. Old World and Clade C New World arenaviruses mimic the molecular mechanism of receptor recognition used by α-dystroglycan's host-derived ligands. J. Virol. 2007, 81, 5685–5695. [Google Scholar] [CrossRef] [PubMed]
- Imperiali, M.; Sporri, R.; Hewitt, J.; Oxenius, A. Post-translational modification of {alpha}-dystroglycan is not critical for lymphocytic choriomeningitis virus receptor function in vivo. J. Gen. Virol. 2008, 89, 2713–2722. [Google Scholar] [CrossRef] [PubMed]
- Kunz, S.; Sevilla, N.; McGavern, D.B.; Campbell, K.P.; Oldstone, M.B. Molecular analysis of the interaction of LCMV with its cellular receptor [alpha]-dystroglycan. J. Cell Biol. 2001, 155, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Kunz, S.; Rojek, J.M.; Perez, M.; Spiropoulou, C.F.; Oldstone, M.B. Characterization of the interaction of Lassa fever virus with its cellular receptor alpha-dystroglycan. J. Virol. 2005, 79, 5979–5987. [Google Scholar] [CrossRef] [PubMed]
- Kunz, S.; Calder, L.; Oldstone, M.B. Electron microscopy of an alpha-dystroglycan fragment containing receptor sites for lymphocytic choriomeningitis virus and laminin, and use of the receptoid body as a reagent to neutralize virus. Virology 2004, 325, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Rojek, J.M.; Campbell, K.P.; Oldstone, M.B.A.; Kunz, S. Old World arenavirus infection interferes with the expression of functional α-dystroglycan in the host cell. Mol. Biol. Cell 2007, 18, 4493–4507. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.; McCormick, J.; Johnson, K.; Webb, P.; Komba-Kono, G.; Elliott, L.; Gardner, J. Pathologic and virologic study of fatal Lassa fever in man. Am. J. Pathol. 1982, 107, 349–356. [Google Scholar] [PubMed]
- Shimojima, M.; Ströher, U.; Ebihara, H.; Feldmann, H.; Kawaoka, Y. Identification of cell surface molecules involved in dystroglycan-independent Lassa virus cell entry. J. Virol. 2012, 86, 2067–2078. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, B.M.; Welch, M.J.; Lemke, G.; Oldstone, M.B. Is the TAM receptor Axl A receptor for lymphocytic choriomeningitis virus? J. Virol. 2013, 87, 4071–4074. [Google Scholar] [CrossRef] [PubMed]
- Beier, J.I.; Jokinen, J.D.; Holz, G.E.; Whang, P.S.; Martin, A.M.; Warner, N.L.; Arteel, G.E.; Lukashevich, I.S. Novel mechanism of arenavirus-induced liver pathology. PloS ONE 2015, 10, e0122839. [Google Scholar] [CrossRef] [PubMed]
- Ibraghimov-Beskrovnaya, O.; Milatovich, A.; Ozcelik, T.; Yang, B.; Koepnick, K.; Francke, U.; Campbell, K.P. Human dystroglycan: Skeletal muscle cDNA, genomic structure, origin of tissue specific isoforms and chromosomal localization. Hum. Mol. Genet. 1993, 2, 1651–1657. [Google Scholar] [CrossRef] [PubMed]
- Lukashevich, I.S. Reproduction of Lassa virus in different cell cultures. Acta Virol. 1983, 27, 282–285. [Google Scholar] [PubMed]
- Jae, L.T.; Raaben, M.; Herbert, A.S.; Kuehne, A.I.; Wirchnianski, A.S.; Soh, T.K.; Stubbs, S.H.; Janssen, H.; Damme, M.; Saftig, P.; et al. Virus entry. Lassa virus entry requires a trigger-induced receptor switch. Science 2014, 344, 1506–1510. [Google Scholar] [CrossRef] [PubMed]
- Radoshitzky, S.R.; Abraham, J.; Spiropoulou, C.F.; Kuhn, J.H.; Nguyen, D.; Li, W.; Nagel, J.; Schmidt, P.J.; Nunberg, J.H.; Andrews, N.C.; et al. Transferrin receptor 1 is a cellular receptor for New World haemorrhagic fever arenaviruses. Nature 2007, 446, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, M.L.; Oldenburg, J.; Reignier, T.; Holt, N.; Hamilton, G.A.; Martin, V.K.; Cannon, P.M. New World Clade B arenaviruses can use transferrin receptor 1 (TfR1)-dependent and -independent entry pathways, and glycoproteins from human pathogenic strains are associated with the use of TfR1. J. Virol. 2008, 82, 938–948. [Google Scholar] [CrossRef] [PubMed]
- Rojek, J.M.; Kunz, S. Cell entry by human pathogenic arenaviruses. Cell. Microbiol. 2008, 10, 828–835. [Google Scholar] [CrossRef] [PubMed]
- Helguera, G.; Jemielity, S.; Abraham, J.; Cordo, S.M.; Martinez, M.G.; Rodriguez, J.A.; Bregni, C.; Wang, J.J.; Farzan, M.; Penichet, M.L.; et al. An antibody recognizing the apical domain of human transferrin receptor 1 efficiently inhibits the entry of all New World hemorrhagic fever arenaviruses. J. Virol. 2012, 86, 4024–4028. [Google Scholar] [CrossRef] [PubMed]
- Radoshitzky, S.R.; Kuhn, J.H.; Spiropoulou, C.F.; Albarino, C.G.; Nguyen, D.P.; Salazar-Bravo, J.; Dorfman, T.; Lee, A.S.; Wang, E.; Ross, S.R.; et al. Receptor determinants of zoonotic transmission of New World hemorrhagic fever arenaviruses. Proc. Natl. Acad. Sci. USA 2008, 105, 2664–2669. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention (CDC). Fatal illnesses associated with a New World arenavirus—California, 1999–2000. MMWR Morb Mortal Wkly Rep. 2000, 49, 709–711. [Google Scholar]
- Enserink, M. Emerging diseases. New arenavirus blamed for recent deaths in California. Science 2000, 289, 842–843. [Google Scholar]
- Milazzo, M.L.; Campbell, G.L.; Fulhorst, C.F. Novel arenavirus infection in humans, United States. Emerg. Infect. Dis. 2011, 17, 1417–1420. [Google Scholar] [CrossRef] [PubMed]
- Hueffer, K.; Parker, J.S.; Weichert, W.S.; Geisel, R.E.; Sgro, J.Y.; Parrish, C.R. The natural host range shift and subsequent evolution of canine parvovirus resulted from virus-specific binding to the canine transferrin receptor. J. Virol. 2003, 77, 1718–1726. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.R.; Schofield, J.J.; Farr, C.J.; Bucan, M. Mouse transferrin receptor 1 is the cell entry receptor for mouse mammary tumor virus. Proc. Natl. Acad. Sci. USA 2002, 99, 12386–12390. [Google Scholar] [CrossRef] [PubMed]
- Demogines, A.; Abraham, J.; Choe, H.; Farzan, M.; Sawyer, S.L. Dual host-virus arms races shape an essential housekeeping protein. PLoS Biol. 2013, 11, e1001571. [Google Scholar] [CrossRef] [PubMed]
- Choe, H.; Jemielity, S.; Abraham, J.; Radoshitzky, S.R.; Farzan, M. Transferrin receptor 1 in the zoonosis and pathogenesis of New World hemorrhagic fever arenaviruses. Curr. Opin. Microbiol. 2011, 14, 476–482. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.G.; Bialecki, M.A.; Belouzard, S.; Cordo, S.M.; Candurra, N.A.; Whittaker, G.R. Utilization of human DC-SIGN and l-SIGN for entry and infection of host cells by the New World arenavirus, Junin virus. Biochem. Biophys. Res. Commun. 2013, 441, 612–617. [Google Scholar] [CrossRef] [PubMed]
- Eschli, B.; Quirin, K.; Wepf, A.; Weber, J.; Zinkernagel, R.; Hengartner, H. Identification of an N-terminal trimeric coiled-coil core within arenavirus glycoprotein 2 permits assignment to class I viral fusion proteins. J. Virol. 2006, 80, 5897–5907. [Google Scholar] [CrossRef] [PubMed]
- Gallaher, W.R.; DiSimone, C.; Buchmeier, M.J. The viral transmembrane superfamily: Possible divergence of arenavirus and filovirus glycoproteins from a common RNA virus ancestor. BMC Microbiol. 2001, 1, 1. [Google Scholar] [CrossRef] [PubMed]
- York, J.; Nunberg, J.H. Distinct requirements for signal peptidase processing and function in the stable signal peptide subunit of the Junín virus envelope glycoprotein. Virology 2007, 359, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Quirin, K.; Eschli, B.; Scheu, I.; Poort, L.; Kartenbeck, J.; Helenius, A. Lymphocytic choriomeningitis virus uses a novel endocytic pathway for infectious entry via late endosomes. Virology 2008, 378, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Pasqual, G.; Rojek, J.M.; Masin, M.; Chatton, J.-Y.; Kunz, S. Old World arenaviruses enter the host cell via the multivesicular body and depend on the endosomal sorting complex required for transport. PLoS Pathogens 2011, 7, e1002232. [Google Scholar] [CrossRef] [PubMed]
- Hass, M.; Gölnitz, U.; Müller, S.; Becker-Ziaja, B.; Günther, S. Replicon system for Lassa virus. J. Virol. 2004, 78, 13793–13803. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.J.; Novella, I.S.; Teng, M.N.; Oldstone, M.B.A.; de la Torre, J.C. NP and L proteins of lymphocytic choriomeningitis virus (LCMV) are sufficient for efficient transcription and replication of LCMV genomic RNA analogs. J. Virol. 2000, 74, 3470–3477. [Google Scholar] [CrossRef] [PubMed]
- López, N.; Jácamo, R.; Franze-Fernández, M.T. Transcription and RNA replication of Tacaribe virus genome and antigenome analogs require N and L proteins: Z protein is an inhibitor of these processes. J. Virol. 2001, 75, 12241–12251. [Google Scholar] [CrossRef] [PubMed]
- Vieth, S.; Torda, A.E.; Asper, M.; Schmitz, H.; Günther, S. Sequence analysis of L RNA of Lassa virus. Virology 2004, 318, 153–168. [Google Scholar] [CrossRef] [PubMed]
- Brunotte, L.; Lelke, M.; Hass, M.; Kleinsteuber, K.; Becker-Ziaja, B.; Günther, S. Domain structure of Lassa virus L protein. J. Virol. 2011, 85, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Brunotte, L.; Kerber, R.; Shang, W.; Hauer, F.; Hass, M.; Gabriel, M.; Lelke, M.; Busch, C.; Stark, H.; Svergun, D.I.; et al. Structure of the Lassa virus nucleoprotein revealed by X-ray crystallography, small-angle X-ray scattering, and electron microscopy. J. Biol. Chem. 2011, 286, 38748–38756. [Google Scholar] [CrossRef] [PubMed]
- Poch, O.; Sauvaget, I.; Delarue, M.; Tordo, N. Identification of four conserved motifs among the RNA-dependent polymerase encoding elements. EMBO J. 1989, 8, 3867–3874. [Google Scholar] [PubMed]
- Lehmann, M.; Pahlmann, M.; Jerome, H.; Busch, C.; Lelke, M.; Gunther, S. Role of the C terminus of Lassa virus L protein in viral mRNA synthesis. J. Virol. 2014, 88, 8713–8717. [Google Scholar] [CrossRef] [PubMed]
- Morin, B.; Coutard, B.; Lelke, M.; Ferron, F.; Kerber, R.; Jamal, S.; Frangeul, A.; Baronti, C.; Charrel, R.; de Lamballerie, X.; et al. The N-terminal domain of the arenavirus L protein is an RNA endonuclease essential in mRNA transcription. PLoS Pathogens 2010, 6, e1001038. [Google Scholar] [CrossRef] [PubMed]
- Wallat, G.D.; Huang, Q.; Wang, W.; Dong, H.; Ly, H.; Liang, Y.; Dong, C. High-resolution structure of the N-terminal endonuclease domain of the Lassa virus L polymerase in complex with magnesium ions. PLoS ONE 2014, 9, e87577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raju, R.; Raju, L.; Hacker, D.; Garcin, D.; Compans, R.; Kolakofsky, D. Nontemplated bases at the 5' ends of Tacaribe virus mRNAs. Virology 1990, 74, 53–59. [Google Scholar] [CrossRef]
- Qi, X.; Lan, S.; Wang, W.; Schelde, L.M.; Dong, H.; Wallat, G.D.; Ly, H.; Liang, Y.; Dong, C. Cap binding and immune evasion revealed by Lassa nucleoprotein structure. Nature 2010, 468, 779–783. [Google Scholar] [CrossRef] [PubMed]
- Linero, F.; Welnowska, E.; Carrasco, L.; Scolaro, L. Participation of eIF4F complex in Junin virus infection: Blockage of eIF4E does not impair virus replication. Cell. Microbiol. 2013, 15, 1766–1782. [Google Scholar] [PubMed]
- D'Antuono, A.; Loureiro, M.E.; Foscaldi, S.; Marino-Buslje, C.; Lopez, N. Differential contributions of Tacaribe arenavirus nucleoprotein N-terminal and C-terminal residues to nucleocapsid functional activity. J. Virol. 2014, 88, 6492–6505. [Google Scholar] [CrossRef] [PubMed]
- Cornu, T.I.; de la Torre, J.C. Ring finger Z protein of lymphocytic choriomeningitis virus (LCMV) inhibits transcription and RNA replication of an LCMV S-segment minigenome. J. Virol. 2001, 75, 9415–9426. [Google Scholar] [CrossRef] [PubMed]
- Cornu, T.I.; Feldmann, H.; de la Torre, J.C. Cells expressing the ring finger Z protein are resistant to arenavirus infection. J. Virol. 2004, 78, 2979–2983. [Google Scholar] [CrossRef] [PubMed]
- Cornu, T.I.; de la Torre, J.C. Characterization of the arenavirus ring finger Z protein regions required for Z-mediated inhibition of viral RNA synthesis. J. Virol. 2002, 76, 6678–6688. [Google Scholar] [CrossRef] [PubMed]
- Kranzusch, P.J.; Whelan, S.P.J. Arenavirus Z protein controls viral RNA synthesis by locking a polymerase–promoter complex. Proc. Natl. Acad. Sci. USA 2011, 108, 19743–19748. [Google Scholar] [CrossRef] [PubMed]
- Eichler, R.; Lenz, O.; Strecker, T.; Garten, W. Signal peptide of Lassa virus glycoprotein GPC exhibits an unusual length. FEBS Lett. 2003, 538, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Beyer, W.R.; Pöpplau, D.; Garten, W.; von Laer, D.; Lenz, O. Endoproteolytic processing of the lymphocytic choriomeningitis virus glycoprotein by the subtilase SKI-1/S1P. J. Virol. 2003, 77, 2866–2872. [Google Scholar] [CrossRef] [PubMed]
- Burri, D.J.; Pasqual, G.; Rochat, C.; Seidah, N.G.; Pasquato, A.; Kunz, S. Molecular characterization of the processing of arenavirus envelope glycoprotein precursors by subtilisin kexin isozyme-1/site-1 protease. J. Virol. 2012, 86, 4935–4946. [Google Scholar] [CrossRef] [PubMed]
- Lenz, O.; ter Meulen, J.; Klenk, H.-D.; Seidah, N.G.; Garten, W. The Lassa virus glycoprotein precursor GPC is proteolytically processed by subtilase SKI-1/S1P. Proc. Natl. Acad. Sci. USA 2001, 98, 12701–12705. [Google Scholar] [CrossRef] [PubMed]
- Wright, K.E.; Spiro, R.C.; Burns, J.W.; Buchmeier, M.J. Post-translational processing of the glycoproteins of lymphocytic choriomeningitis virus. Virology 1990, 177, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Eichler, R.; Lenz, O.; Garten, W.; Strecker, T. The role of single N-glycans in proteolytic processing and cell surface transport of the Lassa virus glycoprotein GPC. Virol. J. 2006, 3, 41. [Google Scholar] [CrossRef] [PubMed]
- Loureiro, M.E.; Wilda, M.; Levingston Macleod, J.M.; D'Antuono, A.; Foscaldi, S.; Buslje, C.M.; Lopez, N. Molecular determinants of arenavirus Z protein homo-oligomerization and l polymerase binding. J. Virol. 2011, 85, 12304–12314. [Google Scholar] [CrossRef] [PubMed]
- Bieniasz, P.D. Late budding domains and host proteins in enveloped virus release. Virology 2006, 344, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Im, Y.J.; Kuo, L.; Ren, X.; Burgos, P.V.; Zhao, X.Z.; Liu, F.; Burke, T.R., Jr.; Bonifacino, J.S.; Freed, E.O.; Hurley, J.H. Crystallographic and functional analysis of the ESCRT-I/HIV-1 gag PTAP interaction. Structure 2010, 18, 1536–1547. [Google Scholar] [CrossRef] [PubMed]
- Freed, E.O. Viral late domains. J. Virol. 2002, 76, 4679–4687. [Google Scholar] [CrossRef] [PubMed]
- Loureiro, M.E.; D’Antuono, A.; Levingston Macleod, J.M.; López, N. Uncovering viral protein-protein interactions and their role in arenavirus life cycle. Viruses 2012, 4, 1651–1667. [Google Scholar] [CrossRef] [PubMed]
- Capul, A.A.; Perez, M.; Burke, E.; Kunz, S.; Buchmeier, M.J.; de la Torre, J.C. Arenavirus Z-glycoprotein association requires Z myristoylation but not functional RING or late domains. J. Virol. 2007, 81, 9451–9460. [Google Scholar] [CrossRef] [PubMed]
- Casabona, J.C.; Levingston Macleod, J.M.; Loureiro, M.E.; Gomez, G.A.; Lopez, N. The RING domain and the L79 residue of Z protein are involved in both the rescue of nucleocapsids and the incorporation of glycoproteins into infectious chimeric arenavirus-like particles. J. Virol. 2009, 83, 7029–7039. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Riaño, E.; Cheng, B.Y.H.; de la Torre, J.C.; Martínez-Sobrido, L. The C-terminal region of lymphocytic choriomeningitis virus nucleoprotein contains distinct and segregable functional domains involved in NP-Z interaction and counteraction of the type I interferon response. J. Virol. 2011, 85, 13038–13048. [Google Scholar] [CrossRef] [PubMed]
- Levingston Macleod, J.M.; D'Antuono, A.; Loureiro, M.E.; Casabona, J.C.; Gomez, G.A.; Lopez, N. Identification of two functional domains within the arenavirus nucleoprotein. J. Virol. 2011, 85, 2012–2023. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Danzy, S.; Kumar, N.; Ly, H.; Liang, Y. Biological roles and functional mechanisms of arenavirus Z protein in viral replication. J. Virol. 2012, 86, 9794–9801. [Google Scholar] [CrossRef] [PubMed]
- Kentsis, A.; Gordon, R.E.; Borden, K.L.B. Self-assembly properties of a model RING domain. Proc. Natl. Acad. Sci. USA 2002, 99, 667–672. [Google Scholar] [CrossRef] [PubMed]
- Wolff, S.; Ebihara, H.; Groseth, A. Arenavirus budding: A common pathway with mechanistic differences. Viruses 2013, 5, 528–549. [Google Scholar] [CrossRef] [PubMed]
- Pinschewer, D.D.; Perez, M.; de la Torre, J.C. Dual role of the lymphocytic choriomeningitis virus intergenic region in transcription termination and virus propagation. J. Virol. 2005, 79, 4519–4526. [Google Scholar] [CrossRef] [PubMed]
- Smelt, S.C.; Borrow, P.; Kunz, S.; Cao, W.; Tishon, A.; Lewicki, H.; Campbell, K.P.; Oldstone, M.B.A. Differences in affinity of binding of lymphocytic choriomeningitis virus strains to the cellular receptor α-dystroglycan correlate with viral tropism and disease kinetics. J. Virol. 2001, 75, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Andersen, K.G.; Shylakhter, I.; Tabrizi, S.; Grossman, S.R.; Happi, C.T.; Sabeti, P.C. Genome-wide scans provide evidence for positive selection of genes implicated in Lassa fever. Philos. Trans. R. Soc. B: Biol. Sci. 2012, 367, 868–877. [Google Scholar] [CrossRef]
- Sabeti, P.C.; Varilly, P.; Fry, B.; Lohmueller, J.; Hostetter, E.; Cotsapas, C.; Xie, X.; Byrne, E.H.; McCarroll, S.A.; Gaudet, R.; et al. Genome-wide detection and characterization of positive selection in human populations. Nature 2007, 449, 913–918. [Google Scholar] [CrossRef] [PubMed]
- Peters, C.J. Human infection with arenaviruses in the Americas. Curr. Top. Microbiol. Immunol. 2002, 262, 65–74. [Google Scholar] [PubMed]
- Maiztegui, J.I. Clinical and epidemiological patterns of Argentine haemorrhagic fever. Bull World Health Organ 1975, 52, 567–575. [Google Scholar] [PubMed]
- Abraham, J.; Kwong, J.A.; Albariño, C.G.; Lu, J.G.; Radoshitzky, S.R.; Salazar-Bravo, J.; Farzan, M.; Spiropoulou, C.F.; Choe, H. Host-species transferrin receptor 1 orthologs are cellular receptors for nonpathogenic New World Clade B arenaviruses. PLoS Pathogens 2009, 5, e1000358. [Google Scholar] [CrossRef] [PubMed]
- Reignier, T.; Oldenburg, J.; Flanagan, M.L.; Hamilton, G.A.; Martin, V.K.; Cannon, P.M. Receptor use by the Whitewater Arroyo virus glycoprotein. Virology 2008, 371, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Wang, J.; Lan, S.; Danzy, S.; McLay Schelde, L.; Seladi-Schulman, J.; Ly, H.; Liang, Y. Characterization of virulence-associated determinants in the envelope glycoprotein of Pichinde virus. Virology 2012, 433, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Aronson, J.; Herzog, N.; Jerrells, T. Pathological and virological features of arenavirus disease in guinea pigs. Comparison of two Pichinde virus strains. Am. J. Pathol. 1994, 145, 228–235. [Google Scholar]
- Jahrling, P.B.; Hesse, R.A.; Rhoderick, J.B.; Elwell, M.A.; Moe, J.B. Pathogenesis of a Pichinde virus strain adapted to produce lethal infections in guinea pigs. Infect. Immun. 1981, 32, 872–880. [Google Scholar] [PubMed]
- Albariño, C.G.; Bird, B.H.; Chakrabarti, A.K.; Dodd, K.A.; Flint, M.; Bergeron, É.; White, D.M.; Nichol, S.T. The major determinant of attenuation in mice of the Candid1 vaccine for Argentine hemorrhagic fever is located in the G2 glycoprotein transmembrane domain. J. Virol. 2011, 85, 10404–10408. [Google Scholar] [CrossRef] [PubMed]
- Droniou-Bonzom, M.E.; Reignier, T.; Oldenburg, J.E.; Cox, A.U.; Exline, C.M.; Rathbun, J.Y.; Cannon, P.M. Substitutions in the glycoprotein (GP) of the Candid#1 vaccine strain of Junin virus increase dependence on human transferrin receptor 1 for entry and destabilize the metastable conformation of GP. J. Virol. 2011, 85, 13457–13462. [Google Scholar] [CrossRef] [PubMed]
- Lukashevich, I.S.; Carrion, R.; Salvato, M.S.; Mansfield, K.; Brasky, K.; Zapata, J.; Cairo, C.; Goicochea, M.; Hoosien, G.E.; Ticer, A.; et al. Safety, immunogenicity, and efficacy of the ML29 reassortant vaccine for Lassa fever in small non-human primates. Vaccine 2008, 26, 5246–5254. [Google Scholar] [CrossRef] [PubMed]
- Carrion, R.; Patterson, J.L.; Johnson, C.; Gonzales, M.; Moreira, C.R.; Ticer, A.; Brasky, K.; Hubbard, G.B.; Moshkoff, D.; Zapata, J.; et al. A ML29 reassortant virus protects guinea pigs against a distantly related Nigerian strain of Lassa virus and can provide sterilizing immunity. Vaccine 2007, 25, 4093–4102. [Google Scholar] [CrossRef] [PubMed]
- Harnish, D.G.; Dimock, K.; Bishop, D.H.; Rawls, W.E. Gene mapping in Pichinde virus: Assignment of viral polypeptides to genomic L and S RNAs. J. Virol. 1983, 46, 638–641. [Google Scholar] [PubMed]
- Riviere, Y.; Oldstone, M.B. Genetic reassortants of lymphocytic choriomeningitis virus: Unexpected disease and mechanism of pathogenesis. J. Virol. 1986, 59, 363–368. [Google Scholar] [PubMed]
- Lukashevich, I.S.; Carrion, R., Jr.; Salvato, M.S.; Mansfield, K.; Brasky, K.; Zapata, J.; Cairo, C.; Goicochea, M.; Hoosien, G.E.; Ticer, A.; et al. Safety, immunogenicity, and efficacy of the ML29 reassortant vaccine for Lassa fever in small non-human primates. Vaccine 2008, 26, 5246–5254. [Google Scholar] [CrossRef] [PubMed]
- Xing, J.; Ly, H.; Liang, Y. The Z proteins of pathogenic but not nonpathogenic arenaviruses inhibit RIG-i-like receptor-dependent interferon production. J. Virol. 2015, 89, 2944–2955. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Briese, T.; Lipkin, W.I. Z proteins of New World arenaviruses bind RIG-i and interfere with type I interferon induction. J. Virol. 2010, 84, 1785–1791. [Google Scholar] [CrossRef] [PubMed]
- Moshkoff, D.A.; Salvato, M.S.; Lukashevich, I.S. Molecular characterization of a reassortant virus derived from Lassa and Mopeia viruses. Virus Genes 2007, 34, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.M.; McCormick, J.B.; Webb, P.A.; Smith, E.S.; Elliott, L.H.; King, I.J. Clinical virology of Lassa fever in hospitalized patients. J. Infect. Dis. 1987, 155, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Bergthaler, A.; Flatz, L.; Hegazy, A.N.; Johnson, S.; Horvath, E.; Löhning, M.; Pinschewer, D.D. Viral replicative capacity is the primary determinant of lymphocytic choriomeningitis virus persistence and immunosuppression. Proc. Natl. Acad. Sci. USA 2010, 107, 21641–21646. [Google Scholar] [CrossRef] [PubMed]
- Matloubian, M.; Kolhekar, S.R.; Somasundaram, T.; Ahmed, R. Molecular determinants of macrophage tropism and viral persistence: Importance of single amino acid changes in the polymerase and glycoprotein of lymphocytic choriomeningitis virus. J. Virol. 1993, 67, 7340–7349. [Google Scholar] [PubMed]
- Albariño, C.G.; Bird, B.H.; Chakrabarti, A.K.; Dodd, K.A.; Erickson, B.R.; Nichol, S.T. Efficient rescue of recombinant Lassa virus reveals the influence of S segment noncoding regions on virus replication and virulence. J. Virol. 2011, 85, 4020–4024. [Google Scholar] [CrossRef] [PubMed]
- McLay, L.; Lan, S.; Ansari, A.; Liang, Y.; Ly, H. Identification of virulence determinants within the L genomic segment of the Pichinde arenavirus. J. Virol. 2013, 87, 6635–6643. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Sobrido, L.; Zúñiga, E.I.; Rosario, D.; García-Sastre, A.; de la Torre, J.C. Inhibition of the type I interferon response by the nucleoprotein of the prototypic arenavirus lymphocytic choriomeningitis virus. J. Virol. 2006, 80, 9192–9199. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Huang, Q.; Wang, W.; Dong, H.; Ly, H.; Liang, Y.; Dong, C. Structures of arenaviral nucleoproteins with triphosphate dsRNA reveal a unique mechanism of immune suppression. J. Biol. Chem. 2013, 288, 16949–16959. [Google Scholar] [CrossRef] [PubMed]
- Hastie, K.M.; Kimberlin, C.R.; Zandonatti, M.A.; MacRae, I.J.; Saphire, E.O. Structure of the Lassa virus nucleoprotein reveals a dsRNA-specific 3′ to 5′ exonuclease activity essential for immune suppression. Proc. Natl. Acad. Sci. USA 2011, 108, 2396–2401. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Shao, J.; Lan, S.; Zhou, Y.; Xing, J.; Dong, C.; Liang, Y.; Ly, H. In vitro and in vivo characterizations of the Pichinde viral NP exoribonuclease function. J. Virol. 2015. [Google Scholar] [CrossRef]
- Carnec, X.; Baize, S.; Reynard, S.; Diancourt, L.; Caro, V.; Tordo, N.; Bouloy, M. Lassa virus nucleoprotein mutants generated by reverse genetics induce a robust type I interferon response in human dendritic cells and macrophages. J. Virol. 2011, 85, 12093–12097. [Google Scholar] [CrossRef] [PubMed]
- Russier, M.; Reynard, S.; Carnec, X.; Baize, S. The exonuclease domain of Lassa virus nucleoprotein is involved in antigen-presenting-cell-mediated NK cell responses. J. Virol. 2014, 88, 13811–13820. [Google Scholar] [CrossRef] [PubMed]
- Pythoud, C.; Rodrigo, W.W.S.I.; Pasqual, G.; Rothenberger, S.; Martínez-Sobrido, L.; de la Torre, J.C.; Kunz, S. Arenavirus nucleoprotein targets interferon regulatory factor-activating kinase IKKε. J. Virol. 2012, 86, 7728–7738. [Google Scholar] [CrossRef] [PubMed]
- Rodrigo, W.W.S.I.; Ortiz-Riaño, E.; Pythoud, C.; Kunz, S.; de la Torre, J.C.; Martínez-Sobrido, L. Arenavirus nucleoproteins prevent activation of Nuclear factor Kappa b. J. Virol. 2012, 86, 8185–8197. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Cerny, A.M.; Zacharia, A.; Fitzgerald, K.A.; Kurt-Jones, E.A.; Finberg, R.W. Induction and inhibition of type I interferon responses by distinct components of lymphocytic choriomeningitis virus. J. Virology 2010, 84, 9452–9462. [Google Scholar] [CrossRef] [PubMed]
- Mahanty, S.; Hutchinson, K.; Agarwal, S.; Mcrae, M.; Rollin, P.E.; Pulendran, B. Cutting edge: Impairment of dendritic cells and adaptive immunity by Ebola and Lassa viruses. J. Immunol. 2003, 170, 2797–2801. [Google Scholar] [CrossRef] [PubMed]
- Baize, S.; Kaplon, J.; Faure, C.; Pannetier, D.; Georges-Courbot, M.-C.; Deubel, V. Lassa virus infection of human dendritic cells and macrophages is productive but fails to activate cells. J. Immunol. 2004, 172, 2861–2869. [Google Scholar] [CrossRef] [PubMed]
- Lukashevich, I.S.; Maryankova, R.; Vladyko, A.S.; Nashkevich, N.; Koleda, S.; Djavani, M.; Horejsh, D.; Voitenok, N.N.; Salvato, M.S. Lassa and Mopeia virus replication in human monocytes/macrophages and in endothelial cells: Different effects on IL-8 and TNF-alpha gene expression. J. Med. Virol. 1999, 59, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Pannetier, D.; Faure, C.; Georges-Courbot, M.-C.; Deubel, V.; Baize, S. Human macrophages, but not dendritic cells, are activated and produce alpha/beta interferons in response to Mopeia virus infection. J. Virology 2004, 78, 10516–10524. [Google Scholar] [CrossRef] [PubMed]
- Groseth, A.; Hoenen, T.; Weber, M.; Wolff, S.; Herwig, A.; Kaufmann, A.; Becker, S. Tacaribe virus but not Junin virus infection induces cytokine release from primary human monocytes and macrophages. PLoS Negl. Trop. Dis. 2011, 5, e1137. [Google Scholar] [CrossRef] [PubMed]
- Lukashevich, I.S. The search for animal models for Lassa fever vaccine development. Exp. Rev. Vaccines 2013, 12, 71–86. [Google Scholar] [CrossRef]
- Vela, E. Animal models, prophylaxis, and therapeutics for arenavirus infections. Viruses 2012, 4, 1802–1829. [Google Scholar] [CrossRef] [PubMed]
- Safronetz, D.; Geisbert, T.W.; Feldmann, H. Animal models for highly pathogenic emerging viruses. Curr. Opin. Virol. 2013, 3, 205–209. [Google Scholar] [CrossRef] [PubMed]
- Jahrling, P.B.; Smith, S.; Hesse, R.A.; Rhoderick, J.B. Pathogenesis of Lassa virus infection in guinea pigs. Infect. Immun. 1982, 37, 771–778. [Google Scholar] [PubMed]
- Peters, C.J.; Jahrling, P.B.; Liu, C.T.; Kenyon, R.H.; McKee, K.T., Jr.; Barrera Oro, J.G. Experimental studies of arenaviral hemorrhagic fevers. Curr. Top. Microbiol. Immunol. 1987, 134, 5–68. [Google Scholar] [PubMed]
- Carrion, R., Jr.; Brasky, K.; Mansfield, K.; Johnson, C.; Gonzales, M.; Ticer, A.; Lukashevich, I.; Tardif, S.; Patterson, J. Lassa virus infection in experimentally infected marmosets: Liver pathology and immunophenotypic alterations in target tissues. J. Virol. 2007, 81, 6482–6490. [Google Scholar] [CrossRef] [PubMed]
- Scott, E.P.; Aronson, J.F. Cytokine patterns in a comparative model of arenavirus haemorrhagic fever in guinea pigs. J. Gen. Virol. 2008, 89, 2569–2579. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Lan, S.; Ly, H. Molecular determinants of Pichinde virus infection of guinea pigs—A small animal model system for arenaviral hemorrhagic fevers. Ann. N. Y. Acad. Sci. 2009, 1171 Suppl 1, E65–E74. [Google Scholar] [PubMed]
- Jahrling, P.B.; Hesse, R.A.; Eddy, G.A.; Johnson, K.M.; Callis, R.T.; Stephen, E.L. Lassa virus infection of rhesus monkeys: Pathogenesis and treatment with ribavirin. J. Infect. Dis. 1980, 141, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Lange, J.V.; Mitchell, S.W.; McCormick, J.B.; Walker, D.H.; Evatt, B.L.; Ramsey, R.R. Kinetic study of platelets and fibrinogen in Lassa virus-infected monkeys and early pathologic events in Mopeia virus-infected monkeys. Am. J. Trop. Med. Hyg. 1985, 34, 999–1007. [Google Scholar] [PubMed]
- Callis, R.T.; Jahrling, P.B.; DePaoli, A. Pathology of Lassa virus infection in the rhesus monkey. Am. J. Trop. Med. Hyg. 1982, 31, 1038–1045. [Google Scholar] [PubMed]
- Fisher-Hoch, S.P.; Mitchell, S.W.; Sasso, D.R.; Lange, J.V.; Ramsey, R.; McCormick, J.B. Physiological and immunologic disturbances associated with shock in a primate model of Lassa fever. J. Infect. Dis. 1987, 155, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Hensley, L.E.; Smith, M.A.; Geisbert, J.B.; Fritz, E.A.; Daddario-DiCaprio, K.M.; Larsen, T.; Geisbert, T.W. Pathogenesis of Lassa fever in cynomolgus macaques. Virol. J. 2011, 8, 205. [Google Scholar] [CrossRef] [PubMed]
- Safronetz, D.; Strong, J.E.; Feldmann, F.; Haddock, E.; Sogoba, N.; Brining, D.; Geisbert, T.W.; Scott, D.P.; Feldmann, H. A recently isolated Lassa virus from Mali demonstrates atypical clinical disease manifestations and decreased virulence in cynomolgus macaques. J. Infect. Dis. 2013, 207, 1316–1327. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, A.L.; Tchitchek, N.; Safronetz, D.; Carter, V.S.; Williams, C.M.; Haddock, E.; Korth, M.J.; Feldmann, H.; Katze, M.G. Delayed inflammatory and cell death responses are associated with reduced pathogenicity in Lujo virus-infected cynomolgus macaques. J. Virol. 2015, 89, 2543–2552. [Google Scholar] [CrossRef] [PubMed]
- Baize, S.; Marianneau, P.; Loth, P.; Reynard, S.; Journeaux, A.; Chevallier, M.; Tordo, N.; Deubel, V.; Contamin, H. Early and strong immune responses are associated with control of viral replication and recovery in Lassa virus-infected cynomolgus monkeys. J. Virol. 2009, 83, 5890–5903. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shao, J.; Liang, Y.; Ly, H. Human Hemorrhagic Fever Causing Arenaviruses: Molecular Mechanisms Contributing to Virus Virulence and Disease Pathogenesis. Pathogens 2015, 4, 283-306. https://doi.org/10.3390/pathogens4020283
Shao J, Liang Y, Ly H. Human Hemorrhagic Fever Causing Arenaviruses: Molecular Mechanisms Contributing to Virus Virulence and Disease Pathogenesis. Pathogens. 2015; 4(2):283-306. https://doi.org/10.3390/pathogens4020283
Chicago/Turabian StyleShao, Junjie, Yuying Liang, and Hinh Ly. 2015. "Human Hemorrhagic Fever Causing Arenaviruses: Molecular Mechanisms Contributing to Virus Virulence and Disease Pathogenesis" Pathogens 4, no. 2: 283-306. https://doi.org/10.3390/pathogens4020283