Leptospiral Pathogenomics


1
Division of Infectious Diseases, Department of Medicine, University of California San Diego, School of Medicine, La Jolla, CA 92093-0741, USA
2
Instituto de Medicine Tropical "Alexander von Humboldt", Department of Cellular and Molecular Sciences, Faculty of Sciences and Laboratory of Research and Development, Universidad Peruana Cayetano Heredia, Lima 100, Peru
3
Craig Venter Institute, Rockville, MD 20850, USA
*
Author to whom correspondence should be addressed.
Pathogens 2014, 3(2), 280-308; https://doi.org/10.3390/pathogens3020280
Submission received: 18 January 2014
/
Revised: 22 March 2014
/
Accepted: 28 March 2014
/
Published: 10 April 2014
(This article belongs to the Special Issue Bacterial Pathogenomics: From Technology to Application)
Abstract
:Leptospirosis, caused by pathogenic spirochetes belonging to the genus Leptospira, is a zoonosis with important impacts on human and animal health worldwide. Research on the mechanisms of Leptospira pathogenesis has been hindered due to slow growth of infectious strains, poor transformability, and a paucity of genetic tools. As a result of second generation sequencing technologies, there has been an acceleration of leptospiral genome sequencing efforts in the past decade, which has enabled a concomitant increase in functional genomics analyses of Leptospira pathogenesis. A pathogenomics approach, by coupling of pan-genomic analysis of multiple isolates with sequencing of experimentally attenuated highly pathogenic Leptospira, has resulted in the functional inference of virulence factors. The global Leptospira Genome Project supported by the U.S. National Institute of Allergy and Infectious Diseases to which key scientific contributions have been made from the international leptospirosis research community has provided a new roadmap for comprehensive studies of Leptospira and leptospirosis well into the future. This review describes functional genomics approaches to apply the data generated by the Leptospira Genome Project towards deepening our knowledge of virulence factors of Leptospira using the emerging discipline of pathogenomics.
Keywords:
Leptospira; pathogenomics; virulence; genomics; evolution; taxonomy; molecular epidemiology; systems biology
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. Leptospirosis
Leptospirosis, caused by pathogenic spirochetes belonging to the genus Leptospira, is a zoonosis that has important impacts on human and animal health worldwide [1]. While the exact global disease burden remains unknown, recent estimates by the Leptospirosis Burden Epidemiology Reference Group (LERG) at the World Health Organization have set the number of human cases of severe leptospirosis to over 500,000 per year [2]. This number almost certainly represents an under-representation due to poor surveillance and difficult diagnosis [3]. Clinical symptoms range from a self-resolving acute undifferentiated febrile illness to severe, sometimes fatal disease with renal failure, jaundice, hemorrhage (particularly affecting the lungs), and vascular collapse [4]. Leptospirosis has gained attention worldwide due to recent epidemics of fatal leptospirosis-associated severe pulmonary hemorrhage syndrome without jaundice or renal complications that was originally diagnosed following heavy flooding in rural Nicaragua [5,6]. This severe pulmonary hemorrhage syndrome due to leptospirosis is now recognized as a cause of death worldwide. Transmission to mammals occurs via direct contact with leptospire-infected urine or tissues or indirectly through contact with contaminated soil or water. Although infection may take place through unbroken skin after prolonged immersion, Leptospira usually gain entry to the host via abrasions or cuts in the skin or through exposed mucosae (eyes, nose, etc.). Incidence is seasonal, peaking in summer and fall in temperate climates and during the rainy season in tropical areas, mirroring the ability of the bacteria to survive in the external environment. Soil [7,8,9], mud [10], and surface waters [11] contaminated with urine from chronically-infected reservoir hosts remain important sources of human leptospirosis transmission worldwide. Flood-associated epidemics are increasingly reported, ranging globally from Hawaii to the Philippines, to even occasionally continental Europe [12,13,14,15,16]. Whether this is due to improved diagnosis or disease emergence is currently unknown. Clinically apparent disease is more commonly found in urban than rural regions and men have consistently been found to experience more severe disease after infection than women.
The natural host immune response to leptospirosis is mediated largely through humoral mechanisms [17], where protective agglutinating antibodies produced during infection are directed mainly towards leptospiral lipopolysaccharide (LPS). This immunity is protective against a limited number of very closely related homologous serovars. Toll-like receptor TLR2 and TLR4 have also been found to be necessary for effective innate immune control of infection [18]; however, the immunological responses underlying the differences between infection of reservoir and incidental hosts remain a mystery.
Human leptospirosis is more common in developing countries, but globalization and international travel have led to its apparent increased incidence in industrialized countries, usually, but not exclusively, associated with eco-sports, such as white-water rafting and triathalons, as well as military-related activities [19,20,21,22]. Due to limited understanding of leptospiral ecology, disease transmission and pathogenic mechanisms, and a paucity of experimental work to identify and validate factors important for infection or virulence, only limited progress has been made towards implementing effective public health responses. Though killed, whole-cell vaccines are registered for veterinary use (cattle, pig, dogs), no vaccine is registered for humans and challenges in prevention, diagnosis, and clinical management remain.
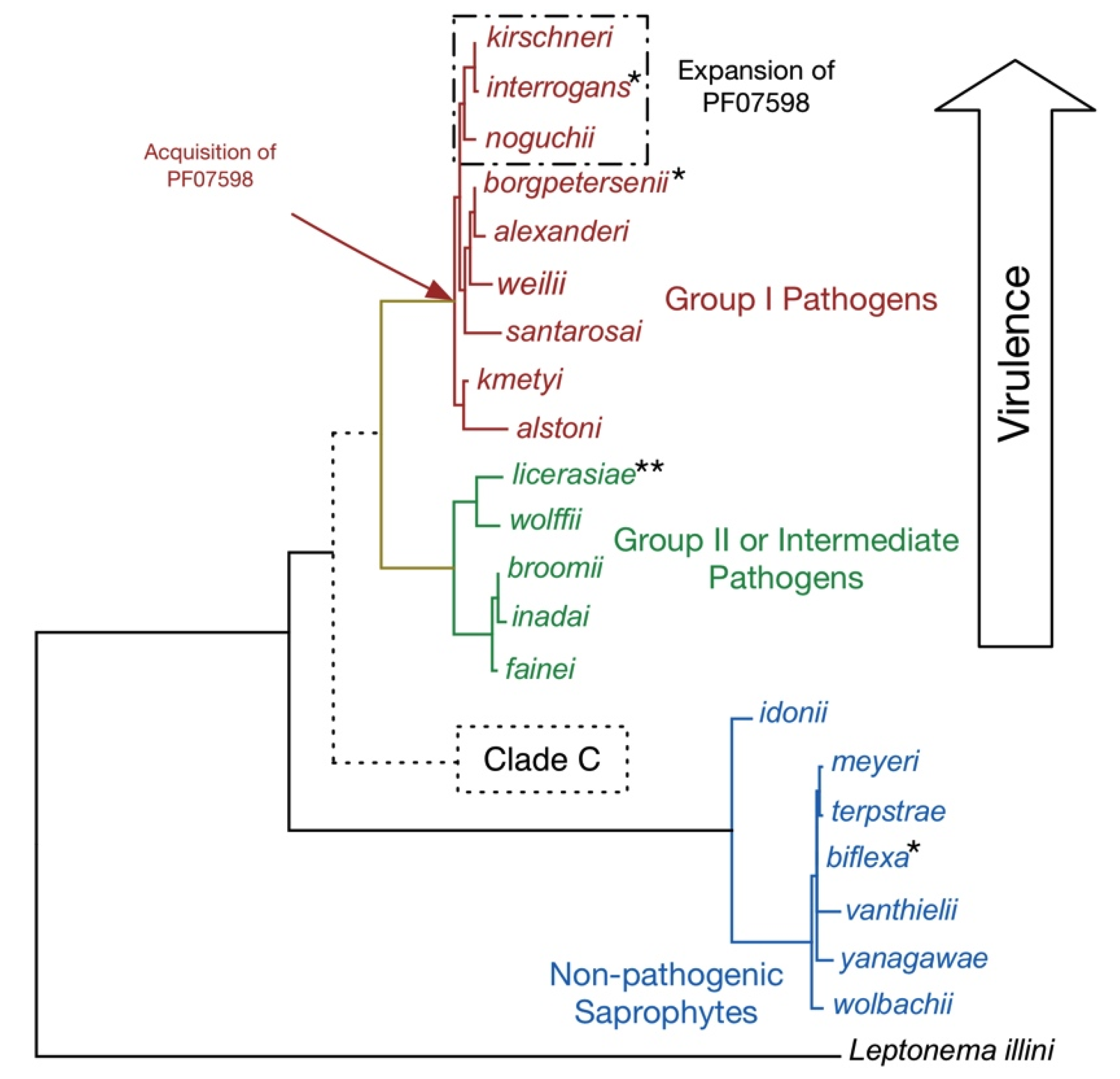
Figure 1.
Taxonomy of the Genus Leptospira. A phylogenetic tree of full-length 16S rRNA sequences showing the relatedness of the 21 recognized leptospiral species. The putative Clade C has been detected in Peruvian surface waters by qPCR [23] and includes strains/species of unknown pathogenicity. For brevity, the Leptospira genus name has been omitted. Key inferred evolutionary events as indicated by whole genome comparisons are shown. Prophages have been detected in L. interrogans, L. licerasiae, and L. biflexa, but not L. borgpetersenii. Of these, only in L. interrogans have the putatively antiviral “clustered regularly interspaced short palindromic repeats” (CRISPR) elements been identified. By contrast, L. licerasiae and L. biflexa have an expanded repertoire of type II and type III toxin-antitoxin systems, which could have anti-phage activity. Of the pathogenic species, only the more virulent Group I pathogens have genes for putative virulence proteins belonging to the paralogous family matching Pfam model PF07598, which we suggest help determine tissue-specific colonization. The outgroup is the closely related spirochete Leptonema illini. For the published genomes, complete genomes are available for six strains of three species (*) and high-quality draft genomes are currently available in GenBank for three strains (**). Genomes representing the remaining Leptospira species are available in GenBank (manuscript in preparation).
Figure 1.
Taxonomy of the Genus Leptospira. A phylogenetic tree of full-length 16S rRNA sequences showing the relatedness of the 21 recognized leptospiral species. The putative Clade C has been detected in Peruvian surface waters by qPCR [23] and includes strains/species of unknown pathogenicity. For brevity, the Leptospira genus name has been omitted. Key inferred evolutionary events as indicated by whole genome comparisons are shown. Prophages have been detected in L. interrogans, L. licerasiae, and L. biflexa, but not L. borgpetersenii. Of these, only in L. interrogans have the putatively antiviral “clustered regularly interspaced short palindromic repeats” (CRISPR) elements been identified. By contrast, L. licerasiae and L. biflexa have an expanded repertoire of type II and type III toxin-antitoxin systems, which could have anti-phage activity. Of the pathogenic species, only the more virulent Group I pathogens have genes for putative virulence proteins belonging to the paralogous family matching Pfam model PF07598, which we suggest help determine tissue-specific colonization. The outgroup is the closely related spirochete Leptonema illini. For the published genomes, complete genomes are available for six strains of three species (*) and high-quality draft genomes are currently available in GenBank for three strains (**). Genomes representing the remaining Leptospira species are available in GenBank (manuscript in preparation).

1.2. Leptospira
The genus Leptospira includes at least 21 species arranged into three large subgroups based on 16S rRNA phylogeny (Figure 1), DNA-DNA hybridization (until recently the gold-standard for defining bacterial species), pathogenicity, virulence, and in vitro growth characteristics. The infectious group (Groups I and II; previously called “pathogens” and “intermediate pathogens”, respectively) includes 14 species (nine in Group I and five in Group II) and the non-infectious group comprised of seven species referred to as “saprophytes”. Group I pathogens [24,25] have been classified into over 250 distinct serotypes and produce disease in people varying in severity, ranging from subclinical infections to severe disease and death; most, if not all, severe disease is caused by serovars belonging to the evolutionarily-related species L. interrogans, L. kirschneri, and L. noguchii. By contrast, Group II pathogens [24,26,27,28,29,30] grow better in culture and cause predominantly mild self-resolving illnesses without fatal complications. Saprophytic Leptospira [24] are free-living environmental microorganisms. A new non-infectious species L. idonii has recently been described [31] and evidence for another subgroup, designated Clade C, comprising species of unknown pathogenicity has been detected by qPCR in the peruvian Amazon [23].
How and why Leptospira evolved from a free-living non-infectious environmental organism is hotly debated. Virulence determinants are poorly understood, as are the mechanisms by which the bacteria produce disease. It is now possible to leverage improved bioinformatics tools to address these and other relevant questions in the field using data from a large leptospiral genome sequencing effort utilizing a hybrid 454/Illumina sequencing strategy (manuscript in preparation) that has provided high-quality draft genomes for representative strains from 20 of the 21 recognized species. “Pathogenomics” approaches coupling high throughput sequencing and bioinformatics comparisons of gene content are indicating specific phenotypic changes that distinguish saprophyte from pathogen. In addition, this approach is resolving questions regarding the pathogenicity of Group II Leptospira species (manuscript in preparation).
1.3. Maintenance Hosts
Leptospira colonize the renal tubules of chronically infected reservoir animals and are shed via urine into the environment. Many mammalian species and amphibians [32,33] may act as reservoirs of Leptospira. Some host species are believed to favor specific serovars (e.g., serovar Copenhageni in rats, Lai in field mice and Hardjo in cattle), but these serovar-host associations are not absolute. High frequency infection by non host-adapted serovars has also been documented (e.g., serovar Ballum in rats [34]). The expectation that reservoir hosts remain asymptomatic during infection has recently come into question considering infrequent outbreaks caused by serovar Pomona that occur among California Sea Lions, which are believed to be reservoirs of this serovar [35]. Perhaps outbreak serovar Pomona strains represent emerging genotypes that differ from benign carriage Pomona strains at other loci? Whole genome comparisons of these with non-outbreak strains could improve our understanding of pathogen emergence. Whereas reservoirs shed Leptospira in urine, often for the lifetime of the animal, in non-reservoir (“incidental”) hosts, such as humans, renal colonization and leptospiruria rarely persist for more than a few months, although chronic renal infections by Group I and Group II strains lasting a year or more have also been documented [36]. Host animals transmit Leptospira to other animals through contact with infected urine, via sexual transmission [37], by vertical transmission from infected mother to susceptible offspring [38,39] and probably indirectly, through contact with contaminated water and soil [40,41]. Rats were the first recognized carriers of Leptospira [42] and remain an important source of transmission in urban areas [43,44]. Other important reservoirs include domesticated dogs, pigs, cattle and horses, and also wild animals (e.g., the spiny rat (Proechimys spp.)) in Latin America and frogs and toads in the Caribbean [32,45]. Curiously, some reservoir hosts for some serovars are also incidental hosts for non-host adapted serovars (e.g., dogs maintain serovar Canicola, but often develop severe disease when infected with serovar Australis [46] or Grippotyphosa [47]). Potentially any vertebrate species can be considered to be susceptible to acute and chronic infection by certain strains, and thus excrete pathogenic Leptospira into the environment [48]. This includes marine mammals such as free-living Californian sea lions (Zalophus californianus) [35,49,50,51] and northern elephant seals (Mirounga angustirostris) [52]. Indeed, new animal sources of carriage, including bats [53], continue to be identified. Host range determinants are unknown, though comparative genomics suggest that the structure of the O-antigen is important. As serovars Lai and Copenhageni, in which the O-antigen gene clusters are 100% identical at the amino acid level [54], have different reservoir hosts; and some species (e.g., rats [28]) can maintain multiple serovars, structural variation of the O-antigen alone cannot explain host predilection.
1.4. Pathogenomics: An Overview
To address the basic question “what makes bacteria pathogenic?” we need to know the functional differences between pathogenic and non-pathogenic strains or species. Early comparative genomics studies, based on Sanger or 454 sequence data, compared up to six isolates of the same or different species [55,56,57,58]. These initial studies focused largely on determining the “core” (i.e., genes found in all strains of a species) versus the “variable” genes of the pan-genome, and whether a particular pan-genome was “open” (and amenable to DNA transfer from other strains or species) or “closed” (closed off to genetic exchange) [59,60]. The variable genome is comprised of genes that are strain specific or are found in some but not all strains. As genome plasticity (gain or loss of genes) allows pathogens to adapt to changing environments, studies of dispensable and strain-specific genes can provide valuable insights into virulence and pathogenesis. In addition, mechanisms mediating genome plasticity (e.g., interstrain or interspecies gene transfer, mutation, and selection) will enhance our understanding of the forces shaping microbial evolution and contexts in which they occur.
As a newly emerged discipline, Pathogenomics seeks to delineate virulence factors and their contributions to overall pathogenesis by comparing gene repertoires of pathogenic and non-pathogenic strains/species (reviewed in [61]). With the advent of higher throughput sequencing technologies and improved bioinformatics tools, it is now possible to sequence and compare dozens of strains of the same or different species. As more strains can be sequenced and compared, new comparative studies use these datasets to address more focused questions (e.g., the evolutionary roles of single nucleotide polymorphisms in core and strain-specific genes, lateral gene transfer (LGT), and prophage lysogenization [62,63]). This review introduces some of the available software for comparing microbial genomes, summarizes current Leptospira genomics and pathogenomics efforts, and illustrates how these data are being used to better understand leptospiral evolution, pathogenicity, and virulence. Finally, we highlight how these approaches can be leveraged to improve leptospirosis prevention, diagnosis and treatment.
2. Software
Several computational tools have been developed to cluster orthologous protein sequences in order to compare predicted functions across multiple isolates (e.g., Sybil [64], OrthoMCL [65], InParanoid [66]). These programs also include paralogs within clusters, but ignore information about expansion and contraction of paralogous protein families, which are important factors in phenotype and genome evolution. They also do not consider genomic neighborhoods of orthologous genes, which can be important for coordinated gene regulation in bacteria through coupled transcription-translation in operons and may give new insights into how genes are acquired and spread. Lastly, none of these programs detect frame-shifted genes, which could represent dispensable genes or sequencing errors. To address these issues, the Pan-Genome Ortholog Clustering Tool (PanOCT) was developed [67]. PanOCT is a graph-based tool written in PERL that utilizes the BLAST score ratio (BSR) [68], conserved gene neighborhood (CGN) and frame-shift detection in a weighted scoring scheme to generate non-paralogous ortholog gene clusters from multiple bacterial genomes. PanOCT was used in a recent publication on the pathogenomics of L. interrogans [69].
A number of bioinformatics tools have been created to identify putative prophage regions within bacterial genomes [70,71,72,73,74]. Phage_Finder automates the once laborious process of prophage finding and is open source. Phage_Finder is a heuristic program written in PERL that uses input BLASTP matches to a database of known phage sequences and also to phage-specific Hidden Markov Models (HMMs) to locate regions of the genome enriched with phage-like genes. Using a set of heuristics, each initial region is extended outward based on the annotation of neighboring genes until housekeeping genes are found. Attachment (att) sites are specific sequences used for phage integration. Phage_Finder is one of the only programs that predict att sites, resembling direct repeats, using local alignment algorithms. The site of phage insertion is then predicted based on the location of the predicted att sites. Insertion can occur at intergenic sites or within tRNA, tmRNA or protein-coding genes. Due to genetic exchange with other mobile elements, Phage_Finder can also identify genomic islands (GIs) and integrated plasmids. In addition to identification of prophage and GIs, Phage_Finder can classify regions based on homology to phage family-specific proteins—version 2.0 of the package, which utilizes HMMER3 [75,76], is publicly available [77]. Other computational tools have been developed to predict GIs utilizing either sequence composition bias or comparative genome approaches. One such tool, IslandViewer [78], is a web accessible application that predict GIs using uploaded sequence data [79] combining output from several accurate independently developed methods for GI prediction: IslandPick [80], IslandPath-DIMOB [81] and SIGI-HMM [81]. Whereas most GI prediction tools utilize either sequence composition bias or comparative genomics, IslandViewer integrates both approaches, resulting in enhanced sensitivity and specificity. Data can be retrieved at both the chromosome and gene level for method-specific or consistent GI prediction.
3. Leptospira Genomics and Pathogenomics Studies
3.1. Mobile Genetic Elements
Differences among bacterial strains include mobile DNA elements (e.g., in Leptospira include prophages, transposons, insertion sequence (IS) elements, plasmids, genomic islands [55,56,57,58,82,83]). Many of these DNAs encode proteins involved in cell surface structures (i.e., O-antigen, capsular polysaccharides (CPS), teichoic acid, S-layer, flagella, pili, and porins), toxins and/or resource utilization. Regions that vary between strains have been referred to as “flexible” GIs [84] and are of particular interest in microbial studies because these mobile genetic elements (MGEs) may introduce virulence factors into a new host genome.
3.1.1. Bacteriophages
Predation by bacteriophages, which use specific bacterial cell surface structures as receptors, is thought to be a major force driving bacterial evolution. Natural selection by bacteriophage rather than the host immune system could be important in bacterial O-antigen evolution, though this has never been tested in Leptospira. Temperate bacteriophages, those that can integrate into the host chromosome, can alter the phenotype of their host (i.e., lysogenic conversion) via delivery of genes involved in adaptation of the host to new environments and can mediate serotype conversion of the O-antigen [85]. Pathogenomics inquiries indicate that in Leptospira, lysogenization might have played an important role in leptospiral evolution since prophages, some of which encode proteins with important functions, have been found in representative species from each of the three major branches (Figure 1). For example, in the intermediate pathogen L. licerasiae, a novel LE1-like prophage, called vB-LliZ_VAR010-LE1 (using systematic bacteriophage nomenclature [86]), encodes efflux pumps sharing homology with chromosomally encoded proteins in pathogenic species [87]. As these pumps are absent from the saprophyte, L. biflexa, they may function in adaptation to the mammalian host. A second prophage region identified in L. licerasiae is adjacent to a cryptic prophage (LA0186 to LA0219) expressed in L. interrogans serovar Lai 56601, which is thought to be associated with pathogenicity since expression is down regulated in the avirulent L. interrogans serovar Lai IPAV [88]. Of note, although it took several years for the LE1 phage [89,90] and a cryptic prophage region [88,91] to be identified and sequenced, the LE1-like phages were quickly identified using high throughput sequencing and bioinformatics. Comparative approaches also independently identified both LE1 and cryptic prophage regions [87].
3.1.2. Evidence for Plasmid Transfer
Type II and type III toxin-antitoxin systems (TASs) belong to the class of bacterial MGEs as they are extensively, if not preferentially, spread via plasmid-mediated LGT [92]. Several roles have been proposed, including non-functional roles such as “junk DNA” and chromosomal remnants from transposons and bacteriophages, and functional roles including gene regulation [93], programmed cell death [94] and anti-phage activity [95,96]. Like many, if not most MGEs, TASs are not simply mobile, but appear to behave like selfish elements, as they contribute to the stable maintenance and dissemination of plasmids and genomic islands in bacterial populations, seemingly despite associated fitness costs. Of the 28 TASs in the L. licerasiae genome, 10 (36%) are associated with putative GIs and a further four are unique amongst Leptospira. 37% of these systems are also located on genomic islands in M. tuberculosis [97]. Identifying genes associated with these MGEs could provide insight into previously unknown ecological differences amongst Leptospira. For instance, three of five putative L. licerasiae type III TASs are unique to the genus. L. interrogans possesses five TASs that are not present in the saprophyte L. biflexa [98,99], four of which have not been found in other pathogenic Leptospira species; therefore, adjacent genes might provide insight into unique virulence mechanisms. Of the completed genomes, only L. biflexa possesses a circular plasmid [100], though a 54-kb GI in Lai 56601 has been shown to excise from the chromosome and exist as a plasmid [101].
3.1.3. Repetitive Elements
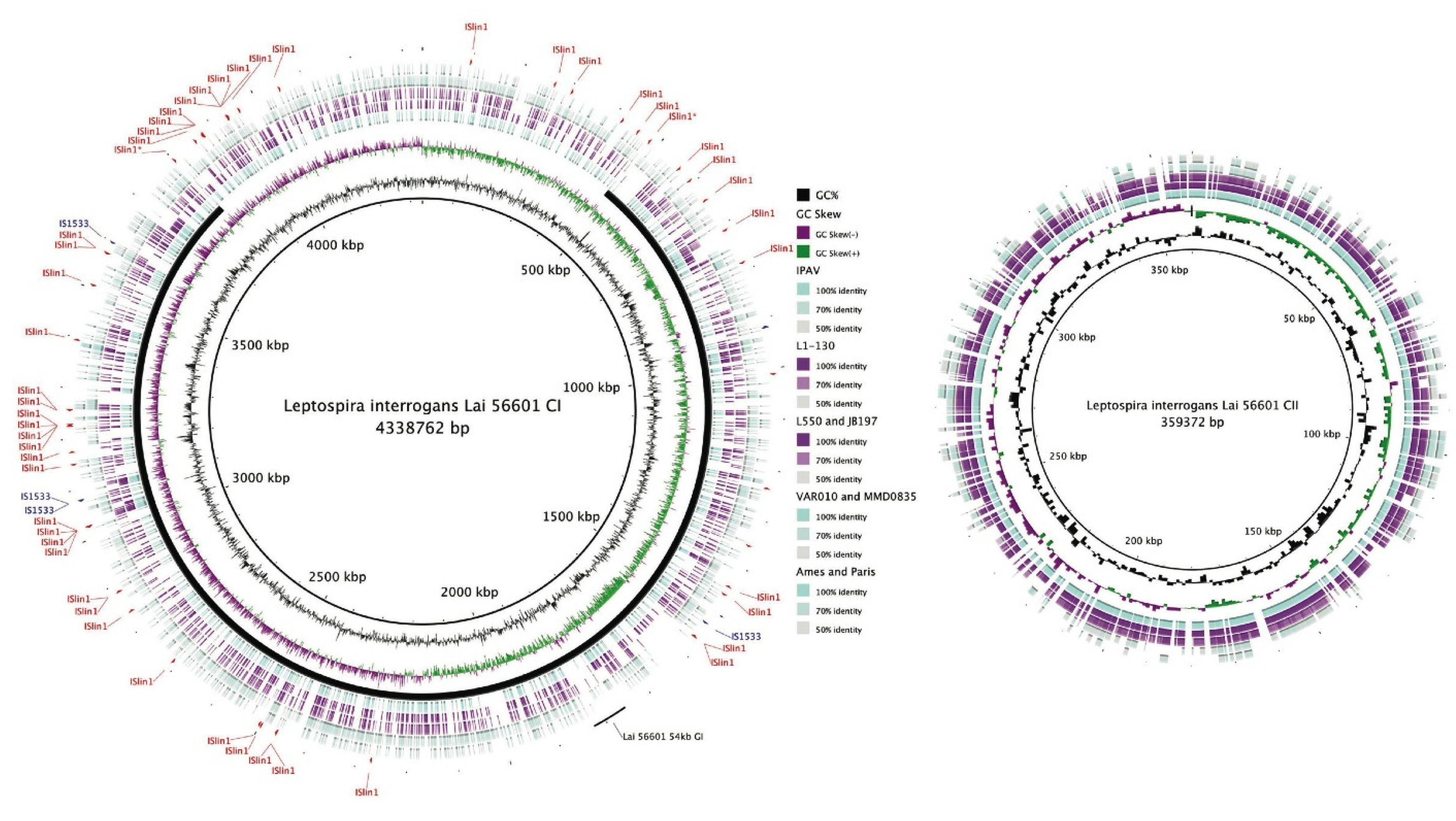
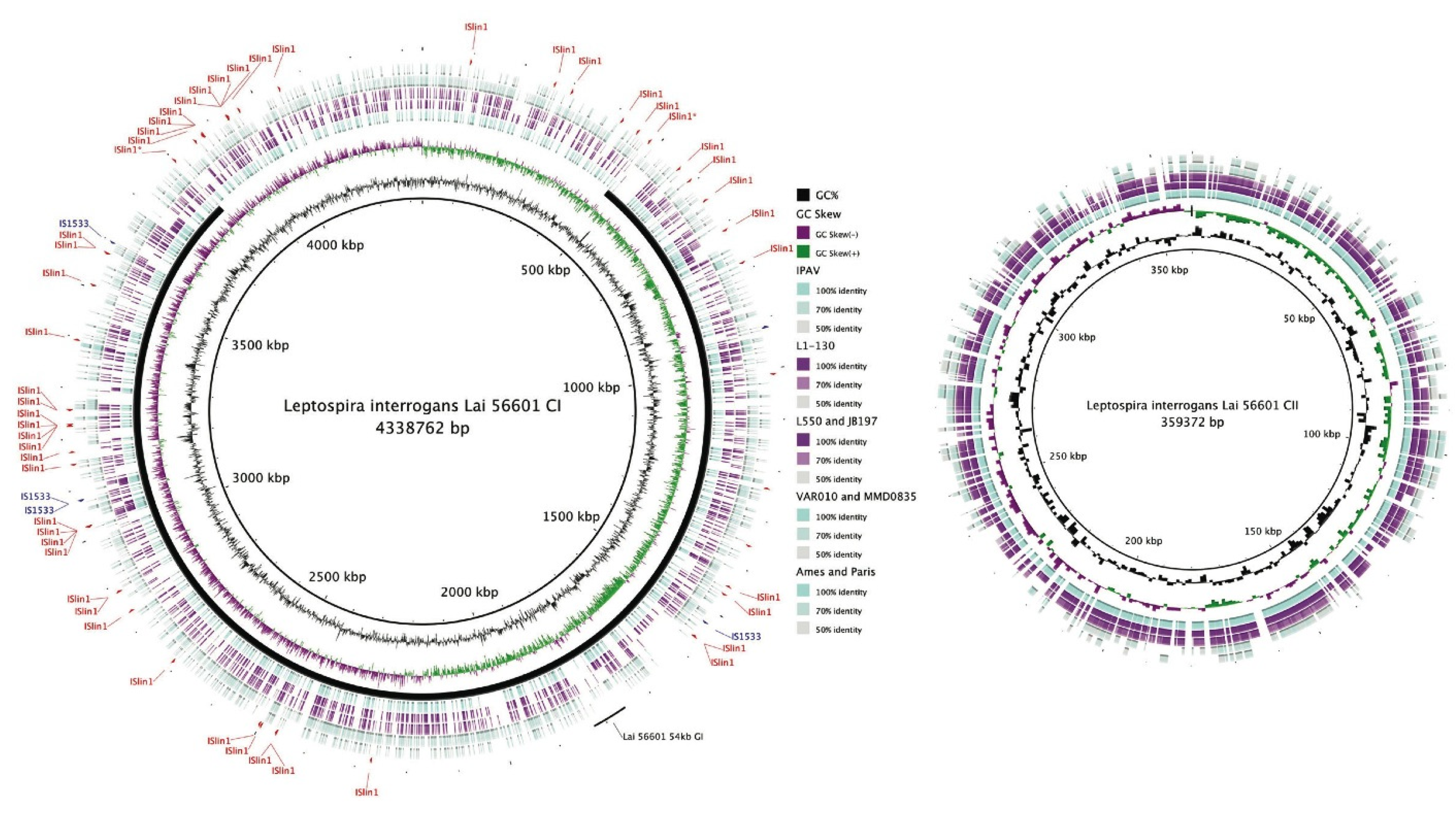
IS elements are short DNA sequences that act as simple transposable elements. They are usually smaller than other transposable elements (700 to 2500 bp in length); and only code for proteins necessary for transposition. Several IS elements have been described in Group I pathogenic Leptospira, including IS1500 [102], IS1501 [103], IS1533 [104], and ISLin1 [54]. By contrast, few have been identified in Group II pathogens [87] or saprophytes [100]. Thus, an expansion of IS elements could be a distinguishing feature of Group I pathogens [105]. As these elements are thought to help mediate gene acquisition [106], inactivation or deletion [107], or large-scale genome rearrangements (Figure 2 and [54]) by transposition or homologous recombination, the genomes of Group I pathogenic Leptospira may be less stable and perhaps more versatile by comparison. For example, L. borgpetersenii serovar Hardjo strains L550 and JB197 contain 77 and 84 complete copies of IS1533, respectively, and roughly 25 partial copies distributed throughout each genome, indicative of frequent transposition and recombination events [107]. IS1533 insertion and subsequent recombination has disrupted a crc-like gene leading to a 41-kb insertion in L. borgpetersenii compared to L. interrogans, and in Lai 56601, several genes appear to have been inactivated by IS elements compared to Copenhageni L1-130 [54]. IS-mediated gene inactivation/deletion may have significant consequences, potentially altering transmission modes. For instance, genome reductions of Hardjo L550/JB197 could have caused a diminished capacity of the strains to grow in artificial media [107]. Due to this poor growth ex vivo, it has been suggested that these strains are becoming obligate parasites that are likely to be transmitted exclusively from animal to animal [102]. A large inversion, presumably due to recombination between ISlin1 elements, has been documented in Lai 56601 [54]. Though the consequences of this inversion are unknown it is unlikely to significantly impact gene expression since the distance of affected genes from the replication fork is unchanged [108,109,110,111,112,113]. IS elements could also facilitate host-range switching. For example, serotype conversion of Copenhageni (carried by rats) to Hardjo (cattle) is thought to be IS-mediated [114].
Figure 2.
Blast Ring Image Generator (BRIG) [115] plot showing whole genome comparison of L. interrogans, L. borgpetersenii, L. licerasiae and L. biflexa. Track 1 (innermost; reference genome): L. interrogans Lai 56601; Track 2: GC%; Track 3: GC Skew; Track 4: L. interrogans Lai IPAV; Track 5: L. interrogans Copenhageni L1-130; Track 6: L. borgpetersenii Hardjo (JB197 and L550); Group II (Track 7): L. licerasiae Varillal (VAR010 and MMD0835); and Track 8: L. biflexa Patoc (Ames and Paris) showing conserved proteins with amino acid similarity of ≥50%. The locations of two common IS elements in Lai 56601: ISlin1 (red) and IS1533 (blue) are shown. The solid black line shows the inverted region of the Lai 56601 genome (with respect to Copenhageni L1-130); the two IS elements believed to have mediated the inversion have been designated with an asterisk (*). The location of 54-kb GI in Lai 56601, capable of excising from the chromosome and replicating as a plasmid [101], is shown. Homologous coding regions have been color-coded based on %identity as indicated by the key. All Leptospira contain both the CI and CII replicons; whereas only L. biflexa contains a large plasmid designated p74 (not shown).
Figure 2.
Blast Ring Image Generator (BRIG) [115] plot showing whole genome comparison of L. interrogans, L. borgpetersenii, L. licerasiae and L. biflexa. Track 1 (innermost; reference genome): L. interrogans Lai 56601; Track 2: GC%; Track 3: GC Skew; Track 4: L. interrogans Lai IPAV; Track 5: L. interrogans Copenhageni L1-130; Track 6: L. borgpetersenii Hardjo (JB197 and L550); Group II (Track 7): L. licerasiae Varillal (VAR010 and MMD0835); and Track 8: L. biflexa Patoc (Ames and Paris) showing conserved proteins with amino acid similarity of ≥50%. The locations of two common IS elements in Lai 56601: ISlin1 (red) and IS1533 (blue) are shown. The solid black line shows the inverted region of the Lai 56601 genome (with respect to Copenhageni L1-130); the two IS elements believed to have mediated the inversion have been designated with an asterisk (*). The location of 54-kb GI in Lai 56601, capable of excising from the chromosome and replicating as a plasmid [101], is shown. Homologous coding regions have been color-coded based on %identity as indicated by the key. All Leptospira contain both the CI and CII replicons; whereas only L. biflexa contains a large plasmid designated p74 (not shown).
3.1.4. O-Antigen Diversity
Host range determinants are unknown as are the mechanisms of cross-species transmission, host range expansion or host shifts, though comparisons of O-antigen (rfb) loci are providing important insights. For instance, two genetically distinct, but serologically indistinguishable subtypes of the bovine-adapted serovar, Hardjo, have been described: L. borgpetersenii serovar Hardjo subtype Hardjobovis and L. interrogans serovar Hardjo subtype Hardjoprajitno. The rfb loci of both subtypes share considerable similarity and have been divided into four gene clusters based on sequence similarity to other leptospiral rfb loci. Two of the four clusters in subtype Hardjoprajitno are more similar to corresponding gene clusters of subtype Hardjobovis, while two (orfJ15-orfJ20 and orfJ23-orfJ31) are almost identical to gene clusters in the rat-borne L. interrogans serovar Copenhageni [114]. This suggests that the ancestral subtype Hardjoprajitno strain was most likely a Copenhageni strain that acquired orf 1–14 and orf 21–22 from subtype Hardjobovis, resulting in serologically indistinguishable Hardjo subtypes. Inspection of these gene clusters (and their encoded functions) could provide important insight into host-range determinants. It’s tempting to speculate that evolutionary pressures such as the aforementioned phage predation that affect O-antigen diversity, whether they occur in vivo or ex vivo, are likely to affect host range and/or pathogen emergence (e.g., evolution and emergence of El Tor O1 and O139 serotypes in Vibrio cholera appear to be phage mediated [116] or a result of large fragment DNA transformation [117]). Disentangling the mechanisms of O-antigen evolution and the contexts in which such evolution occurs could improve our understanding of transmission cycles and mechanisms of serotype emergence.
Sandwiched within the rfb locus of L. interrogans serovars Copenhageni and Lai are several genes predicted to be involved in the biosynthesis of the sialic acid, legionaminic acid [118], a known virulence determinant in Legionella pneumophila [119]. As sialic acids may have important consequences for survival within macrophages [119] and complement resistance [120], two key leptospiral virulence traits, it could explain why these strains are among the most virulent Leptospira. The presence of legionaminic acid—likely by convergent evolution—tempts the speculation that Leptospira and Legionella may share fundamental features of host adaptation.
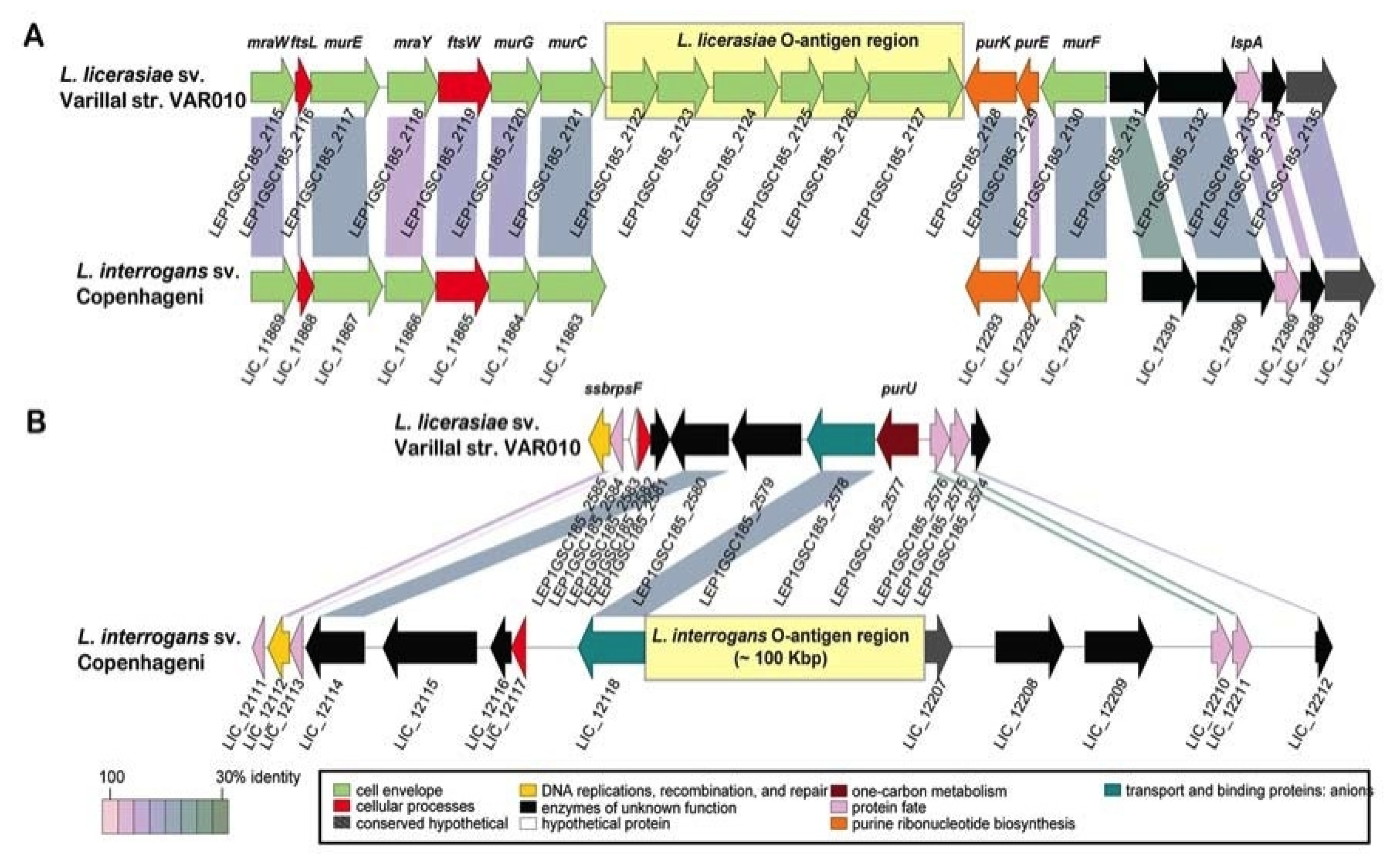
Figure 3.
Genetic organization and alternate location of L. licerasiae rfb locus. (A) Comparison of the rfb locus of L. licerasiae strain VAR010 and homologous region in L. interrogans serovar Copenhageni; (B) Comparison of the rfb locus of L. interrogans Copenhageni and homologous region in L. licerasiae strain VAR10. Yellow shaded boxes mark the locations of the O-antigen regions. CDSs are labeled by locus identifier and colored by functional role categories as noted in the boxed key. Gene symbols, when present, are noted above their respective genes in bold italics. Reproduced from [87].
Figure 3.
Genetic organization and alternate location of L. licerasiae rfb locus. (A) Comparison of the rfb locus of L. licerasiae strain VAR010 and homologous region in L. interrogans serovar Copenhageni; (B) Comparison of the rfb locus of L. interrogans Copenhageni and homologous region in L. licerasiae strain VAR10. Yellow shaded boxes mark the locations of the O-antigen regions. CDSs are labeled by locus identifier and colored by functional role categories as noted in the boxed key. Gene symbols, when present, are noted above their respective genes in bold italics. Reproduced from [87].
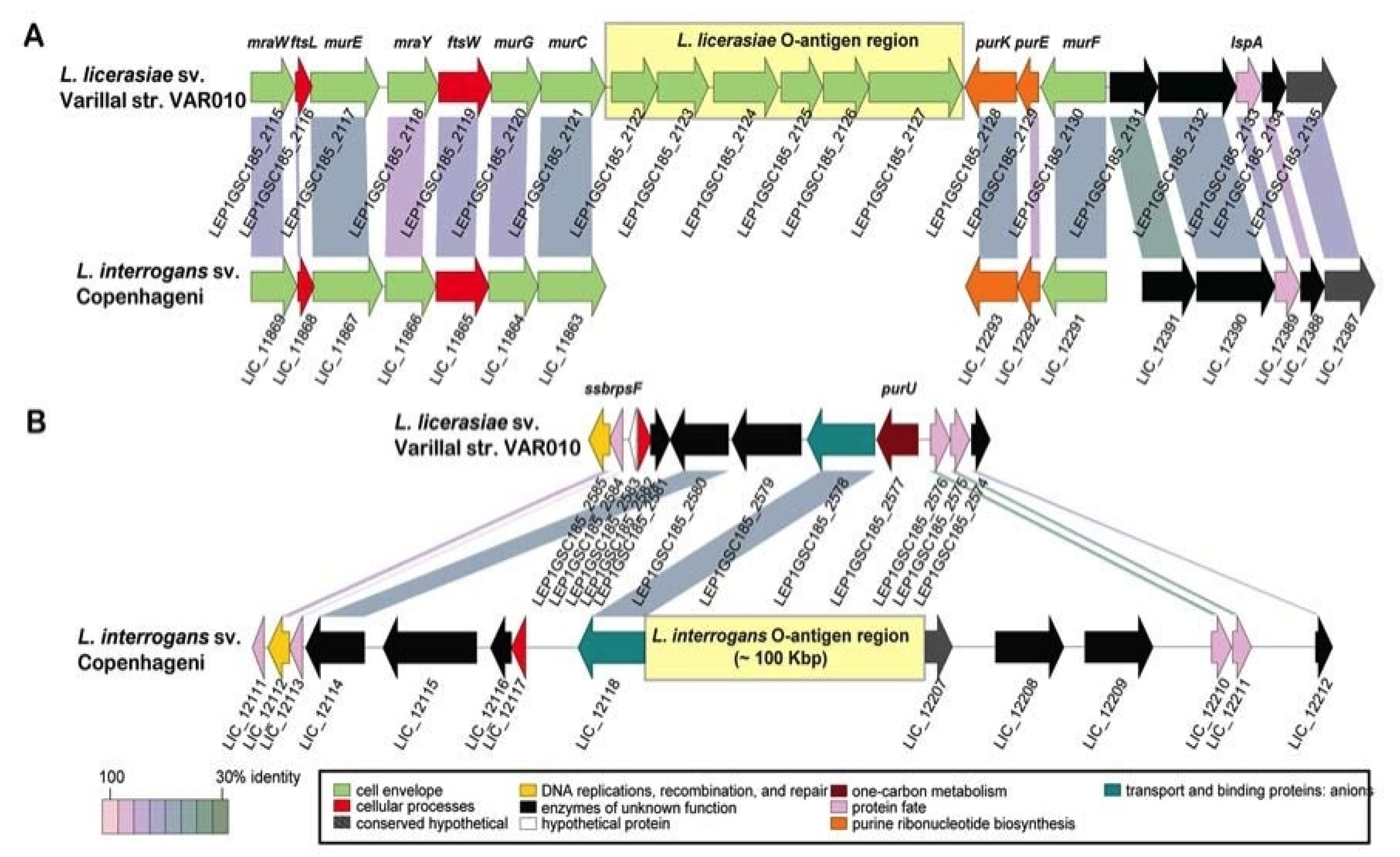
Compared to other Leptospira, the L. licerasiae O-antigen biosynthesis region is unusual, consisting of a short six-gene cassette [87] that includes three glycosyltransferases inserted between two normally adjacent, convergently transcribed genes: the murC gene of cell wall biosynthesis and purK gene of purine biosynthesis. Indeed, the glycosyltransferase, LEP1GSC185_2122 (GenBank: |EIE02925) in L. licerasiae str. VAR010, serves as a good marker for absence of the typical (long) O-antigen biosynthesis region and replacement by a six-gene cassette between murC and purK (Figure 3). Genes in these regions have no close homologs in any other Leptospira, in the O-antigen region or anywhere else, supporting the notion that these cassettes were acquired by LGT and provide unique carbohydrate chemistry and serology. These and other less dramatic differences in rfb loci underlying serological diversity present an opportunity for development of DNA-based typing tools for serological classification that could replace the tedious and sometimes unreliable cross-adsorption agglutination reactions traditionally used to serotype Leptospira [121].
The reasons for the unusual arrangement of the L. licerasiae rfb locus are unknown, but could underpin key differences in host adaptation or other ecological relationships between this and other leptospiral species.
3.2. Virulence Mechanisms and Pathogenesis
Pathogens possess a number of virulence factors, and delineating and understanding the function of these determinants is paramount to understanding the pathogenesis of the diseases they cause. The combinatorial effect of these factors enables microbes to efficiently invade and colonize various tissue niches, obtain nutrients, as well as evade and suppress the immune response of the host. In contrast to other pathogens, where experimental genetic inquiry has defined a number of virulence factors, the mechanisms by which pathogenic Leptospira cause disease remain largely unknown; mostly due to the recalcitrance of pathogenic Leptospira to genetic manipulation. There have been reports of site-directed homologous recombination being used for the deletion of chromosomal genes; however, these reports are exceptions rather than the norm. Targeted gene knockouts still remain out of grasp for most Leptospira researchers; and no replicative plasmid vector is available for pathogenic Leptospira. Alternative approaches to discover virulence related genes in Leptospira have included evaluating transcriptional responses during exposure to “host-like” conditions in vitro [122,123,124,125,126,127]; however, global gene regulation in response to the combination of all extracellular cues in vivo remains to be investigated. The known leptospiral virulence factors have been extensively reviewed [128,129,130], and include LPS (a general virulence factor of Gram-negative bacteria), flagella, heme-oxygenase, the OmpA-like Loa22, and adhesion molecules. In addition, hemolysins and sphingomyelinases may play a role during infection, although there are conflicting reports regarding their true contributions to overall virulence [130].
3.2.1. Genetic Manipulation
As targeted knockouts remain difficult, genetic manipulation of pathogenic Leptospira has been limited to random transposon-based mutagenesis in two of the more virulent serovars Lai [131] and Manilae [132]. This approach has led to the generation of mutant libraries that can then be screened for defects in pathogenesis, and in fact has identified many of the limited number of leptospiral virulence factors known [122,133,134,135]. However, identifying attenuated mutants requires a large commitment of time and resources, and represents the major drawback of this approach. As currently designed, these gene knockout experiments qualitatively (+/– colonization, fatal/non-fatal, etc.) evaluate the involvement of a particular gene towards virulence. Comparisons of knockout mutants to wild-type parental strains are by definition binary assessments. While of great value for characterizing the involvement of single determinants to virulence, this approach cannot determine the relative contributions of multiple genes to the total virulence phenotype. Pathogenomic-based approaches provide an unbiased global view of genome-level differences among pathogenic and non-pathogenic strains that can be used as a complementary alternative to transposon-mediated mutagenesis studies.
3.2.2. Attenuation Studies in L. interrogans Serovar Lai
The genome of a virulence-attenuated L. interrogans serovar Lai strain IPAV derived by prolonged laboratory passage from a highly virulent ancestral strain was recently sequenced and annotated [105]. Comparisons of this strain and a virulent related strain, L. interrogans serovar Lai strain 56601, showed a mostly conserved genome (structure and gene order), but identified 33 insertions, 53 deletions and 301 single-nucleotide variations affecting 101 genes in strain IPAV compared to strain 56601 [105]. Genes involved in signal transduction, stress response, transmembrane transport and nitrogen metabolism comprised the majority of the 44 affected functionally annotated genes. Subsequent comparative proteomic analysis of 1627 orthologs revealed that 174 genes in strain IPAV were upregulated in vitro, with enrichment mainly in energy production and lipid metabolism functions. By contrast, 228 strain 56601 genes, primarily involved in protein translation and DNA replication/repair, were upregulated compared to strain IPAV [105]. As protein expression comparisons were done in vitro, these data provide limited insight into the relevance of these genes during mammalian infection. Moreover, since the ancestral strain was not used in these analyses, it is not clear which of the many differences observed between the two strains resulted in the attenuation phenotype complicating future mechanistic studies. Despite these limitations, this study illustrated that altered expression or mutations in critical genes could account for virulence attenuation in strain IPAV.
In long-term in vitro attenuation studies, the only factors likely to be under selective pressure are resource utilization, proliferation and survival in culture media. As virulence associated genes are dispensable in vitro, these genes or their regulatory systems could be lost during long-term in vitro culture. If stocks are kept of each serial passage (generation), it becomes possible to test the relevance of individual mutations by comparing generations immediately preceding the appearance of the mutation and generations in which the mutation first appeared. A more recent study used this type of study design to identify candidate virulence genes by genomic comparison of a culture-attenuated serovar Lai strain 56601_p18 with its virulent, isogenic parent strain 56601_p1 [69]. Of significance, among the genes inactivated by serial in vitro passage were several previously unstudied putative virulence genes. The first, a predicted adenylate guanylate cyclase (LA_4008), was found to elevate cyclic AMP activity of in vitro cultured monocytes [69]. Increases in the intracellular concentration of cAMP in innate immune cells have been shown to impair phagocytosis, free radical generation, as well as promote the release of anti-inflammatory cytokines. Although modulation of host cAMP levels has been long accepted as a mechanism used by other pathogens to blunt the immune response [136], this is the first evidence of a likely contributor to virulence for Leptospira. This is especially important considering evidence that persistence within and killing of macrophages once phagocytosed might be a fundamental difference between reservoir and human macrophages with important implications for the evolution of disease [137].
LA_1056, a hypothetical protein of unknown function, was also identified in our attenuation study. In silico sequence analysis revealed a phage tail protein (PHA00965 conserved domain) that shares homology with the phage tape measure protein pblA from Streptococcus mitis. Studies exploring pblA in the pathogenesis of S. mitis have demonstrated that bacteria can co-opt tape measure proteins to act as adhesion-type molecules. Specifically, S. mitis decorates their outer surface with PblA and use it to bind α2-8 linked sialic acid residues on platelet membrane gangliosides [138,139,140]. Thrombocytopenia is a well-documented complication of severe leptospirosis. A prospective study from Barbados found thrombocytopenia in more than 50% of patients hospitalized with the disease, while studies from Brazil found rates of 65% in children and 86% in adults [141,142,143]. Despite this high prevalence, the exact pathogenic mechanisms behind it remain unclear. It is possible that Leptospira, through mechanisms similar to those found in S. mitis, employ phage-derived proteins like LA_1056 for use as adhesins for attachment to platelets. These data shed light on a poorly understood pathogenic mechanism of leptospirosis that needs experimental validation.
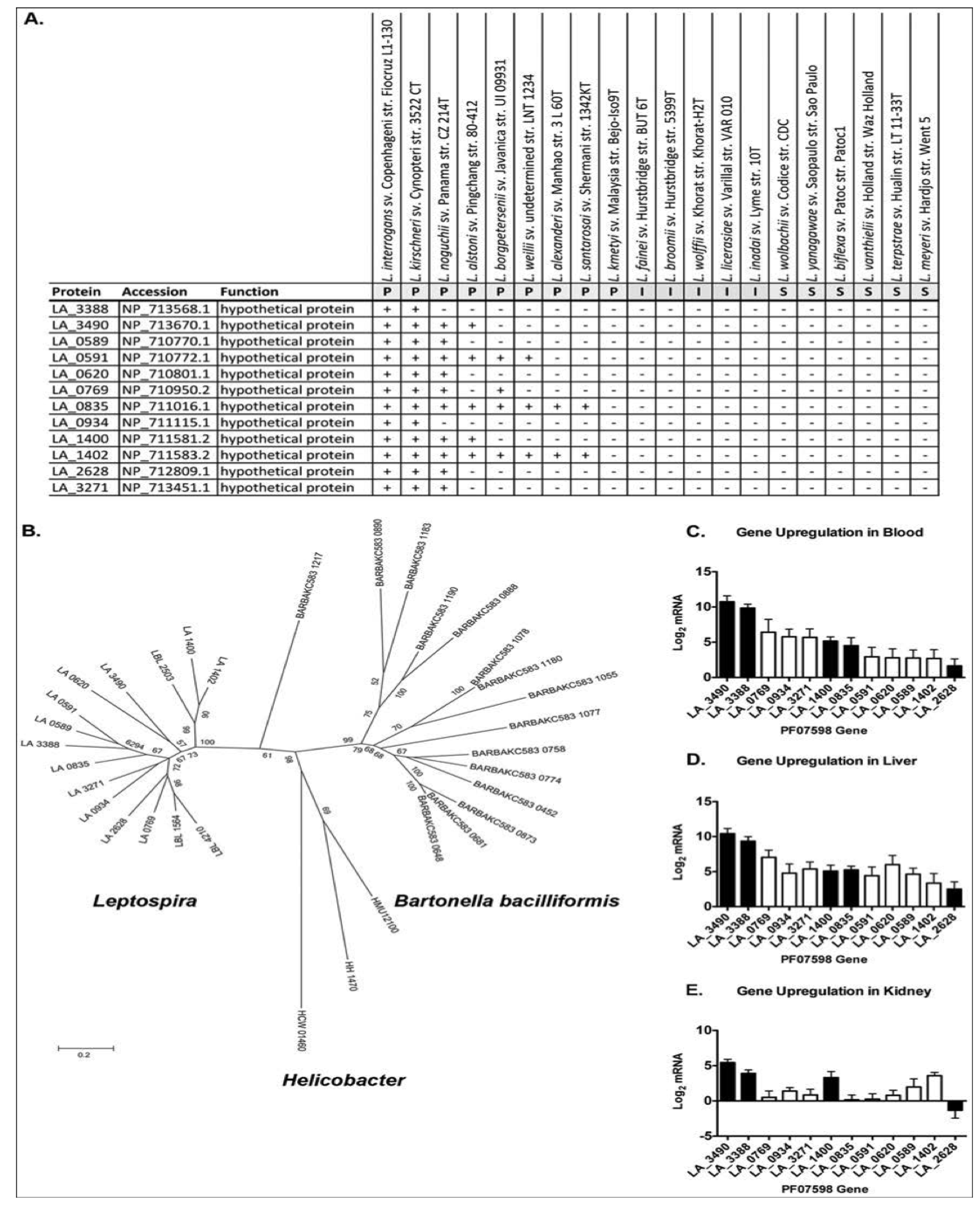
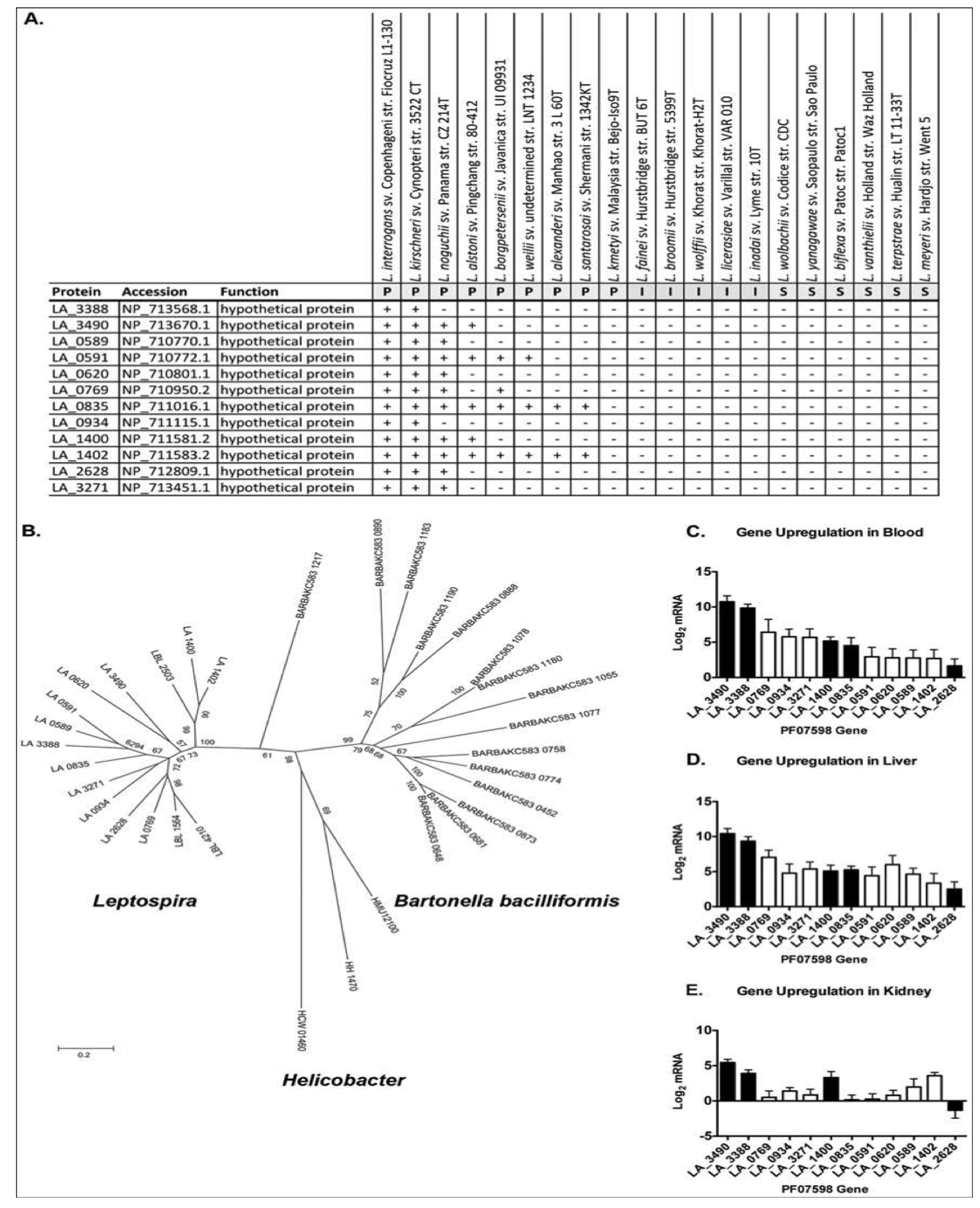
A fascinating discovery that emerged from the same study was the identification of two virulence-associated genes belonging to a paralogous (PF07598) gene family shared by pathogenic Leptospira, but absent in Group I pathogens and saprophytic species (Figure 4A). Paralogs exist in all Group I pathogenic species and are highly up-regulated during infection (Figure 4C,D and [69]). Paralog counts vary amongst the species, ranging from 2 in L. santarosai to 12 in L. interrogans and L. kirschneri. Experimental evidence suggests that members of these proteins may be important for kidney colonization, as a genetic knockout of a PF07598 family member in L. interrogans led to reduced renal colonization [144], suggesting that PF07598 paralogs are not functionally redundant. This hypothesis is supported by the fact that different paralogs are differentially expressed in blood, liver and kidney (Figure 3C–E). Why these proteins are absent from Group II pathogens is unknown, but could explain their reduced capacity to cause symptomatic infections. Curiously, PF07598 protein family has been found in the unrelated α-proteobacteria species: Bartonella bacilliformis and B. australis. B. bacilliformis has 15 paralogs and B. australis has nearly the same [145]. Single gene copies were also found in three animal-infecting ε-proteobacteria: Helicobacter hepaticus, H. mustelae, and H. cetorum. The reasons for the expansion of the family in the related L. interrogans, L. kirshcneri, and L. noguchii species and B. bacilliformis and B. australis is unknown, but could indicate a common pathogenic mechanism or larger scale mechanisms of mammalian host/transmission adaptations.
Figure 4.
Distribution and expression profile of PF07598 gene family (encoding putatively named virulence modulating or VM proteins). (A) Distribution of PF07598 across the genus Leptospira; P, pathogen; I, intermediate; S, saprophyte. (B) Unrooted phylogenetic tree of the VM protein family including sequences from Helicobacter spp. and Bartonella bacilliformis. Node labels represent support from 500 bootstrap replicates. (C–E) Transcript levels of each PF07598 paralog were assessed by real time, reverse transcriptase quantitative PCR of blood, liver and kidney extracts four days after hamster infection and compared to log phase in vitro cultured Leptospira; expressed as the log2 of the fold change between the two conditions. Solid bars indicate proteins potentially extracellular proteins. Data represented are the mean ± SEM of three independent experiments (n = 7 animals). Reproduced from reference [69].
Figure 4.
Distribution and expression profile of PF07598 gene family (encoding putatively named virulence modulating or VM proteins). (A) Distribution of PF07598 across the genus Leptospira; P, pathogen; I, intermediate; S, saprophyte. (B) Unrooted phylogenetic tree of the VM protein family including sequences from Helicobacter spp. and Bartonella bacilliformis. Node labels represent support from 500 bootstrap replicates. (C–E) Transcript levels of each PF07598 paralog were assessed by real time, reverse transcriptase quantitative PCR of blood, liver and kidney extracts four days after hamster infection and compared to log phase in vitro cultured Leptospira; expressed as the log2 of the fold change between the two conditions. Solid bars indicate proteins potentially extracellular proteins. Data represented are the mean ± SEM of three independent experiments (n = 7 animals). Reproduced from reference [69].
Despite the amount of useful data derived from the study, it was not without its limitations. Notably, unlike the previous attenuation study, this study did not determine gene expression of derivative strains, focused on only protein coding genes and because the genomes were not closed, could not report on the impact of genomic rearrangements in vitro. In vitro attenuation has been the basis for many viral and bacterial vaccines (e.g., Yellow fever, measles, mumps, rubella, polio, Bacillus Calmette Guerin (BCG; Mycobacterium bovis)). Why is there gene functional loss and mutation during serial passage in vitro? Are virulence genes superfluous compared to the energy cost of maintaining expression of these genes during a stage of the organism where pathogen-host interactions are not present, or do expressed virulence genes have a directly toxic effect on the organism?
3.2.3. Comparisons of L. borgpetersenii, L. interrogans, L. licerasiae, and L. biflexa
Recent studies underscore the importance of pathogenomics to the study of leptospiral evolution [87,100]. Initial comparisons of L. borgpetersenii, L. interrogans and L. biflexa suggested that infectious species evolved from a non-infectious “free-living” spirochete. As the genome of L. biflexa has fewer transposable elements, it is believed to be much more stable than are the genomes of Group I pathogens, which appear to undergo frequent rearrangements, often involving recombination between insertion sequences. For example, significant differences in organization of the L. borgpetersenii and L. interrogans genomes appear to be IS-mediated. Whereas most putative operons are intact, their order has been shuffled in L. borgpetersenii compared to L. interrogans [107]. Comparisons of the gene repertoires of L. interrogans Lai and Copenhageni and L. borgpetersenii Hardjo indicate that genes absent from L. borgpetersenii include those involved in signal transduction, which could impair adaptation to and thus survival in, diverse environments [107]. In addition, diminished metabolic capacity and reduced solute transport functions in L. borgpetersenii (relative to L. interrogans) would likely limit the range of nutrients that can be utilized by L. borgpetersenii. By contrast, L. interrogans has more signal transduction systems, transcriptional regulatory factors, and metabolic and solute transport functions, consistent with its improved survival ex vivo. These data suggest that L. borgpetersenii has a reduced ability to survive outside a mammalian host and is likely to be restricted to direct animal-animal transmission, whereas L. interrogans has retained environmental sensory functions that facilitate disease transmission through water [107].
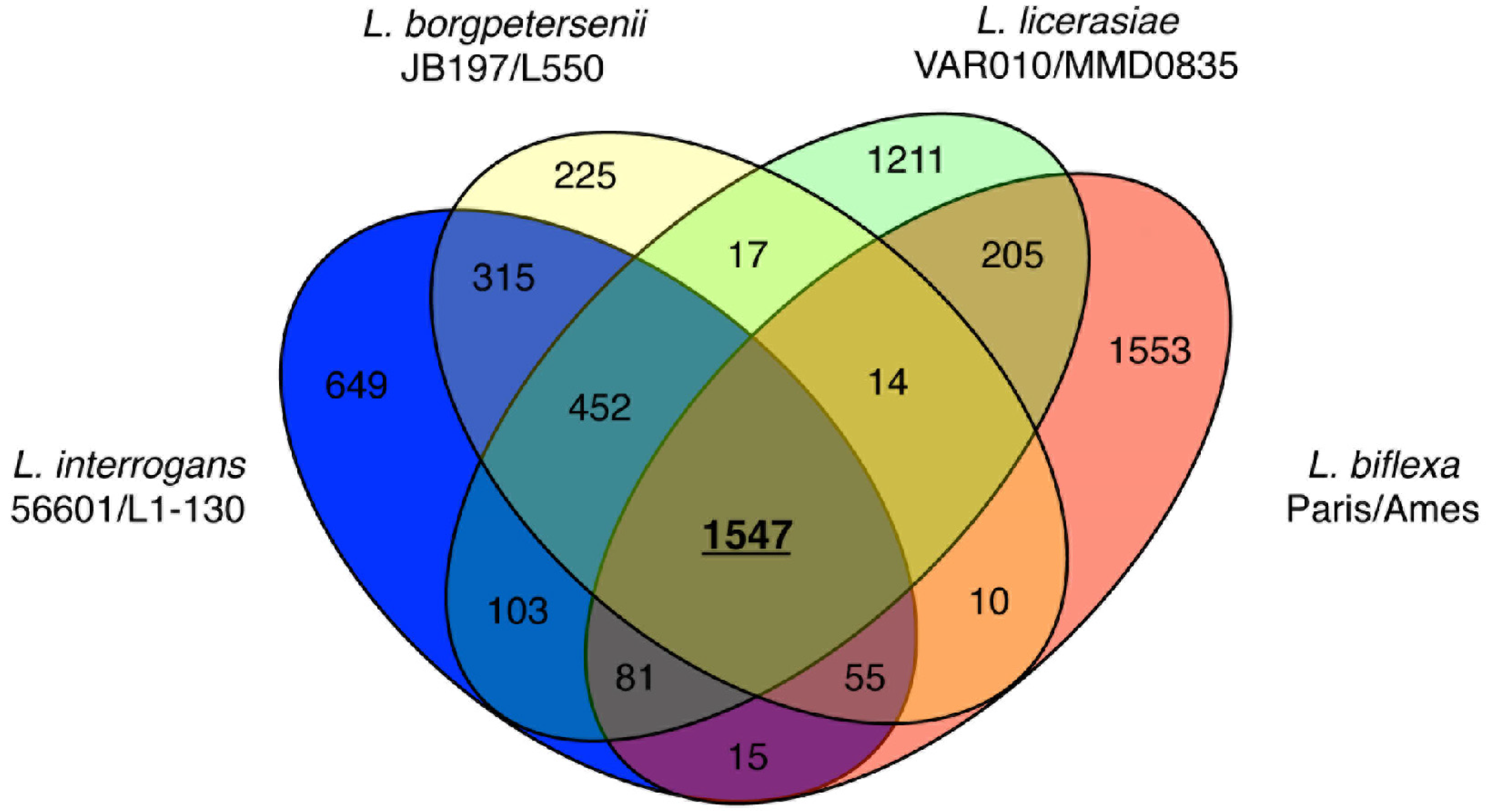
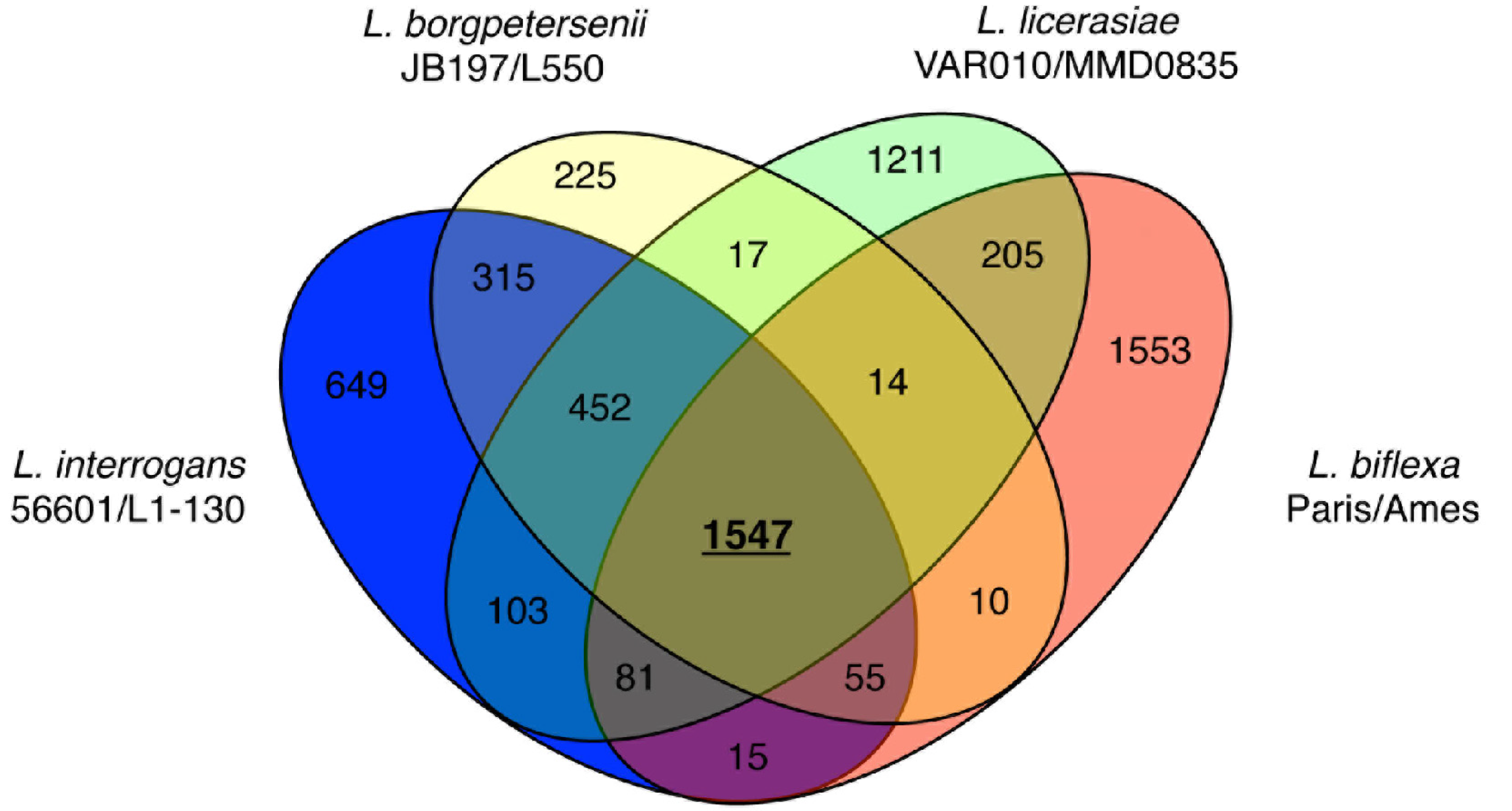
More recent comparisons of eight genomes: L. borgpetersenii (2), L. interrogans (2), L. licerasiae (2) and L. biflexa (2) have extended these early observations, deepening our understanding of leptospiral evolution. The existence of a core leptospiral genome comprising 1547 genes—reduced from the 2052 originally reported based on three strains; 452 conserved genes restricted to pathogenic species and likely to be pathogenicity-related (Figure 5); 649 genes in L. interrogans that could enhance understanding of virulence mechanisms; and 103 genes common to L. interrogans and L. licerasiae, but absent from L. borgpetersenii that could improve understanding of Leptospira environmental sensing and signal transduction systems. Comparisons of the functional content of the genomes suggests that Group II pathogens retain several proteins related to nitrogen, amino acid, and carbohydrate metabolism, which might help to explain why these species grow well in artificial media compared with pathogenic species. Furthermore, several putative GIs are present, suggestive of antecedent LGT with implications for in vivo growth and host preference. Indeed, a 54-kb GI in Lai (Figure 2) could explain altered virulence characteristics and host preference when compared to Copenhageni. How Leptospira became naturally competent for transformation remains to be determined, but considering the phylogenetic origins of the genes comprising the O-antigen cluster and other putative laterally transferred genes, L. licerasiae—and perhaps other pathogenic strains—must be able to exchange genetic material with non-invasive environmental bacteria. Based on predicted functional content and amino acid sequence identity (Figure 2), these results also demonstrated that Group II Leptospira are more closely related genetically to pathogenic than to saprophytic Leptospira. The diminished ability of Group II pathogens to colonize or cause disease in mammals compared to Group I may indicate these strains are adapted to non-mammalian reservoir hosts, but this hypothesis has not been formally tested.
Figure 5.
Venn diagram showing the distribution of shared and unique proteins separated by leptospiral species. Two strains from each species were used for these comparisons. Only proteins present in both strains of a given species are shown. Reproduced from [87].
Figure 5.
Venn diagram showing the distribution of shared and unique proteins separated by leptospiral species. Two strains from each species were used for these comparisons. Only proteins present in both strains of a given species are shown. Reproduced from [87].
3.3. Vaccine Candidates
In addition to providing insight into leptospiral virulence and pathogenic mechanisms by connecting clinical metadata, animal model studies and genomic data, pathogenomics approaches have identified several universal anti-leptospirosis vaccine candidates. Despite decades of research, there is still no licensed human vaccine; and veterinary vaccines provide only transient protection. Livestock must be revaccinated every 6–12 months, which is unrealistic in low-income endemic regions. A hitherto insurmountable problem confronting human or veterinary vaccine development arises from the large geographic differences in transmitted serovars. Dominant serotypes in China and India are not found in Europe or The Americas; and most livestock-infecting strains in the US are uncommon in South America.
The pathogen pan-genome is staggeringly large, comprising some 13,000 predicted genes, of which only ~1500 are part of the core leptospiral genome. Pathogenic species can possess between 200–1,200 species-specific genes [87]. In addition, some genes are conserved in specific, but not all pathogenic species, some are sporadically distributed, and some are restricted to only a few closely related species. Thus, it has been exceedingly difficult to identify broadly reacting leptospiral-specific antigens.
Recent pathogenomics inquiries have narrowed the list of potential candidates considerably. The 452 conserved pathogen-specific genes (Figure 5) include LigB, LipL32, LipL41, four LipL45/FecR-related proteins and LruB. LigA and LigB, which are involved in leptospiral adhesion to extracellular matrix proteins and plasma proteins, including collagens I and IV, laminin, fibronectin, and fibrinogen are induced at physiological osmolarity [146]. Other conserved pathogen-specific outer membrane proteins predicted to mediate attachment to host cells include a putative fibronectin-binding protein, Lfb1 [147] and Lsa66 for Leptospiral surface adhesin of 66 kDa shown to bind laminin and plasma fibronectin [148]. As fibronectin-binding proteins are important adhesins that play an important role in certain bacterial infections, these widely conserved pathogen-specific antigens represent important vaccine candidates. Recent experiments in a hamster model have demonstrated that LigB is a valuable protective antigen that could be used in future subunit anti-leptospirosis vaccines [149]. The function of LruB is unknown, but serology suggests this protein is expressed in vivo [150]. As more infectious and non-infectious species and strains are compared, this list will be winnowed further. Prioritizing these or secreted proteins will result in several good potential candidates.
3.4. Metabolism
As in other bacteria, the availability of different nutrients inside and outside the mammalian host may select for changes in the metabolic capacity of Leptospira. Pathogenomics can provide insight into the unique metabolic capacity of pathogenic microbes. This is important for two reasons: the design of improved growth media and novel therapeutics. Pathogenic strains appear to have developed several strategies to obtain essential nutrients in vivo. For example, LIC13209, which encodes bacterioferritin-associated ferridoxin (BFD), is conserved among infectious species and is upregulated at physiological osmolarity [127]. The function of BFD is not known, but it may be a general redox and/or regulatory component involved in iron storage or mobilization functions of bacterioferritin in bacteria. Iron is essential for bacterial pathogenesis and is required for processes ranging from the tricarboxylic acid cycle to electron transport, DNA metabolism, and response to oxidative stress; however, iron is poorly soluble at physiological pH in the presence of oxygen and not readily bioavailable, especially as it is sequestered from pathogenic organisms as an innate immune mechanism. Thus, during infection, increased expression of this protein might be required to mobilize cellular iron stores to provide a source of iron, which is present in growth-limiting amounts in the host. Similarly pathogenomics analyses suggest that, unlike the saprophytes, pathogenic Leptospira are able to synthesize the essential nutrient B12 [87]; though this has not been tested experimentally. Three genes involved in glutathione metabolism are putatively associated with pathogenicity based on comparisons with other spirochetes. In the oral spirochete, T. denticola, glutathione metabolism is believed to play a significant role in colonization [90]. The potential biological significance of glutathione metabolism by infectious Leptospira may be twofold: H2S production may be critical for hemoxidative, hemolytic, and other toxic activities that could occur in vivo [90], and pyruvate and L-Glutamate, products of glutathione metabolism, can be utilized as nutrients to support bacterial growth and for the synthesis of the essential cofactor vitamin B12, respectively.
4. Future Directions
Leptospirosis, caused by more than 250 different serovars of the genus Leptospira, is the most common and widespread zoonotic disease worldwide. Infection is primarily spread through contact with water contaminated by urine of infected carrier animals. Leptospirosis is clearly an emerging and reemerging infectious disease. There are newly discovered leptospirosis-related syndromes (pulmonary hemorrhage) and newly discovered species of Leptospira that cause human leptospirosis. Accessible and geographically useful leptospirosis diagnostics remain unavailable to diagnose disease efficiently, which prevents the accurate assessment of the burden of disease. Obtaining whole genome sequences of a diverse and representative set of globally-significant Leptospira is a major priority of the leptospirosis community that will directly facilitate these goals of improving public health through the judicious and well-considered application of fundamental scientific discovery. Future directions of Leptospira genome sequencing will include the following:
- To obtain whole genome information for all known Leptospira species. Currently there are nine named pathogenic Leptospira species, five intermediate Leptospira species, and six saprophytic Leptospira species; more have recently been informally reported and inevitably these numbers will grow over coming years. This information will provide the basis for identifying a minimal number of molecular markers for multilocus sequence typing that can differentiate infecting leptospires directly from human samples without the need for bacterial isolation. Conserved protein markers that are the targets of antibody recognition or antigen detection will be identified. Accomplishing these goals depends on obtaining whole genome sequence of a globally diverse and representative set of Leptospira strains.
- To delineate taxonomic and phylogenetic relationships among Leptospira species. Current methods to classify new species or serovars and to identify the emergence of new leptospiral causes of human disease are cumbersome and insufficiently informative. Genomic-level information will allow us to determine serovar without the need for serological typing, will provide fresh insights into the utility of serovar as a tool for strain identification, and if shown to be robust, will facilitate the development of molecular-based serovar typing. Complete genome sequence of reference strains used for serological diagnosis is critical for refining and optimizing efficient diagnosis.
- To understand the mechanisms of leptospirosis pathogenesis and determinants of clinical outcome. Correlations of genetic polymorphisms and virulence will be identified between isolates at the species, serovar, and strain level. This will require sequencing of isolates associated with distinct clinical presentations and outcomes. This data will provide the fundamental basis for hypothesis-driven research to determine virulence factors and for vaccine development.
Key to these efforts will be using pathogenomics tools to comprehensively study Leptospira with the following major goals: understanding the global diversity of Leptospira and the differences in biological behavior of members of this genus; and determining the sequence of geographically and molecularly diverse set of isolates that represent all current Leptospira spp. and diverse serovars. Key outcomes will include a true phylogenetic picture based on global isolates and whole genome data that will lend new insights into taxonomy, evolution and possible global phylogeographic trends. This information will also aid in selection of appropriate targets for diagnostic assays, vaccine development and the development of tools for molecular epidemiology studies.
Acknowledgments
This work was supported by NIH grants T32 GM008666 (NIH predoctoral training grant), R25GM083275 (“Mentoring Young Minds to Increase Diversity in the Biomedical Research”), R21AI064466 (“Microarray Analysis of Leptospiral Genomes”), RO1TW05860 (“Leptospirosis Transmission in the Peruvian Amazon”), 1R01AI108276-01 (“Predicting Risk of Human Leptospirosis Through Environmental Surveillance”) and 1D43TW007120 (Fogarty Global Infectious Diseases Training Grant, “Endemic Infectious Diseases of the Peruvian Amazon”). This project was also funded in part with federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services under contract number HHSN272200900007C. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors would like to acknowledge members of the global leptospirosis community whose work has contributed to completion of this review. Of note, Ben Adler and Dieter Bulach, who identified and characterized the leptospiral rfb locus; Shuang-Xi Ren for ushering in the Leptospira genomics era; Richard Zuerner for his earlier work on leptospiral IS elements, and his contributions to the L. borgpetersenii genome sequencing effort; Mathieu Picardeau who led the L. biflexa genome sequencing effort, sequenced the first Leptospira bacteriophage (LE1) genome and has spearheaded attempts to develop tools for the genetic manipulation of Leptospira; Anna Nascimento and co-authors who published the first Leptospira comparative genomics paper, and Yi Zhong and co-authors for their work comparing the virulent Lai 56601 and avirulent Lai IPAV genomes.
The authors also thank Douglas Berg for helpful suggestions and critical review of the manuscript.
Author Contributions
Jason S. Lehmann, Michael A. Matthias, Joseph M. Vinetz and Derrick E. Fouts wrote the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bharti, A.R.; Nally, J.E.; Ricaldi, J.N.; Matthias, M.A.; Diaz, M.M.; Lovett, M.A.; Levett, P.N.; Gilman, R.H.; Willig, M.R.; Gotuzzo, E. Leptospirosis: a zoonotic disease of global importance. Lancet Infect. Dis. 2003, 3, 757–771. [Google Scholar] [CrossRef]
- Abela-Ridder, B.; Sikkema, R.; Hartskeerl, R.A. Estimating the burden of human leptospirosis. Int. J. Antimicrob. Agents 2010, 36 (Suppl. 1), S5–S7. [Google Scholar] [CrossRef]
- Ko, A.I.; Galvao Reis, M.; Ribeiro Dourado, C.M.; Johnson, W.D., Jr.; Riley, L.W. Urban epidemic of severe leptospirosis in Brazil. Salvador Leptospirosis Study Group. Lancet 1999, 354, 820–825. [Google Scholar] [CrossRef]
- Gouveia, E.L.; Metcalfe, J.; de Carvalho, A.L.; Aires, T.S.; Villasboas-Bisneto, J.C.; Queirroz, A.; Santos, A.C.; Salgado, K.; Reis, M.G.; Ko, A.I. Leptospirosis-associated severe pulmonary hemorrhagic syndrome, Salvador, Brazil. Emerg. Infect. Dis. 2008, 14, 505–508. [Google Scholar] [CrossRef]
- Zaki, S.R.; Shieh, W.J.; The Epidemic Working Group at Ministry of Health in Nicaragua. Leptospirosis associated with outbreak of acute febrile illness and pulmonary haemorrhage, Nicaragua, 1995. Lancet 1996, 347, 535–536. [Google Scholar] [CrossRef]
- Trevejo, R.T.; Rigau-Perez, J.G.; Ashford, D.A.; McClure, E.M.; Jarquin-Gonzalez, C.; Amador, J.J.; de los Reyes, J.O.; Gonzalez, A.; Zaki, S.R.; Shieh, W.J.; et al. Epidemic leptospirosis associated with pulmonary hemorrhage-Nicaragua, 1995. J. Infect. Dis. 1998, 178, 1457–1463. [Google Scholar] [CrossRef]
- Golubev, M.V.; Litvin, V. Mechanism of the maintenance of the infective capacity of soil in a natural focus of leptospirosis. Zh. Mikrobiol. Epidemiol. Immunobiol. 1983, 43–46. [Google Scholar]
- Tonge, J.L.; Smith, D.J. Leptospirosis acquired from soil. Med. J. Aust. 1961, 48, 711–712. [Google Scholar]
- Zaitsev, S.V.; Chernukha Iu, G.; Evdokimova, O.A.; Belov, A.S. Survival rate of Leptospira pomona in the soil at a natural leptospirosis focus. Zh. Mikrobiol. Epidemiol. Immunobiol. 1989, 64–68. [Google Scholar]
- Caldas, E.M.; Sampaio, M.B. Leptospirosis in the city of Salvador, Bahia, Brazil: a case-control seroepidemiologic study. Int. J. Zoonoses 1979, 6, 85–96. [Google Scholar]
- Monahan, A.M.; Miller, I.S.; Nally, J.E. Leptospirosis: risks during recreational activities. J. Appl. Microbiol. 2009, 107, 707–716. [Google Scholar] [CrossRef]
- Gaynor, K.; Katz, A.R.; Park, S.Y.; Nakata, M.; Clark, T.A.; Effler, P.V. Leptospirosis on Oahu: an outbreak associated with flooding of a university campus. Am. J. Trop. Med. Hyg. 2007, 76, 882–885. [Google Scholar]
- Pellizzer, P.; Todescato, A.; Benedetti, P.; Colussi, P.; Conz, P.; Cinco, M. Leptospirosis following a flood in the Veneto area, North-east Italy. Ann. Ig. 2006, 18, 453–456. [Google Scholar]
- Brief report: Leptospirosis after flooding of a university campus--Hawaii, 2004. MMWR Morb. Mortal. Wkly. Rep. 2006, 55, 125–127.
- Vanasco, N.B.; Fusco, S.; Zanuttini, J.C.; Manattini, S.; Dalla Fontana, M.L.; Prez, J.; Cerrano, D.; Sequeira, M.D. Outbreak of human leptospirosis after a flood in Reconquista, Santa Fe, 1998. Rev. Argent Microbiol. 2002, 34, 124–131. [Google Scholar]
- Easton, A. Leptospirosis in Philippine floods. BMJ 1999, 319, 212. [Google Scholar] [CrossRef]
- Adler, B.; Faine, S. Host immunological mechanisms in the resistance of mice to leptospiral infections. Infect. Immun. 1977, 17, 67–72. [Google Scholar]
- Chassin, C.; Picardeau, M.; Goujon, J.M.; Bourhy, P.; Quellard, N.; Darche, S.; Badell, E.; d'Andon, M.F.; Winter, N.; Lacroix-Lamande, S.; et al. TLR4- and TLR2-mediated B cell responses control the clearance of the bacterial pathogen, Leptospira interrogans. J. Immunol. 2009, 183, 2669–2677. [Google Scholar] [CrossRef]
- Outbreak of leptospirosis among white-water rafters--Costa Rica, 1996. MMWR Morb. Mortal. Wkly. Rep. 1997, 46, 577–579.
- Boland, M.; Sayers, G.; Coleman, T.; Bergin, C.; Sheehan, N.; Creamer, E.; O'Connell, M.; Jones, L.; Zochowski, W. A cluster of leptospirosis cases in canoeists following a competition on the River Liffey. Epidemiol. Infect. 2004, 132, 195–200. [Google Scholar] [CrossRef]
- Morgan, J.; Bornstein, S.L.; Karpati, A.M.; Bruce, M.; Bolin, C.A.; Austin, C.C.; Woods, C.W.; Lingappa, J.; Langkop, C.; Davis, B.; et al. Outbreak of leptospirosis among triathlon participants and community residents in Springfield, Illinois, 1998. Clin. Infect. Dis. 2002, 34, 1593–1599. [Google Scholar] [CrossRef]
- Sejvar, J.; Bancroft, E.; Winthrop, K.; Bettinger, J.; Bajani, M.; Bragg, S.; Shutt, K.; Kaiser, R.; Marano, N.; Popovic, T.; et al. Leptospirosis in "Eco-Challenge" athletes, Malaysian Borneo, 2000. Emerg. Infect. Dis. 2003, 9, 702–707. [Google Scholar] [CrossRef]
- Ganoza, C.A.; Matthias, M.A.; Collins-Richards, D.; Brouwer, K.C.; Cunningham, C.B.; Segura, E.R.; Gilman, R.H.; Gotuzzo, E.; Vinetz, J.M. Determining risk for severe leptospirosis by molecular analysis of environmental surface waters for pathogenic Leptospira. PLoS Med. 2006, 3, e308. [Google Scholar] [CrossRef]
- Brenner, D.J.; Kaufmann, A.F.; Sulzer, K.R.; Steigerwalt, A.G.; Rogers, F.C.; Weyant, R.S. Further determination of DNA relatedness between serogroups and serovars in the family Leptospiraceae with a proposal for Leptospira alexanderi sp. nov., and four new Leptospira genomospecies. Int. J. Syst. Bacteriol. 1999, 49, Pt. 2. 839–858. [Google Scholar] [CrossRef]
- Slack, A.T.; Khairani-Bejo, S.; Symonds, M.L.; Dohnt, M.F.; Galloway, R.L.; Steigerwalt, A.G.; Bahaman, A.R.; Craig, S.; Harrower, B.J.; Smythe, L.D. Leptospira kmetyi sp. nov., isolated from an environmental source in Malaysia. Int. J. Syst. Evol. Microbiol. 2009, 59, 705–708. [Google Scholar] [CrossRef]
- Schmid, G.P.; Steere, A.C.; Kornblatt, A.N.; Kaufmann, A.F.; Moss, C.W.; Johnson, R.C.; Hovind-Hougen, K.; Brenner, D.J. Newly recognized Leptospira species ("Leptospira inadai" serovar lyme) isolated from human skin. J. Clin. Microbiol. 1986, 24, 484–486. [Google Scholar]
- Petersen, A.M.; Boye, K.; Blom, J.; Schlichting, P.; Krogfelt, K.A. First isolation of Leptospira fainei serovar Hurstbridge from two human patients with Weil’s syndrome. J. Med. Microbiol. 2001, 50, 96–100. [Google Scholar]
- Matthias, M.A.; Ricaldi, J.N.; Cespedes, M.; Diaz, M.M.; Galloway, R.L.; Saito, M.; Steigerwalt, A.G.; Patra, K.P.; Ore, C.V.; Gotuzzo, E.; et al. Human leptospirosis caused by a new, antigenically unique Leptospira associated with a Rattus species reservoir in the Peruvian Amazon. PLoS Negl. Trop. Dis. 2008, 2, e213. [Google Scholar] [CrossRef]
- Levett, P.N.; Morey, R.E.; Galloway, R.L.; Steigerwalt, A.G. Leptospira broomii sp. nov., isolated from humans with leptospirosis. Int. J. Syst. Evol. Microbiol. 2006, 56, 671–673. [Google Scholar] [CrossRef]
- Slack, A.T.; Kalambaheti, T.; Symonds, M.L.; Dohnt, M.F.; Galloway, R.L.; Steigerwalt, A.G.; Chaicumpa, W.; Bunyaraksyotin, G.; Craig, S.; Harrower, B.J.; et al. Leptospira wolffii sp. nov., isolated from a human with suspected leptospirosis in Thailand. Int. J. Syst. Evol. Microbiol. 2008, 58, 2305–2308. [Google Scholar] [CrossRef]
- Saito, M.; Villanueva, S.Y.; Kawamura, Y.; Iida, K.I.; Tomida, J.; Kanemaru, T.; Kohno, E.; Miyahara, S.; Umeda, A.; Amako, K.; et al. Leptospira idonii sp. nov., isolated from an environmental water in Fukuoka, Japan. Int. J. Syst. Evol. Microbiol. 2012, 63, 2457–2462. [Google Scholar]
- Gravekamp, C.; Korver, H.; Montgomery, J.; Everard, C.O.; Carrington, D.; Ellis, W.A.; Terpstra, W.J. Leptospires isolated from toads and frogs on the Island of Barbados. Zentralbl. Bakteriol. 1991, 275, 403–411. [Google Scholar] [CrossRef]
- Athanazio, D.A.; Silva, E.F.; Santos, C.S.; Rocha, G.M.; Vannier-Santos, M.A.; McBride, A.J.; Ko, A.I.; Reis, M.G. Rattus norvegicus as a model for persistent renal colonization by pathogenic Leptospira interrogans. Acta. Tropica. 2008, 105, 176–180. [Google Scholar] [CrossRef]
- Hathaway, S.C.; Blackmore, D.K. Ecological aspects of the epidemiology of infection with leptospires of the Ballum serogroup in the black rat (Rattus rattus) and the brown rat (Rattus norvegicus) in New Zealand. J. Hyg. (Lond) 1981, 87, 427–436. [Google Scholar] [CrossRef]
- Lloyd-Smith, J.O.; Greig, D.J.; Hietala, S.; Ghneim, G.S.; Palmer, L.; St Leger, J.; Grenfell, B.T.; Gulland, F.M. Cyclical changes in seroprevalence of leptospirosis in California sea lions: endemic and epidemic disease in one host species? BMC Infect. Dis. 2007, 7, 125. [Google Scholar] [CrossRef]
- Ganoza, C.A.; Matthias, M.A.; Saito, M.; Cespedes, M.; Gotuzzo, E.; Vinetz, J.M. Asymptomatic renal colonization of humans in the peruvian Amazon by Leptospira. PLoS Negl. Trop. Dis. 2010, 4, e612. [Google Scholar] [CrossRef]
- Day, T.D.; O'Connor, C.E.; Waas, J.R.; Pearson, A.J.; Matthews, L.R. Transmission of Leptospira interrogans serovar Balcanica infection among socially housed brushtail possums in New Zealand. J. Wildl. Dis. 1998, 34, 576–581. [Google Scholar] [CrossRef]
- Ellis, W.A.; Songer, J.G.; Montgomery, J.; Cassells, J.A. Prevalence of Leptospira interrogans serovar hardjo in the genital and urinary tracts of non-pregnant cattle. Vet. Rec. 1986, 118, 11–13. [Google Scholar]
- Ellis, W.A.; Logan, E.F.; O'Brien, J.J.; Neill, S.D.; Ferguson, H.W.; Hanna, J. Antibodies to Leptospira in the sera of aborted bovine fetuses. Vet. Rec. 1978, 103, 237–239. [Google Scholar]
- Baker, M.F.; Baker, H.J. Pathogenic Leptospira in Malaysian surface waters. I. A method of survey for Leptospira in natural waters and soils. Am. J. Trop. Med. Hyg. 1970, 19, 485–492. [Google Scholar]
- Karaseva, E.V.; Chernukha Yu, G.; Sakhartseva, T.F. Results of the investigation of soil for contamination with pathogenic leptospires. Folia. Parasitol. (Praha) 1977, 24, 301–304. [Google Scholar]
- Ido, Y.; Hoki, R.; Ito, H.; Wani, H. The Rat as a Carrier of Spirochaeta Icterohaemorrhagiae, the Causative Agent of Weil's Disease (Spirochaetosis Icterohaemorrhagica). J. Exp. Med. 1917, 26, 341–353. [Google Scholar] [CrossRef]
- Thiermann, A.B.; Frank, R.R. Human leptospirosis in Detroit and the role of rats as chronic carriers. Int. J. Zoonoses 1980, 7, 62–72. [Google Scholar]
- Vinetz, J.M.; Glass, G.E.; Flexner, C.E.; Mueller, P.; Kaslow, D.C. Sporadic urban leptospirosis. Ann. Intern. Med. 1996, 125, 794–798. [Google Scholar] [CrossRef]
- Everard, C.O.R.; Tikasing, E.S. Ecology of Rodents, Proechimys-Guyannensis-Trinitatis and Oryzomys-Capito-Velutinus, on Trinida. J. Mammal. 1973, 54, 875–886. [Google Scholar] [CrossRef]
- Miller, R.I.; Ross, S.P.; Sullivan, N.D.; Perkins, N.R. Clinical and epidemiological features of canine leptospirosis in North Queensland. Aust. Vet. J. 2007, 85, 13–19. [Google Scholar] [CrossRef]
- Birnbaum, N.; Barr, S.C.; Center, S.A.; Schermerhorn, T.; Randolph, J.F.; Simpson, K.W. Naturally acquired leptospirosis in 36 dogs: serological and clinicopathological features. J. Small Anim. Pract. 1998, 39, 231–236. [Google Scholar] [CrossRef]
- Levett, P.N. Leptospirosis. Clin. Microbiol. Rev. 2001, 14, 296–326. [Google Scholar] [CrossRef]
- Acevedo-Whitehouse, K.; de la Cueva, H.; Gulland, F.M.; Aurioles-Gamboa, D.; Arellano-Carbajal, F.; Suarez-Guemes, F. Evidence of Leptospira interrogans infection in California sea lion pups from the Gulf of California. J. Wildl. Dis. 2003, 39, 145–151. [Google Scholar] [CrossRef]
- Cameron, C.E.; Zuerner, R.L.; Raverty, S.; Colegrove, K.M.; Norman, S.A.; Lambourn, D.M.; Jeffries, S.J.; Gulland, F.M. Detection of pathogenic Leptospira bacteria in pinniped populations via PCR and identification of a source of transmission for zoonotic leptospirosis in the marine environment. J. Clin. Microbiol. 2008, 46, 1728–1733. [Google Scholar] [CrossRef]
- Zuerner, R.L.; Cameron, C.E.; Raverty, S.; Robinson, J.; Colegrove, K.M.; Norman, S.A.; Lambourn, D.; Jeffries, S.; Alt, D.P.; Gulland, F. Geographical dissemination of Leptospira interrogans serovar Pomona during seasonal migration of California sea lions. Vet. Microbiol. 2009, 137, 105–110. [Google Scholar] [CrossRef]
- Colegrove, K.M.; Lowenstine, L.J.; Gulland, F.M. Leptospirosis in northern elephant seals (Mirounga angustirostris) stranded along the California coast. J. Wildl. Dis. 2005, 41, 426–430. [Google Scholar] [CrossRef]
- Matthias, M.A.; Diaz, M.M.; Campos, K.J.; Calderon, M.; Willig, M.R.; Pacheco, V.; Gotuzzo, E.; Gilman, R.H.; Vinetz, J.M. Diversity of bat-associated Leptospira in the Peruvian Amazon inferred by bayesian phylogenetic analysis of 16S ribosomal DNA sequences. Am. J. Trop. Med. Hyg. 2005, 73, 964–974. [Google Scholar]
- Nascimento, A.L.; Ko, A.I.; Martins, E.A.; Monteiro-Vitorello, C.B.; Ho, P.L.; Haake, D.A.; Verjovski-Almeida, S.; Hartskeerl, R.A.; Marques, M.V.; Oliveira, M.C.; et al. Comparative genomics of two Leptospira interrogans serovars reveals novel insights into physiology and pathogenesis. J. Bacteriol. 2004, 186, 2164–2172. [Google Scholar] [CrossRef]
- Fouts, D.E.; Mongodin, E.F.; Mandrell, R.E.; Miller, W.G.; Rasko, D.A.; Ravel, J.; Brinkac, L.M.; DeBoy, R.T.; Parker, C.T.; Daugherty, S.C.; et al. Major structural differences and novel potential virulence mechanisms from the genomes of multiple Campylobacter species. PLoS Biol. 2005, 3, e15. [Google Scholar] [CrossRef] [Green Version]
- Gill, S.R.; Fouts, D.E.; Archer, G.L.; Mongodin, E.F.; Deboy, R.T.; Ravel, J.; Paulsen, I.T.; Kolonay, J.F.; Brinkac, L.; Beanan, M.; et al. Insights on evolution of virulence and resistance from the complete genome analysis of an early methicillin-resistant Staphylococcus aureus strain and a biofilm-producing methicillin-resistant Staphylococcus epidermidis strain. J. Bacteriol. 2005, 187, 2426–2438. [Google Scholar] [CrossRef]
- Nelson, K.E.; Fouts, D.E.; Mongodin, E.F.; Ravel, J.; DeBoy, R.T.; Kolonay, J.F.; Rasko, D.A.; Angiuoli, S.V.; Gill, S.R.; Paulsen, I.T.; et al. Whole genome comparisons of serotype 4b and 1/2a strains of the food-borne pathogen Listeria monocytogenes reveal new insights into the core genome components of this species. Nucleic. Acids. Res. 2004, 32, 2386–2395. [Google Scholar] [CrossRef]
- Chen, Y.; Stine, O.C.; Badger, J.H.; Gil, A.I.; Nair, G.B.; Nishibuchi, M.; Fouts, D.E. Comparative genomic analysis of Vibrio parahaemolyticus: serotype conversion and virulence. BMC Genom. 2011, 12, 294. [Google Scholar] [CrossRef]
- Tettelin, H.; Masignani, V.; Cieslewicz, M.J.; Donati, C.; Medini, D.; Ward, N.L.; Angiuoli, S.V.; Crabtree, J.; Jones, A.L.; Durkin, A.S.; et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. USA 2005, 102, 13950–13955. [Google Scholar] [CrossRef]
- Losada, L.; Ronning, C.M.; DeShazer, D.; Woods, D.; Fedorova, N.; Kim, H.S.; Shabalina, S.A.; Pearson, T.R.; Brinkac, L.; Tan, P.; et al. Continuing evolution of Burkholderia mallei through genome reduction and large-scale rearrangements. Genome. Biol. Evol. 2010, 2, 102–116. [Google Scholar] [CrossRef]
- Pallen, M.J.; Wren, B.W. Bacterial pathogenomics. Nature 2007, 449, 835–842. [Google Scholar] [CrossRef]
- Gardiner, D.M.; McDonald, M.C.; Covarelli, L.; Solomon, P.S.; Rusu, A.G.; Marshall, M.; Kazan, K.; Chakraborty, S.; McDonald, B.A.; Manners, J.M. Comparative pathogenomics reveals horizontally acquired novel virulence genes in fungi infecting cereal hosts. PLoS Pathog. 2012, 8, e1002952. [Google Scholar] [CrossRef]
- Sudheesh, P.S.; Al-Ghabshi, A.; Al-Mazrooei, N.; Al-Habsi, S. Comparative pathogenomics of bacteria causing infectious diseases in fish. Int. J. Evol. Biol. 2012, 2012, 457264. [Google Scholar]
- Crabtree, J.; Angiuoli, S.V.; Wortman, J.R.; White, O.R. Sybil: Methods and Software for Multiple Genome Comparison and Visualization. In Gene Function Analysis; Ochs, M.F., Ed.; Humana Press Inc.: Totowa, NY, USA, 2007; pp. 93–108. [Google Scholar]
- Li, L.; Stoeckert, C.J., Jr.; Roos, D.S. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef]
- Remm, M.; Storm, C.E.; Sonnhammer, E.L. Automatic clustering of orthologs and in-paralogs from pairwise species comparisons. J. Mol. Biol. 2001, 314, 1041–1052. [Google Scholar] [CrossRef]
- Fouts, D.E.; Brinkac, L.; Beck, E.; Inman, J.; Sutton, G. PanOCT: Automated Clustering of Orthologs Using Conserved Gene Neighborhood for Pan-Genomic Analysis of Bacterial Strains and Closely Related Species. Nucleic. Acids. Res. 2012, 40, e172. [Google Scholar] [CrossRef]
- Rasko, D.A.; Myers, G.S.; Ravel, J. Visualization of comparative genomic analyses by BLAST score ratio. BMC Bioinformatics 2005, 6, 2. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, J.S.; Fouts, D.E.; Haft, D.H.; Cannella, A.P.; Ricaldi, J.N.; Brinkac, L.; Harkins, D.; Durkin, S.; Sanka, R.; Sutton, G.; et al. Pathogenomic inference of virulence-associated genes in Leptospira interrogans. PLoS negl. Trop. Dis. 2013, 7, e2468. [Google Scholar] [CrossRef]
- Fouts, D.E. Phage_Finder: automated identification and classification of prophage regions in complete bacterial genome sequences. Nucleic Acids Res. 2006, 34, 5839–5851. [Google Scholar] [CrossRef]
- Srividhya, K.V.; Alaguraj, V.; Poornima, G.; Kumar, D.; Singh, G.P.; Raghavenderan, L.; Katta, A.V.; Mehta, P.; Krishnaswamy, S. Identification of prophages in bacterial genomes by dinucleotide relative abundance difference. PloS One 2007, 2, e1193. [Google Scholar]
- Lima-Mendez, G.; Van Helden, J.; Toussaint, A.; Leplae, R. Prophinder: a computational tool for prophage prediction in prokaryotic genomes. Bioinformatics 2008, 24, 863–865. [Google Scholar] [CrossRef]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A fast phage search tool. Nucleic Acids Res. 2011, 39, W347–W352. [Google Scholar] [CrossRef]
- Akhter, S.; Aziz, R.K.; Edwards, R.A. PhiSpy: A novel algorithm for finding prophages in bacterial genomes that combines similarity- and composition-based strategies. Nucleic Acids Res. 2012, 40, e126. [Google Scholar] [CrossRef]
- Eddy, S.R. A new generation of homology search tools based on probabilistic inference. Genome Inform. 2009, 23, 205–211. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef]
- Phage_Finder version 2.1. Available online: http://sourceforge.net/projects/phage-finder/files/phage_finder_v2.1/ (Accessed on 2 April 2014).
- Langille, M.G.; Brinkman, F.S. IslandViewer: An integrated interface for computational identification and visualization of genomic islands. Bioinformatics 2009, 25, 664–665. [Google Scholar] [CrossRef]
- Langille, M.G.; Hsiao, W.W.; Brinkman, F.S. IslandViewer: An integrated interface for computational identification and visualization of genomic island predictors using a comparative genomics approachislands. BMC Bioinformatics 2009, 925, 329664–329665. [Google Scholar]
- Langille, M.G.; Hsiao, W.W.; Brinkman, F.S. Evaluation of genomic island predictors using a comparative genomics approach. BMC Bioinformatics 2008, 9, 329. [Google Scholar] [CrossRef]
- Waack, S.; Keller, O.; Asper, R.; Brodag, T.; Damm, C.; Fricke, W.F.; Surovcik, K.; Meinicke, P.; Merkl, R. Score-based prediction of genomic islands in prokaryotic genomes using hidden Markov models. BMC Bioinformatics 2006, 7, 142. [Google Scholar] [CrossRef] [Green Version]
- Aydanian, A.; Tang, L.; Morris, J.G.; Johnson, J.A.; Stine, O.C. Genetic diversity of O-antigen biosynthesis regions in Vibrio cholerae. Appl Environ. Microbiol. 2011, 77, 2247–2253. [Google Scholar] [CrossRef]
- Jacobsen, A.; Hendriksen, R.S.; Aaresturp, F.M.; Ussery, D.W.; Friis, C. The Salmonella enterica pan-genome. Microb. Ecol. 2011, 62, 487–504. [Google Scholar] [CrossRef]
- Rodriguez-Valera, F.; Ussery, D.W. Is the pan-genome also a pan-seletome? F1000 Research 2012, 1, 16. [Google Scholar]
- Wright, A. Mechanism of Conversion of the Salmonella O antigen by Bacteriophage Epsilon 34. J. Bacteriol. 1971, 105, 927–936. [Google Scholar]
- Kropinski, A.M.; Prangishvili, D.; Lavigne, R. Position paper: the creation of a rational scheme for the nomenclature of viruses of Bacteria and Archaea. Environ. Microbiol. 2009, 11, 2775–2777. [Google Scholar] [CrossRef]
- Ricaldi, J.N.; Fouts, D.E.; Selengut, J.D.; Harkins, D.M.; Patra, K.P.; Moreno, A.; Lehmann, J.S.; Purushe, J.; Sanka, R.; Torres, M.; et al. Whole Genome Analysis of Leptospira licerasiae Provides Insight into Leptospiral Evolution and Pathogenicity. PLoS negl. Trop. Dis. 2012, 6, e1853. [Google Scholar] [CrossRef]
- Qin, J.H.; Zhang, Q.; Zhang, Z.M.; Zhong, Y.; Yang, Y.; Hu, B.Y.; Zhao, G.P.; Guo, X.K. Identification of a novel prophage-like gene cluster actively expressed in both virulent and avirulent strains of Leptospira interrogans serovar Lai. Infect. Immun. 2008, 76, 2411–2419. [Google Scholar] [CrossRef]
- Bourhy, P.; Frangeul, L.; Couve, E.; Glaser, P.; Saint Girons, I.; Picardeau, M. Complete nucleotide sequence of the LE1 prophage from the spirochete Leptospira biflexa and characterization of its replication and partition functions. J. Bacteriol. 2005, 187, 3931–3940. [Google Scholar] [CrossRef]
- Girons, I.S.; Bourhy, P.; Ottone, C.; Picardeau, M.; Yelton, D.; Hendrix, R.W.; Glaser, P.; Charon, N. The LE1 bacteriophage replicates as a plasmid within Leptospira biflexa: construction of an L. biflexa-Escherichia coli shuttle vector. J. Bacteriol. 2000, 182, 5700–5705. [Google Scholar] [CrossRef]
- He, P.; Sheng, Y.Y.; Shi, Y.Z.; Jiang, X.G.; Qin, J.H.; Zhang, Z.M.; Zhao, G.P.; Guo, X.K. Genetic diversity among major endemic strains of Leptospira interrogans in China. BMC Genomics 2007, 8, 204. [Google Scholar] [CrossRef]
- Van Melderen, L. Toxin-antitoxin systems: why so many, what for? Curr. Opin. Microbiol. 2010, 13, 781–785. [Google Scholar] [CrossRef]
- Gerdes, K. Toxin-antitoxin modules may regulate synthesis of macromolecules during nutritional stress. J. Bacteriol. 2000, 182, 561–572. [Google Scholar] [CrossRef]
- Engelberg-Kulka, H.; Amitai, S.; Kolodkin-Gal, I.; Hazan, R. Bacterial programmed cell death and multicellular behavior in bacteria. PLoS Genet. 2006, 2, e135. [Google Scholar] [CrossRef]
- Hazan, R.; Engelberg-Kulka, H. Escherichia coli mazEF-mediated cell death as a defense mechanism that inhibits the spread of phage P1. Mol. Genet. Genom.: MGG 2004, 272, 227–234. [Google Scholar]
- Pecota, D.C.; Wood, T.K. Exclusion of T4 phage by the hok/sok killer locus from plasmid R1. J. Bacteriol. 1996, 178, 2044–2050. [Google Scholar]
- Ramage, H.R.; Connolly, L.E.; Cox, J.S. Comprehensive functional analysis of Mycobacterium tuberculosis toxin-antitoxin systems: implications for pathogenesis, stress responses, and evolution. PLoS Genet. 2009, 5, e1000767. [Google Scholar] [CrossRef]
- Picardeau, M.; Ren, S.; Girons, I.S. Killing effect and antitoxic activity of the Leptospira interrogans toxin-antitoxin system in Escherichia coli. J. Bacteriol. 2001, 183, 6494–6497. [Google Scholar] [CrossRef]
- Picardeau, M.; Le Dantec, C.; Richard, G.F.; Saint Girons, I. The spirochetal chpK-chromosomal toxin-antitoxin locus induces growth inhibition of yeast and mycobacteria. FEMS Microbiol. Lett. 2003, 229, 277–281. [Google Scholar] [CrossRef]
- Picardeau, M.; Bulach, D.M.; Bouchier, C.; Zuerner, R.L.; Zidane, N.; Wilson, P.J.; Creno, S.; Kuczek, E.S.; Bommezzadri, S.; Davis, J.C.; et al. Genome sequence of the saprophyte Leptospira biflexa provides insights into the evolution of Leptospira and the pathogenesis of leptospirosis. PloS One 2008, 3, e1607. [Google Scholar] [CrossRef]
- Bourhy, P.; Salaun, L.; Lajus, A.; Medigue, C.; Boursaux-Eude, C.; Picardeau, M. A genomic island of the pathogen Leptospira interrogans serovar Lai can excise from its chromosome. Infect. Immun. 2007, 75, 677–683. [Google Scholar] [CrossRef]
- Boursaux-Eude, C.; Saint Girons, I.; Zuerner, R. IS1500, an IS3-like element from Leptospira interrogans. Microbiology 1995, 141, 2165–2173. [Google Scholar] [CrossRef]
- Zuerner, R.L.; Trueba, G.A. Characterization of IS1501 mutants of Leptospira interrogans serovar pomona. FEMS Microbiol. Lett. 2005, 248, 199–205. [Google Scholar] [CrossRef]
- Zuerner, R.L. Nucleotide sequence analysis of IS1533 from Leptospira borgpetersenii: identification and expression of two IS-encoded proteins. Plasmid 1994, 31, 1–11. [Google Scholar] [CrossRef]
- Zhong, Y.; Chang, X.; Cao, X.J.; Zhang, Y.; Zheng, H.; Zhu, Y.; Cai, C.; Cui, Z.; Zhang, Y.; Li, Y.Y.; et al. Comparative proteogenomic analysis of the Leptospira interrogans virulence-attenuated strain IPAV against the pathogenic strain 56601. Cell. Res. 2011, 21, 1210–1229. [Google Scholar] [CrossRef]
- Bulach, D.M.; Kalambaheti, T.; de la Pena-Moctezuma, A.; Adler, B. Lipopolysaccharide biosynthesis in Leptospira. J. Mol. Microbiol. Biotechnol. 2000, 2, 375–380. [Google Scholar]
- Bulach, D.M.; Zuerner, R.L.; Wilson, P.; Seemann, T.; McGrath, A.; Cullen, P.A.; Davis, J.; Johnson, M.; Kuczek, E.; Alt, D.P.; et al. Genome reduction in Leptospira borgpetersenii reflects limited transmission potential. Proc. Natl. Acad. Sci. USA 2006, 103, 14560–14565. [Google Scholar] [CrossRef]
- Chandler, M.G.; Pritchard, R.H. The effect of gene concentration and relative gene dosage on gene output in Escherichia coli. Molecular & general genetics: MGG 1975, 138, 127–141. [Google Scholar]
- Schmid, M.B.; Roth, J.R. Gene location affects expression level in Salmonella typhimurium. J. Bacteriol. 1987, 169, 2872–2875. [Google Scholar]
- Sousa, C.; de Lorenzo, V.; Cebolla, A. Modulation of gene expression through chromosomal positioning in Escherichia coli. Microbiology 1997, 143, 2071–2078. [Google Scholar] [CrossRef]
- Thompson, A.; Gasson, M.J. Location effects of a reporter gene on expression levels and on native protein synthesis in Lactococcus lactis and Saccharomyces cerevisiae. Appl. Environ. Microbiol. 2001, 67, 3434–3439. [Google Scholar]
- Dryselius, R.; Izutsu, K.; Honda, T.; Iida, T. Differential replication dynamics for large and small Vibrio chromosomes affect gene dosage, expression and location. BMC Genomics 2008, 9, 559. [Google Scholar] [CrossRef]
- Block, D.H.; Hussein, R.; Liang, L.W.; Lim, H.N. Regulatory consequences of gene translocation in bacteria. Nucleic Acids Res. 2012, 40, 8979–8992. [Google Scholar] [CrossRef]
- de la Pena-Moctezuma, A.; Bulach, D.M.; Kalambaheti, T.; Adler, B. Comparative analysis of the LPS biosynthetic loci of the genetic subtypes of serovar Hardjo: Leptospira interrogans subtype Hardjoprajitno and Leptospira borgpetersenii subtype Hardjobovis. FEMS Microbiol. Lett. 1999, 177, 319–326. [Google Scholar] [CrossRef]
- Alikhan, N.F.; Petty, N.K.; Ben Zakour, N.L.; Beatson, S.A. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 2011, 12, 402. [Google Scholar] [CrossRef]
- Faruque, S.M.; Sack, D.A.; Sack, R.B.; Colwell, R.R.; Takeda, Y.; Nair, G.B. Emergence and evolution of Vibrio cholerae O139. Proc. Natl. Acad. Sci. USA 2003, 100, 1304–1309. [Google Scholar] [CrossRef]
- Blokesch, M.; Schoolnik, G.K. Serogroup conversion of Vibrio cholerae in aquatic reservoirs. PLoS Pathog. 2007, 3, e81. [Google Scholar] [CrossRef]
- Ricaldi, N.J.; Matthias, M.A.; Vinetz, J.M.; Lewis, A.L. Expression of sialic acids and other nonulosonic acids in Leptospira. BMC Microbiol. 2012, 12, 161. [Google Scholar] [CrossRef]
- Fernandez-Moreira, E.; Helbig, J.H.; Swanson, M.S. Membrane vesicles shed by Legionella pneumophila inhibit fusion of phagosomes with lysosomes. Infect. Immun. 2006, 74, 3285–3295. [Google Scholar] [CrossRef]
- Meri, T.; Murgia, R.; Stefanel, P.; Meri, S.; Cinco, M. Regulation of complement activation at the C3-level by serum resistant leptospires. Microb. Pathog. 2005, 39, 139–147. [Google Scholar] [CrossRef]
- Hathaway, S.C.; Marshall, R.B.; Little, T.W.; Headlam, S.A.; Winter, P.J. Differentiation of reference strains of leptospires of the Pomona serogroup by cross-agglutination absorption and restriction endonuclease analysis. Res. Vet. Sci. 1985, 39, 145–150. [Google Scholar]
- Lo, M.; Bulach, D.M.; Powell, D.R.; Haake, D.A.; Matsunaga, J.; Paustian, M.L.; Zuerner, R.L.; Adler, B. Effects of temperature on gene expression patterns in Leptospira interrogans serovar Lai as assessed by whole-genome microarrays. Infect. Immun. 2006, 74, 5848–5859. [Google Scholar] [CrossRef]
- Xue, F.; Dong, H.; Wu, J.; Wu, Z.; Hu, W.; Sun, A.; Troxell, B.; Yang, X.F.; Yan, J. Transcriptional responses of Leptospira interrogans to host innate immunity: significant changes in metabolism, oxygen tolerance, and outer membrane. PLoS Neglect. Trop. Dis. 2010, 4, e857. [Google Scholar] [CrossRef]
- Wozniak, R.A.; Fouts, D.E.; Spagnoletti, M.; Colombo, M.M.; Ceccarelli, D.; Garriss, G.; Dery, C.; Burrus, V.; Waldor, M.K. Comparative ICE Genomics: Insights into the Evolution of the SXT/R391 Family of ICEs. PLoS Genet. 2009, 5, e1000786. [Google Scholar] [CrossRef]
- Patarakul, K.; Lo, M.; Adler, B. Global transcriptomic response of Leptospira interrogans serovar Copenhageni upon exposure to serum. BMC Microbiol. 2010, 10, 31. [Google Scholar] [CrossRef]
- Lo, M.; Murray, G.L.; Khoo, C.A.; Haake, D.A.; Zuerner, R.L.; Adler, B. Transcriptional response of Leptospira interrogans to iron limitation and characterization of a PerR homolog. Infect. Immun. 2010, 78, 4850–4859. [Google Scholar] [CrossRef]
- Matsunaga, J.; Lo, M.; Bulach, D.M.; Zuerner, R.L.; Adler, B.; Haake, D.A. Response of Leptospira interrogans to physiologic osmolarity: relevance in signaling the environment-to-host transition. Infect. Immun. 2007, 75, 2864–2874. [Google Scholar] [CrossRef]
- Ko, A.I.; Goarant, C.; Picardeau, M. Leptospira: the dawn of the molecular genetics era for an emerging zoonotic pathogen. Nat. Rev. Microbiol. 2009, 7, 736–747. [Google Scholar] [CrossRef]
- Adler, B.; Lo, M.; Seemann, T.; Murray, G.L. Pathogenesis of leptospirosis: The influence of genomics. Vet. Microbiol. 2011, 153, 73–81. [Google Scholar] [CrossRef]
- Narayanavari, S.A.; Sritharan, M.; Haake, D.A.; Matsunaga, J. Multiple leptospiral sphingomyelinases (or are there?). Microbiology 2012, 158, 1137–1146. [Google Scholar] [CrossRef]
- Bourhy, P.; Louvel, H.; Girons, I.S.; Picardeau, M. Random insertional mutagenesis of Leptospira interrogans, the agent of leptospirosis, using a mariner transposon. J. Bacteriol. 2005, 187, 3255–3258. [Google Scholar] [CrossRef]
- Murray, G.L.; Srikram, A.; Henry, R.; Hartskeerl, R.A.; Sermswan, R.W.; Adler, B. Mutations affecting Leptospira interrogans lipopolysaccharide attenuate virulence. Mol. Microbiol. 2010, 78, 701–709. [Google Scholar] [CrossRef]
- Nally, J.E.; Whitelegge, J.P.; Bassilian, S.; Blanco, D.R.; Lovett, M.A. Characterization of the outer membrane proteome of Leptospira interrogans expressed during acute lethal infection. Infect. Immun. 2007, 75, 766–773. [Google Scholar] [CrossRef]
- Lin, Y.P.; Chang, Y.F. A domain of the Leptospira LigB contributes to high affinity binding of fibronectin. Biochem. Biophys. Res. Comm. 2007, 362, 443–448. [Google Scholar] [CrossRef]
- Murray, G.L.; Morel, V.; Cerqueira, G.M.; Croda, J.; Srikram, A.; Henry, R.; Ko, A.I.; Dellagostin, O.A.; Bulach, D.M.; Sermswan, R.W.; et al. Genome-wide transposon mutagenesis in pathogenic Leptospira species. Infect. Immun. 2009, 77, 810–816. [Google Scholar] [CrossRef]
- Ahuja, N.; Kumar, P.; Bhatnagar, R. The adenylate cyclase toxins. Crit. Rev. Microbiol. 2004, 30, 187–196. [Google Scholar] [CrossRef]
- Li, L.; Ojcius, D.M.; Yan, J. Comparison of invasion of fibroblasts and macrophages by high- and low-virulence Leptospira strains: colonization of the host-cell nucleus and induction of necrosis by the virulent strain. Arch. Microbiol. 2007, 188, 591–598. [Google Scholar] [CrossRef]
- Bensing, B.A.; Rubens, C.E.; Sullam, P.M. Genetic loci of Streptococcus mitis that mediate binding to human platelets. Infect. Immun. 2001, 69, 1373–1380. [Google Scholar] [CrossRef]
- Bensing, B.A.; Siboo, I.R.; Sullam, P.M. Proteins PblA and PblB of Streptococcus mitis, which promote binding to human platelets, are encoded within a lysogenic bacteriophage. Infect. Immun. 2001, 69, 6186–6192. [Google Scholar] [CrossRef]
- Mitchell, J.; Sullam, P.M. Streptococcus mitis phage-encoded adhesins mediate attachment to {alpha}2–8-linked sialic acid residues on platelet membrane gangliosides. Infect. Immun. 2009, 77, 3485–3490. [Google Scholar] [CrossRef]
- Edwards, C.N.; Nicholson, G.D.; Hassell, T.A.; Everard, C.O.; Callender, J. Thrombocytopenia in leptospirosis: the absence of evidence for disseminated intravascular coagulation. Am. J. Trop. Med. Hyg. 1986, 35, 352–354. [Google Scholar]
- Marotto, P.C.; Marotto, M.S.; Santos, D.L.; Souza, T.N.; Seguro, A.C. Outcome of leptospirosis in children. Am. J. Trop. Med. Hyg. 1997, 56, 307–310. [Google Scholar]
- Nicodemo, A.C.; Del Negro, G.; Amato Neto, V. Thrombocytopenia and leptospirosis. Rev. Inst. Med. Trop. Sao Paulo 1990, 32, 252–259. [Google Scholar]
- Marcsisin, R.A.; Bartpho, T.; Bulach, D.M.; Srikram, A.; Sermswan, R.W.; Adler, B.; Murray, G.L. Use of a high-throughput screen to identify Leptospira mutants unable to colonize the carrier host or cause disease in the acute model of infection. J. Med. Microbiol. 2013, 62, 1601–1608. [Google Scholar] [CrossRef]
- Guy, L.; Nystedt, B.; Toft, C.; Zaremba-Niedzwiedzka, K.; Berglund, E.C.; Granberg, F.; Naslund, K.; Eriksson, A.S.; Andersson, S.G. A gene transfer agent and a dynamic repertoire of secretion systems hold the keys to the explosive radiation of the emerging pathogen Bartonella. PLoS Genet 2013, 9, e1003393. [Google Scholar] [CrossRef]
- Choy, H.A.; Kelley, M.M.; Chen, T.L.; Moller, A.K.; Matsunaga, J.; Haake, D.A. Physiological osmotic induction of Leptospira interrogans adhesion: LigA and LigB bind extracellular matrix proteins and fibrinogen. Infect. Immun. 2007, 75, 2441–2450. [Google Scholar] [CrossRef]
- Perez, J.; Goarant, C. Rapid Leptospira identification by direct sequencing of the diagnostic PCR products in New Caledonia. BMC Microbiol. 2010, 10, 325. [Google Scholar] [CrossRef]
- Oliveira, R.; de Morais, Z.M.; Goncales, A.P.; Romero, E.C.; Vasconcellos, S.A.; Nascimento, A.L. Characterization of novel OmpA-like protein of Leptospira interrogans that binds extracellular matrix molecules and plasminogen. PloS One 2011, 6, e21962. [Google Scholar]
- Yan, W.; Faisal, S.M.; McDonough, S.P.; Divers, T.J.; Barr, S.C.; Chang, C.F.; Pan, M.J.; Chang, Y.F. Immunogenicity and protective efficacy of recombinant Leptospira immunoglobulin-like protein B (rLigB) in a hamster challenge model. Microbes Infect. 2009, 11, 230–237. [Google Scholar] [CrossRef]
- Verma, A.; Rathinam, S.R.; Priya, C.G.; Muthukkaruppan, V.R.; Stevenson, B.; Timoney, J.F. LruA and LruB antibodies in sera of humans with leptospiral uveitis. Clin. Vaccine. Immunol. CVI 2008, 15, 1019–1023. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Lehmann, J.S.; Matthias, M.A.; Vinetz, J.M.; Fouts, D.E. Leptospiral Pathogenomics. Pathogens 2014, 3, 280-308. https://doi.org/10.3390/pathogens3020280
AMA Style
Lehmann JS, Matthias MA, Vinetz JM, Fouts DE. Leptospiral Pathogenomics. Pathogens. 2014; 3(2):280-308. https://doi.org/10.3390/pathogens3020280
Chicago/Turabian StyleLehmann, Jason S., Michael A. Matthias, Joseph M. Vinetz, and Derrick E. Fouts. 2014. "Leptospiral Pathogenomics" Pathogens 3, no. 2: 280-308. https://doi.org/10.3390/pathogens3020280