An Emerging Tick-Borne Disease of Humans Is Caused by a Subset of Strains with Conserved Genome Structure

Abstract
:
1. Introduction
2. Results and Discussion
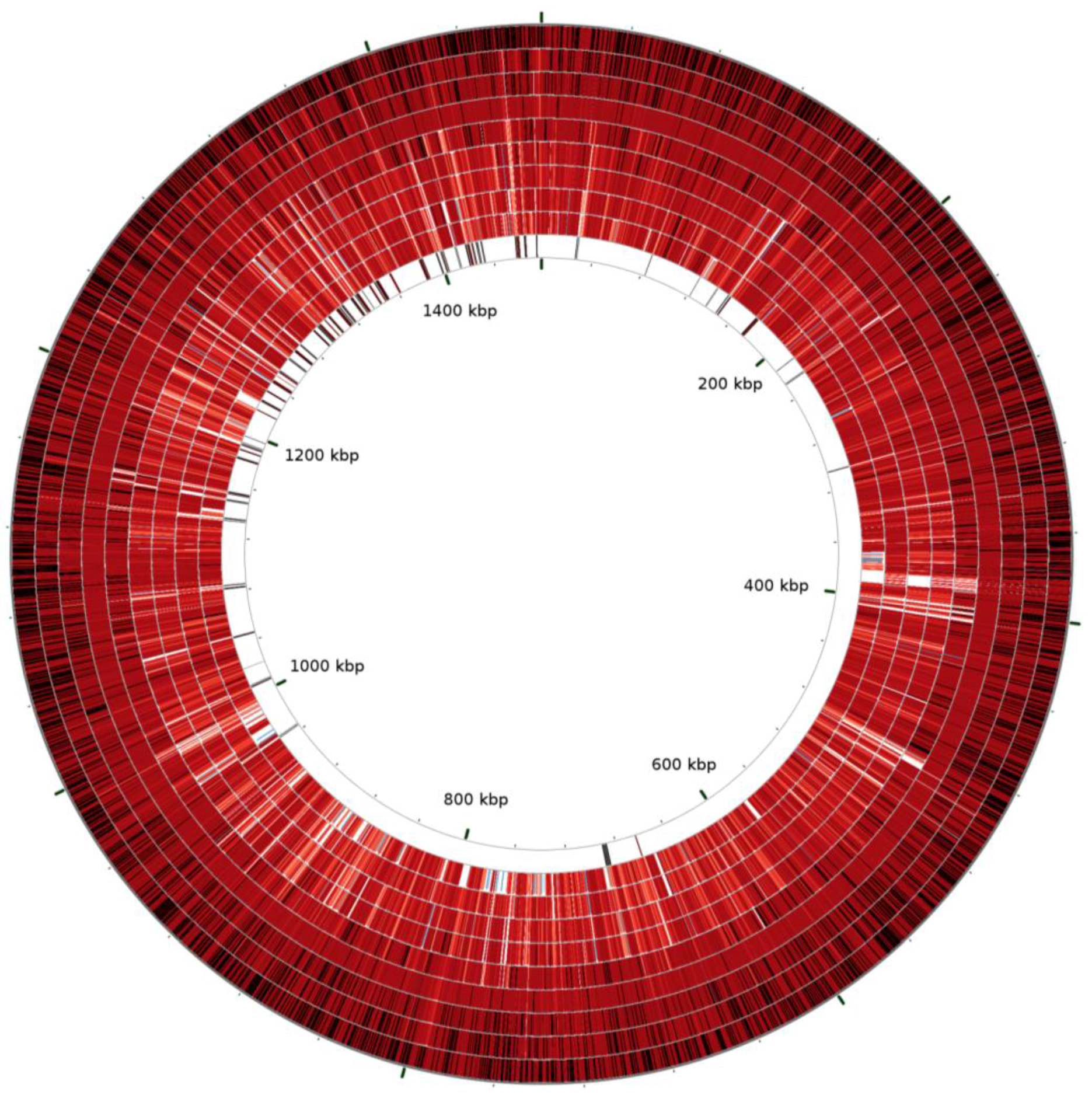
2.1. Comparative Genomics of Nine Strains of A. phagocytophilum
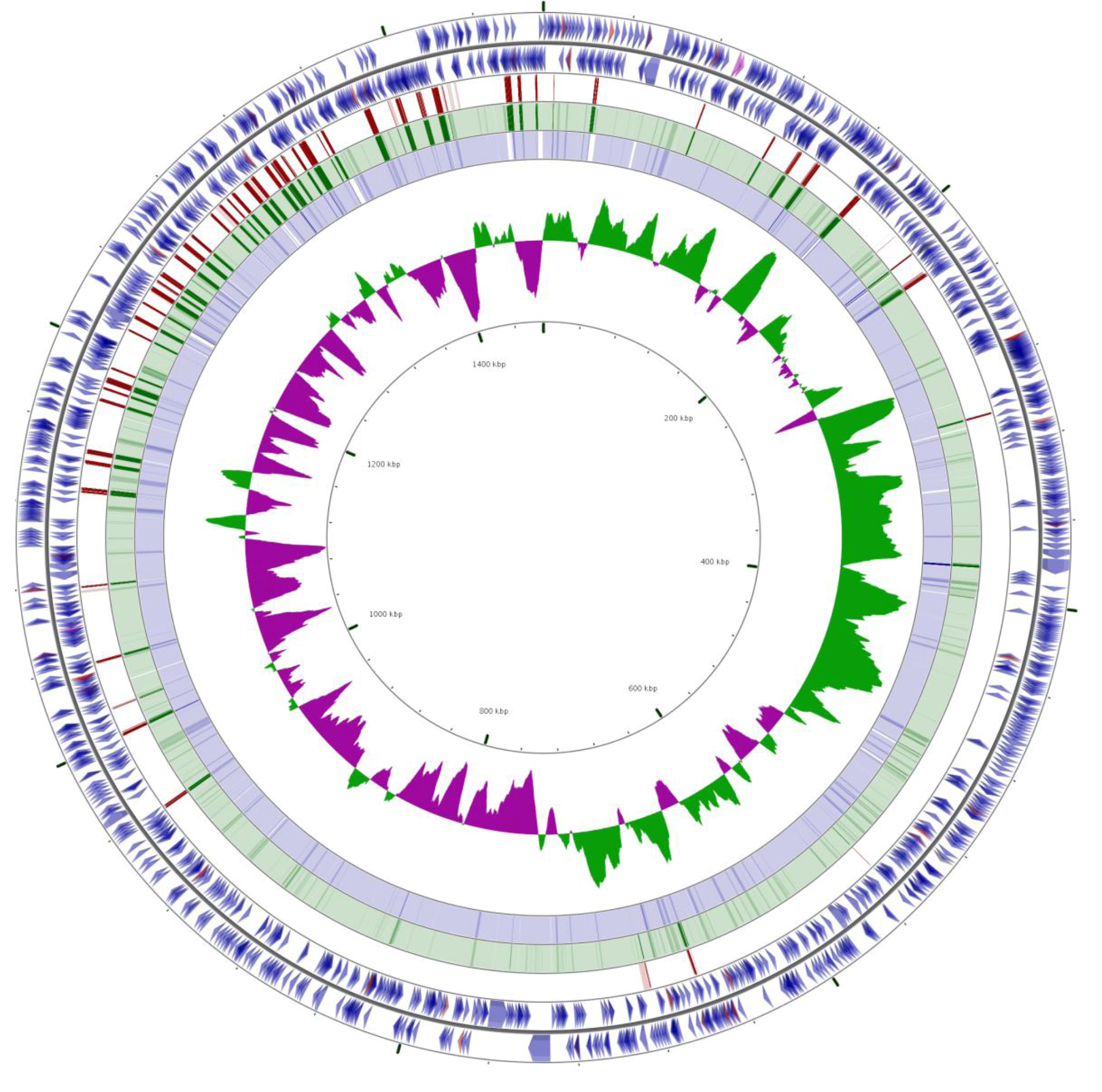
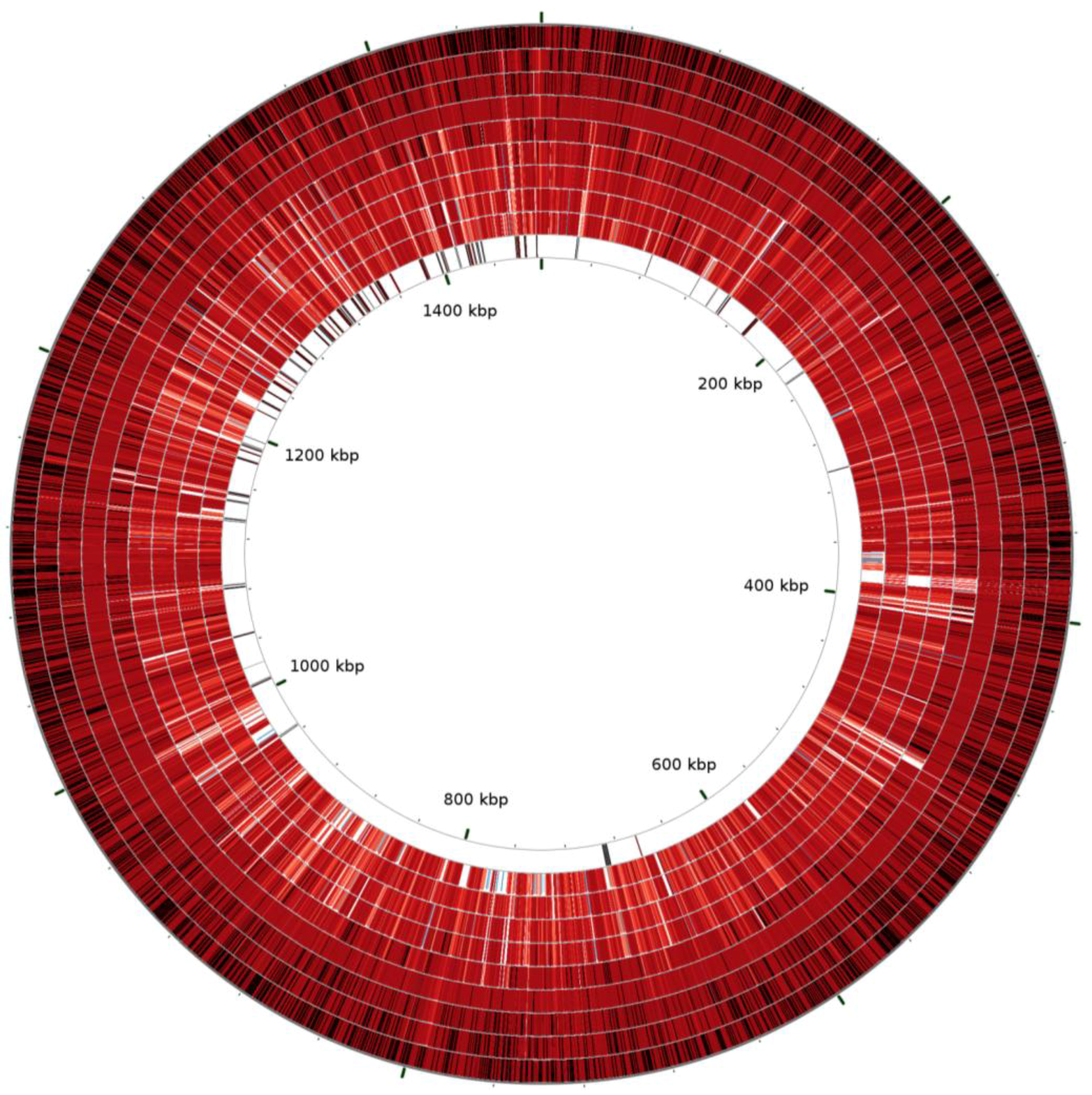

{kind=link}
{kind=link}
{kind=link}
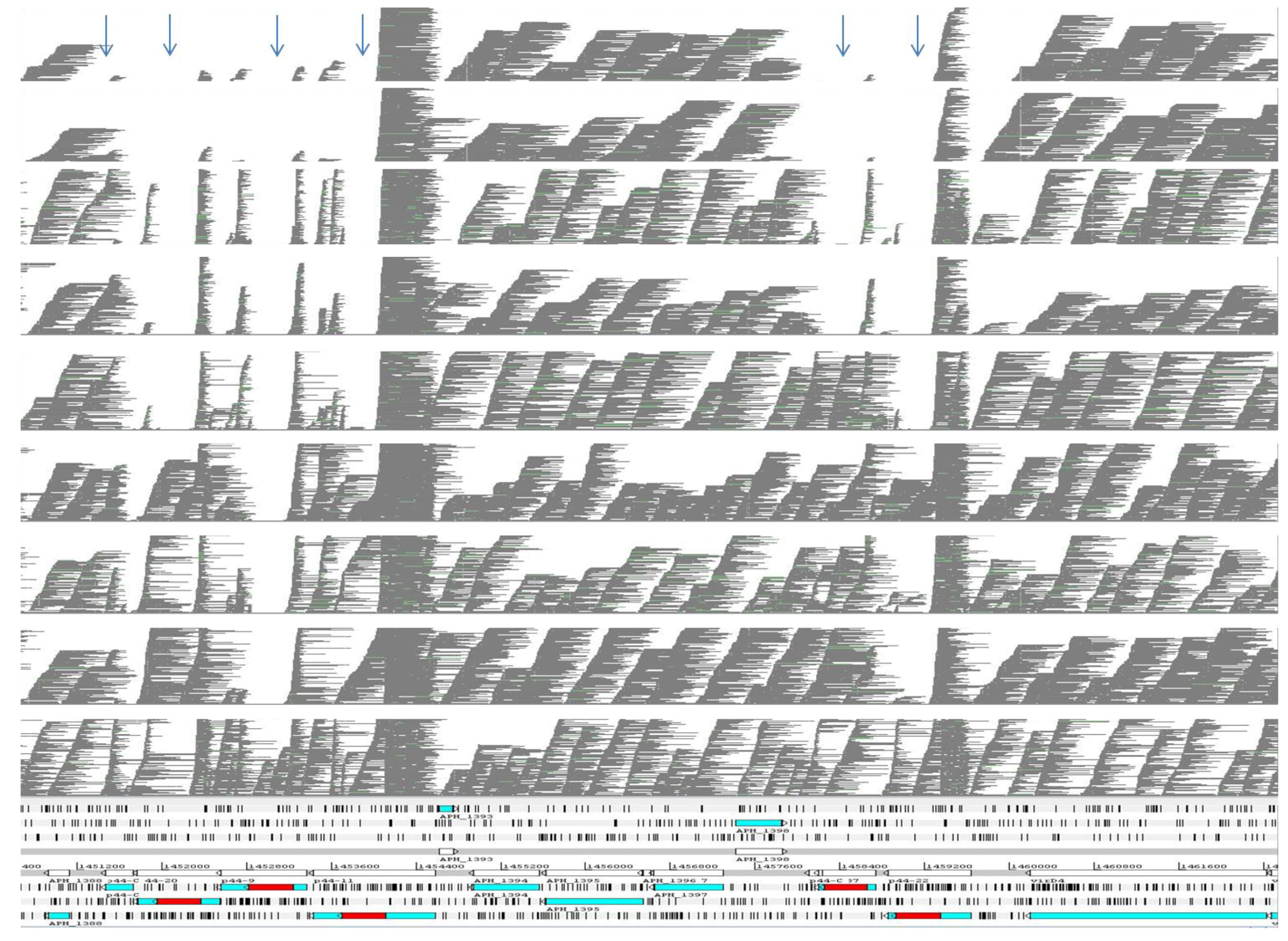{kind=link}
| Strain | ApHZ | ApHGE1 | ApJM | ApDog | ApMRK | ApCRT35 | ApCRT38 | ApNorV1 | ApNorV2 |
|---|---|---|---|---|---|---|---|---|---|
| Different msp2/p44 pseudogenes from ApHZ | 0/95 | 8/95 | 8/95 | 9/95 | 49/95 | 71/95 | 75/95 | 92/95 | 89/95 |
| ANIm | 100 | 98.81 | 98.84 | 98.79 | 97.76 | 96.28 | 96.21 | 94.92 | 95.87 |
| Tetra | 1.000 | 0.999 | 0.999 | 0.999 | 0.998 | 0.996 | 0.996 | 0.995 | 0.996 |
| Strain | AmFL | AmStM | AcIs | ApHZ |
|---|---|---|---|---|
| ANIm | 100 | 98.86 | 88.89 | 77.82 |
| Tetra | 1.000 | 0.999 | 0.989 | 0.821 |
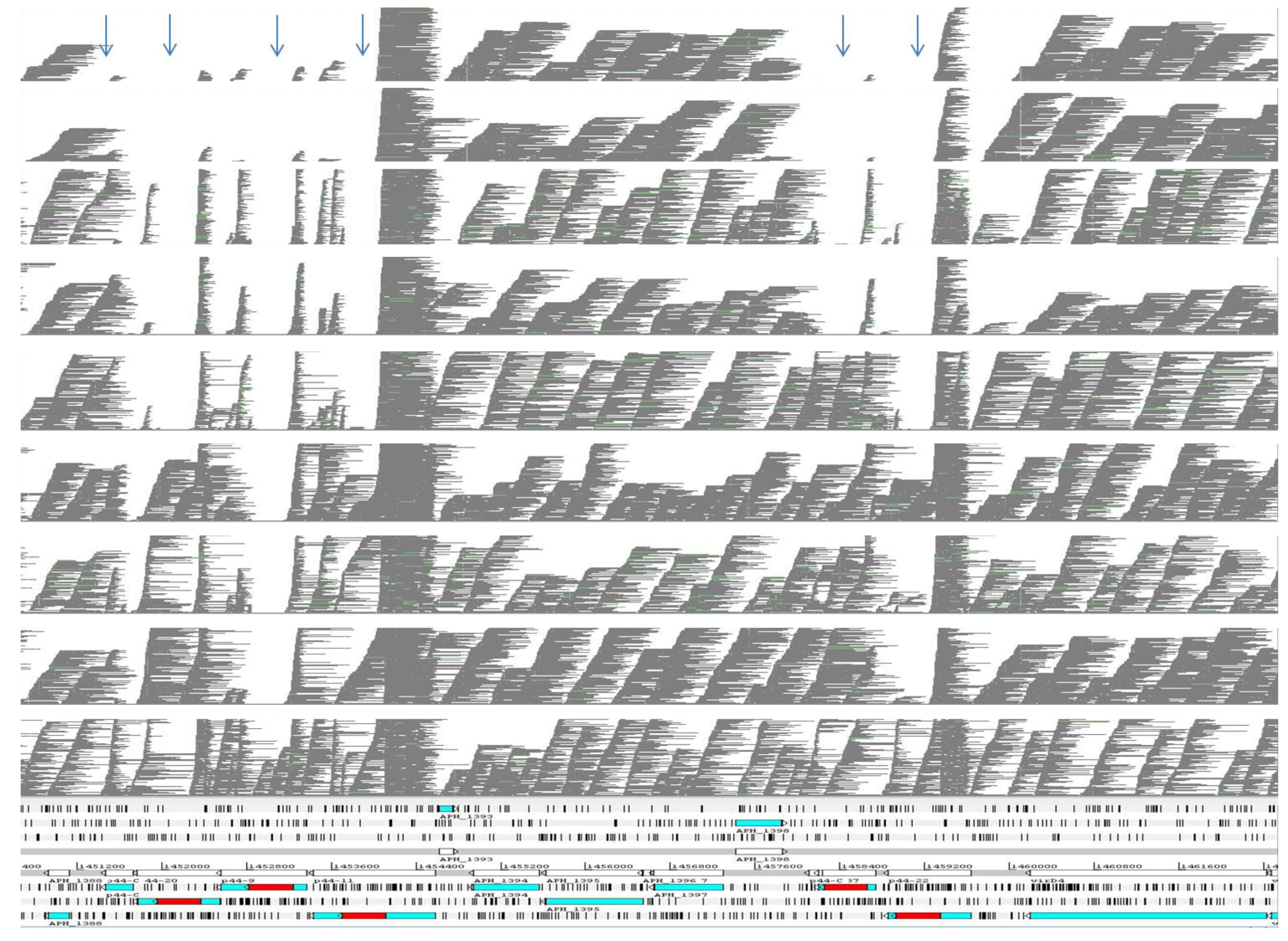
2.2. Comparison of the msp2/p44 Family among Strains of A. phagocytophilum
| Country | Source | Number of msp2/p44 Variants Analyzed | Mean Maximum % a.a. Identity with ApHZ (+/−Std.Dev.) | Significantly different from U.S. Human * |
|---|---|---|---|---|
| U.S.A. | Human | 91 | 97.2 (7.0) | |
| U.S.A. | Dog | 27 | 94.8 (9.8) | No |
| U.S.A. | Horse | 29 | 94.2 (7.3) | No |
| U.S.A. | Bear | 4 | 92.4 (4.1) | N/A |
| U.S.A. | Woodrat | 88 | 78.4 (13.8) | Yes |
| U.S.A. | Ruminant, tick (Ap-variant 1) | 34 | 86.5 (14.7) | Yes |
| Norway | Sheep | 54 | 66.6 (6.4) | Yes |
| Sweden | Dog | 19 | 64.3 (7.9) | Yes |
| U.K. | Goat | 20 | 67.2 (8.2) | Yes |
| U.K. | Sheep | 19 | 65.1 (6.7) | Yes |
| Czech Republic | Human, Roe deer, Perdix, Ixodes ricinus | 6 | 86.6 (14.5) | N/A |
| [Human only] | [2] | [97.8][(2.0)] | N/A | |
| Japan | Sika deer | 17 | 39.3 (3.0) | Yes |
| Japan | Ixodes persulcatus | 87 | 69.0 (6.9) | Yes |
| Japan | Ixodes ovatus | 22 | 71.8 (15.5) | Yes |
| Japan | Haemaphysalis formosensis | 9 | 75.6 (14) | N/A |
| Japan | Human | 27 | 98.1 (7.2) | No |
| China | Human | 2 | 100 (0) | N/A |
3. Experimental
3.1. Origin of A. phagocytophilum Strains
3.2. Ethics Statement
3.3. Genome Sequencing and Bioinformatics
3.4. Analysis of msp2/p44 Repertoires
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Patz, J.A.; Olson, S.H.; Uejio, C.K.; Gibbs, H.K. Disease emergence from global climate and land use change. Med. Clin. North Am. 2008, 92, 1473–1491. [Google Scholar] [CrossRef]
- Dumler, J.S.; Choi, K.S.; Garcia-Garcia, J.C.; Barat, N.S.; Scorpio, D.G.; Garyu, J.W.; Grab, D.J.; Bakken, J.S. Human granulocytic anaplasmosis and Anaplasma phagocytophilum. Emerg. Infect. Dis. 2005, 11, 1828–1834. [Google Scholar] [CrossRef]
- CDC. Statistics and Epidemiology. Annual Cases of Anaplasmosis in the United States. Available online: www.cdc.gov/anaplasmosis/stats/ (accessed on 5 September 2013).
- Dahlgren, F.S.; Mandel, E.J.; Krebs, J.W.; Massung, R.F.; McQuiston, J.H. Increasing incidence of Ehrlichia chaffeensis and Anaplasma phagocytophilum in the United States, 2000–2007. Am. J. Trop. Med. Hyg. 2011, 85, 124–131. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, G.; Liu, Q.; Chen, C.; Li, J.; Long, B.; Yu, H.; Zhang, Z.; He, J.; Qu, Z.; et al. Molecular analysis of Anaplasma phagocytophilum isolated from patients with febrile diseases of unknown etiology in China. PLoS One 2013, 8, e57155. [Google Scholar]
- Annen, K.; Friedman, K.; Eshoa, C.; Horowitz, M.; Gottschall, J.; Straus, T. Two cases of transfusion-transmitted Anaplasma phagocytophilum. Am. J. Clin. Pathol. 2012, 137, 562–565. [Google Scholar] [CrossRef]
- Jereb, M.; Pecaver, B.; Tomazic, J.; Muzlovic, I.; Avsic-Zupanc, T.; Premru-Srsen, T.; Levicnik-Stezinar, S.; Karner, P.; Strle, F. Severe human granulocytic anaplasmosis transmitted by blood transfusion. Emerg. Infect. Dis. 2012, 18, 1354–1357. [Google Scholar]
- Massung, R.F.; Priestley, R.A.; Miller, N.J.; Mather, T.N.; Levin, M.L. Inability of a variant strain of Anaplasma phagocytophilum to infect mice. J. Infect. Dis. 2003, 188, 1757–1763. [Google Scholar] [CrossRef]
- Massung, R.F.; Mather, T.N.; Levin, M.L. Reservoir competency of goats for the Ap-variant 1 strain of Anaplasma phagocytophilum. Infect. Immun. 2006, 74, 1373–1375. [Google Scholar] [CrossRef]
- Dunning Hotopp, J.C.; Lin, M.; Madupu, R.; Crabtree, J.; Angiuoli, S.V.; Eisen, J.A.; Seshadri, R.; Ren, Q.; Wu, M.; Utterback, T.R.; et al. Comparative genomics of emerging human ehrlichiosis agents. PLoS Genet. 2006, 2, e21. [Google Scholar] [CrossRef]
- Barbet, A.F.; Meeus, P.F.; Bélanger, M.; Bowie, M.V.; Yi, J.; Lundgren, A.M.; Alleman, A.R.; Wong, S.J.; Chu, F.K.; Munderloh, U.G.; et al. Expression of multiple outer membrane protein sequence variants from a single genomic locus of Anaplasma phagocytophilum. Infect. Immun. 2003, 71, 1706–1718. [Google Scholar] [CrossRef]
- Lin, Q.; Rikihisa, Y. Establishment of cloned Anaplasma phagocytophilum and analysis of p44 gene conversion within an infected horse and infected SCID mice. Infect. Immun. 2005, 73, 5106–5114. [Google Scholar] [CrossRef]
- Foley, J.E.; Nieto, N.C.; Barbet, A.F.; Foley, P. Antigen diversity in the parasitic bacterium Anaplasma phagocytophilum arises from selectively-represented, spatially clustered functional pseudogenes. PLoS One 2009, 4, e8265. [Google Scholar]
- Stuen, S. Anaplasma phagocytophilum—The most widespread tick-borne infection in animals in Europe. Vet. Res. Commun. 2007, 31, 79–84. [Google Scholar] [CrossRef]
- Richter, M.; Rosselló-Móra, R. Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. USA 2009, 106, 19126–19131. [Google Scholar] [CrossRef]
- Barbet, A.F.; Lundgren, A.M.; Alleman, A.R.; Stuen, S.; Bjöersdorff, A.; Brown, R.N.; Drazenovich, N.L.; Foley, J.E. Structure of the expression site reveals global diversity in MSP2 (P44) variants in Anaplasma phagocytophilum. Infect. Immun. 2006, 74, 6429–6437. [Google Scholar] [CrossRef]
- Al-Khedery, B.; Lundgren, A.M.; Stuen, S.; Granquist, E.G.; Munderloh, U.G.; Nelson, C.M.; Alleman, A.R.; Mahan, S.M.; Barbet, A.F. Structure of the type IV secretion system in different strains of Anaplasma phagocytophilum. BMC Genomics 2012, 13, e678. [Google Scholar] [CrossRef]
- Grant, J.R.; Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, W181–W184. [Google Scholar] [CrossRef]
- Grant, J.R.; Stothard, P. Comparing thousands of circular genomes using the CGView Comparison Tool. BMC Genomics 2012, 13, e202. [Google Scholar] [CrossRef]
- Dark, M.J.; Al-Khedery, B.; Barbet, A.F. Multistrain genome analysis identifies candidate vaccine antigens of Anaplasma marginale. Vaccine 2011, 29, 4923–4932. [Google Scholar] [CrossRef]
- Casey, A.N.J.; Birtles, R.J.; Radford, A.D.; Bown, K.J.; French, N.P.; Woldehiwet, Z.; Ogden, N.H. Groupings of highly similar major surface protein (p44)-encoding paralogues: A potential index of genetic diversity amongst isolates of Anaplasma phagocytophilum. Microbiology 2004, 150, 727–734. [Google Scholar] [CrossRef]
- Gaowa; Wuritu; Wu, D.; Yoshikawa, Y.; Ohashi, N.; Kawamori, F.; Sugiyama, K.; Ohtake, M.; Ohashi, M.; Yamamoto, S.; et al. Dection and characterization of p44/msp2 transcript variants of Anaplasma phagocytophilum from naturally infected ticks and wild deer in Japan. Jpn. J. Infect. Dis. 2012, 65, 79–83. [Google Scholar]
- Wuritu; Gaowa; Kawamori, F.; Aochi, M.; Masuda, T.; Ohashi, N. Characterization of p44/msp2 multigene family of Anaplasma phagocytophilum from two different tick species, Ixodes persulcatus and Ixodes ovatus, in Japan. Jpn. J. Infect. Dis. 2009, 62, 142–145. [Google Scholar]
- Ohashi, N.; Gaowa; Wuritu; Kawamori, F.; Wu, D.; Yoshikawa, Y.; Chiya, S.; Fukunaga, K.; Funato, T.; Shiojiri, M.; et al. Human granulocytic anaplasmosis, Japan. Emerg. Infect. Dis. 2013, 19, 289–292. [Google Scholar] [CrossRef]
- Caspersen, K.; Park, J.H.; Patil, S.; Dumler, J.S. Genetic variability and stability of Anaplasma phagocytophila msp2 (p44). Infect. Immun. 2002, 70, 1230–1234. [Google Scholar] [CrossRef]
- Campanella, J.J.; Bitincka, L.; Smalley, J. MatGAT: An application that generates similarity/identity matrices using protein or DNA sequences. BMC Bioinformatics 2003, 4, e29. [Google Scholar] [CrossRef] [Green Version]
- Bowman, D.; Little, S.E.; Lorentzen, L.; Shields, J.; Sullivan, M.P.; Carlin, E.P. Prevalence and geographic distribution of Dirofilaria immitis, Borrelia burgdorferi, Ehrlichia canis, and Anaplasma phagocytophilum in dogs in the United States: Results of a national clinic-based serologic survey. Vet. Parasitol. 2009, 160, 138–148. [Google Scholar] [CrossRef]
- Kim, H.Y.; Rikihisa, Y. Characterization of monoclonal antibodies to the 44-kilodalton major outer membrane protein of the human granulocytic ehrlichiosis agent. J. Clin. Microbiol. 1998, 36, 3278–3284. [Google Scholar]
- Wang, X.; Kikuchi, T.; Rikihisa, Y. Two monoclonal antibodies with defined epitopes of P44 major surface proteins neutralize Anaplasma phagocytophilum by distinct mechanisms. Infect. Immun. 2006, 74, 1873–1882. [Google Scholar] [CrossRef]
- Foley, J.E.; Nieto, N.C.; Massung, R.; Barbet, A.; Madigan, J.; Brown, R.N. Distinct ecologically relevant strains of Anaplasma phagocytophilum. Emerg. Infect. Dis. 2009, 15, 842–843. [Google Scholar] [CrossRef]
- The NCBI Bioproject Portal. Available online: www.ncbi.nlm.nih.gov/bioproject (accessed on 5 September 2013).
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Barbet, A.F.; Al-Khedery, B.; Stuen, S.; Granquist, E.G.; Felsheim, R.F.; Munderloh, U.G. An Emerging Tick-Borne Disease of Humans Is Caused by a Subset of Strains with Conserved Genome Structure. Pathogens 2013, 2, 544-555. https://doi.org/10.3390/pathogens2030544
Barbet AF, Al-Khedery B, Stuen S, Granquist EG, Felsheim RF, Munderloh UG. An Emerging Tick-Borne Disease of Humans Is Caused by a Subset of Strains with Conserved Genome Structure. Pathogens. 2013; 2(3):544-555. https://doi.org/10.3390/pathogens2030544
Chicago/Turabian StyleBarbet, Anthony F., Basima Al-Khedery, Snorre Stuen, Erik G. Granquist, Roderick F. Felsheim, and Ulrike G. Munderloh. 2013. "An Emerging Tick-Borne Disease of Humans Is Caused by a Subset of Strains with Conserved Genome Structure" Pathogens 2, no. 3: 544-555. https://doi.org/10.3390/pathogens2030544