Host-Viral Interactions: Role of Pattern Recognition Receptors (PRRs) in Human Pneumovirus Infections

Abstract
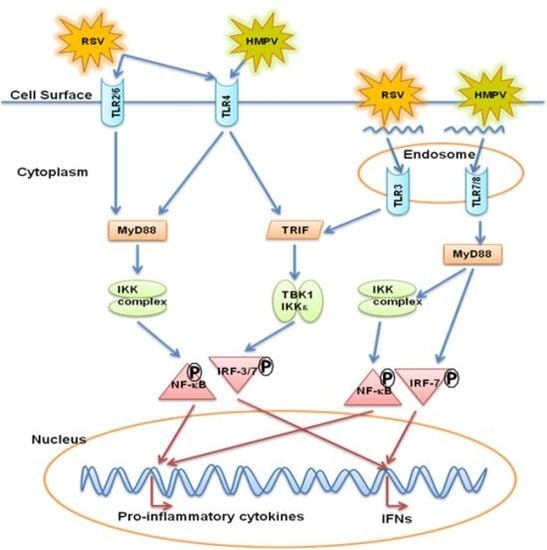
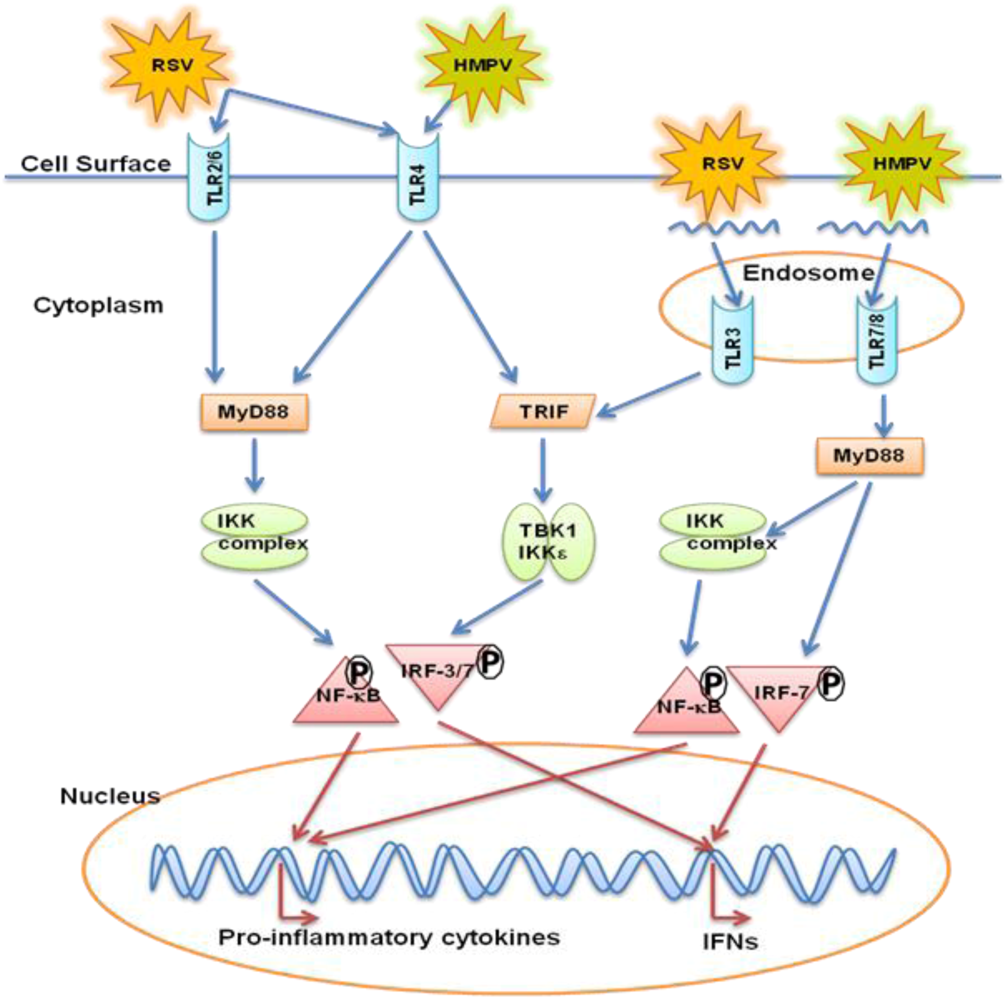
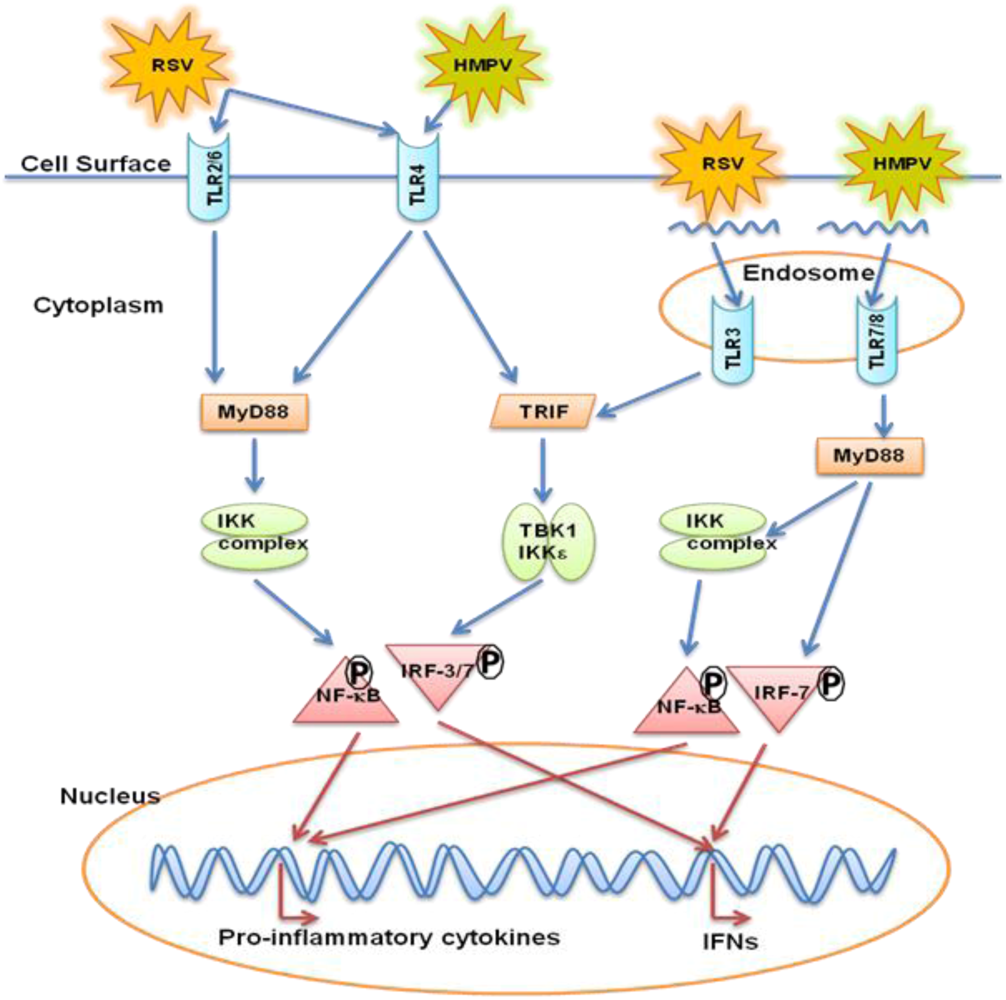
:
1. Introduction
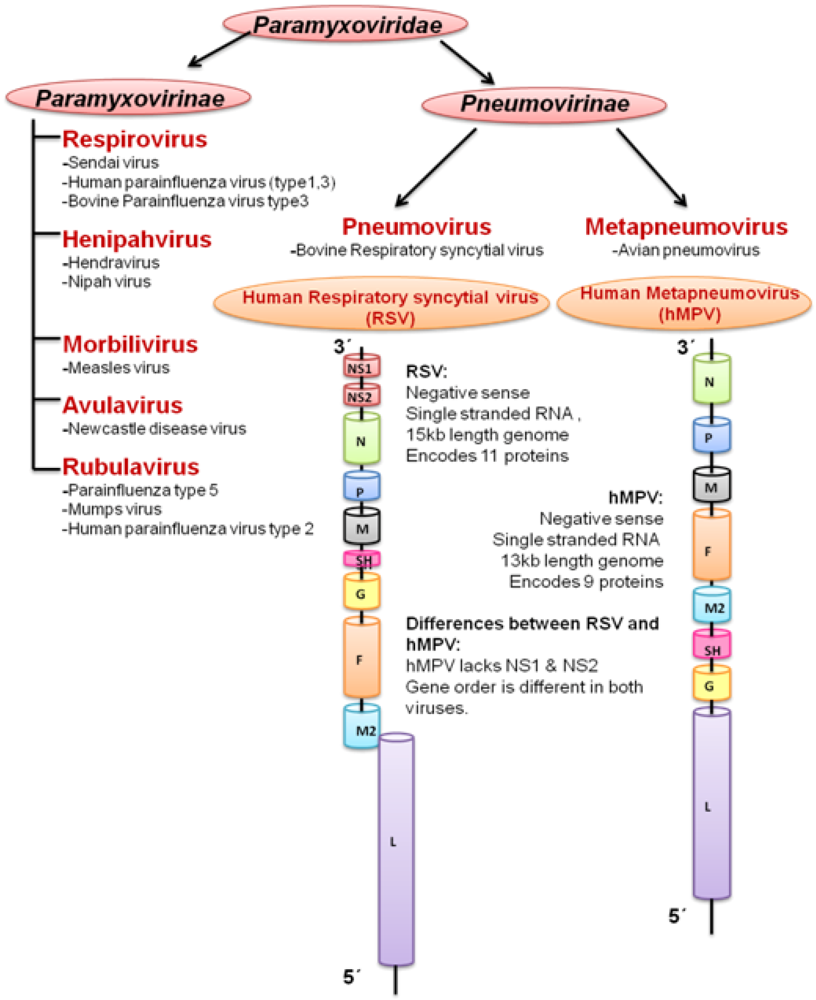
2. Respiratory Syncytial Virus (RSV)

3. Human Metapneumovirus (hMPV)
4. Pattern Recognition Receptors (PRRs)
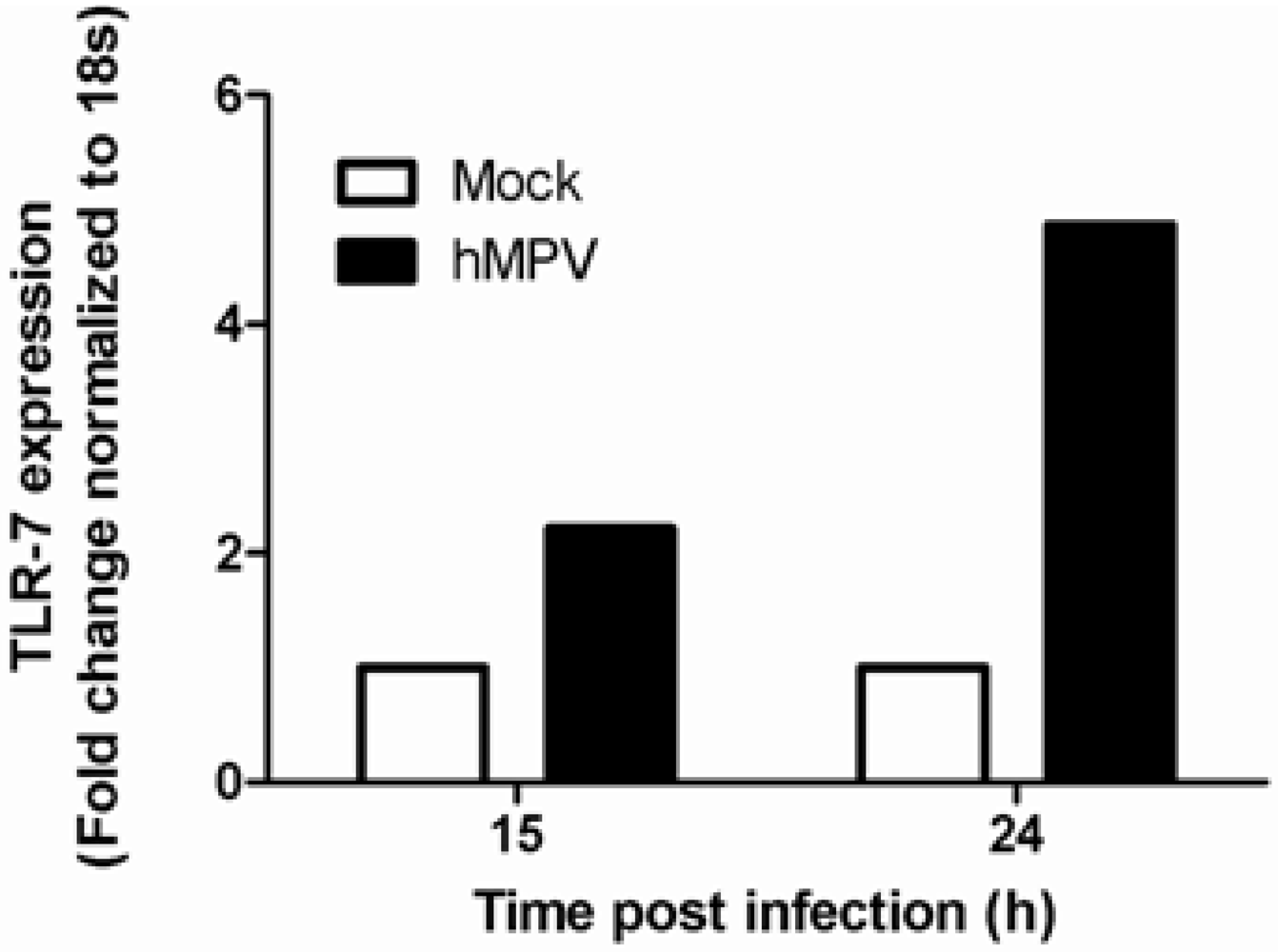
5. Toll Like Receptors (TLRs)


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pattern Recognition Receptors | Virus | Studies | |
|---|---|---|---|
| In vivo / ex vivo (Ref) | In vitro (Ref) | ||
| TLR 1/2,6 | RSV | 61, 71–74 | 74 |
| hMPV | - | - | |
| TLR 3 | RSV | 62, 84–88 | 79–81 |
| hMPV | 65, 90 | - | |
| TLR 4 | RSV | 61, 63, 114–123 | 107–113, 118, 121 |
| hMPV | 65, 134 | - | |
| TLR 7/ 8 | RSV | 64, 89, 136–140 | - |
| hMPV | 66, 65, 77, 90 | 66 | |
| TLR 9 | RSV | 140, 141, 143, 200 | - |
| hMPV | 77 | - | |
| MyD88 | RSV | 166 | - |
| hMPV | - | - | |
| RIG-I | RSV | 166–169 | 33, 81, 164, 165, 168, 170–172 |
| hMPV | 66 | 66, 162, 174, 176 | |
| MDA5 | RSV | - | 172 |
| hMPV | 173 | - | |
| MAVS | RSV | 166, 167 | 176 |
| hMPV | - | 175, 176 | |
| NLRP3 | RSV | 74 | 74 |
| hMPV | - | - | |
| NOD2 | RSV | 32, 169, 200 | 32 |
| hMPV | - | - | |
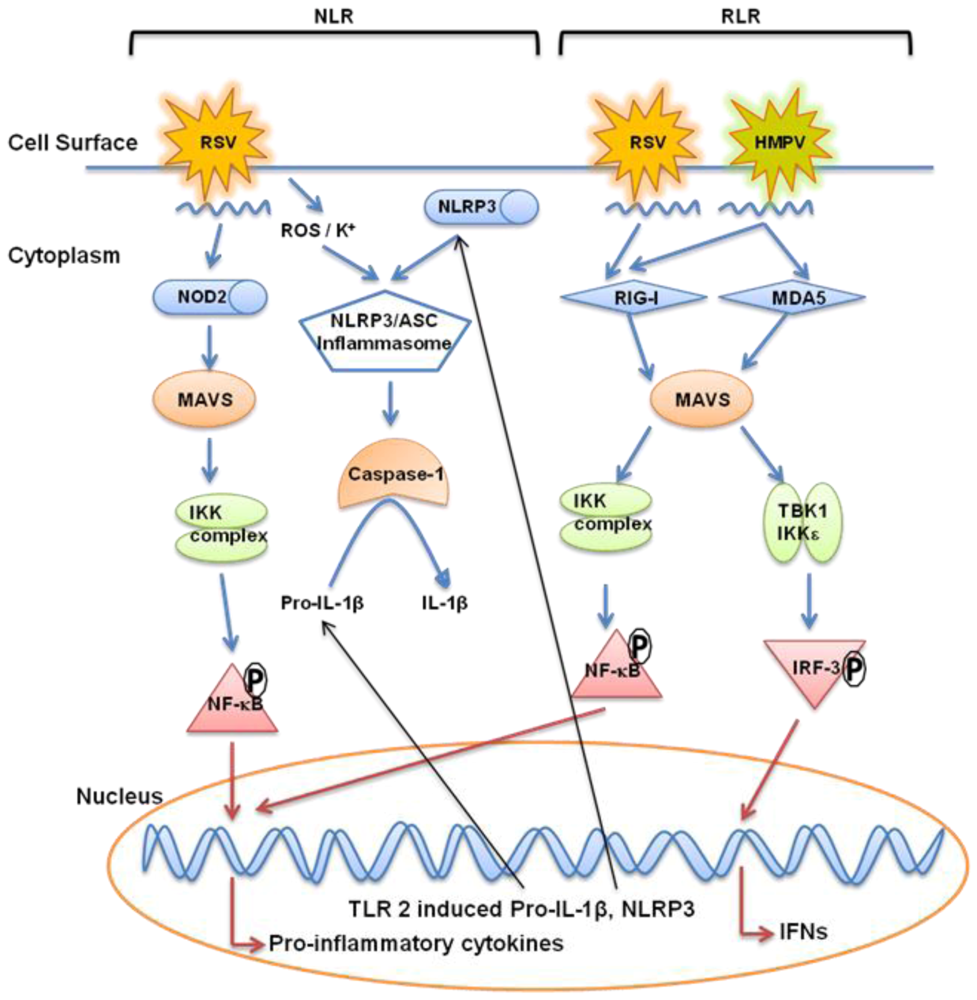
6. Retinoic Acid Inducible Gene like Receptors (RLRs)
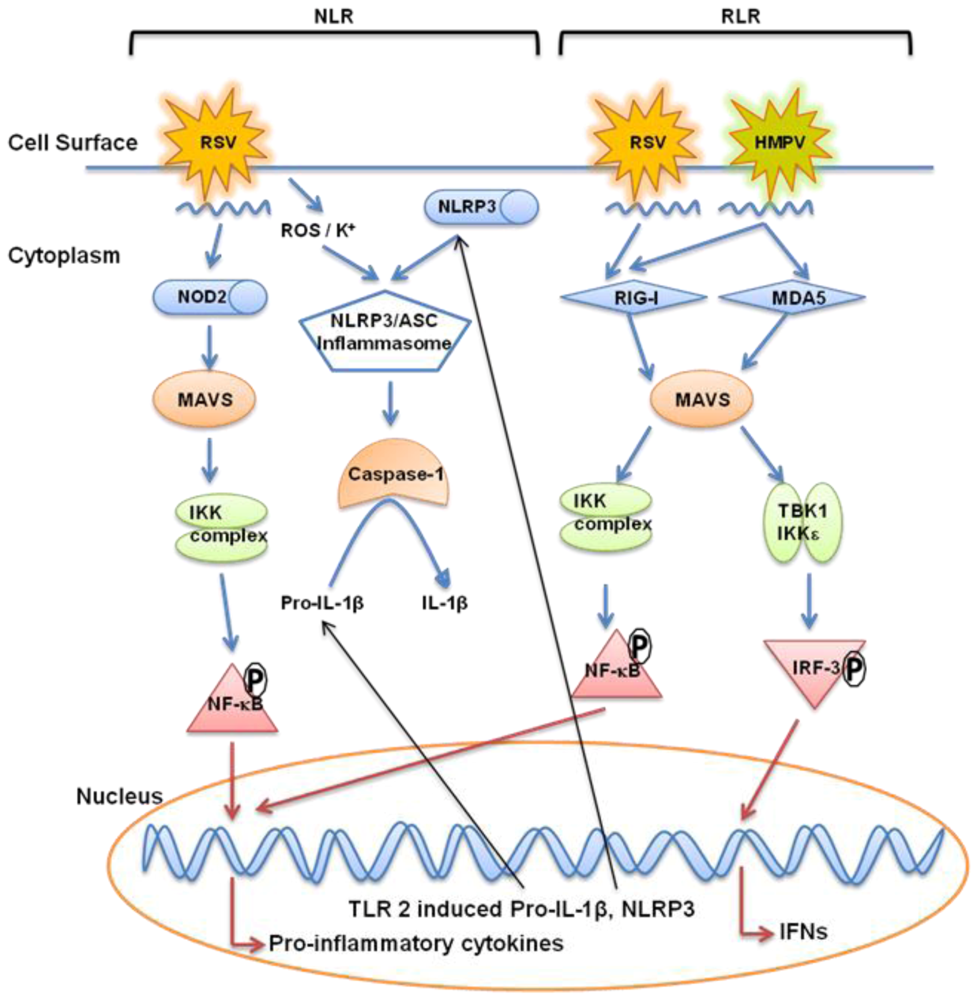
7. Nucleotide Binding Oligomerization Domain-Like Receptor (NLRs)
Acknowledgments
Conflict of Interest
References
- Papadopoulos, N.G.; Skevaki, C.L. Viruses of the Lung. In Encyclopedia of Respiratory Medicine; Geoffrey, J.L., Steven, D.S., Eds.; Academic Press: Oxford, UK, 2006; pp. 483–488. [Google Scholar]
- Lamb, R.A.K.D. Paramyxoviridae: The Viruses and Their Replication. In Fundamental Virology, 4th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott, Williams and Wilkins: Philadelphia, PA, USA, 2001; pp. 689–724. [Google Scholar]
- Blount, R.E., Jr.; Morris, J.A.; Savage, R.E. Recovery of cytopathogenic agent from chimpanzees with coryza. Proc. Soc. Exp. Biol. Med. 1956, 92, 544–549. [Google Scholar]
- Chanock, R.; Roizman, B.; Myers, R. Recovery from infants with respiratory illness of a virus related to chimpanzee coryza agent (CCA). I. Isolation, properties and characterization. Am. J. Hyg. 1957, 66, 281–290. [Google Scholar]
- Chanock, R.; Finberg, L. Recovery from infants with respiratory illness of a virus related to chimpanzee coryza agent (CCA). II. Epidemiologic aspects of infection in infants and young children. Am. J. Hyg. 1957, 66, 291–300. [Google Scholar]
- Glezen, W.P.; Taber, L.H.; Frank, A.L. Risk of primary infection and reinfection with respiratory syncytial virus. Am. J. Dis. Child. 1986, 140, 543–546. [Google Scholar]
- Hall, C.B. Respiratory syncytial virus and parainfluenza virus. N. Engl. J. Med. 2001, 344, 1917–1928. [Google Scholar] [CrossRef]
- Mufson, M.A.; Orvell, C.; Rafnar, B.; Norrby, E. Two distinct subtypes of human respiratory syncytial virus. J. Gen. Virol. 1985, 66 (Pt. 10), 2111–2124. [Google Scholar]
- Sullender, W.M. Respiratory syncytial virus genetic and antigenic diversity. Clin. Microbiol. Rev. 2000, 13, 1–15. [Google Scholar] [CrossRef]
- Beckham, J.D.; Cadena, A.; Lin, J.; Piedra, P.A.; Glezen, W.P.; Greenberg, S.B.; Atmar, R.L. Respiratory viral infections in patients with chronic, obstructive pulmonary disease. J. Infect. 2005, 50, 322–330. [Google Scholar] [CrossRef]
- Atreya, P.L.; Kulkarni, S. Respiratory syncytial virus strain A2 is resistant to the antiviral effects of type I interferons and human MxA. Virology 1999, 261, 227–241. [Google Scholar] [CrossRef]
- Ferris, J.A.; Aherne, W.A.; Locke, W.S. Sudden and unexpected deaths to infants: Histology and virology. Br. Med. J. 1973, 2, 439–449. [Google Scholar] [CrossRef]
- Fiedler, M.A.; Wernke-Dollries, K.; Stark, J.M. Inhibition of viral replication reverses respiratory syncytial virus-induced NF-kB activation and interleukin-8 gene expression in A549 cells. J. Virol. 1996, 70, 9079–9082. [Google Scholar]
- Fiedler, M.A.; Wernke-Dollries, K. Incomplete regulation of NF-kappaB by IkappaBalpha during respiratory syncytial virus infection in A549 cells. J. Virol. 1999, 73, 4502–4507. [Google Scholar]
- Garofalo, R.P.; Sabry, M.; Jamaluddin, M.; Yu, R.K.; Casola, A.; Ogra, P.L.; Brasier, A.R. Transcriptional activation of the interleukin-8 gene by respiratory syncytial virus infection in alveolar epithelial cells: Nuclear translocation of the RelA transcription factor as a mechanism producing airway mucosal inflammation. J. Virol. 1996, 70, 8773–8781. [Google Scholar]
- Jamaluddin, M.; Casola, A.; Garofalo, R.P.; Han, Y.; Elliott, T.; Ogra, P.L.; Brasier, A.R. The major component of IkBa proteolysis occurs independently of the proteasome pathway in Respiratory Syncytial Virus-infected pulmonary epithelial cells. J. Virol. 1998, 72, 4849–4857. [Google Scholar]
- Olszewska-Pazdrak, B.; Casola, A.; Saito, T.; Alam, R.; Crowe, S.E.; Mei, F.; Ogra, P.L.; Garofalo, R.P. Cell-Specific expression of RANTES, MCP-1, and MIP-1alpha by lower airway epithelial cells and eosinophils infected with respiratory syncytial virus. J. Virol. 1998, 72, 4756–4764. [Google Scholar]
- Zhang, Y.; Luxon, B.A.; Casola, A.; Garofalo, R.P.; Jamaluddin, M.; Brasier, A.R. Expression of respiratory syncytial virus-induced chemokine gene networks in lower airway epithelial cells revealed by cDNA microarrays. J. Virol. 2001, 75, 9044–9058. [Google Scholar] [CrossRef]
- Van den Hoogen, B.G.; de Jong, J.C.; Groen, J.; Kuiken, T.; de Groot, R.; Fouchier, R.A.; Osterhaus, A.D. A newly discovered human pneumovirus isolated from young children with respiratory tract disease. Nat. Med. 2001, 7, 719–724. [Google Scholar] [CrossRef]
- Feuillet, F.; Lina, B.; Rosa-Calatrava, M.; Boivin, G. Ten years of human metapneumovirus research. J. Clin. Virol. 2012, 53, 97–105. [Google Scholar] [CrossRef]
- Principi, N.; Bosis, S.; Esposito, S. Human metapneumovirus in paediatric patients. Clin. Microbiol. Infect. 2006, 12, 301–308. [Google Scholar] [CrossRef]
- Kahn, J.S. Epidemiology of human metapneumovirus. Clin. Microbiol. Rev. 2006, 19, 546–557. [Google Scholar] [CrossRef]
- Williams, J.V.; Harris, P.A.; Tollefson, S.J.; Halburnt-Rush, L.L.; Pingsterhaus, J.M.; Edwards, K.M.; Wright, P.F.; Crowe, J.E., Jr. Human metapneumovirus and lower respiratory tract disease in otherwise healthy infants and children. N. Engl. J. Med. 2004, 350, 443–450. [Google Scholar] [CrossRef]
- Crowe, J.E., Jr. Human metapneumovirus as a major cause of human respiratory tract disease. Pediatr. Infect. Dis. J. 2004, 23, S215–S221. [Google Scholar] [CrossRef]
- Biovin, G.; Abed, L.; Pelletier, G.; Ruel, L.; Moisan, D.; Cote', S.; Peret, T.C.; Erdman, D.D.; Anderson, L.J. Virological features and clinical manifestations associated with human metapneumovirus: a new paramyxovirus responsible for acute respiratort-tract infections in all age groups. J Infect Dis 2002, 186, 1330–1334. [Google Scholar] [CrossRef]
- Esper, F.; Boucher, D.; Weibel, C.; Martinello, R.A.; Kahn, J.S. Human metapneumovirus infection in the United States: Clinical manifestations associated with a newly emerging respiratory infection in children. Pediatrics 2003, 111, 1407–1410. [Google Scholar] [CrossRef]
- Guerrero-Plata, A.; Casola, A.; Garofalo, R.P. Human metapneumovirus induces a profile of lung cytokines distinct from that of respiratory syncytial virus. J. Virol. 2005, 79, 14992–14997. [Google Scholar] [CrossRef]
- Guerrero-Plata, A.; Casola, A.; Suarez, G.; Yu, X.; Spetch, L.; Peeples, M.E.; Garofalo, R.P. Differential response of dendritic cells to human metapneumovirus and respiratory syncytial virus. Am. J. Respir. Cell Mol. Biol. 2006, 34, 320–329. [Google Scholar]
- Bao, X.; Liu, T.; Spetch, L.; Kolli, D.; Garofalo, R.P.; Casola, A. Airway epithelial cell response to human metapneumovirus infection. Virology 2007, 368, 91–101. [Google Scholar] [CrossRef]
- Kumar, H.; Kawai, T.; Akira, S. Toll-Like receptors and innate immunity. Biochem. Biophys. Res. Commun. 2009, 388, 621–625. [Google Scholar] [CrossRef]
- Seth, R.B.; Sun, L.; Chen, Z.J. Antiviral innate immunity pathways. Cell Res. 2006, 16, 141–147. [Google Scholar] [CrossRef]
- Sabbah, A.; Chang, T.H.; Harnack, R.; Frohlich, V.; Tominaga, K.; Dube, P.H.; Xiang, Y.; Bose, S. Activation of innate immune antiviral responses by Nod2. Nat. Immunol. 2009, 10, 1073–1080. [Google Scholar] [CrossRef]
- Loo, Y.M.; Fornek, J.; Crochet, N.; Bajwa, G.; Perwitasari, O.; Martinez-Sobrido, L.; Akira, S.; Gill, M.A.; Garcia-Sastre, A.; Katze, M.G.; et al. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J. Virol. 2008, 82, 335–345. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Antiviral signaling through pattern recognition receptors. J. Biochem. (Tokyo) 2006, 141, 137–145. [Google Scholar]
- Leulier, F.; Lemaitre, B. Toll-like receptors--taking an evolutionary approach. Nat. Rev. Genet. 2008, 9, 165–178. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Koblansky, A.A.; Jankovic, D.; Oh, H.; Hieny, S.; Sungnak, W.; Mathur, R.; Hayden, M.S.; Akira, S.; Sher, A.; Ghosh, S. Recognition of profilin by toll-like receptor 12 Is critical for host resistance to toxoplasma gondii. Immunity 2013, 38, 119–130. [Google Scholar] [CrossRef]
- Oldenburg, M.; Kruger, A.; Ferstl, R.; Kaufmann, A.; Nees, G.; Sigmund, A.; Bathke, B.; Lauterbach, H.; Suter, M.; Dreher, S.; et al. TLR13 recognizes bacterial 23S rRNA devoid of erythromycin resistance-forming modification. Science 2012, 337, 1111–1115. [Google Scholar] [CrossRef]
- He, X.; Jia, H.; Jing, Z.; Liu, D. Recognition of pathogen-associated nucleic acids by endosomal nucleic acid-sensing toll-like receptors. Acta Biochim. Biophys. Sin. 2013, 45, 241–258. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. TLR signaling. Cell Death. Differ. 2006, 13, 816–825. [Google Scholar] [CrossRef]
- Takeda, K.; Akira, S. Toll-Like receptors in innate immunity. Int. Immunol. 2005, 17, 1–14. [Google Scholar] [CrossRef]
- Chen, Z.J. Ubiquitin signalling in the NF-kappaB pathway. Nat. Cell Biol. 2005, 7, 758–765. [Google Scholar] [CrossRef]
- Sato, S.; Sanjo, H.; Takeda, K.; Ninomiya-Tsuji, J.; Yamamoto, M.; Kawai, T.; Matsumoto, K.; Takeuchi, O.; Akira, S. Essential function for the kinase TAK1 in innate and adaptive immune responses. Nat. Immunol. 2005, 6, 1087–1095. [Google Scholar] [CrossRef]
- Funami, K.; Matsumoto, M.; Oshiumi, H.; Akazawa, T.; Yamamoto, A.; Seya, T. The cytoplasmic 'linker region' in Toll-like receptor 3 controls receptor localization and signaling. Int. Immunol. 2004, 16, 1143–1154. [Google Scholar] [CrossRef]
- Yamamoto, M.; Sato, S.; Hemmi, H.; Hoshino, K.; Kaisho, T.; Sanjo, H.; Takeuchi, O.; Sugiyama, M.; Okabe, M.; Takeda, K.; et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science 2003, 301, 640–643. [Google Scholar] [CrossRef]
- Meylan, E.; Burns, K.; Hofmann, K.; Blancheteau, V.; Martinon, F.; Kelliher, M.; Tschopp, J. RIP1 is an essential mediator of Toll-like receptor 3-induced NF-kappa B activation. Nat. Immunol. 2004, 5, 503–507. [Google Scholar] [CrossRef]
- Mayer, A.K.; Muehmer, M.; Mages, J.; Gueinzius, K.; Hess, C.; Heeg, K.; Bals, R.; Lang, R.; Dalpke, A.H. Differential recognition of TLR-dependent microbial ligands in human bronchial epithelial cells. J. Immunol. 2007, 178, 3134–3142. [Google Scholar]
- Muir, A.; Soong, G.; Sokol, S.; Reddy, B.; Gomez, M.I.; Van, H.A.; Prince, A. Toll-Like receptors in normal and cystic fibrosis airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2004, 30, 777–783. [Google Scholar]
- Ioannidis, I.; Ye, F.; McNally, B.; Willette, M.; Flano, E. TLR expression and induction of type I and type III interferons in primary airway epithelial cells. J. Virol 2013, 87, 3261–3270. [Google Scholar] [CrossRef]
- Maris, N.A.; Dessing, M.C.; de Vos, A.F.; Bresser, P.; van der Zee, J.S.; Jansen, H.M.; Spek, C.A.; van der Poll, T. Toll-Like receptor mRNA levels in alveolar macrophages after inhalation of endotoxin. Eur. Respir. J. 2006, 28, 622–626. [Google Scholar] [CrossRef]
- Oshikawa, K.; Sugiyama, Y. Gene expression of Toll-like receptors and associated molecules induced by inflammatory stimuli in the primary alveolar macrophage. Biochem. Biophys. Res. Commun. 2003, 305, 649–655. [Google Scholar] [CrossRef]
- Suzuki, K.; Suda, T.; Naito, T.; Ide, K.; Chida, K.; Nakamura, H. Impaired toll-like receptor 9 expression in alveolar macrophages with no sensitivity to CpG DNA. Am. J. Respir. Crit. Care Med. 2005, 171, 707–713. [Google Scholar] [CrossRef]
- Demedts, I.K.; Brusselle, G.G.; Vermaelen, K.Y.; Pauwels, R.A. Identification and characterization of human pulmonary dendritic cells. Am. J. Respir. Cell Mol. Biol. 2005, 32, 177–184. [Google Scholar] [CrossRef]
- Holt, P.G.; Strickland, D.H.; Wikstrom, M.E.; Jahnsen, F.L. Regulation of immunological homeostasis in the respiratory tract. Nat. Rev. Immunol. 2008, 8, 142–152. [Google Scholar] [CrossRef]
- Masten, B.J.; Olson, G.K.; Tarleton, C.A.; Rund, C.; Schuyler, M.; Mehran, R.; Archibeque, T.; Lipscomb, M.F. Characterization of myeloid and plasmacytoid dendritic cells in human lung. J. Immunol. 2006, 177, 7784–7793. [Google Scholar]
- Fan, J.; Frey, R.S.; Malik, A.B. TLR4 signaling induces TLR2 expression in endothelial cells via neutrophil NADPH oxidase. J. Clin. Invest. 2003, 112, 1234–1243. [Google Scholar]
- Li, J.; Ma, Z.; Tang, Z.L.; Stevens, T.; Pitt, B.; Li, S. CpG DNA-mediated immune response in pulmonary endothelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004, 287, L552–L558. [Google Scholar] [CrossRef]
- Brant, K.A.; Fabisiak, J.P. Nickel Alterations of TLR2-Dependent Chemokine Profiles in Lung Fibroblasts Are Mediated by COX-2. Am. J. Respir Cell Mol. Biol. 2008, 38, 591–599. [Google Scholar] [CrossRef]
- Sugiura, H.; Ichikawa, T.; Koarai, A.; Yanagisawa, S.; Minakata, Y.; Matsunaga, K.; Hirano, T.; Akamatsu, K.; Ichinose, M. Activation of toll-like receptor 3 augments myofibroblast differentiation. Am. J. Respir. Cell Mol. Biol. 2009, 40, 654–662. [Google Scholar] [CrossRef]
- Opitz, B.; van, L.V.; Eitel, J.; Suttorp, N. Innate immune recognition in infectious and noninfectious diseases of the lung. Am. J. Respir. Crit. Care Med. 2010, 181, 1294–1309. [Google Scholar] [CrossRef]
- Murawski, M.R.; Bowen, G.N.; Cerny, A.M.; Anderson, L.J.; Haynes, L.M.; Tripp, R.A.; Kurt-Jones, E.A.; Finberg, R.W. Respiratory syncytial virus activates innate immunity through Toll-like receptor 2. J. Virol. 2009, 83, 1492–1500. [Google Scholar] [CrossRef]
- Rudd, B.D.; Smit, J.J.; Flavell, R.A.; Alexopoulou, L.; Schaller, M.A.; Gruber, A.; Berlin, A.A.; Lukacs, N.W. Deletion of TLR3 alters the pulmonary immune environment and mucus production during respiratory syncytial virus infection. J. Immunol. 2006, 176, 1937–1942. [Google Scholar]
- Kurt-Jones, E.A.; Popova, L.; Kwinn, L.; Haynes, L.M.; Jones, L.P.; Tripp, R.A.; Walsh, E.E.; Freeman, M.W.; Golenbock, D.T.; Anderson, L.J.; et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat. Immunol. 2000, 1, 398–401. [Google Scholar] [CrossRef]
- Lukacs, N.W.; Smit, J.J.; Mukherjee, S.; Morris, S.B.; Nunez, G.; Lindell, D.M. Respiratory virus-induced TLR7 activation controls IL-17-associated increased mucus via IL-23 regulation. J. Immunol. 2010, 185, 2231–2239. [Google Scholar] [CrossRef]
- Kolli, D.; Bao, X.; Liu, T.; Hong, C.; Wang, T.; Garofalo, R.P.; Casola, A. Human metapneumovirus glycoprotein G inhibits TLR4-dependent signaling in monocyte-derived dendritic cells. J. Immunol. 2011, 187, 47–54. [Google Scholar] [CrossRef]
- Goutagny, N.; Jiang, Z.; Tian, J.; Parroche, P.; Schickli, J.; Monks, B.G.; Ulbrandt, N.; Ji, H.; Kiener, P.A.; Coyle, A.J.; et al. Cell type-specific recognition of human metapneumoviruses (HMPVs) by retinoic acid-inducible gene I (RIG-I) and TLR7 and viral interference of RIG-I ligand recognition by HMPV-B1 phosphoprotein. J. Immunol. 2010, 184, 1168–1179. [Google Scholar] [CrossRef]
- Imran, M.; Waheed, Y.; Manzoor, S.; Bilal, M.; Ashraf, W.; Ali, M.; Ashraf, M. Interaction of Hepatitis C virus proteins with pattern recognition receptors. Virol. J. 2012, 9, 126. [Google Scholar] [CrossRef]
- Villalba, M.; Hott, M.; Martin, C.; Aguila, B.; Valdivia, S.; Quezada, C.; Zambrano, A.; Concha, M.I.; Otth, C. Herpes simplex virus type 1 induces simultaneous activation of Toll-like receptors 2 and 4 and expression of the endogenous ligand serum amyloid A in astrocytes. Med. Microbiol. Immunol. 2012, 201, 371–379. [Google Scholar] [CrossRef]
- Zhou, S.; Halle, A.; Kurt-Jones, E.A.; Cerny, A.M.; Porpiglia, E.; Rogers, M.; Golenbock, D.T.; Finberg, R.W. Lymphocytic choriomeningitis virus (LCMV) infection of CNS glial cells results in TLR2-MyD88/Mal-dependent inflammatory responses. J. Neuroimmunol. 2008, 194, 70–82. [Google Scholar] [CrossRef]
- Chaudhuri, S.; Lowen, B.; Chan, G.; Davey, A.; Riddell, M.; Guilbert, L.J. Human cytomegalovirus interacts with toll-like receptor 2 and CD14 on syncytiotrophoblasts to stimulate expression of TNFalpha mRNA and apoptosis. Placenta 2009, 30, 994–1001. [Google Scholar] [CrossRef]
- Cyr, S.L.; Jones, T.; Stoica-Popescu, I.; Burt, D.; Ward, B.J. C57Bl/6 mice are protected from respiratory syncytial virus (RSV) challenge and IL-5 associated pulmonary eosinophilic infiltrates following intranasal immunization with Protollin-eRSV vaccine. Vaccine 2007, 25, 3228–3232. [Google Scholar] [CrossRef]
- Hancock, G.E.; Heers, K.M.; Pryharski, K.S.; Smith, J.D.; Tiberio, L. Adjuvants recognized by toll-like receptors inhibit the induction of polarized type 2 T cell responses by natural attachment (G) protein of respiratory syncytial virus. Vaccine 2003, 21, 4348–4358. [Google Scholar] [CrossRef]
- Janssen, R.; Pennings, J.; Hodemaekers, H.; Buisman, A.; van Oosten, M.; de Rond, L.; Ozturk, K.; Dormans, J.; Kimman, T.; Hoebee, B. Host transcription profiles upon primary respiratory syncytial virus infection. J. Virol. 2007, 81, 5958–5967. [Google Scholar] [CrossRef]
- Segovia, J.; Sabbah, A.; Mgbemena, V.; Tsai, S.Y.; Chang, T.H.; Berton, M.T.; Morris, I.R.; Allen, I.C.; Ting, J.P.; Bose, S. TLR2/MyD88/NF-kappaB pathway, reactive oxygen species, potassium efflux activates NLRP3/ASC inflammasome during respiratory syncytial virus infection. PLoS One 2012, 7, e29695. [Google Scholar] [CrossRef]
- Mailaparambil, B.; Krueger, M.; Heinze, J.; Forster, J.; Heinzmann, A. Polymorphisms of toll like receptors in the genetics of severe RSV associated diseases. Dis. Markers 2008, 25, 59–65. [Google Scholar]
- Daley, D.; Park, J.E.; He, J.Q.; Yan, J.; Akhabir, L.; Stefanowicz, D.; Becker, A.B.; Chan-Yeung, M.; Bosse, Y.; Kozyrskyj, A.L.; et al. Associations and interactions of genetic polymorphisms in innate immunity genes with early viral infections and susceptibility to asthma and asthma-related phenotypes. J. Allergy Clin. Immunol. 2012, 130, 1284–1293. [Google Scholar] [CrossRef]
- Kukavica-Ibrulj, I.; Hamelin, M.E.; Prince, G.A.; Gagnon, C.; Bergeron, Y.; Bergeron, M.G.; Boivin, G. Infection with human metapneumovirus predisposes mice to severe pneumococcal pneumonia. J. Virol. 2009, 83, 1341–1349. [Google Scholar] [CrossRef]
- Matsumoto, M.; Funami, K.; Tanabe, M.; Oshiumi, H.; Shingai, M.; Seto, Y.; Yamamoto, A.; Seya, T. Subcellular localization of Toll-like receptor 3 in human dendritic cells. J. Immunol. 2003, 171, 3154–3162. [Google Scholar]
- Rudd, B.D.; Burstein, E.; Duckett, C.S.; Li, X.; Lukacs, N.W. Differential role for TLR3 in respiratory syncytial virus-induced chemokine expression. J. Virol. 2005, 79, 3350–3357. [Google Scholar] [CrossRef]
- Groskreutz, D.J.; Monick, M.M.; Powers, L.S.; Yarovinsky, T.O.; Look, D.C.; Hunninghake, G.W. Respiratory syncytial virus induces TLR3 protein and protein kinase R, leading to increased double-stranded RNA responsiveness in airway epithelial cells. J. Immunol. 2006, 176, 1733–1740. [Google Scholar]
- Liu, P.; Jamaluddin, M.; Li, K.; Garofalo, R.P.; Casola, A.; Brasier, A.R. Retinoic Acid-inducible gene I mediates early antiviral response and toll-like receptor 3 expression in respiratory syncytial virus-infected airway epithelial cells. J. Virol. 2007, 81, 1401–1411. [Google Scholar] [CrossRef]
- Lukacs, N.W.; Tekkanat, K.K.; Berlin, A.; Hogaboam, C.M.; Miller, A.; Evanoff, H.; Lincoln, P.; Maassab, H. Respiratory syncytial virus predisposes mice to augmented allergic airway responses via IL-13-mediated mechanisms. J. Immunol. 2001, 167, 1060–1065. [Google Scholar]
- Tekkanat, K.K.; Maassab, H.F.; Cho, D.S.; Lai, J.J.; John, A.; Berlin, A.; Kaplan, M.H.; Lukacs, N.W. IL-13-Induced airway hyperreactivity during respiratory syncytial virus infection is STAT6 dependent. J. Immunol. 2001, 166, 3542–3548. [Google Scholar]
- Huang, S.; Wei, W.; Yun, Y. Upregulation of TLR7 and TLR3 gene expression in the lung of respiratory syncytial virus infected mice. Wei Sheng Wu Xue Bao 2009, 49, 239–245. [Google Scholar]
- Boukhvalova, M.S.; Sotomayor, T.B.; Point, R.C.; Pletneva, L.M.; Prince, G.A.; Blanco, J.C. Activation of interferon response through toll-like receptor 3 impacts viral pathogenesis and pulmonary toll-like receptor expression during respiratory syncytial virus and influenza infections in the cotton rat Sigmodon hispidus model. J. Interf. Cytokine Res. 2010, 30, 229–242. [Google Scholar]
- Glasser, S.W.; Witt, T.L.; Senft, A.P.; Baatz, J.E.; Folger, D.; Maxfield, M.D.; Akinbi, H.T.; Newton, D.A.; Prows, D.R.; Korfhagen, T.R. Surfactant protein C-deficient mice are susceptible to respiratory syncytial virus infection. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L64–L72. [Google Scholar] [CrossRef]
- Aeffner, F.; Traylor, Z.P.; Yu, E.N.; Davis, I.C. Double-Stranded RNA induces similar pulmonary dysfunction to respiratory syncytial virus in BALB/c mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 301, L99–L109. [Google Scholar] [CrossRef]
- Guerrero-Plata, A.; Baron, S.; Poast, J.S.; Adegboyega, P.A.; Casola, A.; Garofalo, R.P. Activity and regulation of alpha interferon in respiratory syncytial virus and human metapneumovirus experimental infections. J. Virol. 2005, 79, 10190–10199. [Google Scholar]
- Scagnolari, C.; Midulla, F.; Pierangeli, A.; Moretti, C.; Bonci, E.; Berardi, R.; De, A.D.; Selvaggi, C.; Di, M.P.; Girardi, E.; et al. Gene expression of nucleic acid-sensing pattern recognition receptors in children hospitalized for respiratory syncytial virus-associated acute bronchiolitis. Clin. Vaccine Immunol. 2009, 16, 816–823. [Google Scholar] [CrossRef]
- Dou, Y.; Zhao, Y.; Zhang, Z.Y.; Zhao, X.D. Toll-Like receptors expression in the lungs of human metapneumovirus infected mice and the effects of polyI:C on viral infection. Bing Du Xue Bao 2010, 26, 1–7. [Google Scholar]
- Finberg, R.W.; Wang, J.P.; Kurt-Jones, E.A. Toll like receptors and viruses. Rev. Med. Virol. 2007, 17, 35–43. [Google Scholar] [CrossRef]
- O'Mahony, D.S.; Pham, U.; Iyer, R.; Hawn, T.R.; Liles, W.C. Differential constitutive and cytokine-modulated expression of human Toll-like receptors in primary neutrophils, monocytes, and macrophages. Int. J. Med. Sci. 2008, 5, 1–8. [Google Scholar]
- Sabroe, I.; Jones, E.C.; Usher, L.R.; Whyte, M.K.; Dower, S.K. Toll-Like receptor (TLR)2 and TLR4 in human peripheral blood granulocytes: a critical role for monocytes in leukocyte lipopolysaccharide responses. J. Immunol. 2002, 168, 4701–4710. [Google Scholar]
- Shimazu, R.; Akashi, S.; Ogata, H.; Nagai, Y.; Fukudome, K.; Miyake, K.; Kimoto, M. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J. Exp. Med. 1999, 189, 1777–1782. [Google Scholar] [CrossRef]
- Viriyakosol, S.; Kirkland, T.; Soldau, K.; Tobias, P. MD-2 binds to bacterial lipopolysaccharide. J. Endotoxin. Res. 2000, 6, 489–491. [Google Scholar]
- Kim, H.M.; Park, B.S.; Kim, J.I.; Kim, S.E.; Lee, J.; Oh, S.C.; Enkhbayar, P.; Matsushima, N.; Lee, H.; Yoo, O.J.; et al. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell 2007, 130, 906–917. [Google Scholar] [CrossRef]
- Da Costa, C.U.; Wantia, N.; Kirschning, C.J.; Busch, D.H.; Rodriguez, N.; Wagner, H.; Miethke, T. Heat shock protein 60 from Chlamydia pneumoniae elicits an unusual set of inflammatory responses via Toll-like receptor 2 and 4 in vivo. Eur. J. Immunol. 2004, 34, 2874–2884. [Google Scholar]
- Dogan, S.; Zhang, Q.; Pridmore, A.C.; Mitchell, T.J.; Finn, A.; Murdoch, C. Pneumolysin-induced CXCL8 production by nasopharyngeal epithelial cells is dependent on calcium flux and MAPK activation via Toll-like receptor 4. Microbes. Infect. 2011, 13, 65–75. [Google Scholar] [CrossRef]
- Okumura, A.; Pitha, P.M.; Yoshimura, A.; Harty, R.N. Interaction between Ebola virus glycoprotein and host toll-like receptor 4 leads to induction of proinflammatory cytokines and SOCS1. J. Virol. 2010, 84, 27–33. [Google Scholar] [CrossRef]
- Barrera, V.; Skorokhod, O.A.; Baci, D.; Gremo, G.; Arese, P.; Schwarzer, E. Host fibrinogen stably bound to hemozoin rapidly activates monocytes via TLR-4 and CD11b/CD18-integrin: A new paradigm of hemozoin action. Blood 2011, 117, 5674–5682. [Google Scholar] [CrossRef]
- Noh, K.T.; Shin, S.J.; Son, K.H.; Jung, I.D.; Kang, H.K.; Lee, S.J.; Lee, E.K.; Shin, Y.K.; You, J.C.; Park, Y.M. The Mycobacterium avium subsp. paratuberculosis fibronectin attachment protein, a toll-like receptor 4 agonist, enhances dendritic cell-based cancer vaccine potency. Exp. Mol. Med. 2012, 44, 340–349. [Google Scholar]
- Campo, G.M.; Avenoso, A.; D'Ascola, A.; Prestipino, V.; Scuruchi, M.; Nastasi, G.; Calatroni, A.; Campo, S. Hyaluronan differently modulates TLR-4 and the inflammatory response in mouse chondrocytes. Biofactors 2012, 38, 69–76. [Google Scholar] [CrossRef]
- Awasthi, S.; Brown, K.; King, C.; Awasthi, V.; Bondugula, R. A toll-like receptor-4-interacting surfactant protein-A-derived peptide suppresses tumor necrosis factor-alpha release from mouse JAWS II dendritic cells. J. Pharmacol. Exp. Ther. 2011, 336, 672–681. [Google Scholar] [CrossRef]
- Zong, M.; Bruton, J.D.; Grundtman, C.; Yang, H.; Li, J.H.; Alexanderson, H.; Palmblad, K.; Andersson, U.; Harris, H.E.; Lundberg, I.E.; et al. TLR4 as receptor for HMGB1 induced muscle dysfunction in myositis. Ann. Rheum. Dis. 2012, 0, 1–10. [Google Scholar] [CrossRef]
- Tsan, M.F.; Gao, B. Endogenous ligands of Toll-like receptors. J. Leukoc. Biol. 2004, 76, 514–519. [Google Scholar] [CrossRef]
- Yu, H.T.; Jiang, H.; Zhang, Y.; Nan, X.P.; Li, Y.; Wang, W.; Jiang, W.; Yang, D.Q.; Su, W.J.; Wang, J.P.; et al. Hantaan virus triggers TLR4-dependent innate immune responses. Viral. Immunol. 2012, 25, 387–393. [Google Scholar] [CrossRef]
- Rallabhandi, P.; Phillips, R.L.; Boukhvalova, M.S.; Pletneva, L.M.; Shirey, K.A.; Gioannini, T.L.; Weiss, J.P.; Chow, J.C.; Hawkins, L.D.; Vogel, S.N.; Blanco, J.C.G. Respiratory syncytial virus fusion protein-induced Toll-Like Receptor 4 (TLR4) signaling is inhibited by the TLR4 antagonists rhodobacter sphaeroides lipopolysaccharide and Eritoran (E5564) and requires direct interaction with MD-2. mBio 2012, 3, 218–212. [Google Scholar]
- Lizundia, R.; Sauter, K.S.; Taylor, G.; Werling, D. Host species-specific usage of the TLR4-LPS receptor complex. Innate Immun. 2008, 14, 223–231. [Google Scholar] [CrossRef]
- Marr, N.; Turvey, S.E. Role of human TLR4 in respiratory syncytial virus-induced NF-kappaB activation, viral entry and replication. Innate Immun. 2012, 18, 856–865. [Google Scholar] [CrossRef]
- Monick, M.M.; Yarovinsky, T.O.; Powers, L.S.; Butler, N.S.; Carter, A.B.; Gudmundsson, G.; Hunninghake, G.W. Respiratory syncytial virus up-regulates TLR4 and sensitizes airway epithelial cells to endotoxin. J. Biol. Chem. 2003, 278, 53035–53044. [Google Scholar] [CrossRef]
- Xie, X.H.; Law, H.K.; Wang, L.J.; Li, X.; Yang, X.Q.; Liu, E.M. Lipopolysaccharide induces IL-6 production in respiratory syncytial virus-infected airway epithelial cells through the toll-like receptor 4 signaling pathway. Pediatr. Res. 2009, 65, 156–162. [Google Scholar] [CrossRef]
- Xie, X.H.; Liu, E.M.; Yang, X.Q.; Law, H.K.; Li, X.; Wang, L.J.; Liu, W.; Xu, W.F. Toll-Like receptor 4 expression and function of respiratory syncytial virus-infected airway epithelial cells. Zhonghua Jie He He Hu Xi Za Zhi 2008, 31, 213–217. [Google Scholar]
- Marchant, D.; Singhera, G.K.; Utokaparch, S.; Hackett, T.L.; Boyd, J.H.; Luo, Z.; Si, X.; Dorscheid, D.R.; McManus, B.M.; Hegele, R.G. Toll-Like receptor 4-mediated activation of p38 mitogen-activated protein kinase is a determinant of respiratory virus entry and tropism. J. Virol. 2010, 84, 11359–11373. [Google Scholar] [CrossRef]
- Haynes, L.M.; Moore, D.D.; Kurt-Jones, E.A.; Finberg, R.W.; Anderson, L.J.; Tripp, R.A. Involvement of toll-like receptor 4 in innate immunity to respiratory syncytial virus. J. Virol. 2001, 75, 10730–10737. [Google Scholar]
- Ehl, S.; Bischoff, R.; Ostler, T.; Vallbracht, S.; Schulte-Monting, J.; Poltorak, A.; Freudenberg, M. The role of Toll-like receptor 4 versus interleukin-12 in immunity to respiratory syncytial virus. Eur. J. Immunol. 2004, 34, 1146–1153. [Google Scholar] [CrossRef]
- Haeberle, H.; Takizawa, R.; Casola, A.; Brasier, A.R.; Dieterich, H.-J.; van Rooijen, N.; Gatalica, Z.; Garofalo, R.P. Respiratory syncytial virus-induced activation of NF-kB in the lung involves alveolar macrophages and Toll-like receptor 4-dependent pathways. J. Infect. Dis. 2002, 186, 1199–1206. [Google Scholar] [CrossRef]
- Shirey, K.A.; Pletneva, L.M.; Puche, A.C.; Keegan, A.D.; Prince, G.A.; Blanco, J.C.; Vogel, S.N. Control of RSV-induced lung injury by alternatively activated macrophages is IL-4R alpha-, TLR4-, and IFN-beta-dependent. Mucosal Immunol. 2010, 3, 291–300. [Google Scholar]
- Kunzelmann, K.; Sun, J.; Meanger, J.; King, N.J.; Cook, D.I. Inhibition of airway Na+ transport by respiratory syncytial virus. J. Virol. 2007, 81, 3714–3720. [Google Scholar] [CrossRef] [Green Version]
- Cyr, S.L.; Angers, I.; Guillot, L.; Stoica-Popescu, I.; Lussier, M.; Qureshi, S.; Burt, D.S.; Ward, B.J. TLR4 and MyD88 control protection and pulmonary granulocytic recruitment in a murine intranasal RSV immunization and challenge model. Vaccine 2009, 27, 421–430. [Google Scholar] [CrossRef]
- Boukhvalova, M.S.; Prince, G.A.; Soroush, L.; Harrigan, D.C.; Vogel, S.N.; Blanco, J.C.G. The TLR4 agonist, monophosphoryl lipid A, attenuates the cytokine storm associated with respiratory syncytial virus vaccine-enhanced disease. Vaccine 2006, 24, 5027–5035. [Google Scholar] [CrossRef]
- Numata, M.; Chu, H.W.; Dakhama, A.; Voelker, D.R. Pulmonary surfactant phosphatidylglycerol inhibits respiratory syncytial virusGÇôinduced inflammation and infection. Proc. Natl. Acad. Sci. USA 2010, 107, 320–325. [Google Scholar] [CrossRef]
- Gagro, A.; Tominac, M.; Krsulovic-Hresic, V.; Bace, A.; Matic, M.; Drazenovic, V.; Mlinaric- Galinovic, G.; Kosor, E.; Gotovac, K.; Bolanca, I.; et al. Increased Toll-like receptor 4 expression in infants with respiratory syncytial virus bronchiolitis. Clin. Exp. Immunol. 2004, 135, 267–272. [Google Scholar] [CrossRef]
- Halfhide, C.P.; Brearey, S.P.; Flanagan, B.F.; Hunt, J.A.; Howarth, D.; Cummerson, J.; Edwards, S.; Hart, C.A.; Smyth, R.L. Neutrophil TLR4 expression is reduced in the airways of infants with severe bronchiolitis. Thorax 2009, 64, 798–805. [Google Scholar] [CrossRef]
- Arbour, N.C.; Lorenz, E.; Schutte, B.C.; Zabner, J.; Kline, J.N.; Jones, M.; Frees, K.; Watt, J.L.; Schwartz, D.A. TLR4 mutations are associated with endotoxin hyporesponsiveness in humans. Nat. Genet. 2000, 25, 187–191. [Google Scholar]
- Miyairi, I.; Devincenzo, J.P. Human genetic factors and respiratory syncytial virus disease severity. Clin. Microbiol. Rev. 2008, 21, 686–703. [Google Scholar] [CrossRef]
- Tal, G.; Mandelberg, A.; Dalal, I.; Cesar, K.; Somekh, E.; Tal, A.; Oron, A.; Itskovich, S.; Ballin, A.; Houri, S.; et al. Association between common Toll-like receptor 4 mutations and severe respiratory syncytial virus disease. J. Infect. Dis. 2004, 189, 2057–2063. [Google Scholar] [CrossRef]
- Puthothu, B.; Forster, J.; Heinzmann, A.; Krueger, M. TLR-4 and CD14 polymorphisms in respiratory syncytial virus associated disease. Dis. Markers 2006, 22, 303–308. [Google Scholar]
- Awomoyi, A.A.; Rallabhandi, P.; Pollin, T.I.; Lorenz, E.; Sztein, M.B.; Boukhvalova, M.S.; Hemming, V.G.; Blanco, J.C.; Vogel, S.N. Association of TLR4 polymorphisms with symptomatic respiratory syncytial virus infection in high-risk infants and young children. J. Immunol. 2007, 179, 3171–3177. [Google Scholar]
- Mandelberg, A.; Tal, G.; Naugolny, L.; Cesar, K.; Oron, A.; Houri, S.; Gilad, E.; Somekh, E. Lipopolysaccharide hyporesponsiveness as a risk factor for intensive care unit hospitalization in infants with respiratory syncitial virus bronchiolitis. Clin. Exp. Immunol. 2006, 144, 48–52. [Google Scholar] [CrossRef]
- Lofgren, J.; Marttila, R.; Renko, M.; Ramet, M.; Hallman, M. Toll-Like receptor 4 Asp299Gly polymorphism in respiratory syncytial virus epidemics. Pediatr. Pulmonol. 2010, 45, 687–692. [Google Scholar] [CrossRef]
- Lavoie, P.M.; Ladd, M.; Hirschfeld, A.F.; Huusko, J.; Mahlman, M.; Speert, D.P.; Hallman, M.; Lacaze-Masmonteil, T.; Turvey, S.E. Influence of common non-synonymous Toll-like receptor 4 polymorphisms on bronchopulmonary dysplasia and prematurity in human infants. PLoS One 2012, 7, e31351. [Google Scholar] [CrossRef]
- Kresfelder, T.L.; Janssen, R.; Bont, L.; Venter, M. Confirmation of an association between single nucleotide polymorphisms in the VDR gene with respiratory syncytial virus related disease in South African children. J. Med. Virol. 2011, 83, 1834–1840. [Google Scholar] [CrossRef]
- Douville, R.N.; Lissitsyn, Y.; Hirschfeld, A.F.; Becker, A.B.; Kozyrskyj, A.L.; Liem, J.; Bastien, N.; Li, Y.; Victor, R.E.; Sekhon, M.; et al. TLR4 Asp299Gly and Thr399Ile polymorphisms: No impact on human immune responsiveness to LPS or respiratory syncytial virus. PLoS One 2010, 5, e12087. [Google Scholar] [CrossRef]
- Velayutham, T.S.; Kolli, D.; Ivanciuc, T.; Chao, H; Garofal, R.P.; Casola, A. Critical role of TLR4 in human metapneumovirus mediated innate immune responses and disease pathogenesis. Submitted to Plos One..
- McCartney, S.A.; Colonna, M. Viral sensors: diversity in pathogen recognition. Immunol. Rev. 2009, 227, 87–94. [Google Scholar] [CrossRef]
- Lindemans, C.A.; Coffer, P.J.; Schellens, I.M.M.; de Graaff, P.M.A.; Kimpen, J.L.L.; Koenderman, L. Respiratory Syncytial Virus Inhibits Granulocyte Apoptosis through a Phosphatidylinositol 3-Kinase and NF-κB-Dependent Mechanism. J. Immunol. 2006, 176, 5529–5537. [Google Scholar]
- Phipps, S.; Lam, C.E.; Mahalingam, S.; Newhouse, M.; Ramirez, R.; Rosenberg, H.F.; Foster, P.S.; Matthaei, K.I. Eosinophils contribute to innate antiviral immunity and promote clearance of respiratory syncytial virus. Blood 2007, 110, 1578–1586. [Google Scholar] [CrossRef]
- McGill, J.L.; Nonnecke, B.J.; Lippolis, J.D.; Reinhardt, T.A.; Sacco, R.E. Differential chemokine and cytokine production by neonatal bovine gammadelta T cell subsets in response to viral toll-like receptor agonists and in vivo RSV infection. Immunology 2013. [Google Scholar] [CrossRef]
- Bendelja, K.; Vojvoda, V.; Aberle, N.; Cepin-Bogovic, J.; Gagro, A.; Mlinaric-Galinovic, G.; Rabatic, S. Decreased Toll-like receptor 8 expression and lower TNF-alpha synthesis in infants with acute RSV infection. Respir. Res. 2010, 11, 143. [Google Scholar] [CrossRef]
- Johnson, T.R.; Rao, S.; Seder, R.A.; Chen, M.; Graham, B.S. TLR9 agonist, but not TLR7/8, functions as an adjuvant to diminish FI-RSV vaccine-enhanced disease, while either agonist used as therapy during primary RSV infection increases disease severity. Vaccine 2009, 27, 3045–3052. [Google Scholar] [CrossRef]
- Tayyari, F.; Sutton, T.C.; Manson, H.E.; Hegele, R.G. CpG-Oligodeoxynucleotides inhibit RSV-enhanced allergic sensitisation in guinea pigs. Eur. Respir. J. 2005, 25, 295–302. [Google Scholar] [CrossRef]
- Becker, Y. Respiratory syncytial virus (RSV) evades the human adaptive immune system by skewing the Th1/Th2 cytokine balance toward increased levels of Th2 cytokines and IgE, markers of allergy-a review. Virus Genes 2006, 33, 235–252. [Google Scholar]
- Yamaguchi, Y.; Harker, J.A.; Wang, B.; Openshaw, P.J.; Tregoning, J.S.; Culley, F.J. Preexposure to CpG Protects against the Delayed Effects of Neonatal Respiratory Syncytial Virus Infection. J. Virol. 2012, 86, 10456–10461. [Google Scholar]
- Yu, M.; Tong, J.H.; Mao, M.; Kan, L.X.; Liu, M.M.; Sun, Y.W.; Fu, G.; Jing, Y.K.; Yu, L.; Lepaslier, D.; et al. Cloning of a gene (RIG-G) associated with retinoic acid-induced differentiation of acute promyelocytic leukemia cells and representing a new member of a family of interferon-stimulated genes. Proc. Natl. Acad. Sci. USA 1997, 94, 7406–7411. [Google Scholar] [CrossRef]
- Kang, D.C.; Gopalkrishnan, R.V.; Wu, Q.; Jankowsky, E.; Pyle, A.M.; Fisher, P.B. mda-5: An interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proc. Natl. Acad. Sci. USA 2002, 99, 637–642. [Google Scholar]
- Cui, Y.; Li, M.; Walton, K.D.; Sun, K.; Hanover, J.A.; Furth, P.A.; Hennighausen, L. The Stat3/5 locus encodes novel endoplasmic reticulum and helicase-like proteins that are preferentially expressed in normal and neoplastic mammary tissue. Genomics 2001, 78, 129–134. [Google Scholar]
- Saito, T.; Hirai, R.; Loo, Y.M.; Owen, D.; Johnson, C.L.; Sinha, S.C.; Akira, S.; Fujita, T.; Gale, M., Jr. Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc. Natl. Acad. Sci. USA 2007, 104, 582–587. [Google Scholar] [CrossRef]
- Yoneyama, M.; Kikuchi, M.; Matsumoto, K.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Foy, E.; Loo, Y.M.; Gale, M., Jr.; Akira, S.; et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol. 2005, 175, 2851–2858. [Google Scholar]
- Yoneyama, M.; Fujita, T. Function of RIG-I-like receptors in antiviral innate immunity. J. Biol. Chem. 2007, 282, 15315–15318. [Google Scholar] [CrossRef]
- Rothenfusser, S.; Goutagny, N.; DiPerna, G.; Gong, M.; Monks, B.G.; Schoenemeyer, A.; Yamamoto, M.; Akira, S.; Fitzgerald, K.A. The RNA helicase Lgp2 inhibits TLR-independent sensing of viral replication by retinoic acid-inducible gene-I. J. Immunol. 2005, 175, 5260–5268. [Google Scholar]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef]
- Seth, R.B.; Sun, L.; Ea, C.K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef]
- Xu, L.G.; Wang, Y.Y.; Han, K.J.; Li, L.Y.; Zhai, Z.; Shu, H.B. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol. Cell 2005, 19, 727–740. [Google Scholar] [CrossRef]
- Meylan, E.; Curran, J.; Hofmann, K.; Moradpour, D.; Binder, M.; Bartenschlager, R.; Tschopp, J. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 2005, 437, 1167–1172. [Google Scholar] [CrossRef]
- Baril, M.; Racine, M.E.; Penin, F.; Lamarre, D. MAVS dimer is a crucial signaling component of innate immunity and the target of hepatitis C virus NS3/4A protease. J. Virol. 2009, 83, 1299–1311. [Google Scholar] [CrossRef]
- Tang, E.D.; Wang, C.Y. MAVS self-association mediates antiviral innate immune signaling. J. Virol. 2009, 83, 3420–3428. [Google Scholar] [CrossRef]
- Johnson, C.L.; Gale, M., Jr. CARD games between virus and host get a new player. Trends Immunol. 2006, 27, 1–4. [Google Scholar] [CrossRef]
- Maniatis, T.; Falvo, J.V.; Kim, T.H.; Kim, T.K.; Lin, C.H.; Parekh, B.S.; Wathelet, M.G. Structure and function of the interferon-beta enhanceosome. Cold Spring Harb. Symp. Quant. Biol. 1998, 63, 609–620. [Google Scholar] [CrossRef]
- Hiscott, J. Triggering the innate antiviral response through IRF-3 activation. J. Biol. Chem. 2007, 282, 15325–15329. [Google Scholar] [CrossRef]
- Kato, H.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Uematsu, S.; Matsui, K.; Tsujimura, T.; Takeda, K.; Fujita, T.; Takeuchi, O.; et al. Cell type-specific involvement of RIG-I in antiviral response. Immunity. 2005, 23, 19–28. [Google Scholar] [CrossRef]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar]
- Liao, S.; Bao, X.; Liu, T.; Lai, S.; Li, K.; Garofalo, R.P.; Casola, A. Role of retinoic acid inducible gene-I in human metapneumovirus-induced cellular signalling. J. Gen. Virol. 2008, 89, 1978–1986. [Google Scholar] [CrossRef]
- Gitlin, L.; Barchet, W.; Gilfillan, S.; Cella, M.; Beutler, B.; Flavell, R.A.; Diamond, M.S.; Colonna, M. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc. Natl. Acad. Sci. USA 2006, 103, 8459–8464. [Google Scholar] [CrossRef]
- Sasai, M.; Shingai, M.; Funami, K.; Yoneyama, M.; Fujita, T.; Matsumoto, M.; Seya, T. NAK-associated protein 1 participates in both the TLR3 and the cytoplasmic pathways in type I IFN induction. J. Immunol. 2006, 177, 8676–8683. [Google Scholar]
- Liu, P.; Li, K.; Garofalo, R.P.; Brasier, A.R. Respiratory syncytial virus induces RelA release from cytoplasmic 100-kDa NF-kappa B2 complexes via a novel retinoic acid-inducible gene-I{middle dot}NF- kappa B-inducing kinase signaling pathway. J. Biol. Chem. 2008, 283, 23169–23178. [Google Scholar] [CrossRef]
- Bhoj, V.G.; Sun, Q.; Bhoj, E.J.; Somers, C.; Chen, X.; Torres, J.P.; Mejias, A.; Gomez, A.M.; Jafri, H.; Ramilo, O.; et al. MAVS and MyD88 are essential for innate immunity but not cytotoxic T lymphocyte response against respiratory syncytial virus. Proc. Natl. Acad. Sci. USA 2008, 105, 14046–14051. [Google Scholar] [CrossRef]
- Demoor, T.; Petersen, B.C.; Morris, S.; Mukherjee, S.; Ptaschinski, C.; De Almeida Nagata, D.E.; Kawai, T.; Ito, T.; Akira, S.; Kunkel, S.L.; et al. IPS-1 Signaling Has a Nonredundant Role in Mediating Antiviral Responses and the Clearance of Respiratory Syncytial Virus. J. Immunol. 2012, 189, 5942–5953. [Google Scholar] [CrossRef]
- Okabayashi, T.; Kojima, T.; Masaki, T.; Yokota, S.i.; Imaizumi, T.; Tsutsumi, H.; Himi, T.; Fujii, N.; Sawada, N. Type-III interferon, not type-I, is the predominant interferon induced by respiratory viruses in nasal epithelial cells. Virus Res. 2011, 160, 360–366. [Google Scholar] [CrossRef]
- Vissers, M.; Remijn, T.; Oosting, M.; de Jong, D.J.; Diavatopoulos, D.A.; Hermans, P.W.; Ferwerda, G. Respiratory syncytial virus infection augments NOD2 signaling in an IFN-beta-dependent manner in human primary cells. Eur. J. Immunol. 2012, 42, 2727–2735. [Google Scholar] [CrossRef]
- Ling, Z.; Tran, K.C.; Teng, M.N. Human respiratory syncytial virus nonstructural protein NS2 antagonizes the activation of beta interferon transcription by interacting with RIG-I. J. Virol. 2009, 83, 3734–3742. [Google Scholar] [CrossRef]
- Boyapalle, S.; Wong, T.; Garay, J.; Teng, M.; San Juan-Vergara, H.; Mohapatra, S.; Mohapatra, S. Respiratory syncytial virus NS1 protein colocalizes with mitochondrial antiviral signaling protein MAVS following infection. PLoS One 2012, 7, e29386. [Google Scholar]
- Lifland, A.W.; Jung, J.; Alonas, E.; Zurla, C.; Crowe, J.E.; Santangelo, P.J. Human respiratory syncytial virus nucleoprotein and inclusion bodies antagonize the innate immune response mediated by MDA5 and MAVS. J. Virol. 2012, 86, 8245–8258. [Google Scholar] [CrossRef]
- Banos-Lara, Mdel.R.; Ghosh, A.; Guerrero-Plata, A. Critical role of MDA5 in the interferon response induced by human metapneumovirus infection in dendritic cells and in vivo. J. Virol. 2013, 87, 1242–1251. [Google Scholar] [CrossRef]
- Bao, X.; Liu, T.; Shan, Y.; Li, K.; Garofalo, R.P.; Casola, A. Human metapneumovirus glycoprotein G inhibits innate immune responses. PLoS. Pathog. 2008, 4, e1000077. [Google Scholar] [CrossRef]
- Ren, J.; Wang, Q.; Kolli, D.; Prusak, D.J.; Tseng, C.T.; Chen, Z.J.; Li, K.; Wood, T.G.; Bao, X. Human metapneumovirus M2–2 protein inhibits innate cellular signaling by targeting MAVS. J. Virol. 2012, 86, 13049–13061. [Google Scholar]
- Bao, X.; Kolli, D.; Ren, J.; Liu, T.; Garofalo, R.P.; Casola, A. Human metapneumovirus glycoprotein g disrupts mitochondrial signaling in airway epithelial cells. PLoS One 2013. Accepted. [Google Scholar]
- Inohara; Chamaillard; McDonald, C.; Nunez, G. NOD-LRR proteins: Role in host-microbial interactions and inflammatory disease. Annu. Rev. Biochem. 2005, 74, 355–383. [Google Scholar]
- Kufer, T.A.; Fritz, J.H.; Philpott, D.J. NACHT-LRR proteins (NLRs) in bacterial infection and immunity. Trends Microbiol. 2005, 13, 381–388. [Google Scholar] [CrossRef]
- Harton, J.A.; Linhoff, M.W.; Zhang, J.; Ting, J.P. Cutting edge: CATERPILLER: A large family of mammalian genes containing CARD, pyrin, nucleotide-binding, and leucine-rich repeat domains. J. Immunol. 2002, 169, 4088–4093. [Google Scholar]
- Ting, J.P.; Lovering, R.C.; Alnemri, E.S.; Bertin, J.; Boss, J.M.; Davis, B.K.; Flavell, R.A.; Girardin, S.E.; Godzik, A.; Harton, J.A.; et al. The NLR gene family: A standard nomenclature. Immunity 2008, 28, 285–287. [Google Scholar] [CrossRef]
- Ting, J.P.; Davis, B.K. CATERPILLER: A novel gene family important in immunity, cell death, and diseases. Annu. Rev. Immunol. 2005, 23, 387–414. [Google Scholar] [CrossRef]
- Chen, G.; Shaw, M.H.; Kim, Y.G.; Nunez, G. NOD-Like receptors: Role in innate immunity and inflammatory disease. Annu. Rev. Pathol. 2009, 4, 365–398. [Google Scholar] [CrossRef]
- Ye, Z.; Ting, J.P.-Y. NLR, the nucleotide-binding domain leucine-rich repeat containing gene family. Curr. Opin. Immunol. 2008, 20, 3–9. [Google Scholar]
- Pauleau, A.L.; Murray, P.J. Role of Nod2 in the response of macrophages to toll-like receptor agonists. Mol. Cell Biol. 2003, 23, 7531–7539. [Google Scholar] [CrossRef]
- Girardin, S.E.; Tournebize, R.; Mavris, M.; Page, A.L.; Li, X.; Stark, G.R.; Bertin, J.; DiStefano, P.S.; Yaniv, M.; Sansonetti, P.J.; et al. CARD4/Nod1 mediates NF-kappaB and JNK activation by invasive Shigella flexneri. EMBO Rep. 2001, 2, 736–742. [Google Scholar]
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proil-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Srinivasula, S.M.; Poyet, J.L.; Razmara, M.; Datta, P.; Zhang, Z.; Alnemri, E.S. The PYRIN-CARD protein ASC is an activating adaptor for Caspase-1. J. Biol. Chem. 2002, 277, 21119–21122. [Google Scholar]
- Faustin, B.; Lartigue, L.; Bruey, J.M.; Luciano, F.; Sergienko, E.; Bailly-Maitre, B.; Volkmann, N.; Hanein, D.; Rouiller, I.; Reed, J.C. Reconstituted NALP1 inflammasome reveals two-step mechanism of Caspase-1 activation. Mol. Cell 2007, 25, 713–724. [Google Scholar] [CrossRef]
- Kanneganti, T.D.; Body-Malapel, M.; Amer, A.; Park, J.H.; Whitfield, J.; Franchi, L.; Taraporewala, Z.F.; Miller, D.; Patton, J.T.; Inohara, N.; et al. Critical role for Cryopyrin/Nalp3 in activation of Caspase-1 in response to viral infection and double-stranded RNA. J. Biol. Chem. 2006, 281, 36560–36568. [Google Scholar] [CrossRef]
- Bruey, J.M.; Bruey-Sedano, N.; Newman, R.; Chandler, S.; Stehlik, C.; Reed, J.C. PAN1/NALP2/PYPAF2, an inducible inflammatory mediator that regulates NF-kappaB and caspase-1 activation in macrophages. J. Biol. Chem. 2004, 279, 51897–51907. [Google Scholar]
- Conti, B.J.; Davis, B.K.; Zhang, J.; O'Connor, W.; Williams, K.L.; Ting, J.P.Y. CATERPILLER 16.2 (CLR16.2), a Novel NBD/LRR family member that negatively regulates T cell function. J. Biol. Chem. 2005, 280, 18375–18385. [Google Scholar]
- Lich, J.D.; Williams, K.L.; Moore, C.B.; Arthur, J.C.; Davis, B.K.; Taxman, D.J.; Ting, J.P. Monarch-1 suppresses non-canonical NF-kappaB activation and p52-dependent chemokine expression in monocytes. J. Immunol. 2007, 178, 1256–1260. [Google Scholar]
- Masumoto, J.; Dowds, T.A.; Schaner, P.; Chen, F.F.; Ogura, Y.; Li, M.; Zhu, L.; Katsuyama, T.; Sagara, J.; Taniguchi, S.; et al. ASC is an activating adaptor for NF-+¦B and caspase-8-dependent apoptosis. Biochem. Biophys. Res. Commun. 2003, 303, 69–73. [Google Scholar] [CrossRef]
- Mariathasan, S.; Newton, K.; Monack, D.M.; Vucic, D.; French, D.M.; Lee, W.P.; Roose-Girma, M.; Erickson, S.; Dixit, V.M. Differential activation of the inflammasome by caspase-1 adaptors ASC and Ipaf. Nature 2004, 430, 213–218. [Google Scholar]
- Chen, G.; Pedra, J.H. The inflammasome in host defense. Sensors (Basel) 2010, 10, 97–111. [Google Scholar]
- Masumoto, J.; Yang, K.; Varambally, S.; Hasegawa, M.; Tomlins, S.A.; Qiu, S.; Fujimoto, Y.; Kawasaki, A.; Foster, S.J.; Horie, Y.; et al. Nod1 acts as an intracellular receptor to stimulate chemokine production and neutrophil recruitment in vivo. J. Exp. Med. 2006, 203, 203–213. [Google Scholar]
- Allen, I.C.; Scull, M.A.; Moore, C.B.; Holl, E.K.; McElvania-TeKippe, E.; Taxman, D.J.; Guthrie, E.H.; Pickles, R.J.; Ting, J.P.Y. The NLRP3 inflammasome mediates in vivo innate immunity to influenza a virus through recognition of viral RNA. Immunity 2009, 30, 556–565. [Google Scholar] [CrossRef]
- Kanneganti, T.D. Central roles of NLRs and inflammasomes in viral infection. Nat. Rev. Immunol. 2010, 10, 688–698. [Google Scholar] [CrossRef]
- Takeuchi, R.; Tsutsumi, H.; Osaki, M.; Sone, S.; Imai, S.; Chiba, S. Respiratory syncytial virus infection of neonatal monocytes stimulates synthesis of interferon regulatory factor 1 and interleukin-1beta (IL-1beta)-converting enzyme and secretion of IL-1beta. J. Virol. 1998, 72, 837–840. [Google Scholar]
- Shafique, M.; Wilschut, J.; de Haan, A. Induction of mucosal and systemic immunity against respiratory syncytial virus by inactivated virus supplemented with TLR9 and NOD2 ligands. Vaccine 2012, 30, 597–606. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kolli, D.; Velayutham, T.S.; Casola, A. Host-Viral Interactions: Role of Pattern Recognition Receptors (PRRs) in Human Pneumovirus Infections. Pathogens 2013, 2, 232-263. https://doi.org/10.3390/pathogens2020232
Kolli D, Velayutham TS, Casola A. Host-Viral Interactions: Role of Pattern Recognition Receptors (PRRs) in Human Pneumovirus Infections. Pathogens. 2013; 2(2):232-263. https://doi.org/10.3390/pathogens2020232
Chicago/Turabian StyleKolli, Deepthi, Thangam Sudha Velayutham, and Antonella Casola. 2013. "Host-Viral Interactions: Role of Pattern Recognition Receptors (PRRs) in Human Pneumovirus Infections" Pathogens 2, no. 2: 232-263. https://doi.org/10.3390/pathogens2020232