Combined Galvanostatic and Potentiostatic Plasma Electrolytic Oxidation of Titanium in Different Concentrations of H2SO4

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
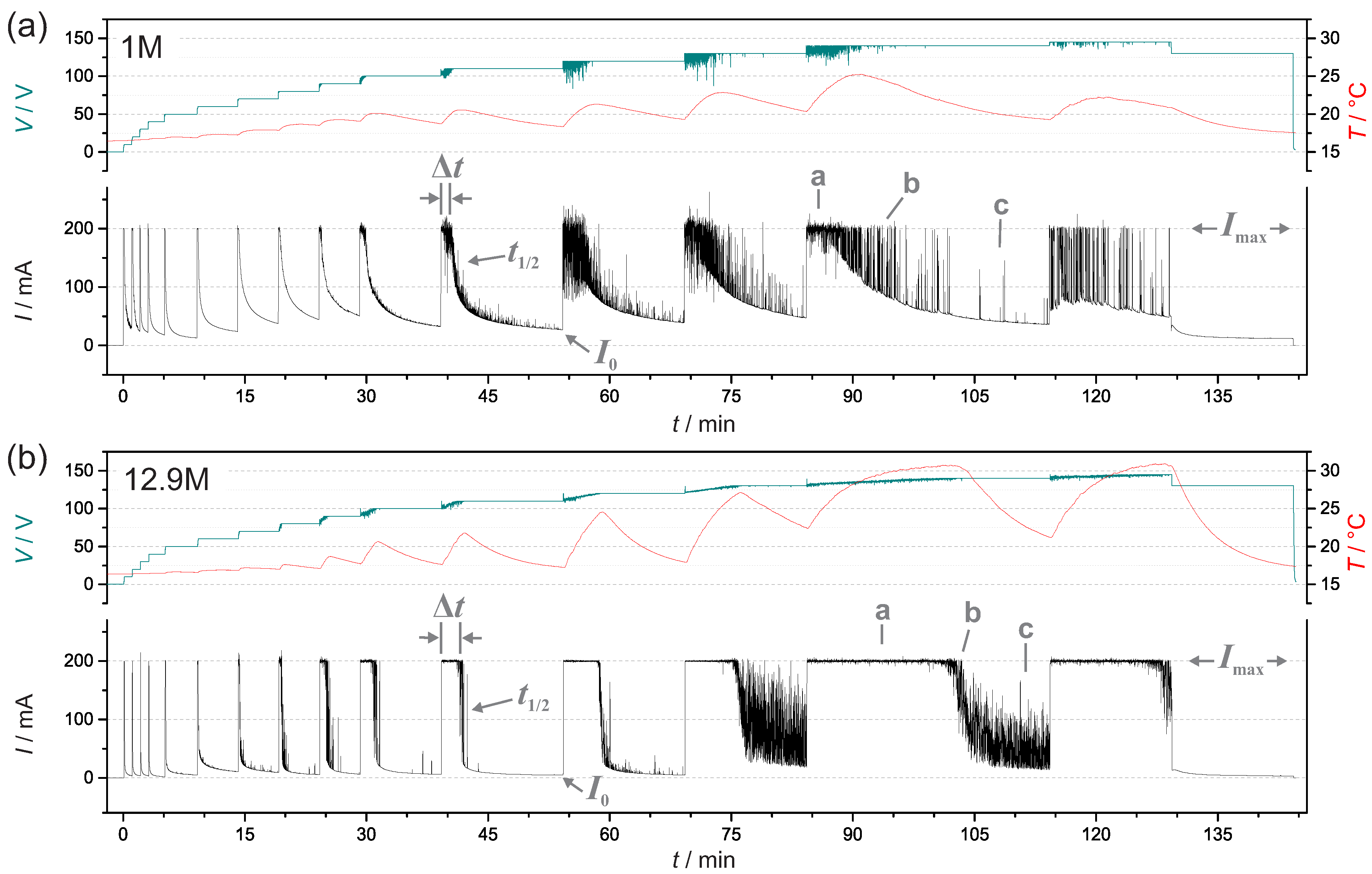
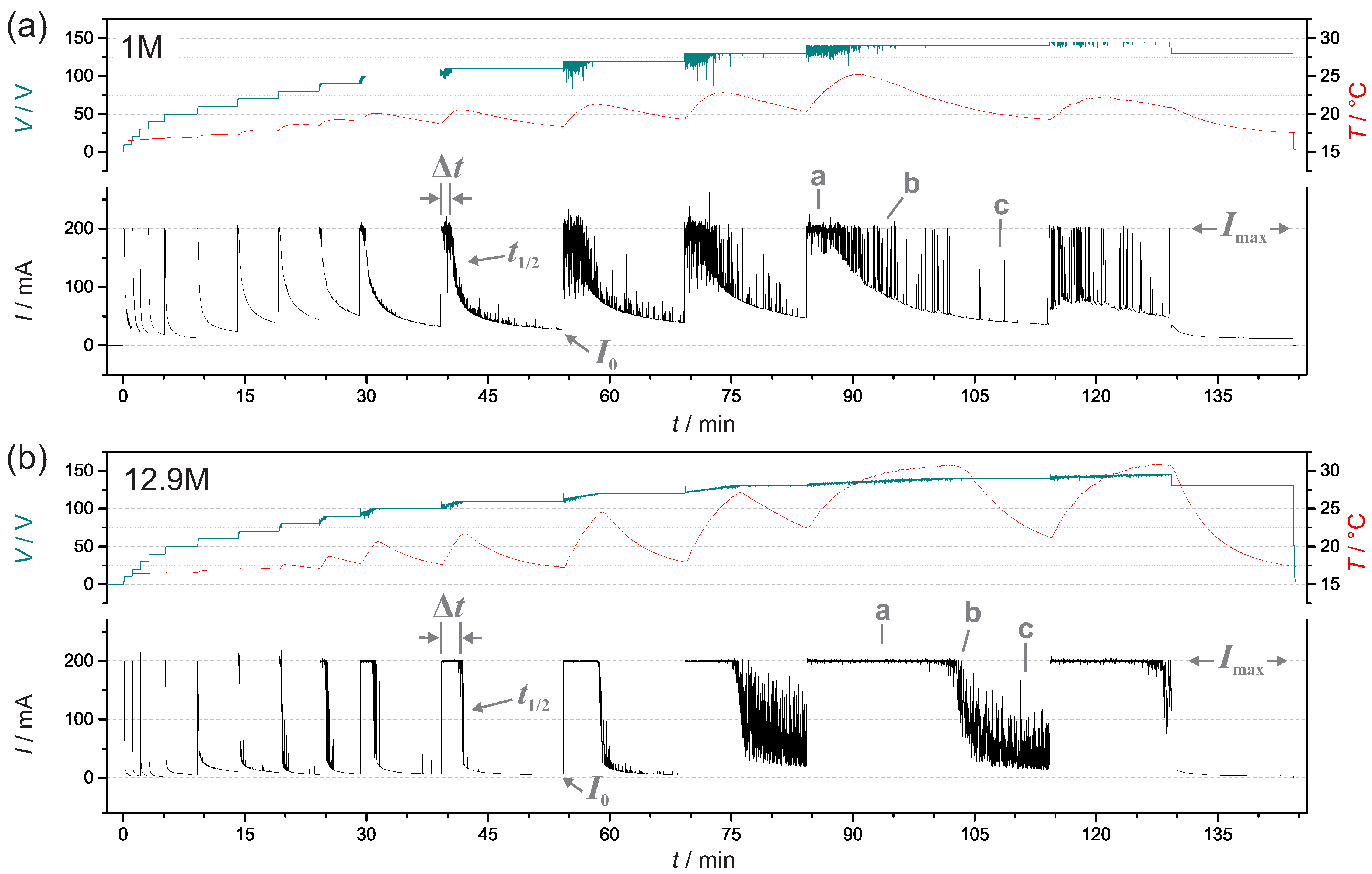
3.1. Current, Voltage and Temperature During PEO
3.2. Microstructure
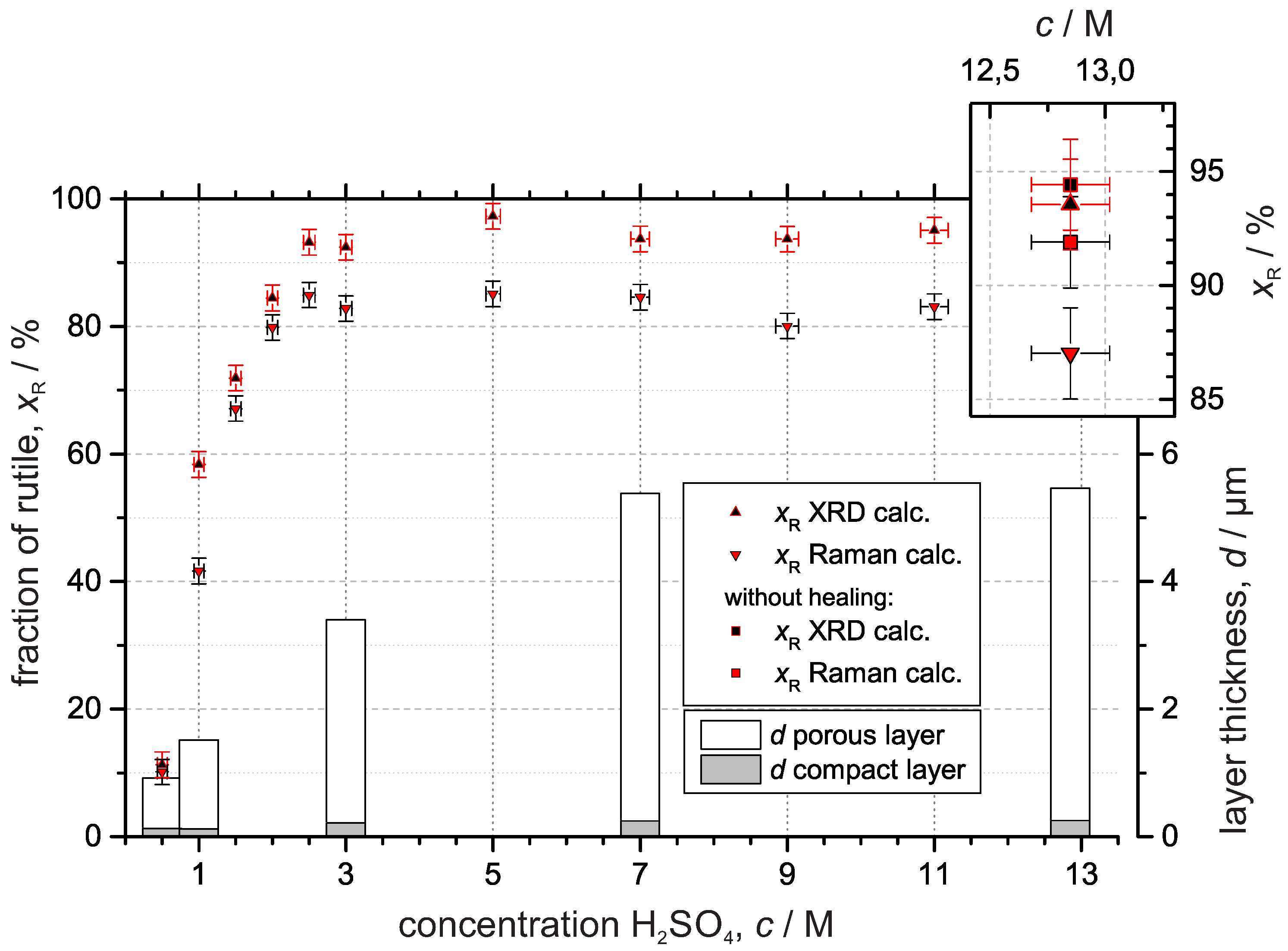
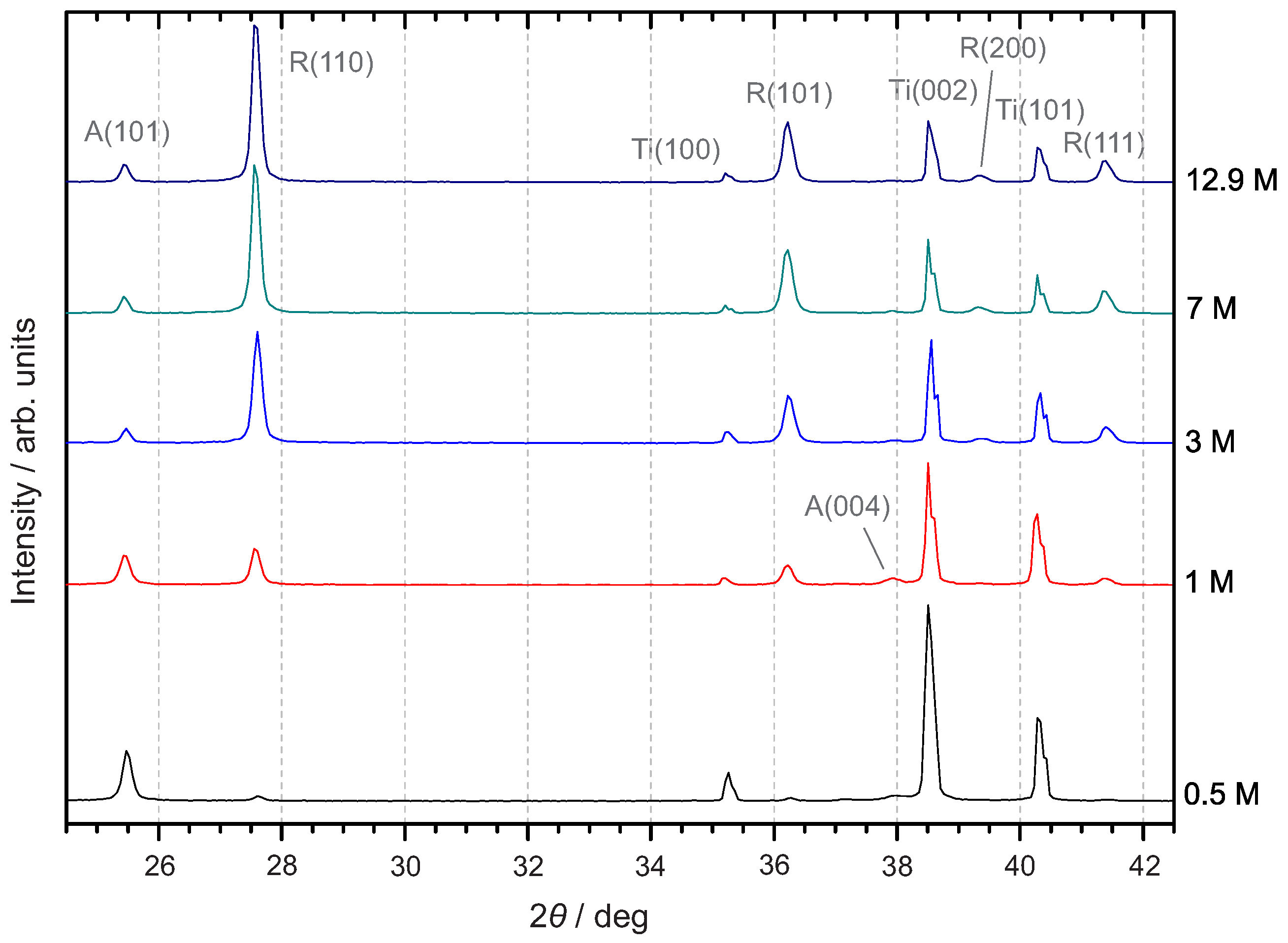
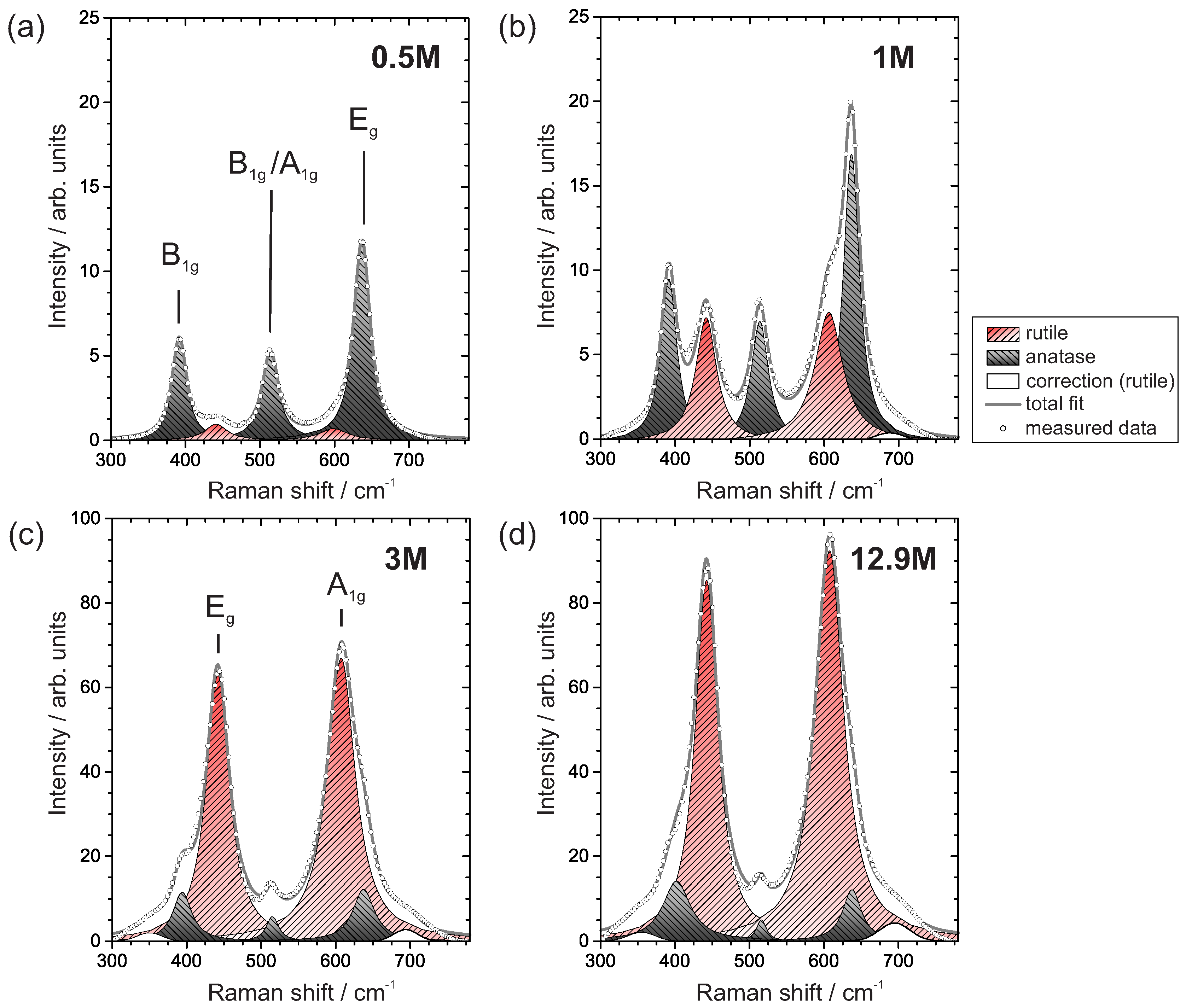
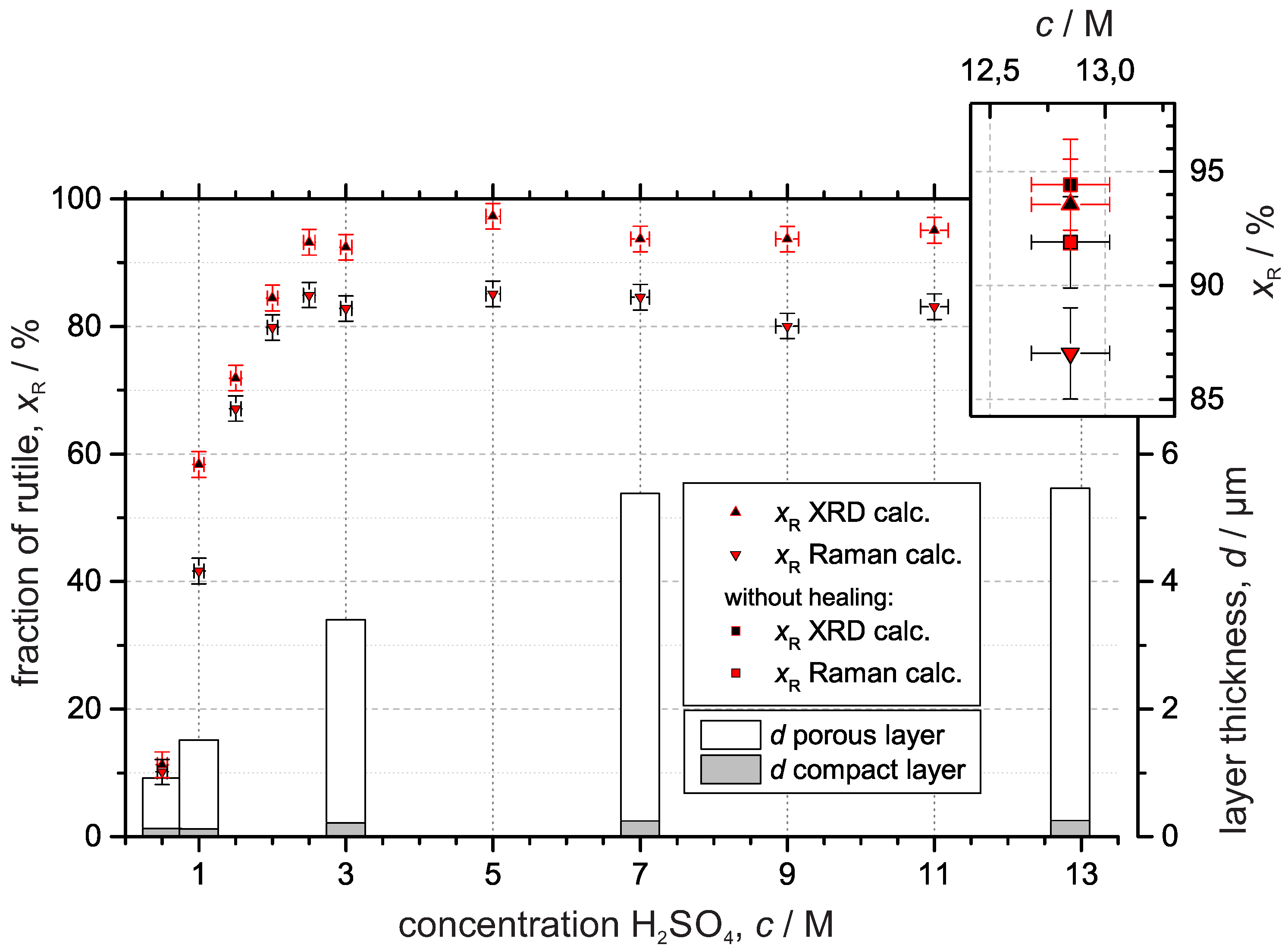
3.3. Crystal Structure
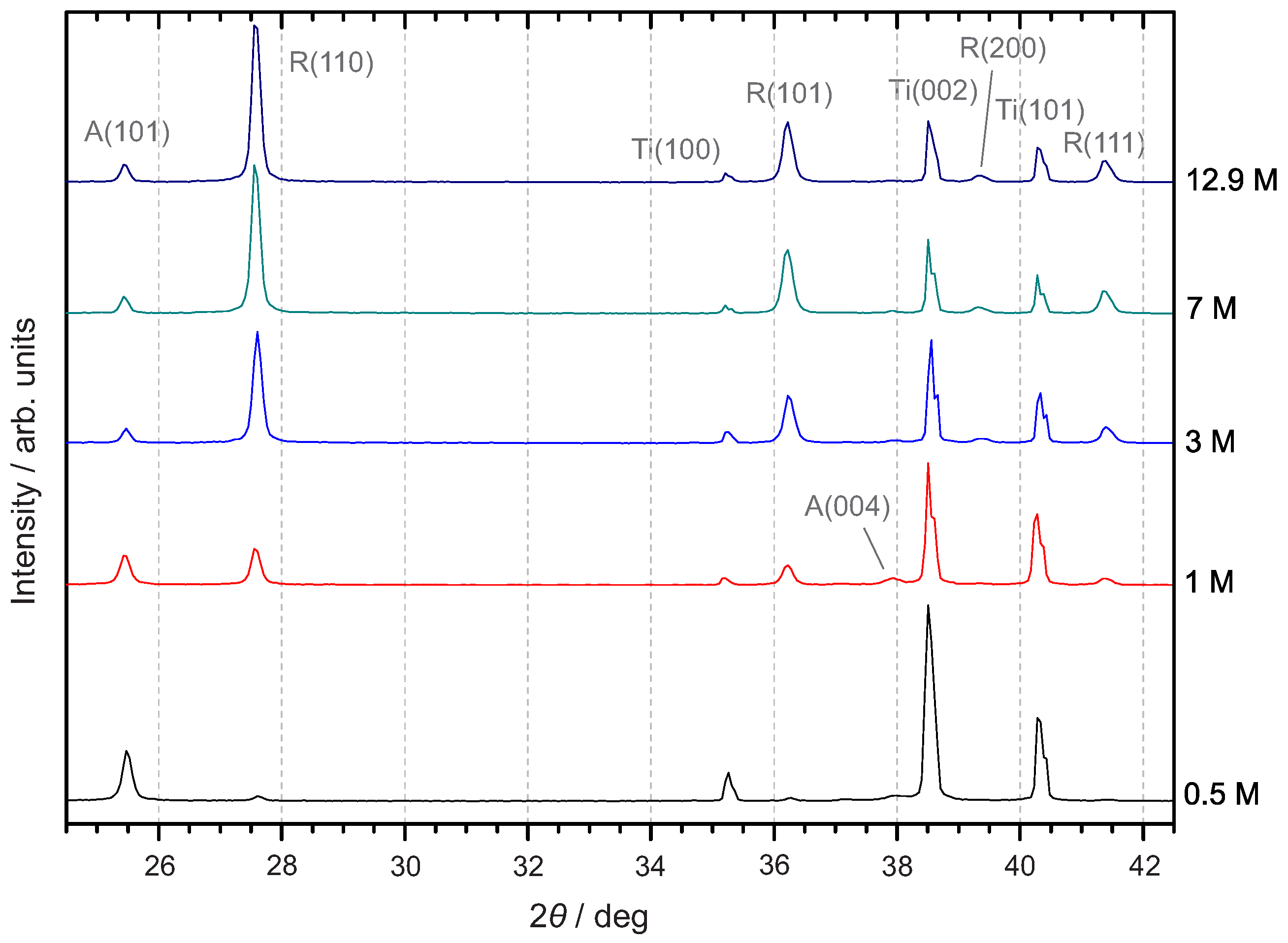
3.3.1. X-ray Diffraction
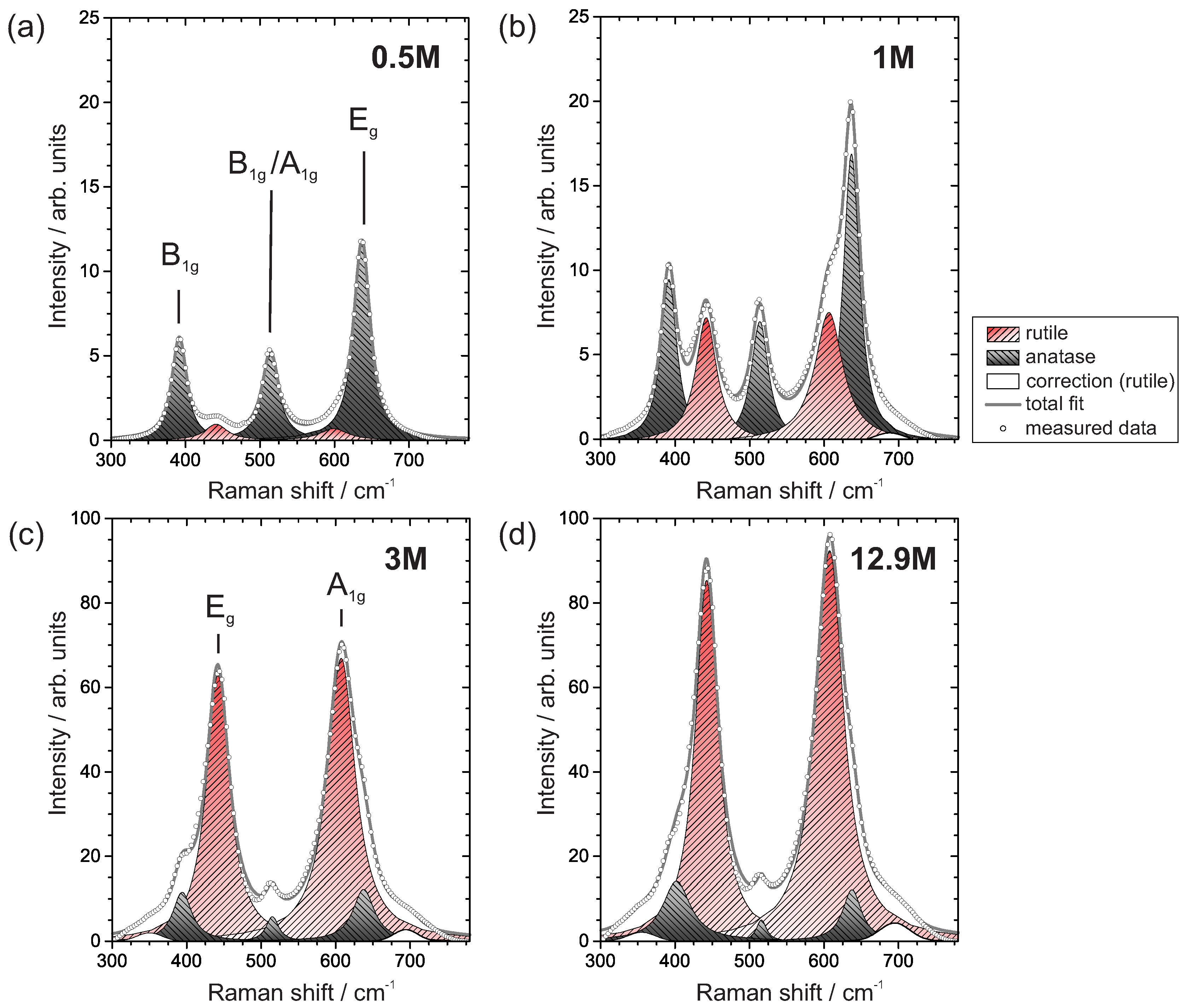
3.3.2. Raman Spectroscopy
4. Discussion
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Diamanti, M.V.; Ormellese, M.; Pedeferri, M. Application-wise nanostructuring of anodic films on titanium: A review. J. Exp. Nanosci. 2015, 10, 1285–1308. [Google Scholar] [CrossRef]
- Diamanti, M.V.; Del Curto, B.; Pedeferri, M. Anodic oxidation of titanium: From technical aspects to biomedical applications. J. Appl. Biomater. Biomech. 2011, 9. [Google Scholar] [CrossRef] [PubMed]
- Schierbaum, K.; El Achhab, M. Generation of an electromotive force by hydrogen-to-water oxidation with Pt-coated oxidized titanium foils. Phys. Status Solidi 2011, 208, 2796–2802. [Google Scholar] [CrossRef]
- Karpov, E.G.; Hashemian, M.A.; Dasari, S.K. Chemistry-Driven Signal Transduction in a Mesoporous Pt/TiO2 System. J. Phys. Chem. C 2013, 117, 15632–15638. [Google Scholar] [CrossRef]
- Cakabay, Ö.; El Achhab, M.; Schierbaum, K. Thermal properties of solid-state Pt/TiO2/Ti emf cells studied by microcalorimetry. Appl. Phys. A 2015, 118, 1127–1132. [Google Scholar] [CrossRef]
- El Achhab, M.; Schierbaum, K. Gas sensors based on plasma-electrochemically oxidized titanium foils. J. Sensors Sensor Syst. 2016, 5, 273–281. [Google Scholar] [CrossRef]
- Vanhumbeeck, J.F.; Proost, J. Current understanding of Ti anodisation: Functional, morphological, chemical and mechanical aspects. Corros. Rev. 2009, 27, 117–204. [Google Scholar] [CrossRef]
- Quintero, D.; Galvis, O.; Calderón, J.; Castaño, J.; Echeverría, F. Effect of electrochemical parameters on the formation of anodic films on commercially pure titanium by plasma electrolytic oxidation. Surf. Coat. Technol. 2014, 258, 1223–1231. [Google Scholar] [CrossRef]
- Sul, Y.T.; Johansson, C.B.; Jeong, Y.; Albrektsson, T. The electrochemical oxide growth behaviour on titanium in acid and alkaline electrolytes. Med. Eng. Phys. 2001, 23, 329–346. [Google Scholar] [CrossRef]
- Yang, B.; Uchida, M.; Kim, H.M.; Zhang, X.; Kokubo, T. Preparation of bioactive titanium metal via anodic oxidation treatment. Biomaterials 2004, 25, 1003–1010. [Google Scholar] [CrossRef]
- Diamanti, M.V.; Pedeferri, M.P. Effect of anodic oxidation parameters on the titanium oxides formation. Corros. Sci. 2007, 49, 939–948. [Google Scholar] [CrossRef]
- Masahashi, N.; Mizukoshi, Y.; Semboshi, S.; Ohtsu, N. Enhanced photocatalytic activity of rutile TiO2 prepared by anodic oxidation in a high concentration sulfuric acid electrolyte. Appl. Catal. B Environ. 2009, 90, 255–261. [Google Scholar] [CrossRef]
- Friedemann, A.; Gesing, T.; Plagemann, P. Electrochemical rutile and anatase formation on (PEO) surfaces. Surf. Coat. Technol. 2017, 315, 139–149. [Google Scholar] [CrossRef]
- Fadl-Allah, S.A.; El-Sherief, R.M.; Badawy, W.A. Electrochemical formation and characterization of porous titania (TiO2) films on Ti. J. Appl. Electrochem. 2008, 38, 1459. [Google Scholar] [CrossRef]
- Sun, C.; Hui, R.; Qu, W.; Yick, S.; Sun, C.; Qian, W. Effects of processing parameters on microstructures of TiO2 coatings formed on titanium by plasma electrolytic oxidation. J. Mater. Sci. 2010, 45, 6235–6241. [Google Scholar] [CrossRef]
- Yerokhin, A.; Nie, X.; Leyland, A.; Matthews, A.; Dowey, S. Plasma electrolysis for surface engineering. Surf. Coat. Technol. 1999, 122, 73–93. [Google Scholar] [CrossRef]
- Li, Q.; Liang, J.; Wang, Q. Plasma Electrolytic Oxidation Coatings on Lightweight Metals. In Modern Surface Engineering Treatments; Aliofkhazraei, M., Ed.; InTech: Rijeka, Croatia, 2013; Chapter 4. [Google Scholar]
- El Achhab, M.; Erbe, A.; Koschek, G.; Hamouich, R.; Schierbaum, K. A microstructural study of the structure of plasma electrolytically oxidized titanium foils. Appl. Phys. A 2014, 116, 2039–2044. [Google Scholar] [CrossRef]
- Hussein, R.O.; Nie, X.; Northwood, D.O.; Yerokhin, A.; Matthews, A. Spectroscopic study of electrolytic plasma and discharging behaviour during the plasma electrolytic oxidation (PEO) process. J. Phys. D Appl. Phys. 2010, 43, 105203. [Google Scholar] [CrossRef]
- Spurr, R.A.; Myers, H. Quantitative analysis of anatase-rutile mixtures with an X-ray diffractometer. Anal. Chem. 1957, 29, 760–762. [Google Scholar] [CrossRef]
- Zanatta, A. A fast-reliable methodology to estimate the concentration of rutile or anatase phases of TiO2. AIP Adv. 2017, 7, 075201. [Google Scholar] [CrossRef]
- Hanaor, D.A.H.; Sorrell, C.C. Review of the anatase to rutile phase transformation. J. Mater. Sci. 2011, 46, 855–874. [Google Scholar] [CrossRef]
- Balachandran, U.; Eror, N.G. Raman spectra of titanium dioxide. J. Solid State Chem. 1982, 42, 276–282. [Google Scholar] [CrossRef]
- Frank, O.; Zukalova, M.; Laskova, B.; Kürti, J.; Koltai, J.; Kavan, L. Raman spectra of titanium dioxide (anatase, rutile) with identified oxygen isotopes (16, 17, 18). Phys. Chem. Chem. Phys. 2012, 14, 14567–14572. [Google Scholar] [CrossRef] [PubMed]
- Van Santen, R.A. The Ostwald step rule. J. Phys. Chem. 1984, 88, 5768–5769. [Google Scholar] [CrossRef]
- Klapkiv, M.D.; Nykyforchyn, H.M.; Posuvailo, V.M. Spectral analysis of an electrolytic plasma in the process of synthesis of aluminum oxide. Mater. Sci. 1995, 30, 333–343. [Google Scholar] [CrossRef]
- Albella, J.; Montero, I.; Martinez-Duart, J. A theory of avalanche breakdown during anodic oxidation. Electrochimica Acta 1987, 32, 255–258. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 0.5 M | 1 M | 3 M | 7 M | 12.9 M | |
|---|---|---|---|---|---|
| Pore size/m | 0.20 ± 0.09 | 0.25 ± 0.07 | 0.29 ± 0.08 | 0.35 ± 0.06 | 0.38 ± 0.07 |
| Thickness porous layer/m | 0.79 ± 0.06 | 1.39 ± 0.09 | 3.18 ± 0.20 | 5.14 ± 0.20 | 5.21 ± 0.62 |
| Thickness compact layer/m | 0.13 ± 0.03 | 0.12 ± 0.03 | 0.22 ± 0.04 | 0.25 ± 0.03 | 0.25 ± 0.03 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Engelkamp, B.; El Achhab, M.; Fischer, B.; Kökçam-Demir, Ü.; Schierbaum, K. Combined Galvanostatic and Potentiostatic Plasma Electrolytic Oxidation of Titanium in Different Concentrations of H2SO4. Metals 2018, 8, 386. https://doi.org/10.3390/met8060386
Engelkamp B, El Achhab M, Fischer B, Kökçam-Demir Ü, Schierbaum K. Combined Galvanostatic and Potentiostatic Plasma Electrolytic Oxidation of Titanium in Different Concentrations of H2SO4. Metals. 2018; 8(6):386. https://doi.org/10.3390/met8060386
Chicago/Turabian StyleEngelkamp, Bernd, Mhamed El Achhab, Björn Fischer, Ülkü Kökçam-Demir, and Klaus Schierbaum. 2018. "Combined Galvanostatic and Potentiostatic Plasma Electrolytic Oxidation of Titanium in Different Concentrations of H2SO4" Metals 8, no. 6: 386. https://doi.org/10.3390/met8060386