Preparation of Vanadium Nitride Using a Thermally Processed Precursor with Coating Structure
by
Jingli Han
1,2,3, 
Yimin Zhang
1,2,3,4,*,
Tao Liu
1,2,3,
Jing Huang
1,2,3,
Nannan Xue
1,3 and
Pengcheng Hu
1,2,3 1
College of Resource and Environment Engineering, Wuhan University of Science and Technology, Wuhan 430081, China
2
Hubei Collaborative Innovation Center of High Efficient Utilization for Vanadium Resources, Wuhan 430081, China
3
Hubei Provincial Engineering Technology Research Center of High Efficient Cleaning Utilization for Shale Vanadium Resource, Wuhan 430081, China
4
College of Resource and Environment Engineering, Wuhan University of Technology, Wuhan 430070, China
*
Author to whom correspondence should be addressed.
Metals 2017, 7(9), 360; https://doi.org/10.3390/met7090360
Submission received: 30 July 2017
/
Revised: 22 August 2017
/
Accepted: 5 September 2017
/
Published: 11 September 2017
Abstract
:A new effective method is proposed to prepare vanadium nitride (VN) via carbothermal reduction–nitridation (CRN) of the precursor, obtained by adding carbon black (C) to the stripping solution during the vanadium recovery from black shale. VN was successfully prepared at a low temperature of 1150 °C for only 1 h with a C/V2O5 mass ratio of 0.30 in N2 atmosphere, but a temperature of 1300–1500 °C is required for several hours in the traditional CRN method. The low synthesis temperature and short period for the preparation of VN was due to the vanadium-coated carbon structure of the precursor, which enlarged the contact area between reactants significantly and provided more homogeneous chemical composition. In addition, the simultaneous direct reduction and indirect reduction of the interphase caused by the coating structure obviously accelerated the reaction. The phase evolution of the precursor was as follows: (NH4)2V6O16·1.5H2O → V2O5 → V6O13 → VO2 → V4O7 → V2O3 → VC → VN. The precursor converted to V6O13 and VO2 completely after being calcined at 550 °C, indicating that the pre-reduction of V2O5 in the traditional CRN method can be omitted. This method combined the synthesis of VN with the vanadium extraction creatively, having the advantages of simple reaction conditions, low cost and short processing time.
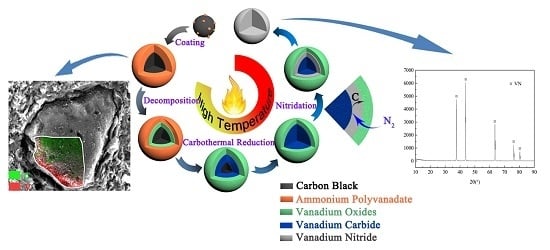
1. Introduction
Vanadium nitride (VN) has received increasing attention in recent years due to its typical properties including extreme hardness, high melting point, wear and corrosion resistance, good electric and thermal conductivity, high-temperature stability, as well as good catalytic activity. It can be widely used as iron and steel additive [1,2], electrode [3,4], catalyst [5,6], superconductor [7], and coating [8,9]. The frequent application of VN, as an important steel additive, can be ascribed to its beneficial effect on fine grain and precipitation strengthening, which can improve the comprehensive performance of steel and reduce the cost of the smelting process by using nitrogen to reduce the vanadium content of steel [10].
The traditional procedure for the synthesis of VN is the process of carbothermal reduction–nitridation (CRN) of V2O5 in N2 at a high temperature of 1500 °C for 3 h with a N2 flow rate of 13.3 × 10−6 m3·s−1 [11], and at 1400 °C with a N2 flow rate of 50 L·h−1, using a microwave method [12]. The low melting point (670 °C) and the high saturation vapor pressure of V2O5 will result in a volatile loss of V2O5 near its melting point. Hence, the pre-reduction during transformation from V2O5 to a high melting point and low-valent vanadium oxides below the melting point of V2O5 is necessary [13], resulting inevitably in a long reaction period. However, using the low-valent vanadium oxides will increase the production cost due to the higher price compared to V2O5. In addition to the traditional CRN method, VN was prepared by thermal liquid-solid reaction [14]: Ammonolysis of precursor compounds of metal [7], self-propagating high-temperature synthesis [15], sol-gel method [16], chemical vapor deposition [17], mechanical alloying [18] and other methods [19,20]. However, most methods are plagued by the presence of air-sensitive or toxic reagents, expensive equipment and raw materials, elevated temperatures and long reaction time, which limit its large-scale production and application. Therefore, there is a need for a simple synthesis route of VN. Thermal processing precursor, as a novel method to prepare metal nitrides, has gradually become a research hotspot. Zhao et al. [21] synthesized VN nanopowders by thermal nitridation of the precursor obtained by physically drying an aqueous solution of ammonium vanadate (NH4VO3) and nanometer carbon black. (Cr,V)2(C,N) solid solution powders were synthesized by CRN of the precursor, prepared by heating admixtures of ammonium dichromate ((NH4)2Cr2O7), NH4VO3, and carbon black in distilled water with continuous stirring [22]. These methods can reduce the synthesizing temperature greatly, but the production cost remained high due to the use of de-ionized water, NH4VO3 and nanometer carbon black, indicating the strong demand of a simple, low-cost and low-temperature approach to synthesize VN. Moreover, the formation process and microstructure of the precursor and its promoting effect on the preparation of metal nitrides were not discussed.
In China, most of the vanadium resources exist in the form of black shale, so the utilization and exploitation of black shale is an essential way to recover vanadium [23]. The vanadium recovery from black shale was investigated using a pyro-hydrometallurgical process specifically including roasting, acid leaching, purification, and chemical precipitation [24,25,26]. Solvent extraction was the most commonly adopted process to purify the acid leaching solution for better selectivity and economy [27,28]. After solvent extraction and stripping, vanadium can be precipitated from the stripping solution with acidic ammonium salt [29]. In view of the preparation method of precursor [7,21,22] in the synthesis of VN, and the application of the precipitation method [30] to synthesize Pb(Mg1/3Nb2/3)O3 ceramic, it may be feasible to prepare the precursor during the vanadium precipitation from the stripping solution in the process of vanadium extraction from black shale, to synthesize VN. This method revealed the merits of a low reaction temperature, short period, and a widely available, cheap vanadium source, which make it more practical, economical and efficient in industrial applications.
In this study, we combined the synthesis of VN with vanadium extraction from black shale for the first time by adding carbon black (C) to the stripping solution. First, the precursor containing the vanadium source and reducing material formed gradually during the vanadium precipitation, then the precursor was reduced and nitrided in the N2 atmosphere to yield VN. The effects of the main reaction conditions on the nitrogen content (N content) in VN product were investigated. In order to explain the lower temperature and shorter time for the preparation of VN in comparison with the conventional CRN method: The phase and microstructure of the precursor were analyzed. In addition, the mechanism of preparing VN from the precursor was discussed through phase analysis, thermodynamic calculation, and thermogravimetric (TG) experiment.
2. Experimental Section
2.1. Materials
The stripping solution was obtained by blank roasting, acid leaching, solvent extraction and stripping of black shale obtained from Tongshan, China. The main chemical composition of the stripping solution is given in Table 1, the concentration of V in the stripping solution is 20.63 g·L−1, while the major impurity ion is Al with the content of 5.78 g·L−1. The pH of the stripping solution is 0.03. Carbon black containing 94.80% of carbon with the particle size of less than 0.074 mm was used as the reducing agent.
2.2. Typical Procedure for the Preparation of VN
The process flow sheet to prepare VN is shown in Figure 1. Typically, sodium chlorate (NaClO3, AR (analytical reagent), 2.95 g) was added to the stripping solution (200 mL) to oxidize vanadium (IV) to vanadium (V) in a temperature-controlled magnetic stirrer (SZCL-2A, Wuhan Keer Instrument Co., Ltd., Wuhan, China) at room temperature. After complete oxidation, a certain amount of carbon black was added to the solution and dispersed by stirring. The pH of the solution was adjusted to 1.8 with ammonia (AR), and then the solution was stirred to precipitate ammonium polyvanadate (APV) at 90 °C for 1 h. After solid-liquid separation by vacuum filtration (model SHB-III, Wuhan Keer Instrument Co., Ltd., Wuhan, China), the precursor was washed by water, dried in an oven, and then compressed into cylindrical blocks of 30 mm diameter at a pressure of 14 MPa. The cylindrical block was placed into the tubular atmosphere furnace (SGL-1700, Shanghai Institute of Optics and Fine Mechanics, Shanghai, China) with a certain flow rate of nitrogen (99.999%). The sample was calcined to deaminize at 550 °C for 20 min and pre-reduced at 650 °C for 2.5 h, and then heated to the desired temperature for reduction and nitridation for a certain of time. Subsequently, the product was cooled to room temperature in nitrogen atmosphere.
2.3. Material Characterization
The nitrogen content (N content) was determined employing distillation-acid base titration analysis. The X-ray diffraction (XRD) patterns were obtained by using a Rigaku D/MAX-RB x-ray diffractometer (Rigaku, Akishima, Japan) with Cu Kα radiation to analyze the phase compositions in the products. Microscopic observation and elemental analysis (SEM–EDS; SEM: Scanning electron microscope; EDS: Energy-dispersive X-Ray spectroscope) was conducted using a JSM-IT300 scanning electron microscope (JEOL, Tokyo, Japan), equipped with an X-ACT energy dispersive spectrometer (Oxford Instruments, Oxford, UK). The thermogravimetric (TG) experiment was carried out by utilizing a STA449C analyzer (Netzsch, Selb, Germany) to change from room temperature to 1400 °C with a linear heating rate of 10 °C·min−1 under N2, and a gas flow rate of 50 mL·min−1.
3. Results and Discussion
3.1. Effect of the Main Reaction Conditions on the N Content in VN Product
3.1.1. Effect of C/V2O5 Mass Ratio
The effect of C/V2O5 mass ratio (m(C:V2O5)) on the N content in VN product is shown in Figure 2, obtained at a reaction temperature of 1250 °C, a reaction time of 3 h, and a N2 flow rate of 300 mL·min−1.
As evident from Figure 2a, the m(C:V2O5) has a pronounced effect on the N content in VN, as the N content increased from 15.37% to 17.03% when m(C:V2O5) increased from 0.27 to 0.30. Beyond 0.30, the N content decreased from 17.03% to 10.17%. The low N content was a possible result of the incomplete reduction, due to the shortage of carbon. This may be confirmed by the appearance of V2O3 (Figure 2b) at an m(C:V2O5) value of 0.27. Conversely, when the m(C:V2O5) exceeded 0.30, the excess carbon and the formation of vanadium carbonitride solid solution led to the decrease of the N content. However, there were no peaks that could be indexed to C or VC in Figure 2b, which was due to the amorphous state of carbon black and very similar diffraction patterns of VC and VN [31], respectively. Therefore, 0.30 was considered as the most suitable C/V2O5 mass ratio for the reaction.
3.1.2. Effect of Reaction Temperature
To investigate the effect of the reaction temperature on the N content in VN product, several experiments were performed at a C/V2O5 mass ratio of 0.30, a reaction time of 3 h and a N2 flow rate of 300 mL·min−1. The experimental results are depicted in Figure 3.
As shown in Figure 3a, the N content in VN product increased rapidly from 4.69% to 16.70% as the reaction temperature increased from 950 °C to 1150 °C; the N content then stayed at a constant level over the elevated reaction temperature. Figure 3b exhibits XRD patterns of the products obtained at different temperatures. At 950 °C, the peaks were identified as V2O3 and VN, indicating that the reduction and nitridation of the precursor were not complete at a temperature as low as 950 °C. The XRD patterns were identical and all diffraction peaks corresponded to VN at 1150 °C and 1250 °C, revealing that the precursor was reduced and nitrided almost completely. Hence, a reaction temperature of 1150 °C was chosen as the optimal condition in this experiment.
3.1.3. Effect of Reaction Time
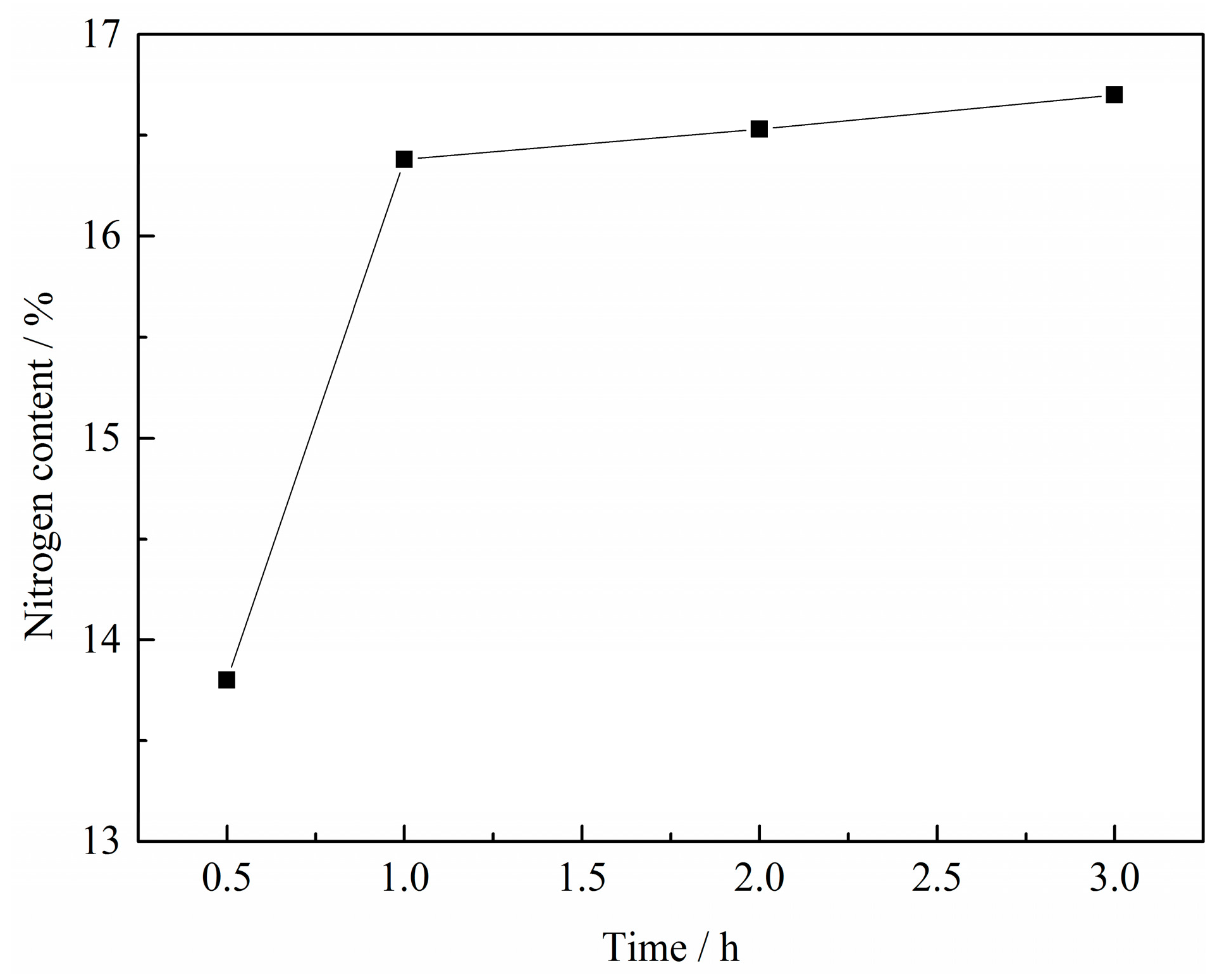
The effect of reaction time on the N content in VN product was investigated under the conditions of a C/V2O5 mass ratio of 0.30, a reaction temperature of 1150 °C and a N2 flow rate of 300 mL·min−1. The results are shown in Figure 4.
It can be observed that the N content in VN product increased strongly from 13.80% to 16.38% as the reaction time increased from 0.5 h to 1 h and then remained almost constant beyond 1 h, which indicated that the reduction and nitridation of the precursor was almost complete at 1 h. Thus, the optimum reaction time was determined to be 1 h.
3.1.4. Effect of N2 Flow Rate
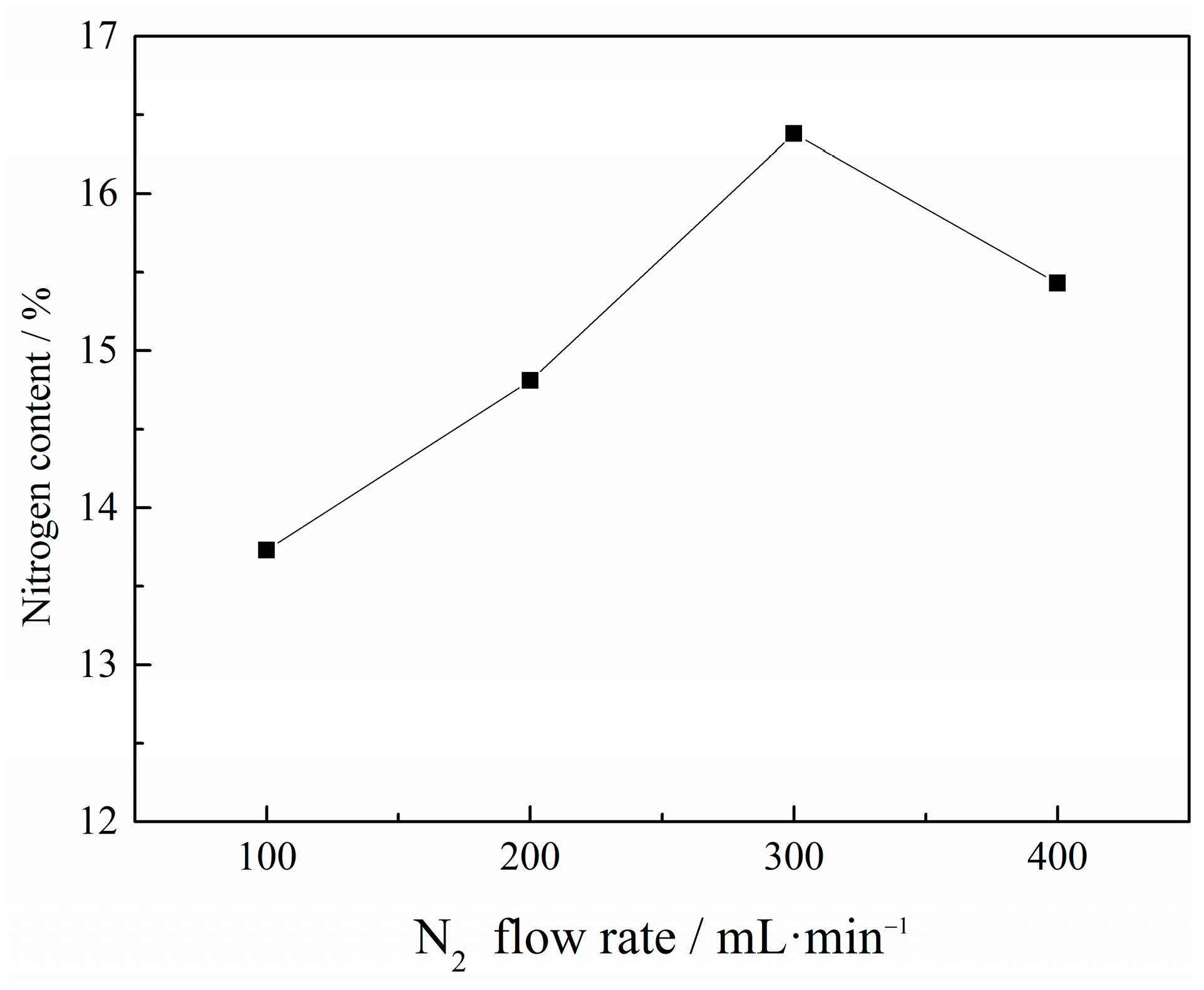
The effect of N2 flow rate on the N content in VN product was investigated under the conditions of a C/V2O5 mass ratio of 0.30, a reaction temperature of 1150 °C and a reaction time of 1 h. The results are shown in Figure 5.
Figure 5 indicates that the N2 flow rate substantially influenced the N content in VN product. The N content first rose from 13.73% to 16.38% and then decreased to 15.43% with an increasing N2 flow rate. There was an optimum N2 flow rate of 300 mL·min−1, at which a maximum N content was achieved. The flowing nitrogen can increase N2 partial pressure and decrease the CO partial pressure, which might accelerate the reduction and nitridation reactions. However, the rapid flowing nitrogen could not be sufficiently heated, and the contact time with vanadium oxides was too short to react adequately, resulting in decreased N content as the flow rate increased further. Therefore, 300 mL·min−1 was the optimal N2 flow rate.
3.2. Phase and Microstructure Analyses of the Precursor
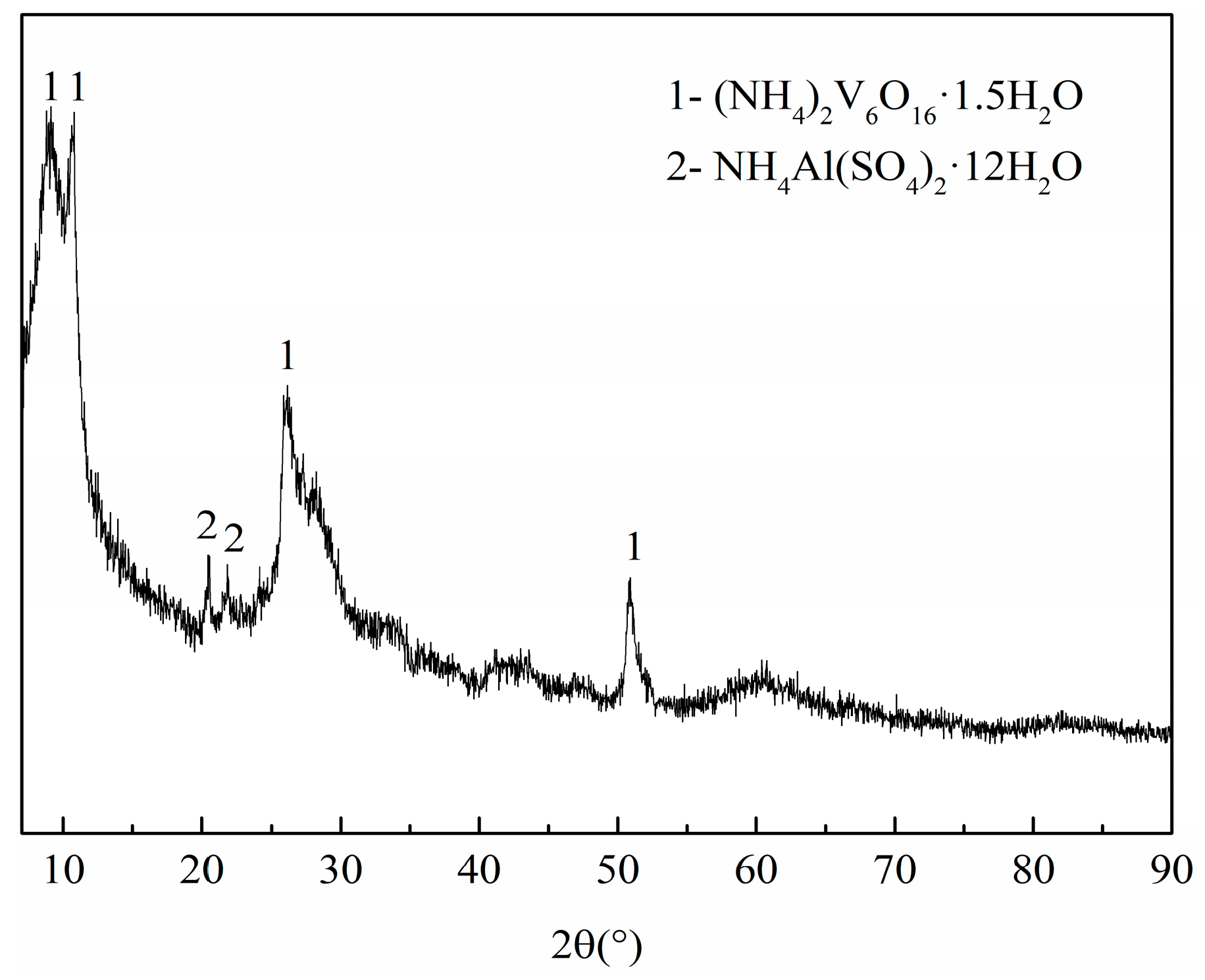
Although an appropriate flow rate of nitrogen can promote the synthesis of VN, high reaction temperature and long reaction time were still required in the synthesis of VN with the flow of nitrogen in the tubular furnace [11,12]. Compared with a temperature of 1300–1500 °C required to prepare VN by the traditional CRN method, this study permitted a lower temperature and a shorter time by as much as several hundred degrees centigrade and several hours, respectively. The traditional raw materials were a mixture of V2O5 and C, while a precursor with V-coated C structure was applied in our research, a fact we consider an essential difference. Hence, we had to investigate the precursor. To understand the phase compositions of the precursor, XRD analysis was conducted. The result shown in Figure 6 revealed that the precursor consists of (NH4)2V6O16·1.5H2O with a small amount of NH4Al(SO4)2·12H2O. No peaks can be indexed to C, due to the amorphous state of carbon black.
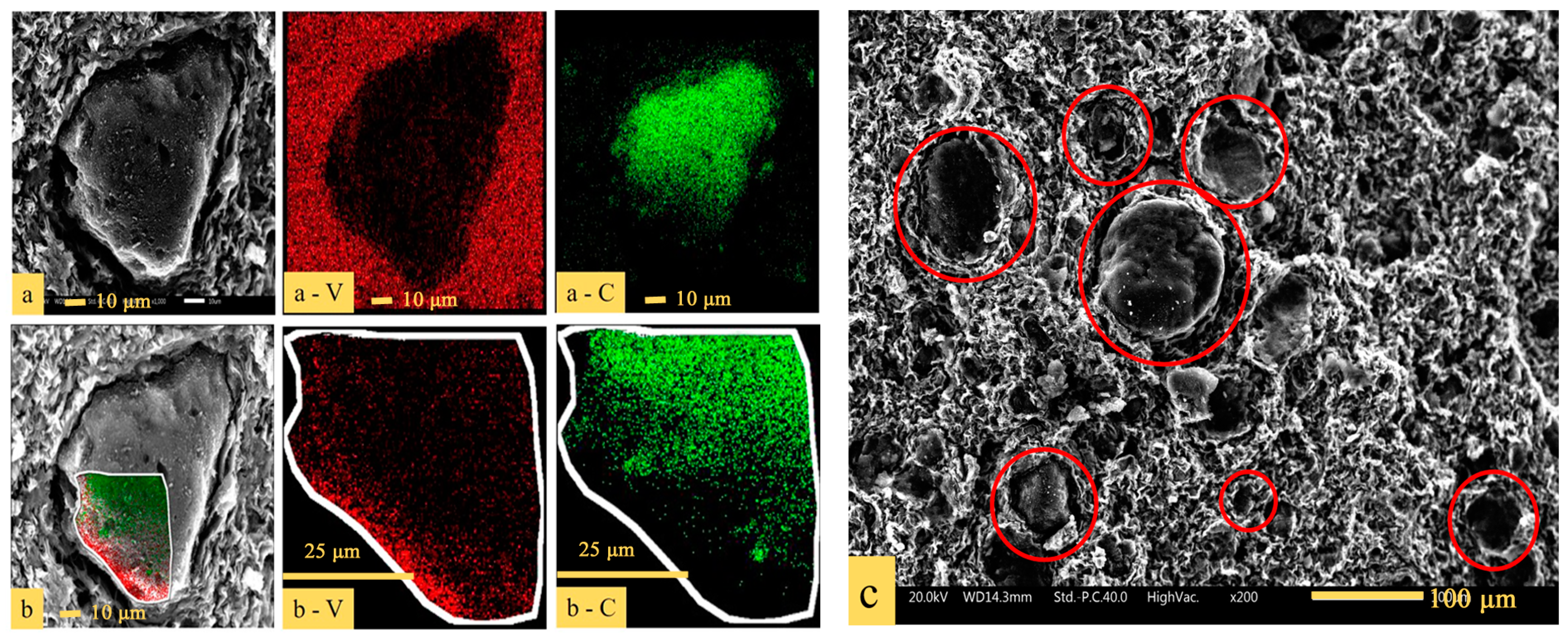
To have a visualized comprehension of the distribution of (NH4)2V6O16·1.5H2O and carbon black in the precursor, SEM–EDS analysis was conducted and the results are presented in Figure 7. As shown in Figure 7a, there was C in the center and V was plentiful around C in a single particle, which indicated that the carbon black was surrounded by vanadium during the process of vanadium precipitation. Figure 7b is a partially enlarged view of the edge of the particle shown in Figure 7a. It can be clearly seen that V is located at the edge of the particle, with C being in the interior. There were many vanadium-coated carbon structures (V-coated C structures), as shown in the red circles in Figure 7c.
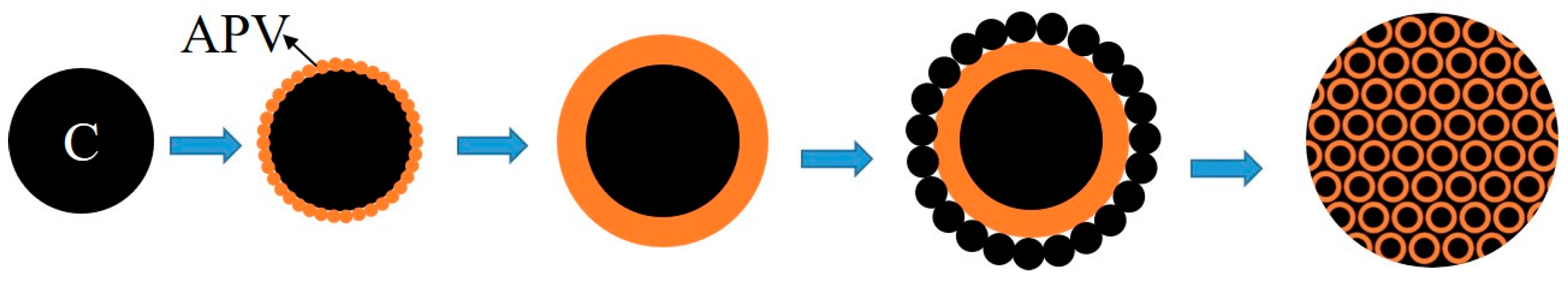
The formation process of the V-coated C structure is schematically illustrated in Figure 8, where the black parts and orange parts represent carbon black and APV, respectively. Due to the presence of the dispered carbon black, the nuclei of the vanadium precipitates in the slurry grow on the surface of carbon black, which served as the heterogeneous nucleation site, instead of forming discrete precipitates. As the precipitation proceeded, APV particles grew gradually and coated carbon black completely, the typical coated structure formed with the core and the coating layer was carbon black and APV, respectively; the uncoated carbon black located around APV. The overall distribution of the precursor was the V-coated C structure which was surrounded by the uncoated carbon black.
The V-coated C structure evidently promoted the reaction between vanadium oxide and carbon black due to the following reasons. First, on the internal interphase of the V-coated C structure, CO produced by direct reduction of vanadium oxides by carbon black could not escape due to the coated structure; hence, it continued to react with vanadium oxide by indirect reduction. Subsequently, CO2 produced by indirect reduction will react with carbon black to produce CO at high temperature. That is to say, inside the vanadium oxides, the direct reduction and indirect reduction occur simultaneously at the internal interphase. Secondly, outside the vanadium oxides, the uncoated C reacted with vanadium oxides simultaneously on the external interphase. Lastly, the V-coated C structure increased the contact area between carbon black and vanadium oxides remarkably and provided a more homogeneous chemical composition than traditional mechanical blending. APV decomposed into V2O5 and V2O5 was reduced and nitrided gradually during the synthesis of VN. The coating structure always existed with the coating layer changing from APV to lower-valent vanadium oxides, vanadium carbide and vanadium nitride, until the precursor converted into VN completely. Hence, the V-coated C structure obtained by adding carbon black to the stripping solution before the precipitation of vanadium significantly reduced the reaction temperature and shortened the reaction time.
3.3. Phase Evolution of Preparing VN from the Precursor
The progress of the phase formation during the preparation of VN was monitored by interrupting the reaction intermittently at different temperatures and then the samples were analyzed by XRD analysis. Figure 9 shows the XRD patterns of the precursor and products at different temperatures.
The precursor was composed of (NH4)2V6O16·1.5H2O and a small amount of NH4Al(SO4)2·12H2O as shown in curve (a). The diffraction peaks were identified as V6O13 and VO2 [32,33,34,35] in curve (b), attributed to the decomposition of (NH4)2V6O16·1.5H2O into V2O5 and NH3; then, the V2O5 was initially reduced to V6O13 and VO2 by NH3 under 550 °C. As shown in curve (c), all peaks corresponded to V4O7, which indicated that the further reduction of V6O13 and VO2 to V4O7 may be by the progressive reduction of V6O13 to VO2 and VO2 to V4O7 below 650 °C. The appearance of V2O3 and VN in curve (d) illustrates that V4O7 was reduced to V2O3 continuously and the VN phase was formed from V2O3 below 950 °C. There was only a single phase of VN at 1150 °C. Based on the above recorded phase changes, the possible mechanism of preparing VN could be formulated as follows:
According to the standard Gibbs free energy changes (ΔGθ (T)) of Reactions (8)–(10), calculated by the “Reaction” module of Factsage 7.1 software (Factsage 7.1, Thermfact/CRCT, Montreal, QC, Canada; GTT-Technologies, Aachen, Germany), the VN phase can be formed at 16,575 °C via V2O3 reacting directly with nitrogen under standard state conditions, while the VC phase can be formed at a lower temperature of about 1092 °C. The transformation of VC to VN was spontaneous below 1257 °C. This means that intermediate formation of VC can occur and subsequently convert to VN when the temperature varies between 1092 °C and 1257 °C under standard state conditions and in an extended range of temperatures in flowing nitrogen atmosphere, indicating that 1150 °C satisfies the thermodynamic conditions. Therefore, the phase evolution of (NH4)2V6O16·1.5H2O to VN took place in the following sequential order:
(NH4)2V6O16·1.5H2O → V2O5 → V6O13 → VO2 → V4O7 → V2O3 → VC → VN
It is worth noting that after being heated at 550 °C for 20 min, the precursor converted completely to low-valent vanadium oxides V6O13 and VO2 without the appearance of residual unreacted V2O5, which has a low melting point (670 °C) and high saturation vapor pressure. V6O13 can decompose to VO2 at 700 °C, and VO2 has a high melting point of 1542 °C [36]. Thus, the pre-reduction at 650 °C for 2.5 h can be eliminated in this process.
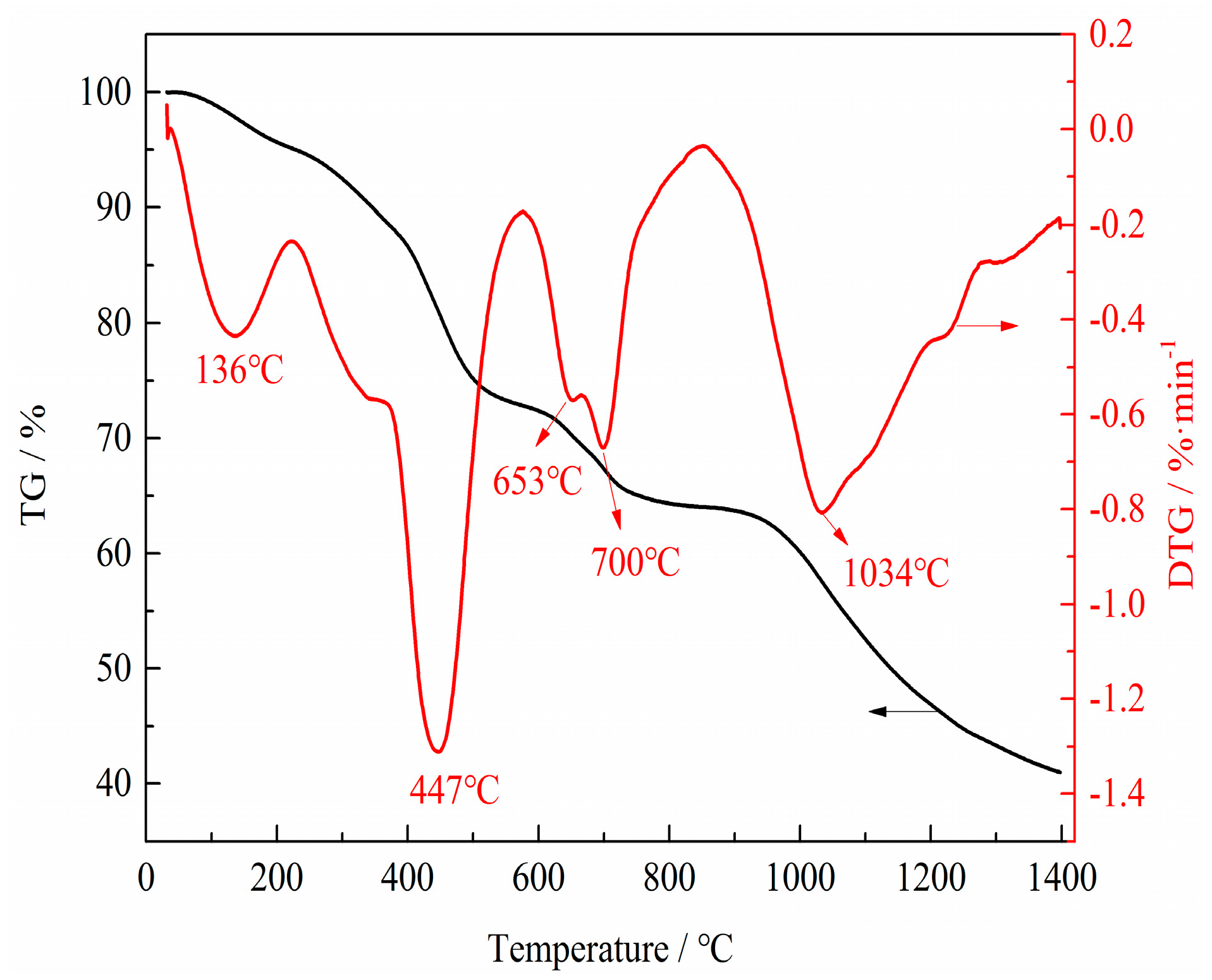
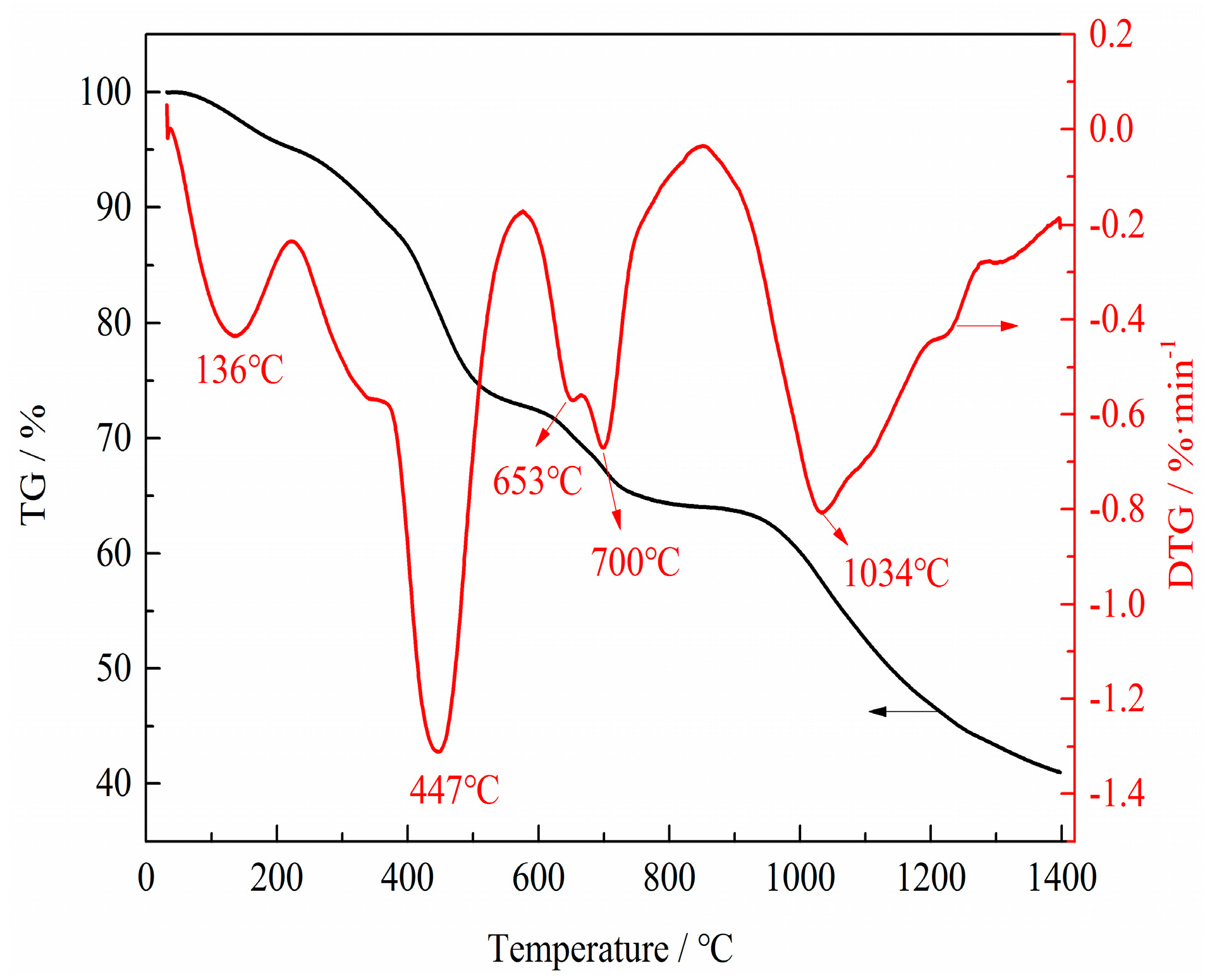
TG experiments were carried out under N2 atmosphere to reflect the physical and chemical phenomena in the preparation process of VN. TG and DTG (differential thermogravimetric) curves of the precursor are shown in Figure 10. There were four major weight losses indicated by four DTG peaks in the DTG curve. The first major weight loss between room temperature and 136 °C was attributed to the evaporation of physically absorbed and crystalline water. The second major weight loss from 380 °C to 447 °C was due to the decomposition of APV and the initial reduction of V2O5 to V6O13 and VO2, which corresponded to the Reactions (1)–(3). The third weight loss corresponded to the further reduction of V6O13 and VO2 to V4O7 according to Reactions (4)–(6). In this stage, the absolute value of weight loss rates first increased, then decreased and eventually increased again at the temperatures of 600 °C–653 °C, 653 °C–660 °C and 660 °C–700 °C, respectively. The absolute value of weight loss rate escalated with increasing temperature because the reduction reaction was endothermic and increasing the temperature promoted the reduction. The absolute value of weight loss rate decreased as the reaction progressed. The increase of the weight loss rate was due to the decomposition of V6O13 to VO2; the maximum, absolute value of the weight loss rate at 700 °C corresponded to the decomposition temperature of V6O13. The last weight loss was assigned to the phase transformation from V4O7 to V2O3 and V2O3 to the desired phase VN, which corresponded to Reactions (7), (9) and (10).
4. Conclusions
A novel synthesis method of a (NH4)2V6O16·1.5H2O precursor to prepare VN was proposed, characterized by adding carbon black to the stripping solution during the vanadium recovery from black shale. VN with an N content of 16.38% was successfully prepared by thermal processing of the precursor at 1150 °C for 1 h with a C/V2O5 mass ratio of 0.30 and a N2 flow rate of 300 mL·min−1. Low synthesis temperature and short period for the preparation of VN was due to the V-coated C structure of the precursor, which enlarged the contact area between reactants significantly and provided a more homogeneous chemical composition. In addition, the simultaneous direct reduction and indirect reduction at the interphase caused by the coating structure evidently accelerated the reaction. XRD patterns and thermodynamic analysis indicated that the phase evolution from the precursor to VN took place in the following order (NH4)2V6O16·1.5H2O → V2O5 → V6O13 → VO2 → V4O7 → V2O3 → VC → VN. The precursor converted to V6O13 and VO2 completely after being calcined at 550 °C, indicating that the pre-reduction of V2O5 in the traditional CRN method can be dispensed with. Therefore, this method is an effective, simple and low-cost route to prepare VN.
Acknowledgments
This research was funded by the National Natural Science Foundation of China (No. 51404174 and No. 51474162) and the Project in the National Science & Technology Pillar Program of China (No. 2015BAB18B01).
Author Contributions
Jingli Han and Yimin Zhang conceived and designed the experiments; Jingli Han performed the experiments and analyzed the data; Yimin Zhang, Tao Liu and Jing Huang contributed reagents, materials and analysis tools; Nannan Xue and Pengcheng Hu provided valuable scientific advice for the study; Jingli Han wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Huang, J.; Peng, H.; Xia, G. Microwave synthesis of vanadium nitride for industrial applications. Ironmak. Steelmak. 2009, 36, 110–114. [Google Scholar] [CrossRef]
- Yu, S.; Li, W.; Ji, Z.; Fu, N.; Sui, Z. Effect of technical parameters on preparing for vanadium nitride. Adv. Mater. Res. 2010, 150–151, 480–483. [Google Scholar] [CrossRef]
- Morel, A.; Piron, Y.B.; Porto, R.L.; Brousse, T.; Belanger, D. Suitable conditions for the use of vanadium nitride as an electrode for electrochemical capacitor. J. Electrochem. Soc. 2016, 163, A1077–A1082. [Google Scholar] [CrossRef]
- Zhou, X.; Chen, H.; Shu, D.; He, C.; Nan, J. Study on the electrochemical behavior of vanadium nitride as a promising supercapacitor material. J. Phys. Chem. Solids 2009, 70, 495–500. [Google Scholar] [CrossRef]
- Wu, M.; Guo, H.; Lin, Y.; Wu, K.; Ma, T.; Hagfeldt, A. Synthesis of highly effective vanadium nitride (VN) peas as a counter electrode catalyst in dye-sensitized solar cells. J. Phys. Chem. C 2014, 118, 12625–12631. [Google Scholar] [CrossRef]
- Krawiec, P.; Cola, P.L.D.; Gläser, R.; Weitkamp, J.; Weidenthaler, C.; Kaskel, S. Oxide foams for the synthesis of high-surface-area vanadium nitride catalysts. Adv. Mater. 2006, 18, 506–508. [Google Scholar] [CrossRef]
- Gajbhiye, N.S.; Ningthoujam, R.S. Low temperature synthesis, crystal structure and thermal stability studies of nanocrystalline VN particles. Mater. Res. Bull. 2006, 41, 1612–1621. [Google Scholar] [CrossRef]
- Smolik, J.; Mazurkiewicz, A.; Słomka, Z.; Bujak, J.; Gołacka, J.K.; Garbacz, H.; Wieciński, P. Nanomultilayer coatings based on vanadium nitride. Solid State Phenom. 2015, 237, 15–20. [Google Scholar] [CrossRef]
- Caicedo, J.C.; Zambrano, G.; Aperador, W.; Escobar-Alarcon, L.; Camps, E. Mechanical and electrochemical characterization of vanadium nitride (VN) thin films. Appl. Surf. Sci. 2011, 258, 312–320. [Google Scholar] [CrossRef]
- Duan, X.; Srinivasakannan, C.; Zhang, H.; Zhang, Y. Process optimization of the preparation of vanadium nitride from vanadium pentoxide. Arab. J. Sci. Eng. 2015, 40, 2133–2139. [Google Scholar]
- Tripathy, P.K.; Sehra, J.C.; Kulkarni, A.V. On the carbonitrothermic reduction of vanadium pentoxide. J. Mater. Chem. 2001, 11, 691–695. [Google Scholar] [CrossRef]
- Pan, H.; Zhang, Z.; Peng, J.; Zhang, L.; Li, W. Densification of vanadium nitride by microwave-assisted carbothermal nitridation. Adv. Mater. Res. 2011, 201–203, 1787–1792. [Google Scholar] [CrossRef]
- Chen, Z.; Xue, Z.; Wang, W.; Yu, Y.; Liu, Q.; Li, P. One-step method of carbon thermal reduction and nitride to produce vanadium nitrogen alloy. Adv. Mater. Res. 2012, 476–478, 194–198. [Google Scholar] [CrossRef]
- Cai, P.; Yang, Z.; Wang, C.; Xia, P.; Qian, Y. Synthesis of nanocrystalline VN via thermal liquid-solid reaction. Mater. Lett. 2006, 60, 410–413. [Google Scholar] [CrossRef]
- Yeh, C.L.; Chen, Y.D. Combustion synthesis of vanadium carbonitride from V-C powder compacts under nitrogen pressure. Ceram. Int. 2007, 33, 365–371. [Google Scholar] [CrossRef]
- Cheng, F.; He, C.; Shu, D.; Chen, H.; Zhang, J.; Tang, S.; Finlow, D.E. Preparation of nanocrystalline VN by the melamine reduction of V2O5 xerogel and its supercapacitive behavior. Mater. Chem. Phys. 2011, 131, 268–273. [Google Scholar] [CrossRef]
- Azargohar, R.; Dalai, A.K. Production of activated carbon from luscar char: Experimental and modeling studies. Microporous Mesoporous Mater. 2005, 85, 219–225. [Google Scholar] [CrossRef]
- Zhang, B.; Li, Z. Synthesis of vanadium carbide by mechanical alloying. J. Alloys Compd. 2005, 392, 183–186. [Google Scholar] [CrossRef]
- Yao, W.; Makowski, P.; Giordano, C.; Goettmann, F. Synthesis of early-transition-metal carbide and nitride nanoparticles through the urea route and their use as alkylation catalysts. Chem. Eur. J. 2009, 15, 11999–12004. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Gu, Y.; Shi, L.; Yang, Z.; Ma, J.; Qian, Y. A room-temperature synthesis of nanocrystalline vanadium nitride. J. Eur. Ceram. Soc. 2010, 30, 2099–2107. [Google Scholar] [CrossRef]
- Zhao, Z.; Liu, Y.; Cao, H.; Ye, J.; Gao, S.; Tu, M. Synthesis of VN nanopowders by thermal nitridation of the precursor and their characterization. J. Alloys Compd. 2008, 464, 75–80. [Google Scholar] [CrossRef]
- Liu, A.; Liu, Y.; Ma, S.; Qiu, Y.; Rong, P.; Ye, J. Synthesis of (Cr,V)2(C,N) solid solution powders by thermal processing precursors. Mater. Chem. Phys. 2017, 193, 196–202. [Google Scholar] [CrossRef]
- Cai, Z.; Zhang, Y.; Liu, T.; Huang, J. Mechanisms of vanadium recovery from stone coal by novel BaCO3/BaO composite additive roasting and acid leaching technology. Minerals 2016, 6, 26. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, K.; Tian, X.; Qin, W. Vanadium leaching from carbonaceous shale using fluosilicic acid. Int. J. Miner. Process. 2011, 100, 184–187. [Google Scholar] [CrossRef]
- Zhang, Y.; Bao, S.; Liu, T.; Huang, J. The technology of extracting vanadium from stone coal in China: History, current status and future prospects. Hydrometallurgy 2011, 109, 116–124. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, C.; Li, P.; Li, S. A new process of extracting vanadium from stone coal. Int. J. Miner. Metall. Mater. 2010, 17, 381–388. [Google Scholar] [CrossRef]
- Nguyen, T.; Lee, M. Solvent extraction of vanadium (V) from sulfate solutions using LIX 63 and PC 88A. J. Ind. Eng. Chem. 2015, 31, 118–123. [Google Scholar] [CrossRef]
- Li, X.; Wei, C.; Deng, Z.; Li, M.; Li, C.; Fan, G. Selective solvent extraction of vanadium over iron from a stone coal/black shale acid leach solution by D2EHPA/TBP. Hydrometallurgy 2011, 105, 359–363. [Google Scholar] [CrossRef]
- Li, X.; Wei, C.; Deng, Z.; Li, C.; Fan, G.; Li, M.; Huang, H. Recovery of vanadium from H2SO4-HF acidic leaching solution of black shale by solvent extraction and precipitation. Metals 2016, 6, 63. [Google Scholar] [CrossRef]
- Gu, H.M.; Shih, W.Y.; Shih, W.H. Single-calcination synthesis of pyrochlore-free 0.9Pb(Mg1/3Nb2/3)O3–0.1PbTiO3 and Pb(Mg1/3Nb2/3)O3 Ceramics Using a Coating Method. J. Am. Ceram. Soc. 2003, 86, 217–221. [Google Scholar] [CrossRef]
- Ivashchenko, V.I.; Turhci, P.E.A. Phonon softening and the phase transition in VN. Phys. Rev. B 2008, 78, 224113. [Google Scholar] [CrossRef]
- Vernardou, D.; Apostolopoulou, M.; Louloudakis, D.; Spanakis, E.; Katsarakis, N.; Koudoumas, E.; McGrath, J.; Pemble, M.E. Electrochemical properties of opal-V6O13 composites. J. Alloys Compd. 2014, 586, 621–626. [Google Scholar] [CrossRef]
- Vernardou, D.; Pemble, M.E.; Sheel, D.W. In-situ FTIR studies of the growth of vanadium dioxide coatings on glass by atmospheric pressure chemical vapour deposition for VCl4 and H2O system. Thin Solid Films 2007, 515, 8768–8770. [Google Scholar] [CrossRef]
- Vernardou, D.; Louloudakis, D.; Spanakis, E.; Katsarakis, N.; Koudoumas, E. Functional properties of APCVD VO2 layers. Int. J. Thin Films Sci. Technol. 2015, 4, 187–191. [Google Scholar]
- Vernardou, D.; Bei, A.; Louloudakis, D.; Katsarakis, N.; Koudoumas, E. Oxygen source-oriented control of atmospheric pressure chemical vapor deposition of VO2 for capacitive applications. J. Electrochem. Sci. Eng. 2016, 6, 165–173. [Google Scholar]
- Bennett, L.H.; Massalski, T.B.; Giessen, B.C. Alloy Phase Diagrams. Mater. Res. Soc. Symp. Proc. 1983, 19. [Google Scholar]
Figure 1.
Process flow sheet for preparation of vanadium nitride (VN).
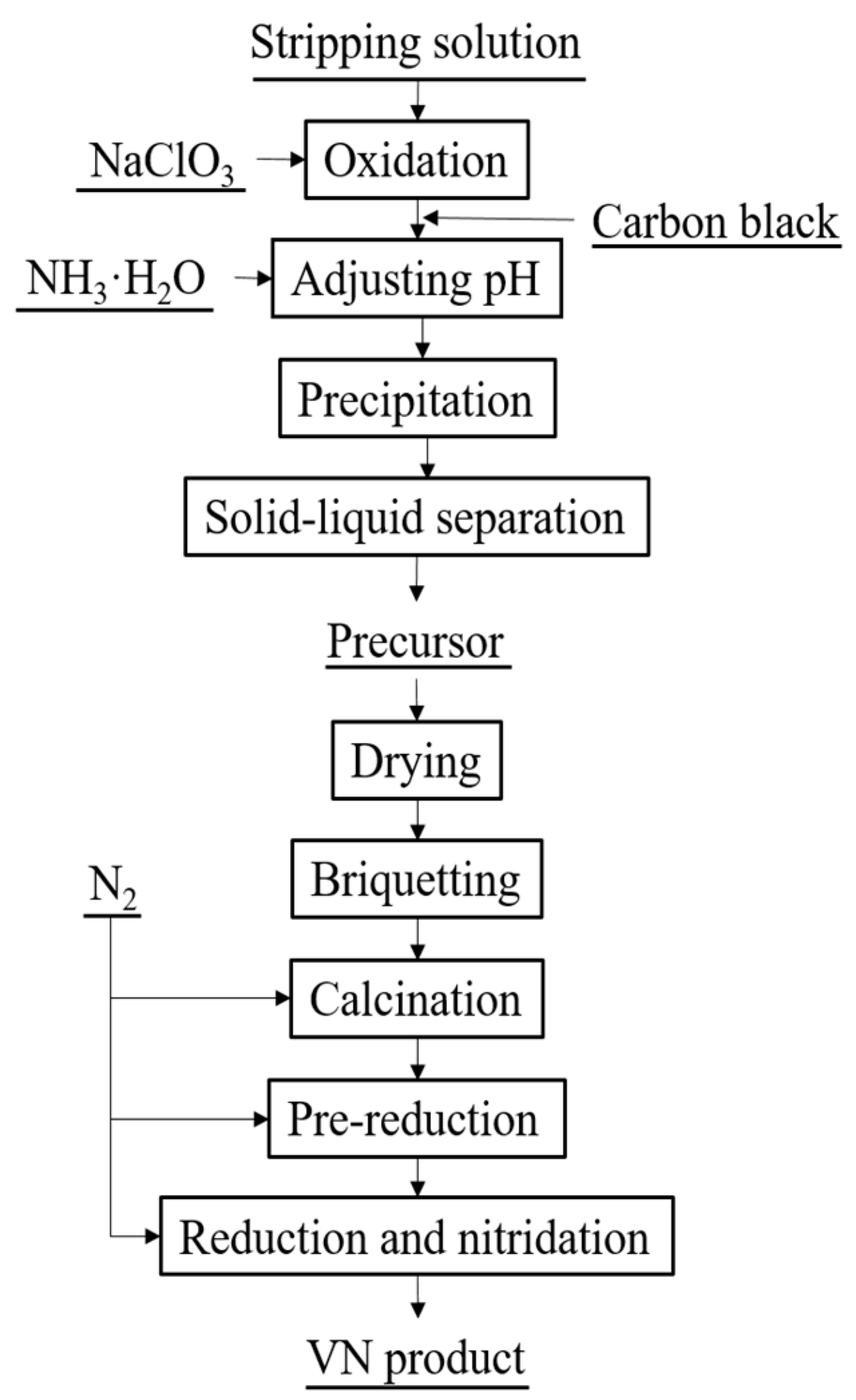
Figure 2.
(a) Effect of C/V2O5 mass ratio on the N content in VN product with a reaction temperature of 1250 °C, reaction time of 3 h and N2 flow rate of 300 mL·min−1; (b) XRD (X-ray diffraction) patterns of the products obtained at different C/V2O5 mass ratios with a reaction temperature of 1250 °C, reaction time of 3 h and N2 flow rate of 300 mL·min−1.
Figure 2.
(a) Effect of C/V2O5 mass ratio on the N content in VN product with a reaction temperature of 1250 °C, reaction time of 3 h and N2 flow rate of 300 mL·min−1; (b) XRD (X-ray diffraction) patterns of the products obtained at different C/V2O5 mass ratios with a reaction temperature of 1250 °C, reaction time of 3 h and N2 flow rate of 300 mL·min−1.
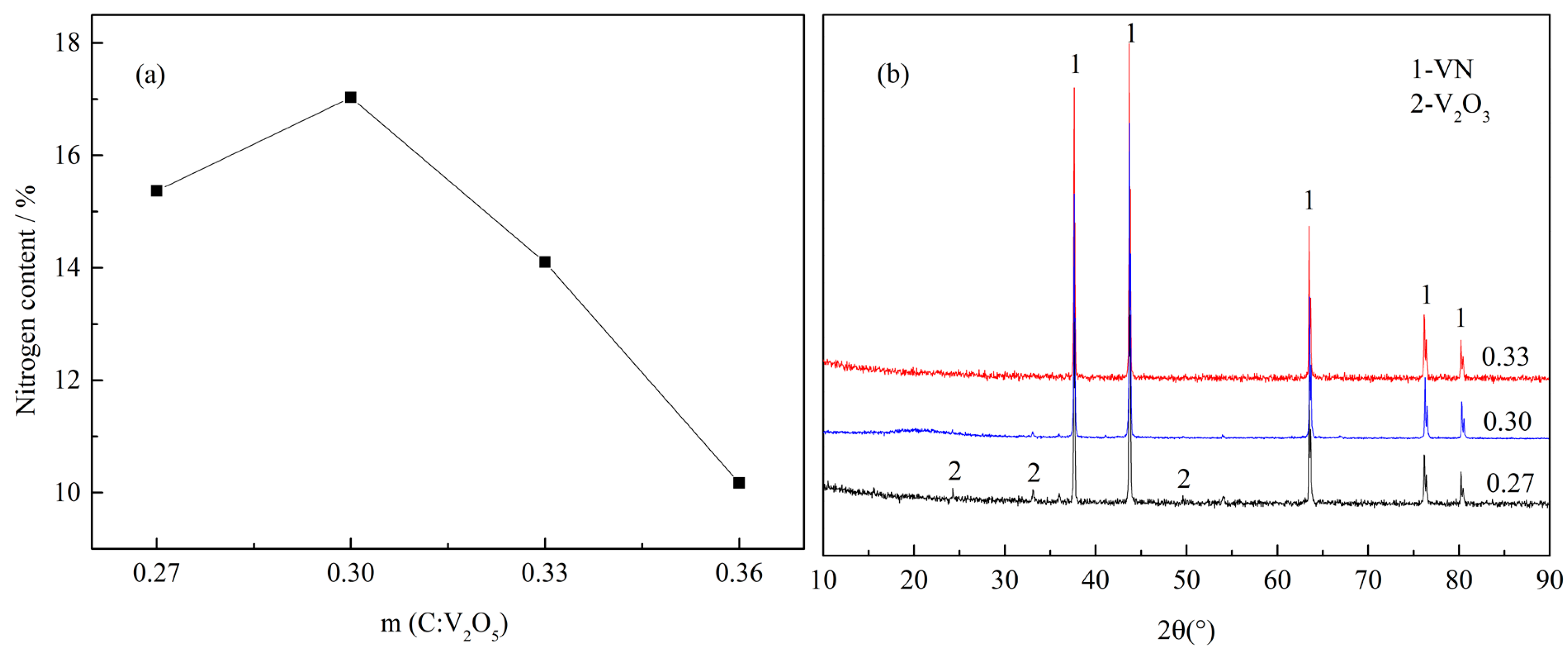
Figure 3.
(a) Effect of reaction temperature on the N content in VN product with a C/V2O5 mass ratio of 0.30, reaction time of 3 h and N2 flow rate of 300 mL·min−1; (b) XRD patterns of the products obtained at different temperatures with a C/V2O5 mass ratio of 0.30, reaction time of 3 h and N2 flow rate of 300 mL·min−1.
Figure 3.
(a) Effect of reaction temperature on the N content in VN product with a C/V2O5 mass ratio of 0.30, reaction time of 3 h and N2 flow rate of 300 mL·min−1; (b) XRD patterns of the products obtained at different temperatures with a C/V2O5 mass ratio of 0.30, reaction time of 3 h and N2 flow rate of 300 mL·min−1.
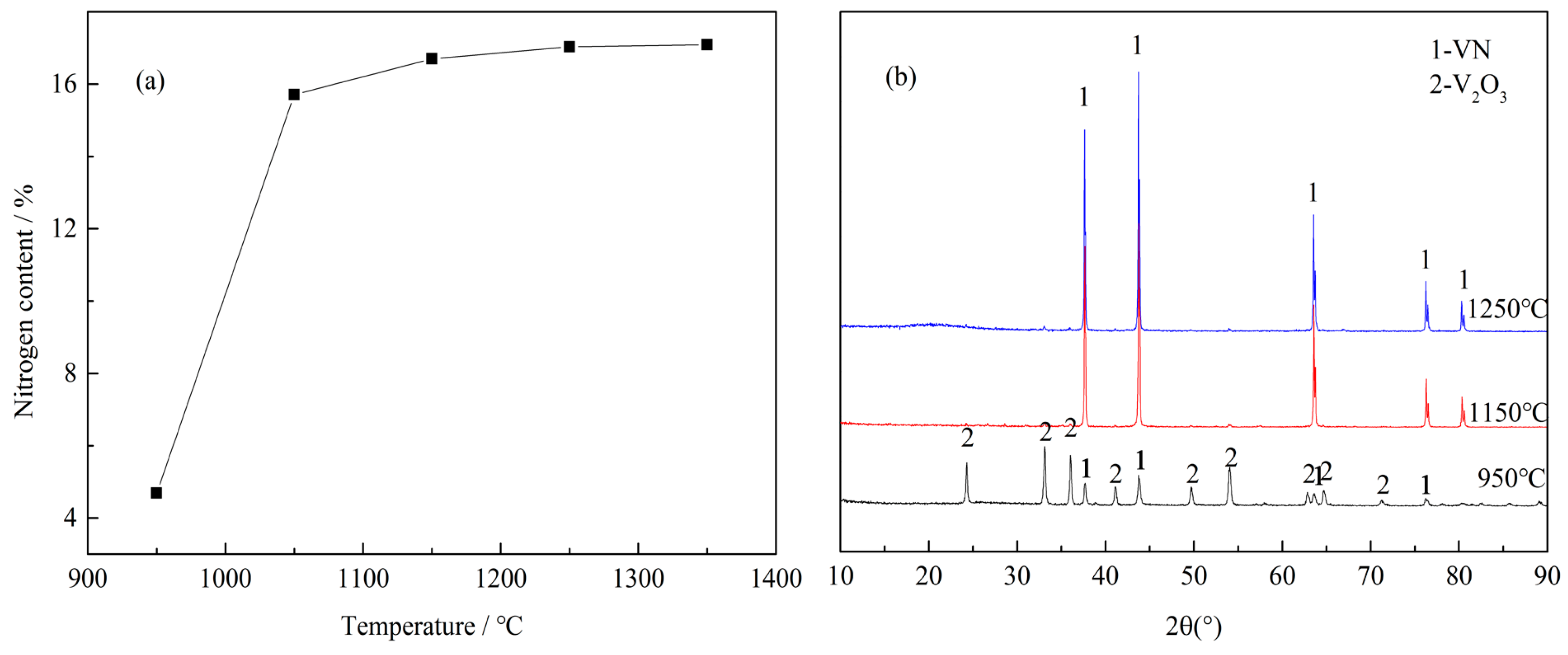
Figure 4.
Effect of reaction time on the N content in VN product with a C/V2O5 mass ratio of 0.30, reaction temperature of 1150 °C and N2 flow rate of 300 mL·min−1.
Figure 4.
Effect of reaction time on the N content in VN product with a C/V2O5 mass ratio of 0.30, reaction temperature of 1150 °C and N2 flow rate of 300 mL·min−1.
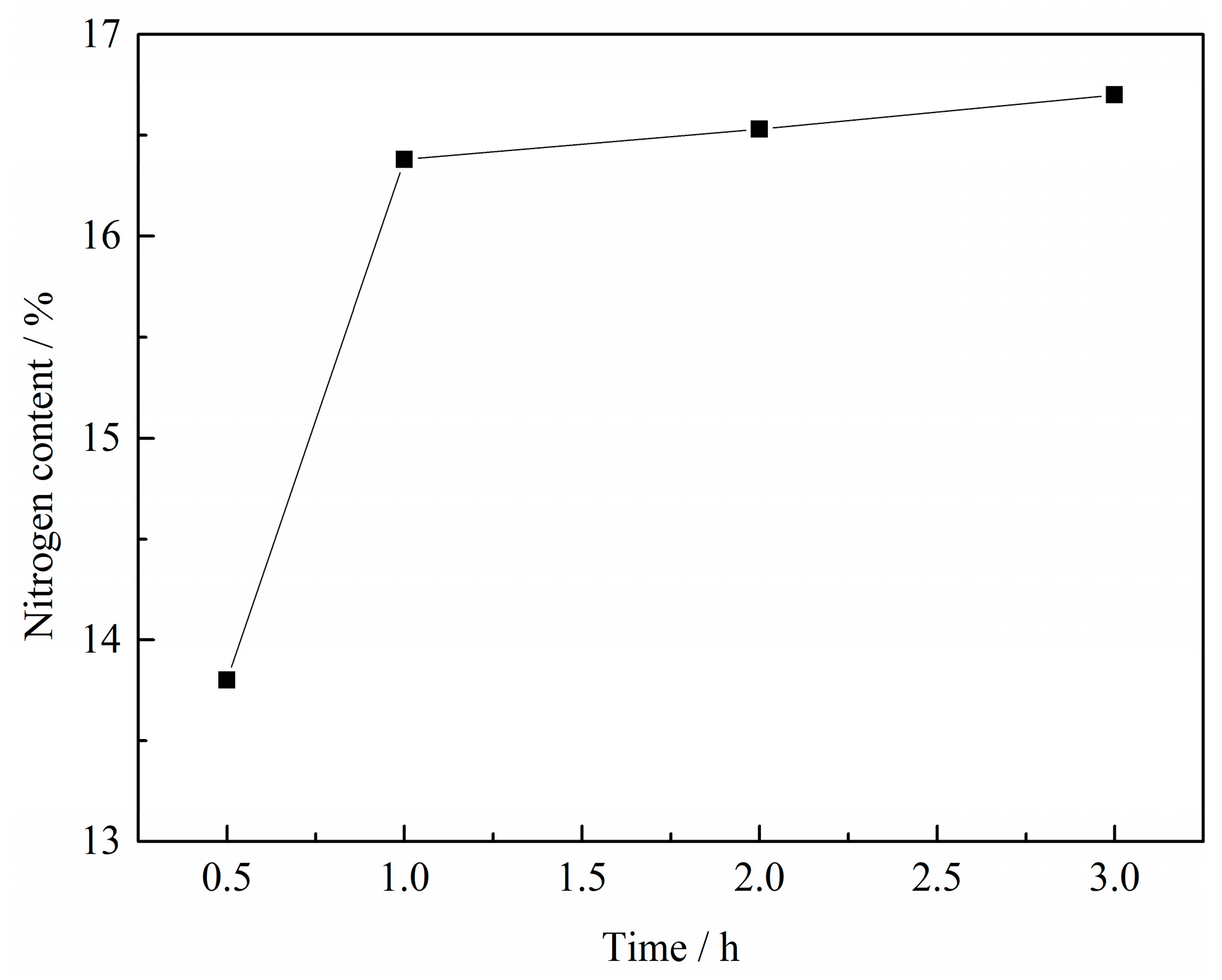
Figure 5.
Effect of N2 flow rate on the N content in VN product with a C/V2O5 mass ratio of 0.30, reaction temperature of 1150 °C and reaction time of 1 h.
Figure 5.
Effect of N2 flow rate on the N content in VN product with a C/V2O5 mass ratio of 0.30, reaction temperature of 1150 °C and reaction time of 1 h.
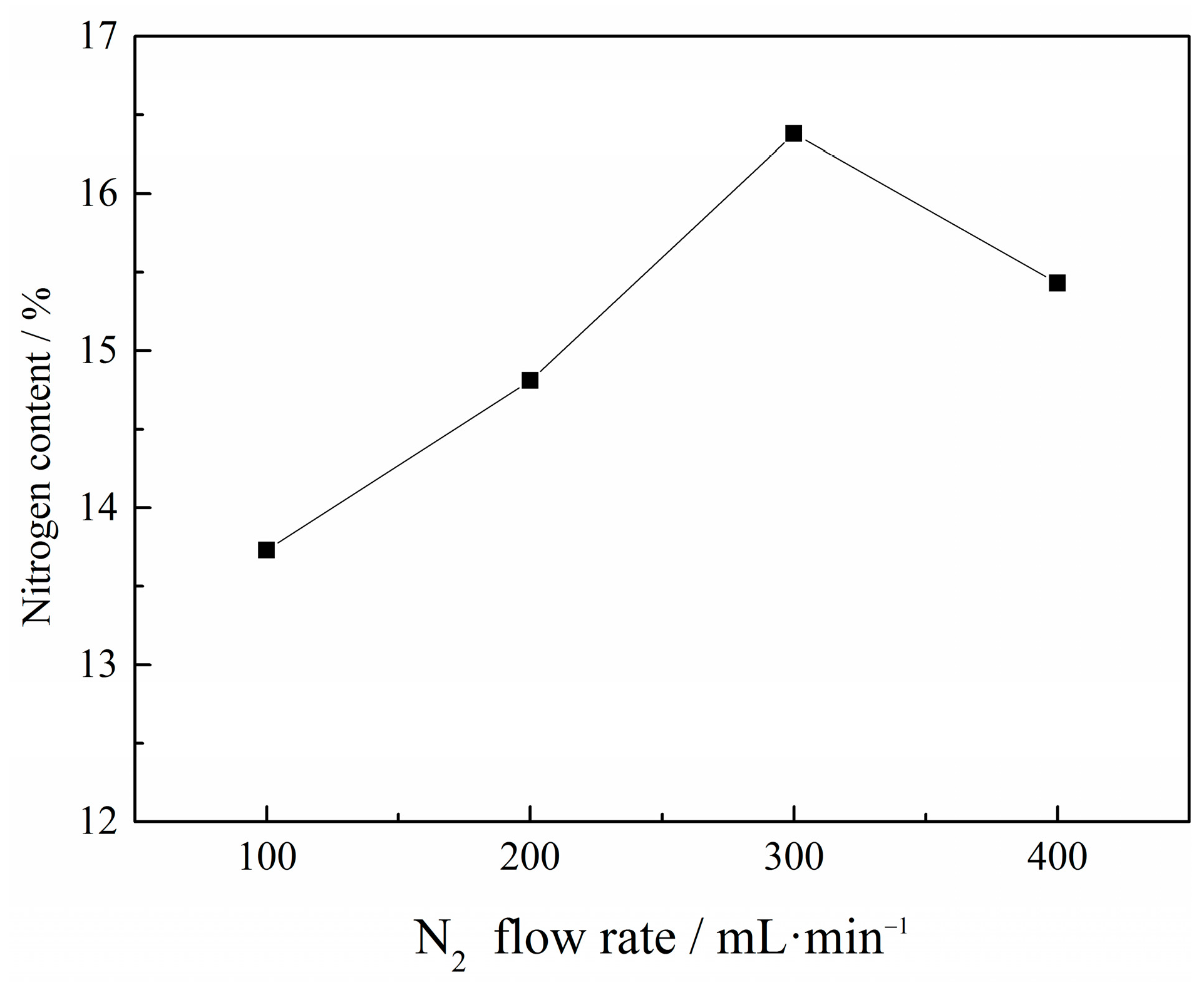
Figure 6.
The XRD pattern of the precursor.
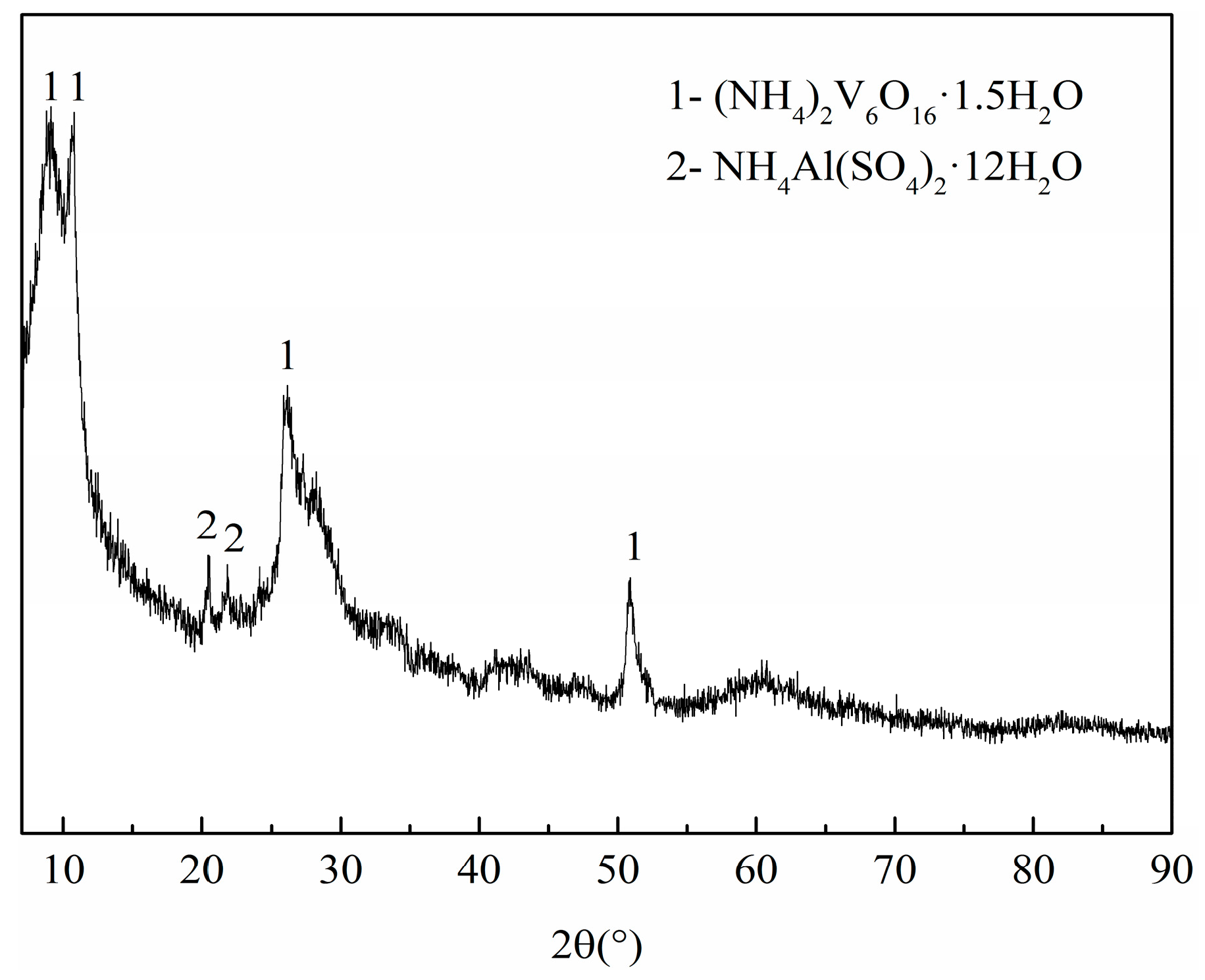
Figure 7.
SEM (scanning electron microscope) images with EDS (energy-dispersive X-ray spectroscope) element mapping of: (a) The precursor; (b) the partial enlarged view of the edge of the particle; (c) SEM image of the overall distribution of the precursor.
Figure 7.
SEM (scanning electron microscope) images with EDS (energy-dispersive X-ray spectroscope) element mapping of: (a) The precursor; (b) the partial enlarged view of the edge of the particle; (c) SEM image of the overall distribution of the precursor.
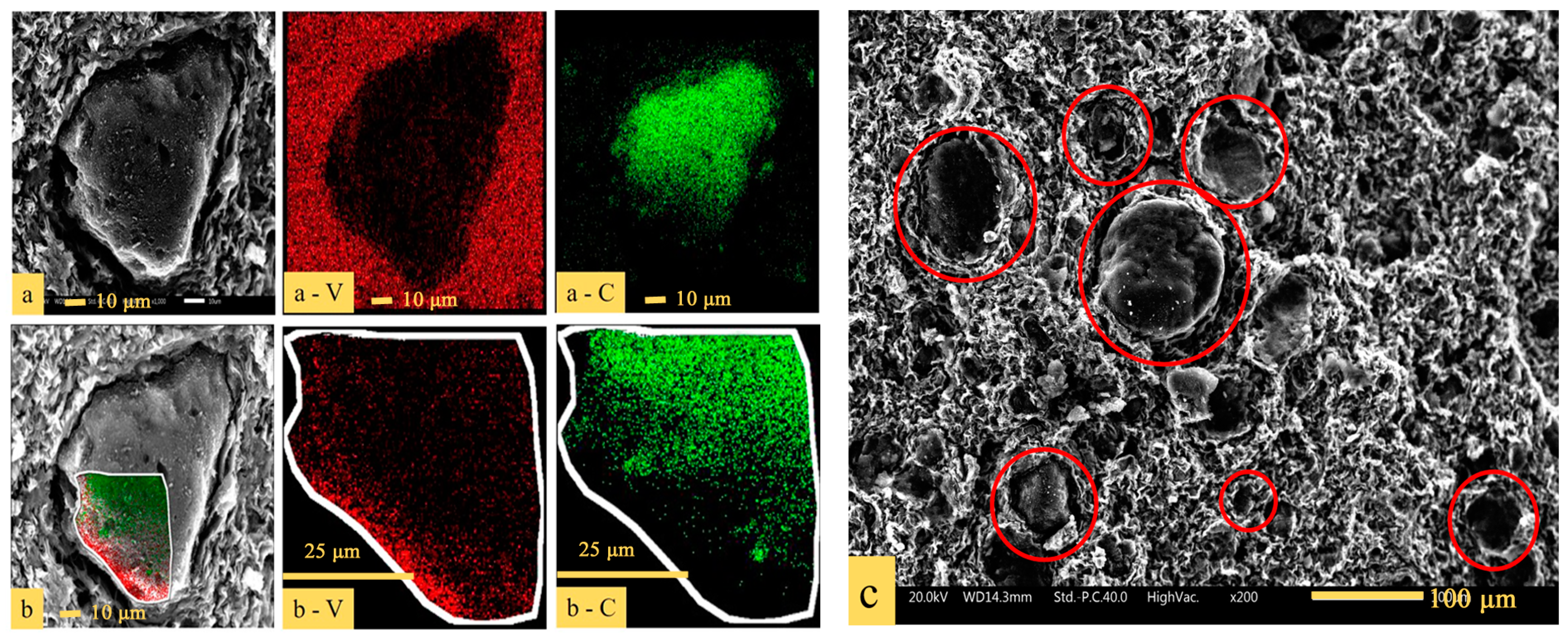
Figure 8.
Formation process sketch of the precursor. (APV: Ammonium polyvanadate.)
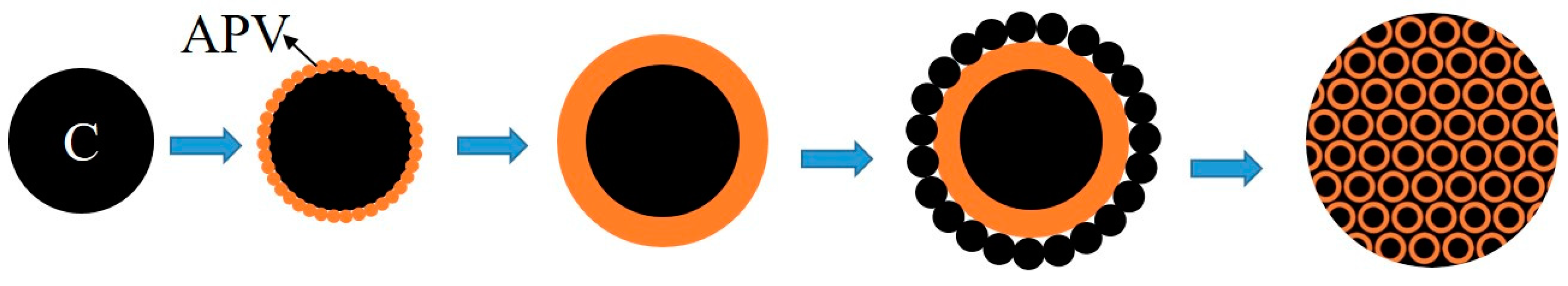
Figure 9.
XRD patterns of the precursor and products at different temperatures with a C/V2O5 mass ratio of 0.30: (a) The precursor; (b) 550 °C; (c) 650 °C; (d) 950 °C; (e) 1150 °C.
Figure 9.
XRD patterns of the precursor and products at different temperatures with a C/V2O5 mass ratio of 0.30: (a) The precursor; (b) 550 °C; (c) 650 °C; (d) 950 °C; (e) 1150 °C.
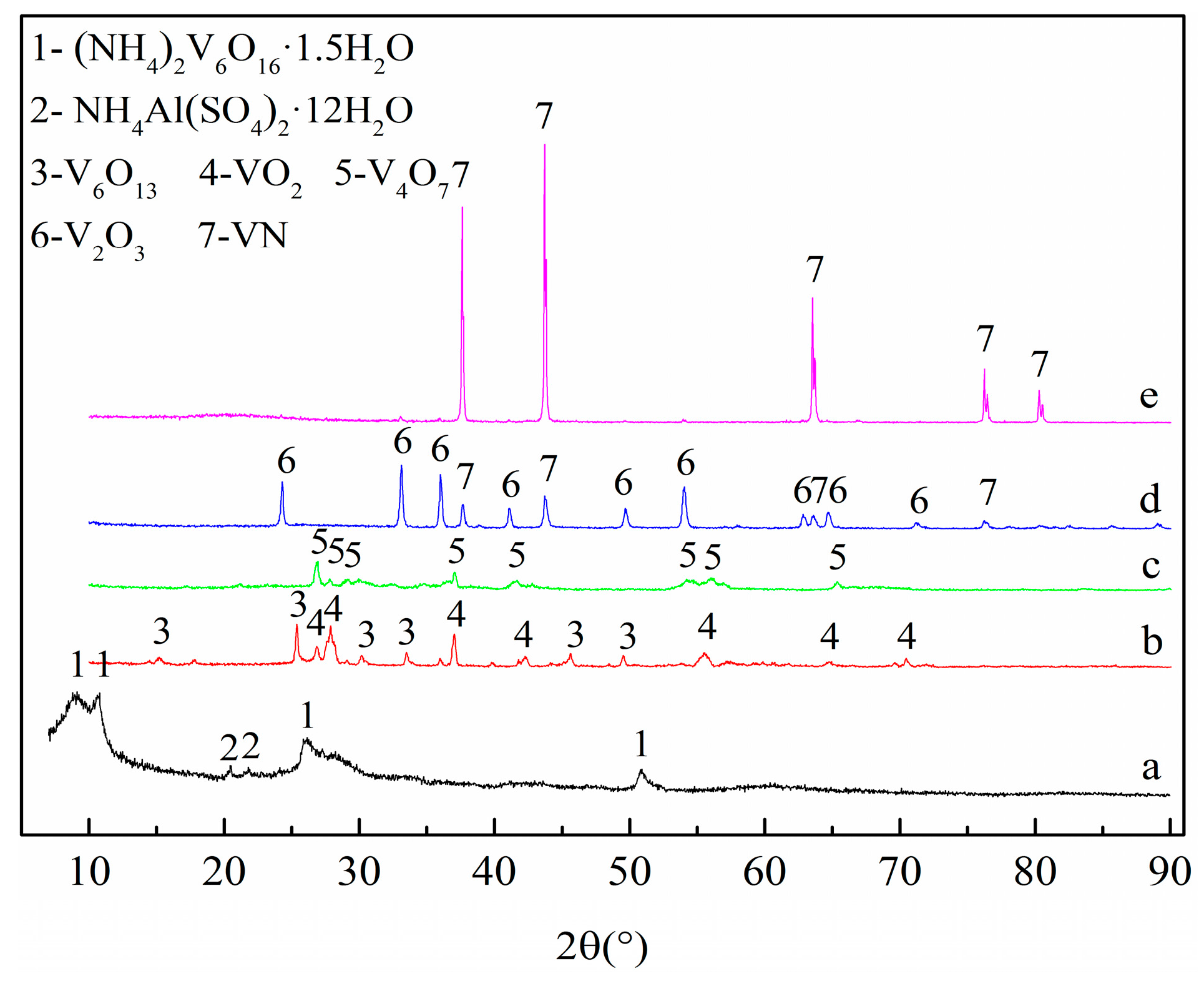
Figure 10.
TG (thermogravimetric) and DTG (differential thermogravimetric) curves of the precursor from 30 °C to 1400 °C with a linear heating rate of 10 °C·min−1 and N2 flow rate of 50 mL·min−1.
Figure 10.
TG (thermogravimetric) and DTG (differential thermogravimetric) curves of the precursor from 30 °C to 1400 °C with a linear heating rate of 10 °C·min−1 and N2 flow rate of 50 mL·min−1.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Main chemical composition of the stripping solution.
| Element | V | Al | Fe | K | Na | P |
|---|---|---|---|---|---|---|
| Concentration (g·L−1) | 20.63 | 5.78 | 0.06 | 0.34 | 0.42 | 0.1 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Han, J.; Zhang, Y.; Liu, T.; Huang, J.; Xue, N.; Hu, P. Preparation of Vanadium Nitride Using a Thermally Processed Precursor with Coating Structure. Metals 2017, 7, 360. https://doi.org/10.3390/met7090360
AMA Style
Han J, Zhang Y, Liu T, Huang J, Xue N, Hu P. Preparation of Vanadium Nitride Using a Thermally Processed Precursor with Coating Structure. Metals. 2017; 7(9):360. https://doi.org/10.3390/met7090360
Chicago/Turabian StyleHan, Jingli, Yimin Zhang, Tao Liu, Jing Huang, Nannan Xue, and Pengcheng Hu. 2017. "Preparation of Vanadium Nitride Using a Thermally Processed Precursor with Coating Structure" Metals 7, no. 9: 360. https://doi.org/10.3390/met7090360
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.