
Synthesis, Characterization, and Toxicity Evaluation of Dextran-Coated Iron Oxide Nanoparticles



 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Dextran-Coated Iron Oxide Nanoparticles
2.3. Characterization
2.4. Preparation of Water-Nanoparticle Suspension
2.5. Hydrodynamic Size and Zeta Potential
2.6. Measurement of Relaxivity (r1 and r2)
2.7. Cell Line and Treatment
2.8. Cell Viability Assay
2.9. Membrane Integrity Evaluation
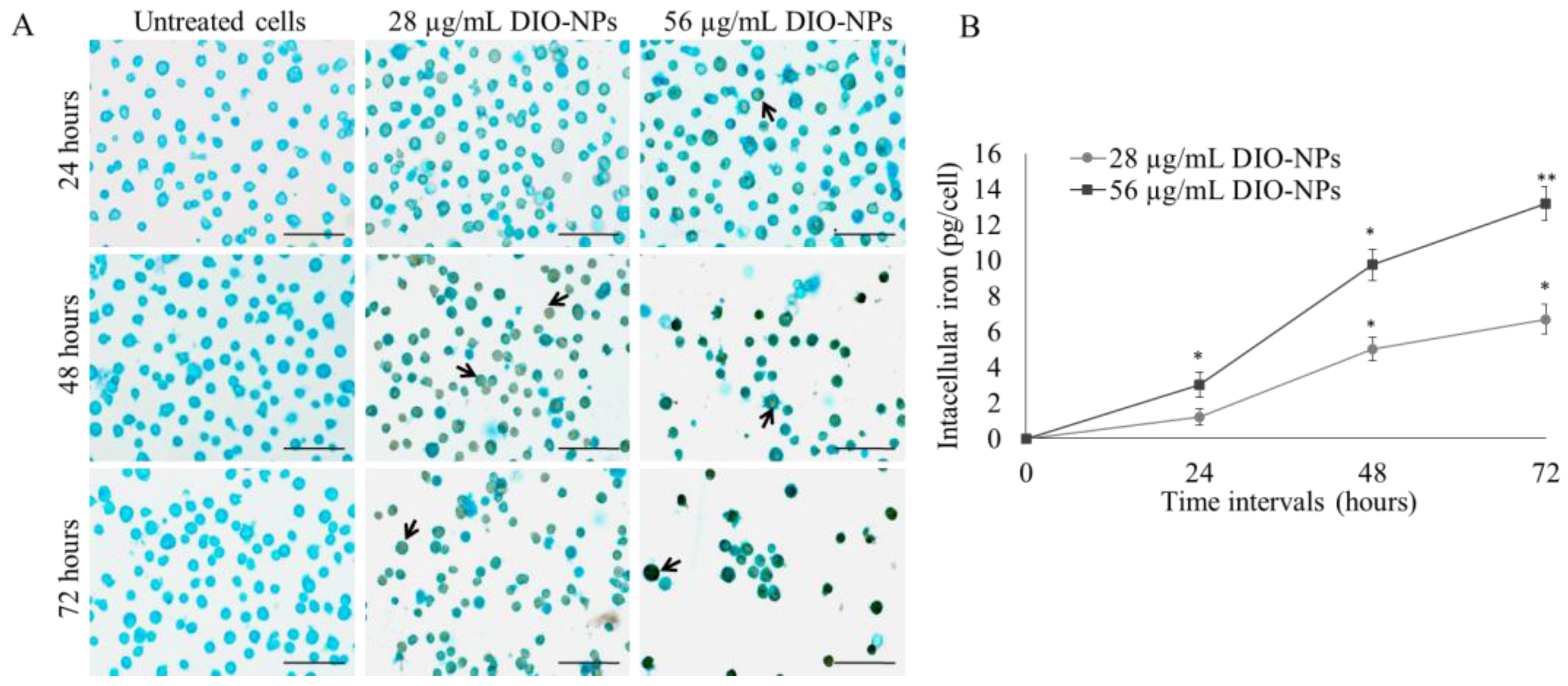
2.10. Intracellular Iron Quantification
2.10.1. DAB-Enhanced Prussian Blue Staining
2.10.2. Relaxometry Technique
2.11. Lysosome Labeling
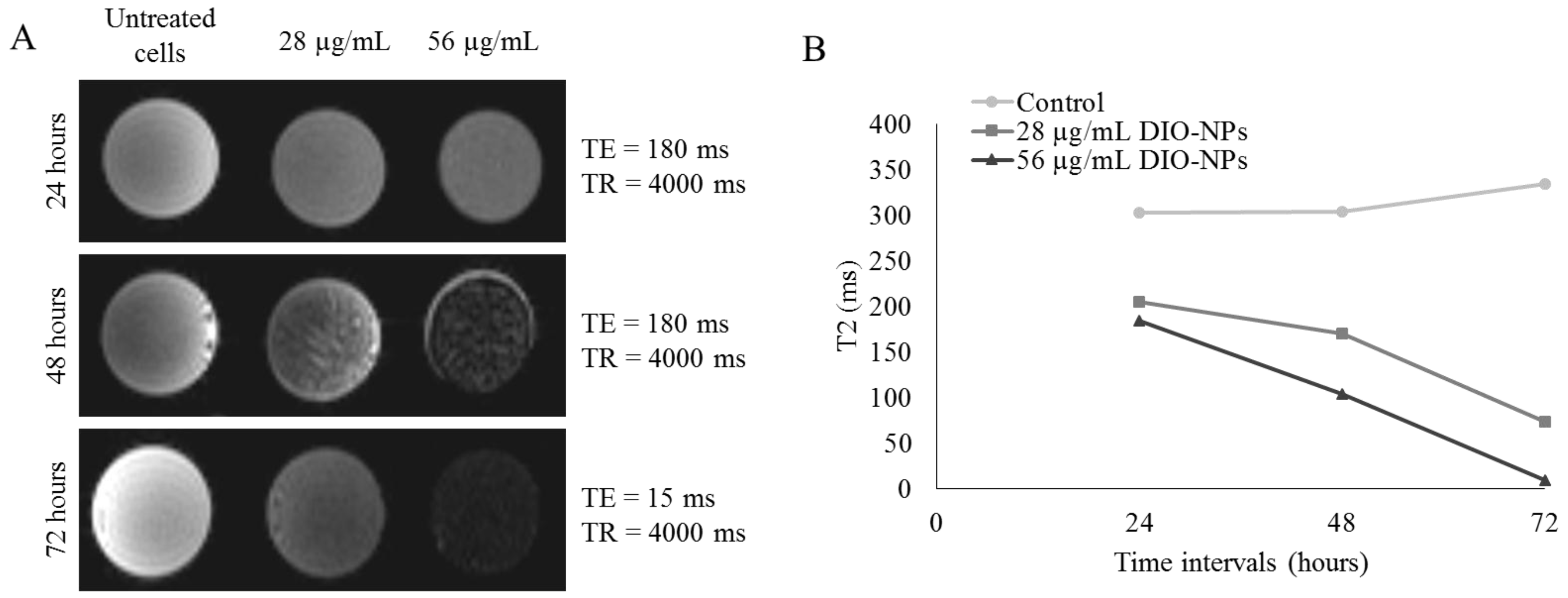
2.12. MRI Analysis
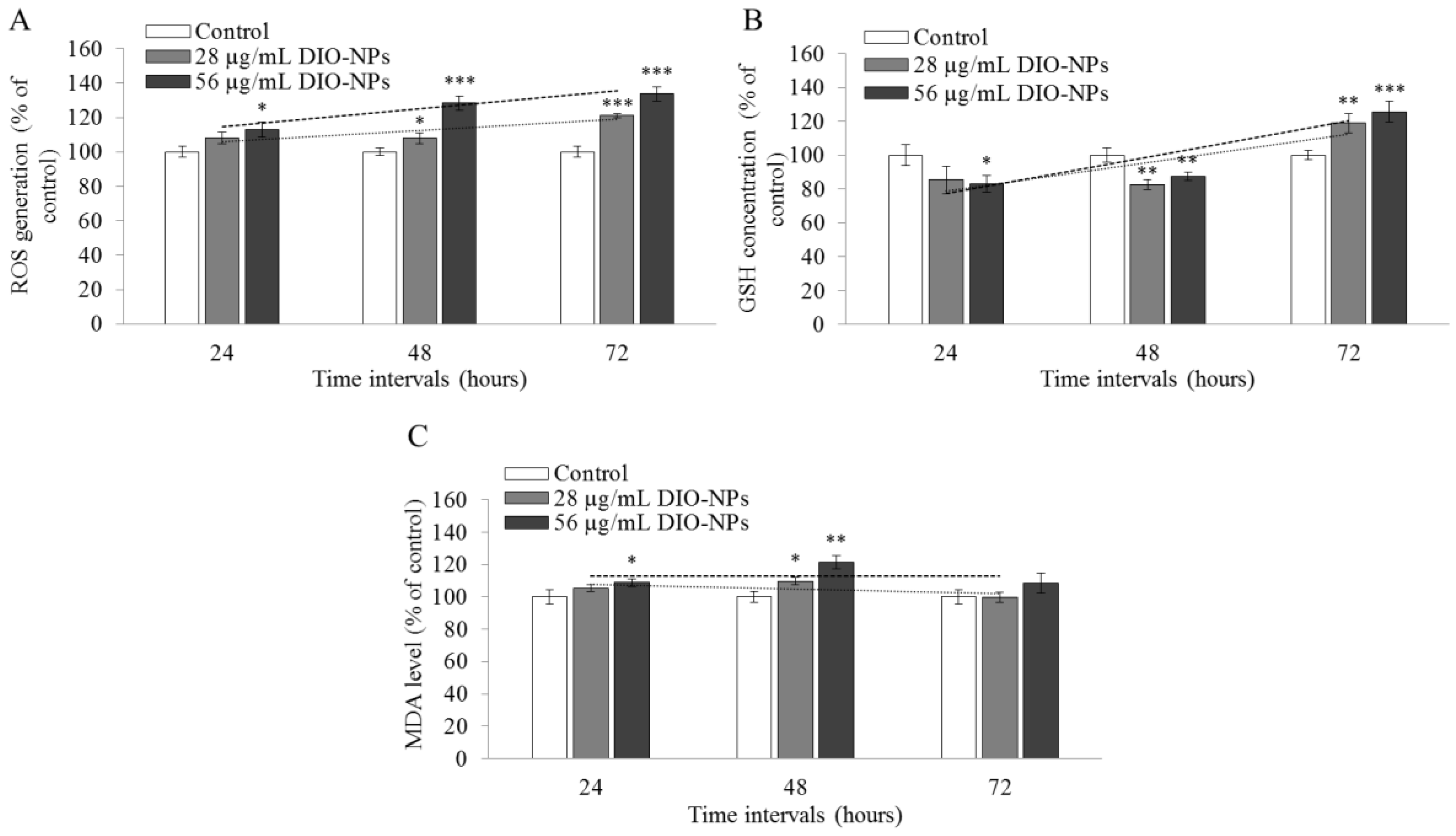
2.13. Measurement of ROS Content
2.14. Measurement of GSH Content
2.15. Measurement of Lipid Peroxidation Level
2.16. Data Analysis
3. Results
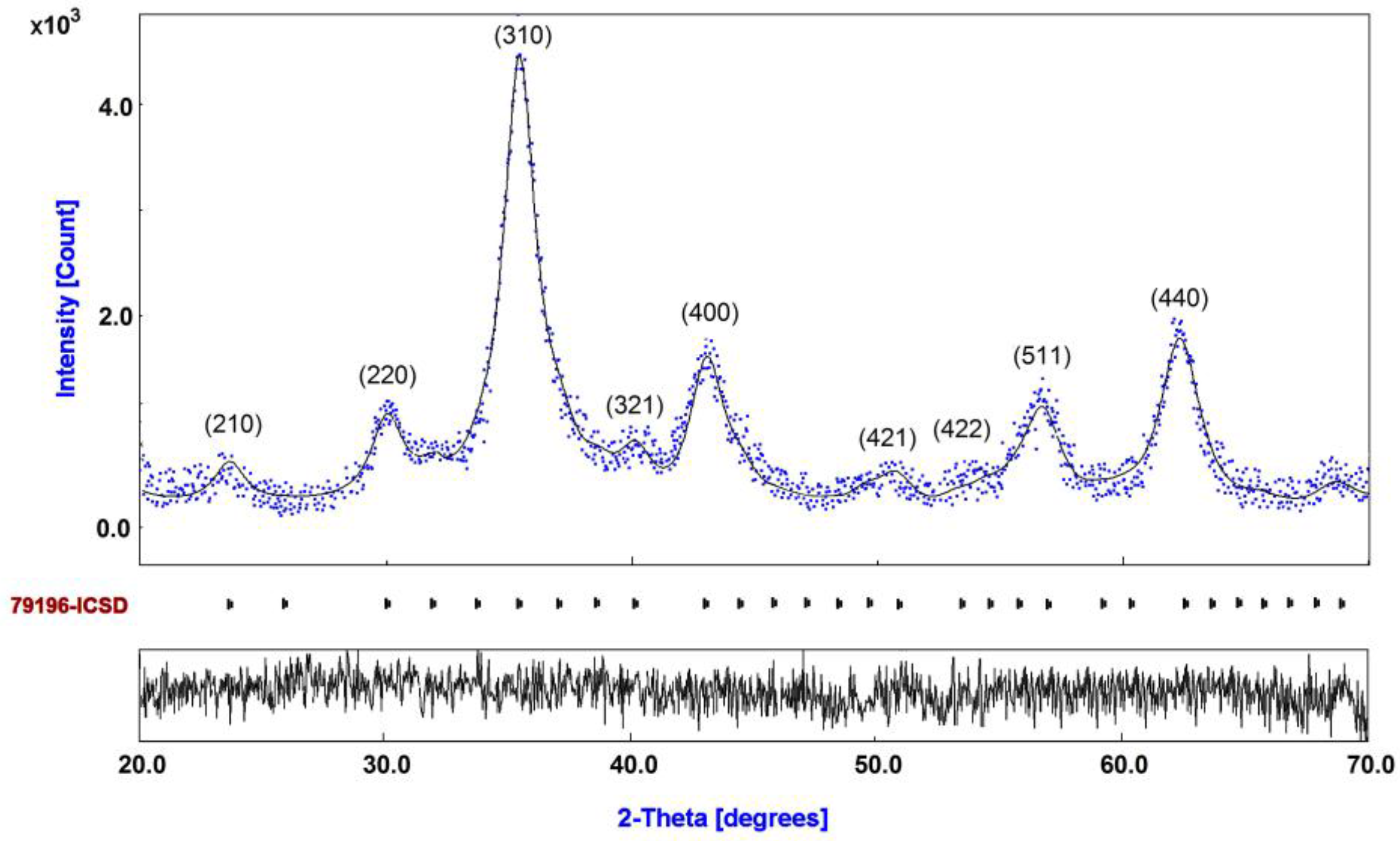
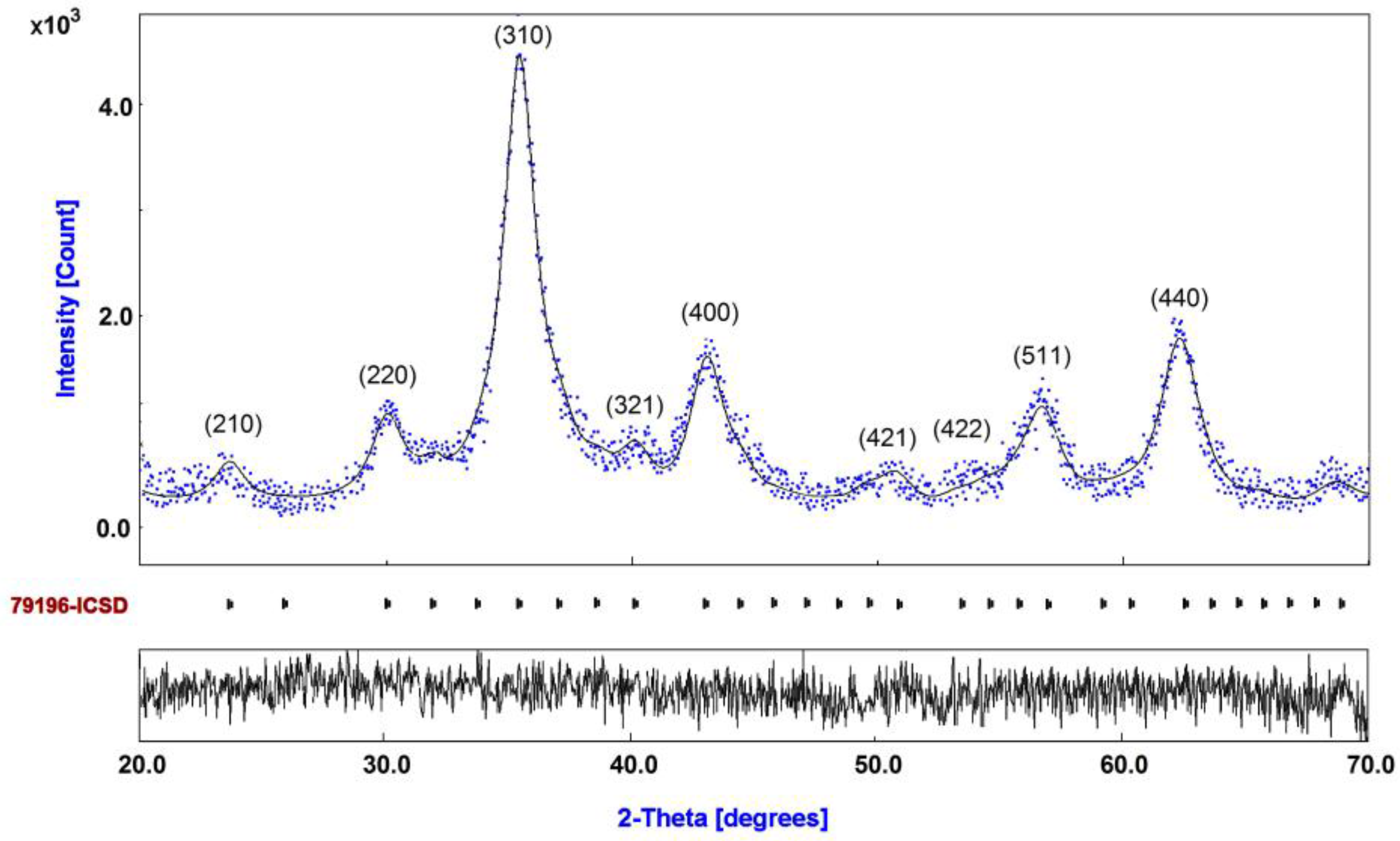
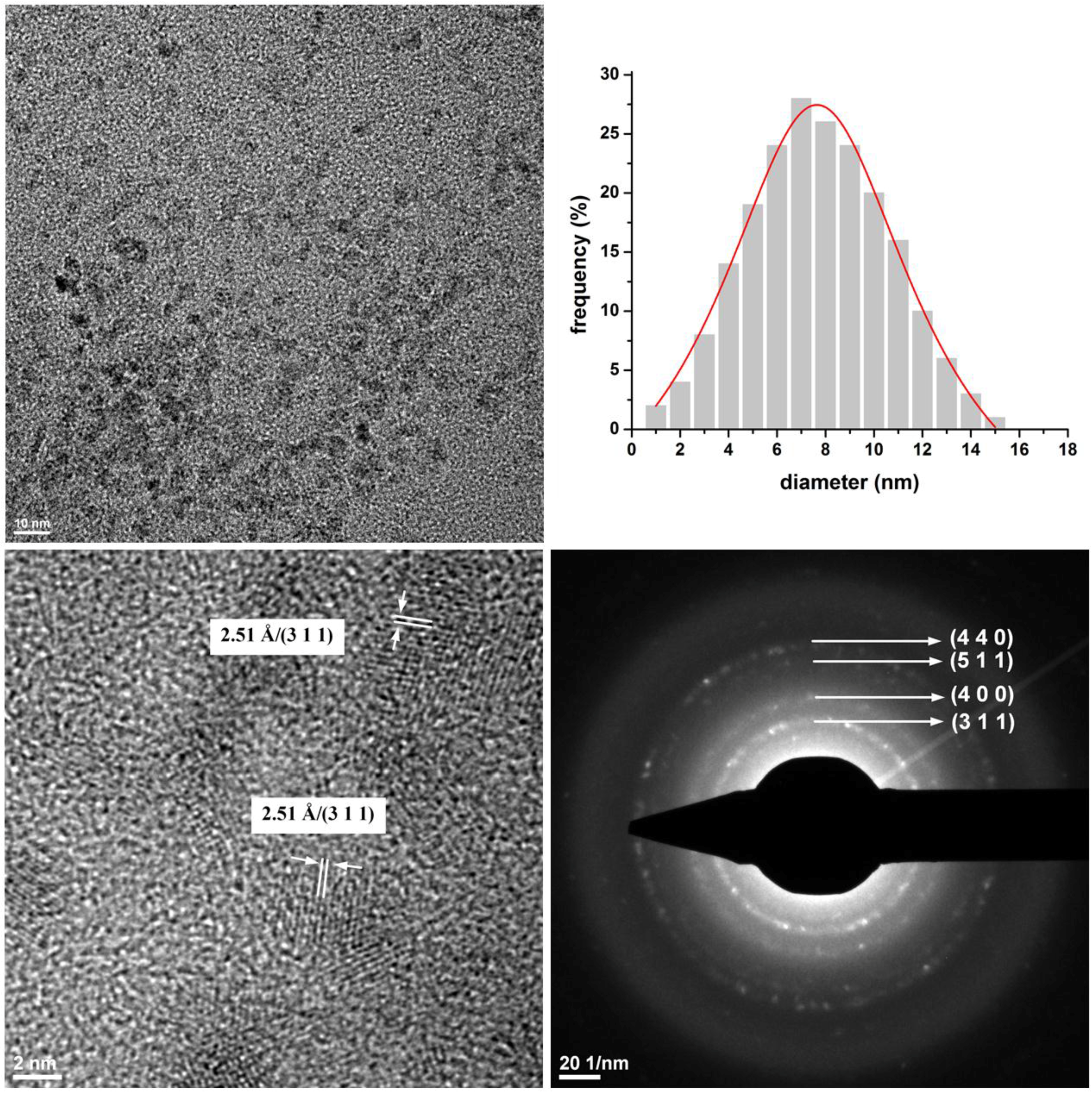
3.1. Diffraction Pattern and Size Distribution of DIO-NPs
3.2. The Colloidal Stability
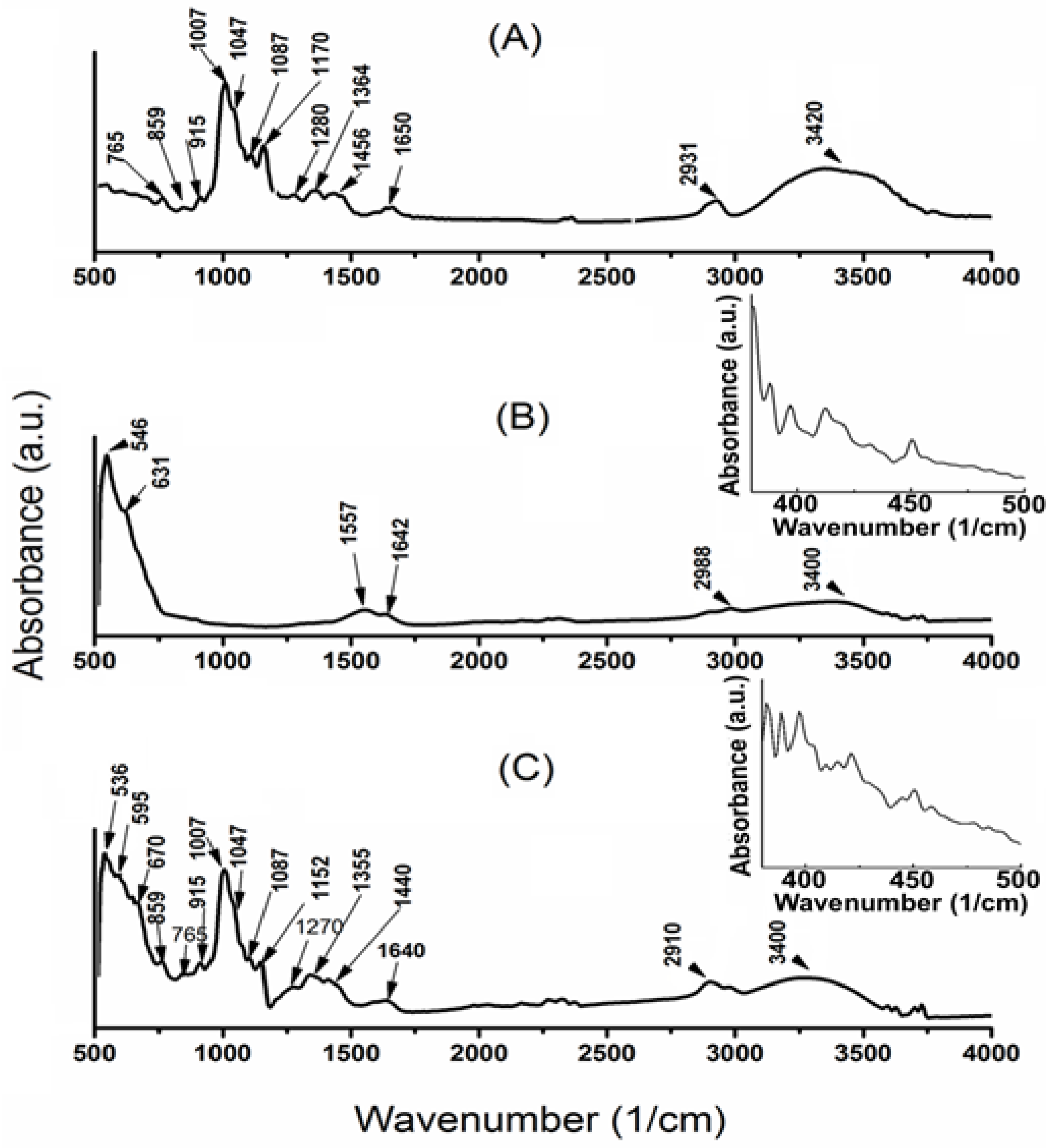
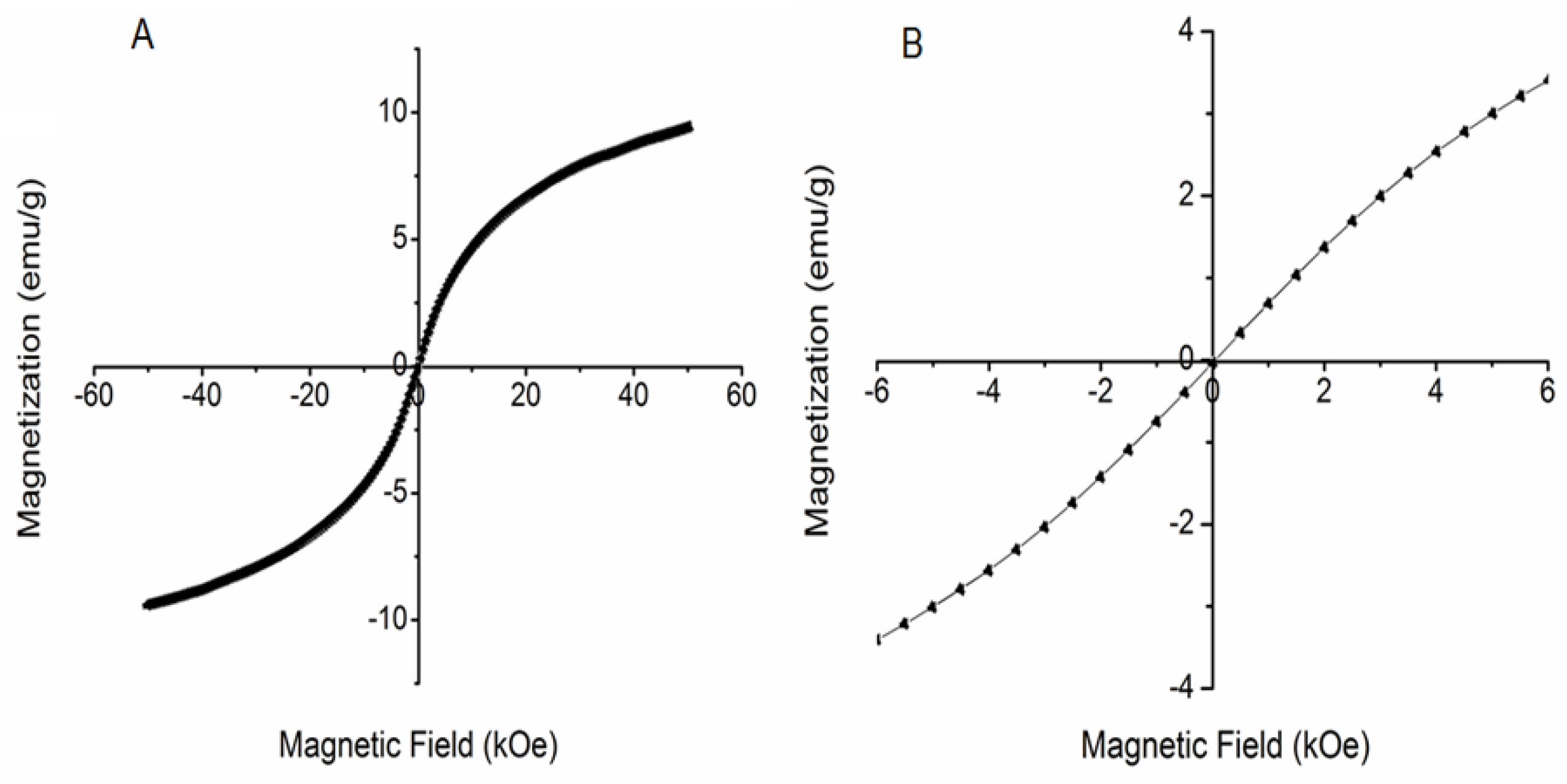
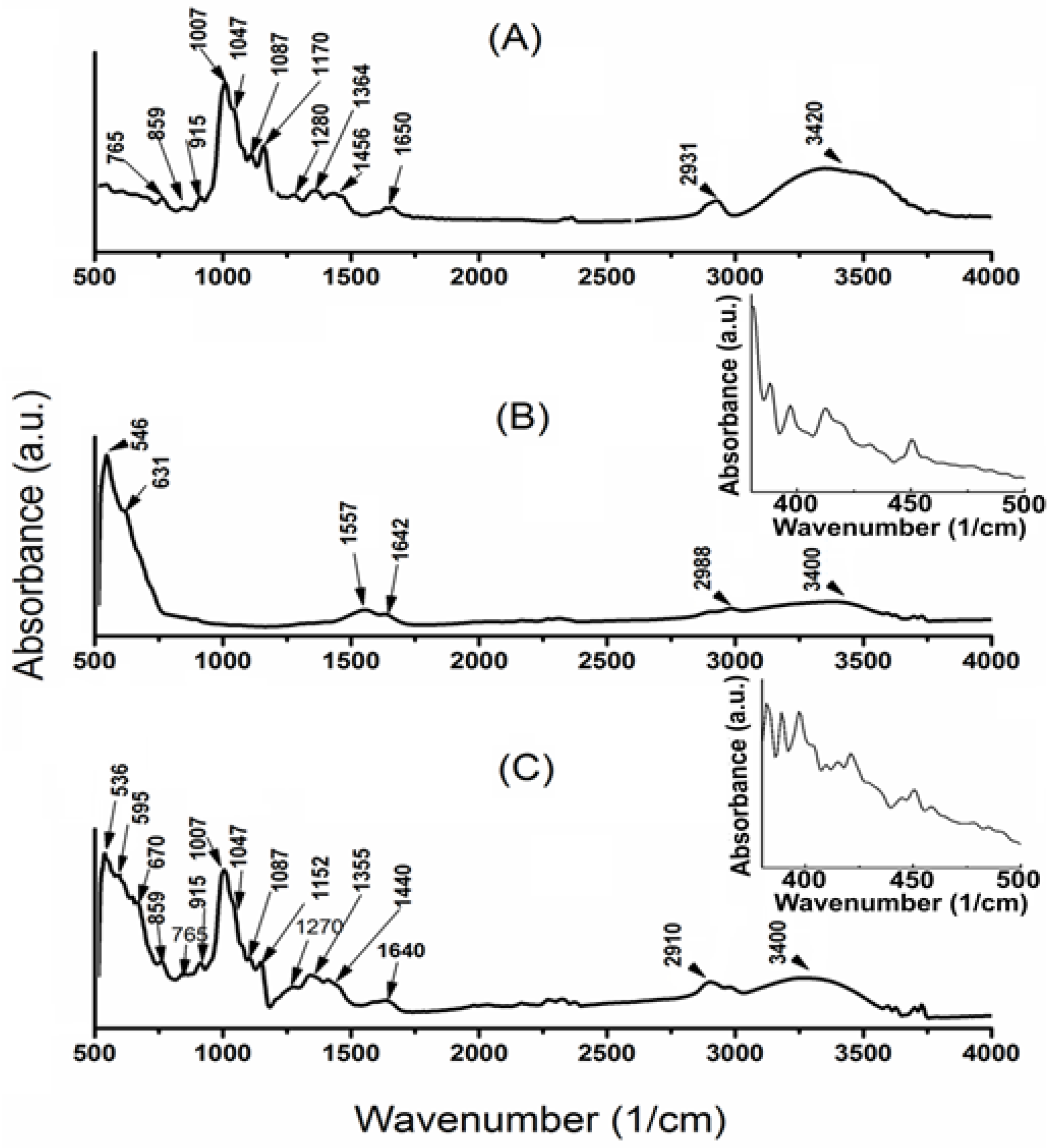
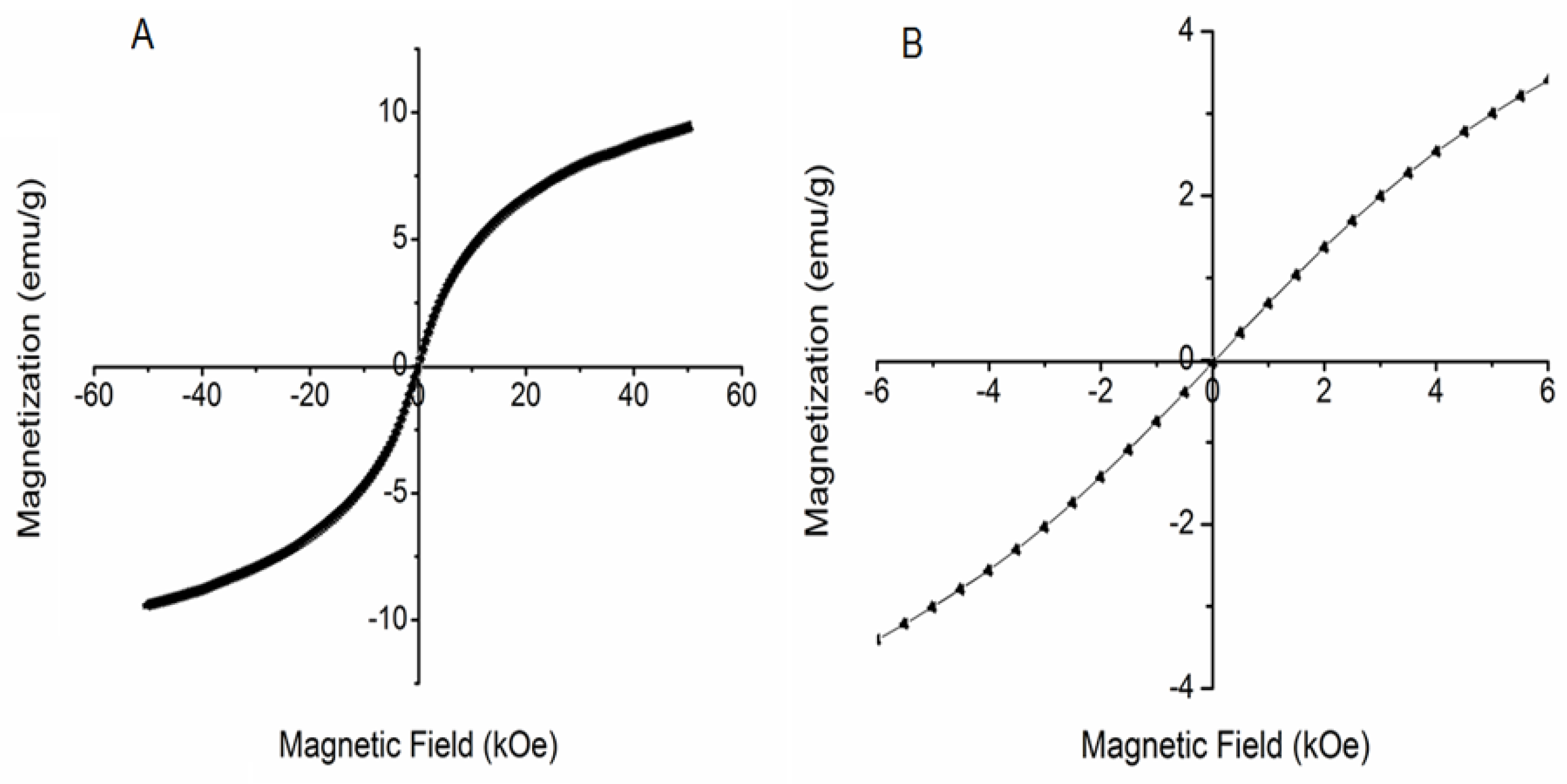
3.3. ATR–FTIR Spectra and Magnetic Behavior of DIO-NPs
3.4. DIO-NPs Relaxivity
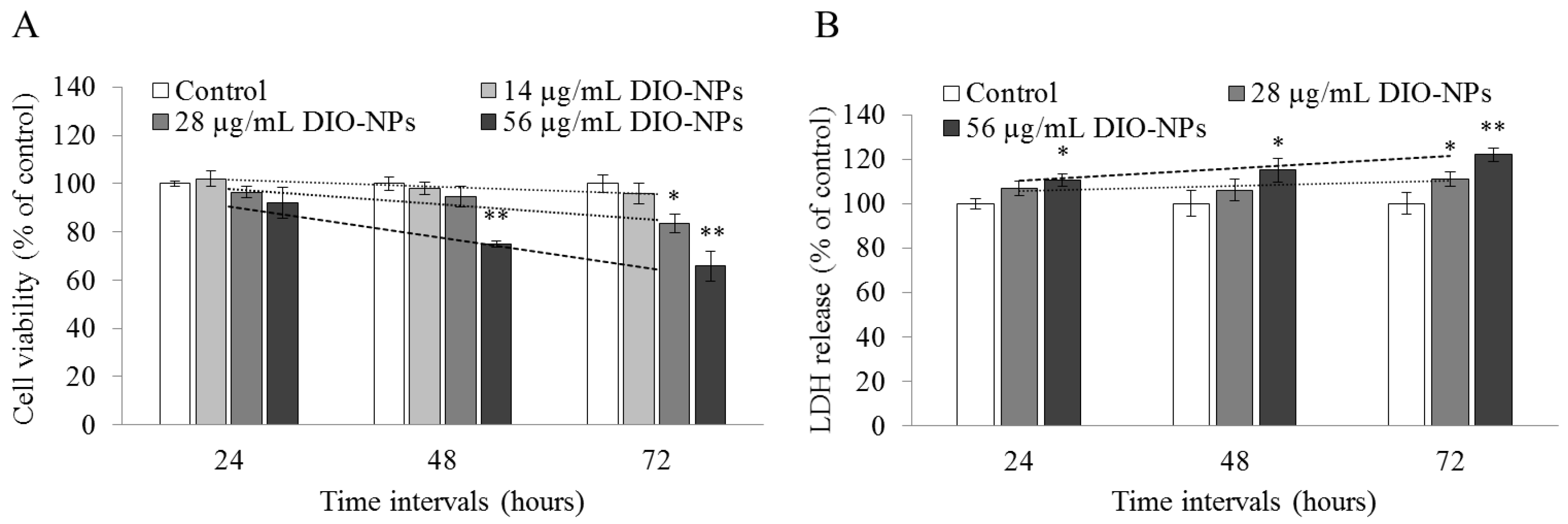
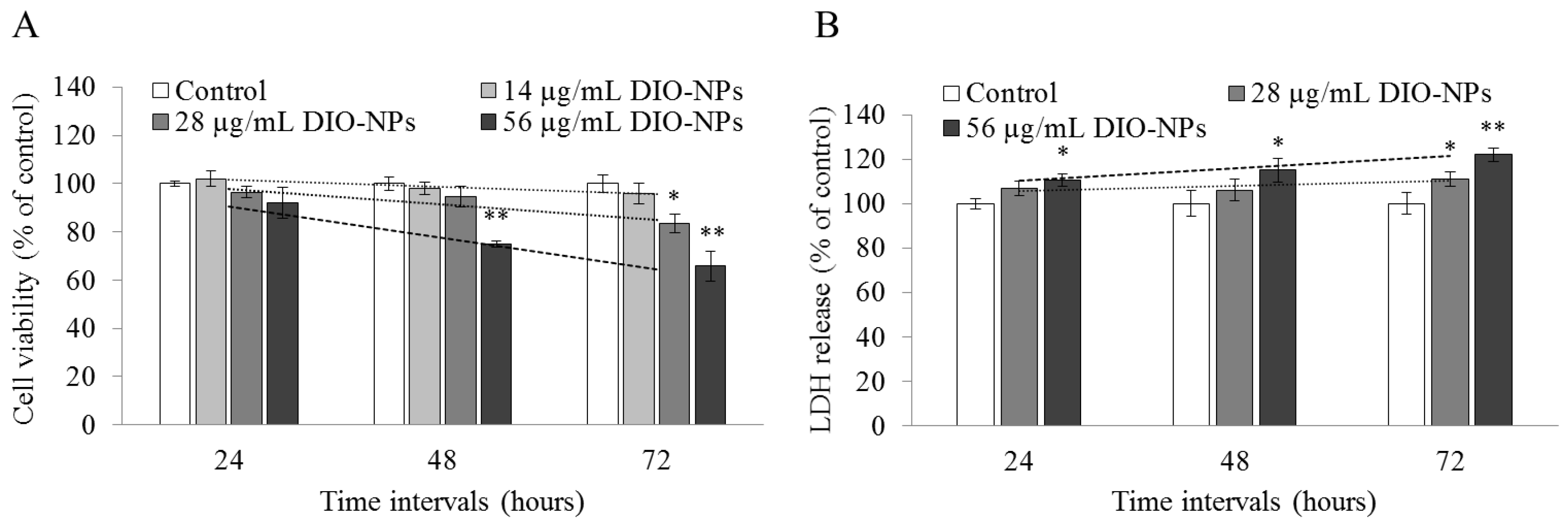
3.5. DIO-NPs Cytotoxicity
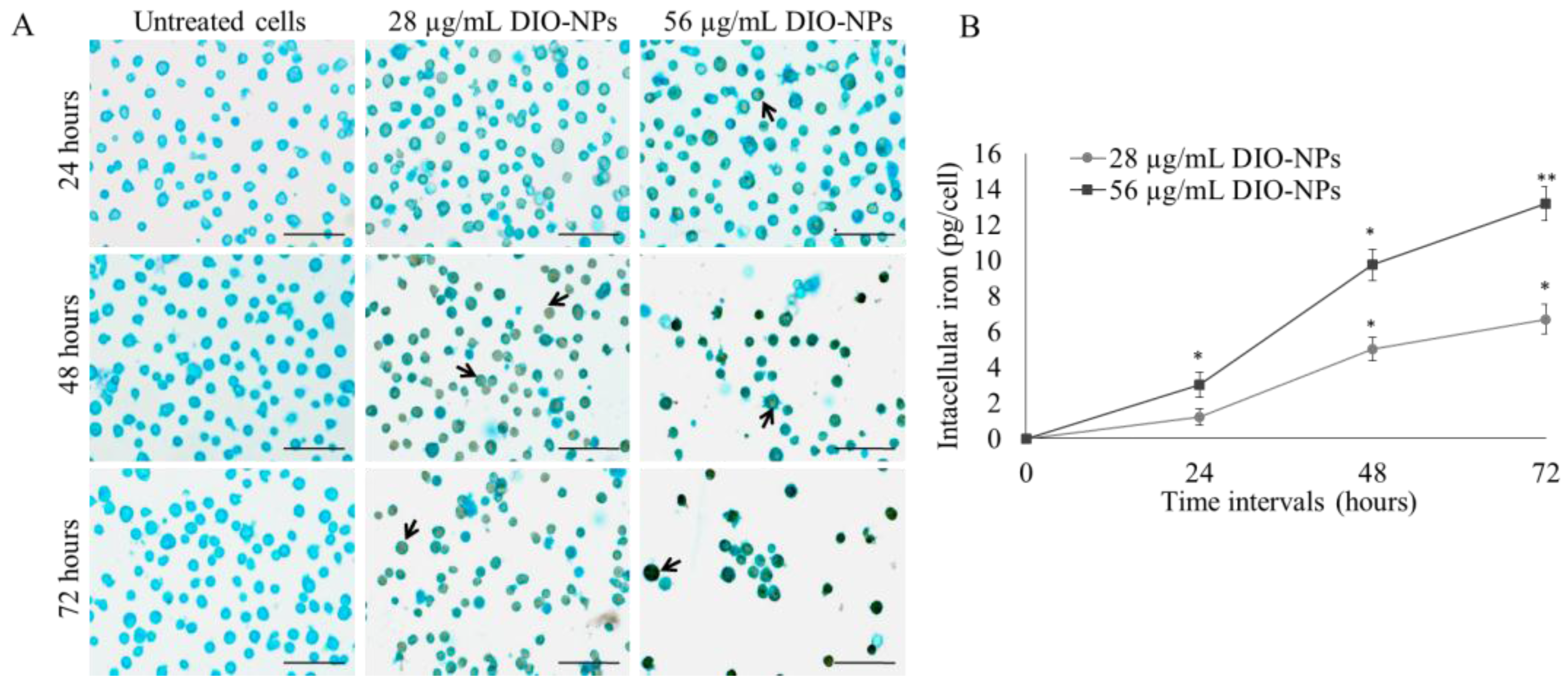
3.6. Iron Accumulation in Jurkat Cells
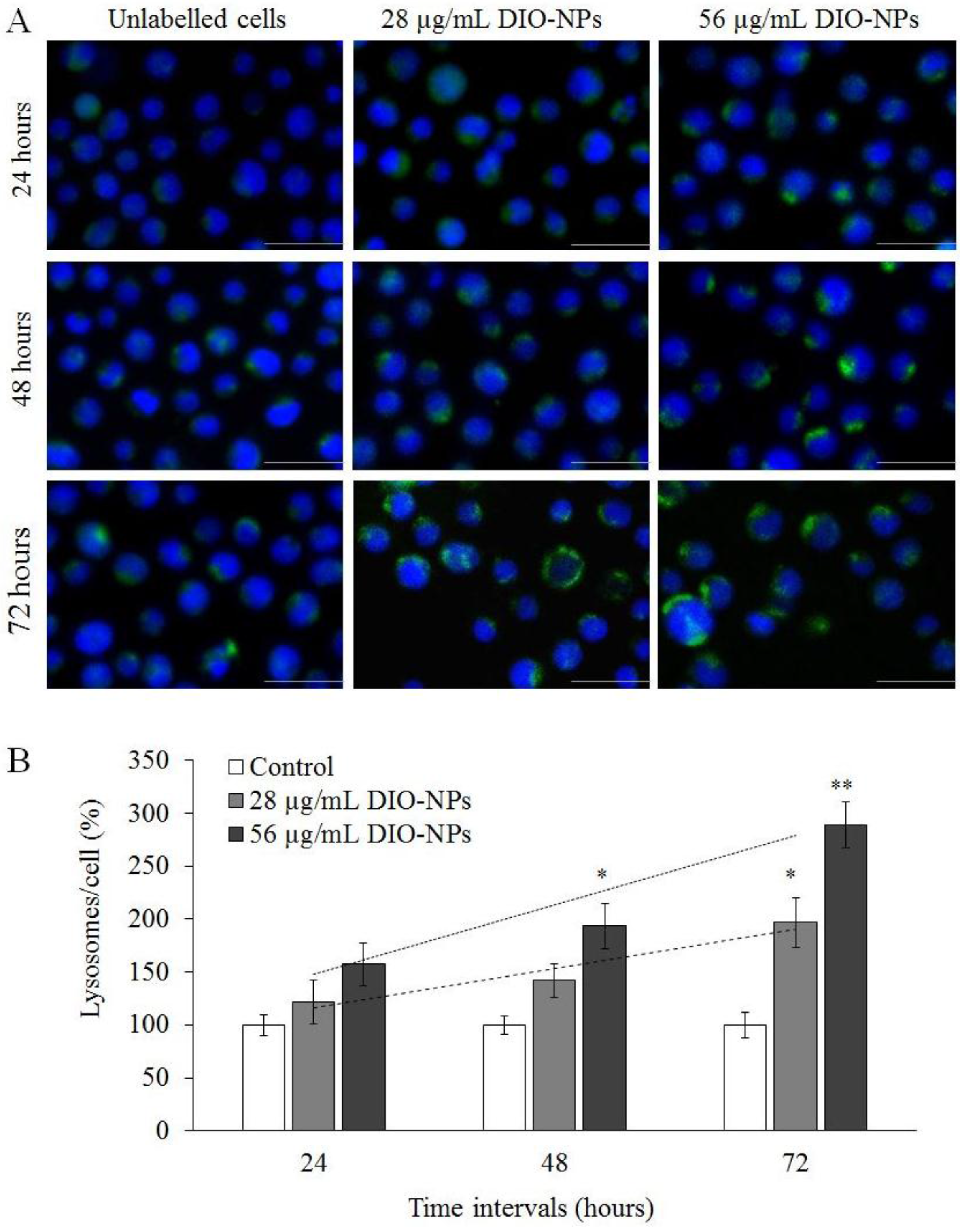
3.7. Intracellular Distribution of Lysosomes
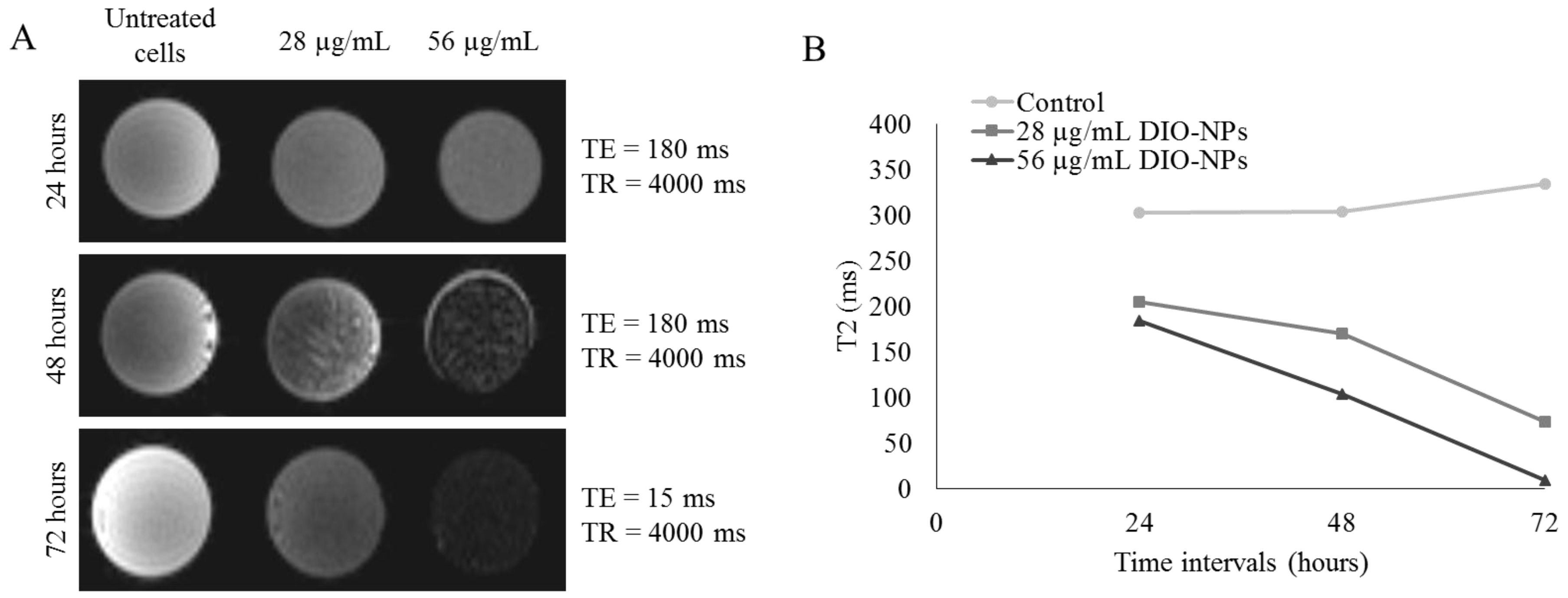
3.8. MRI and Relaxivity of NPs-Labeled Jurkat Cells
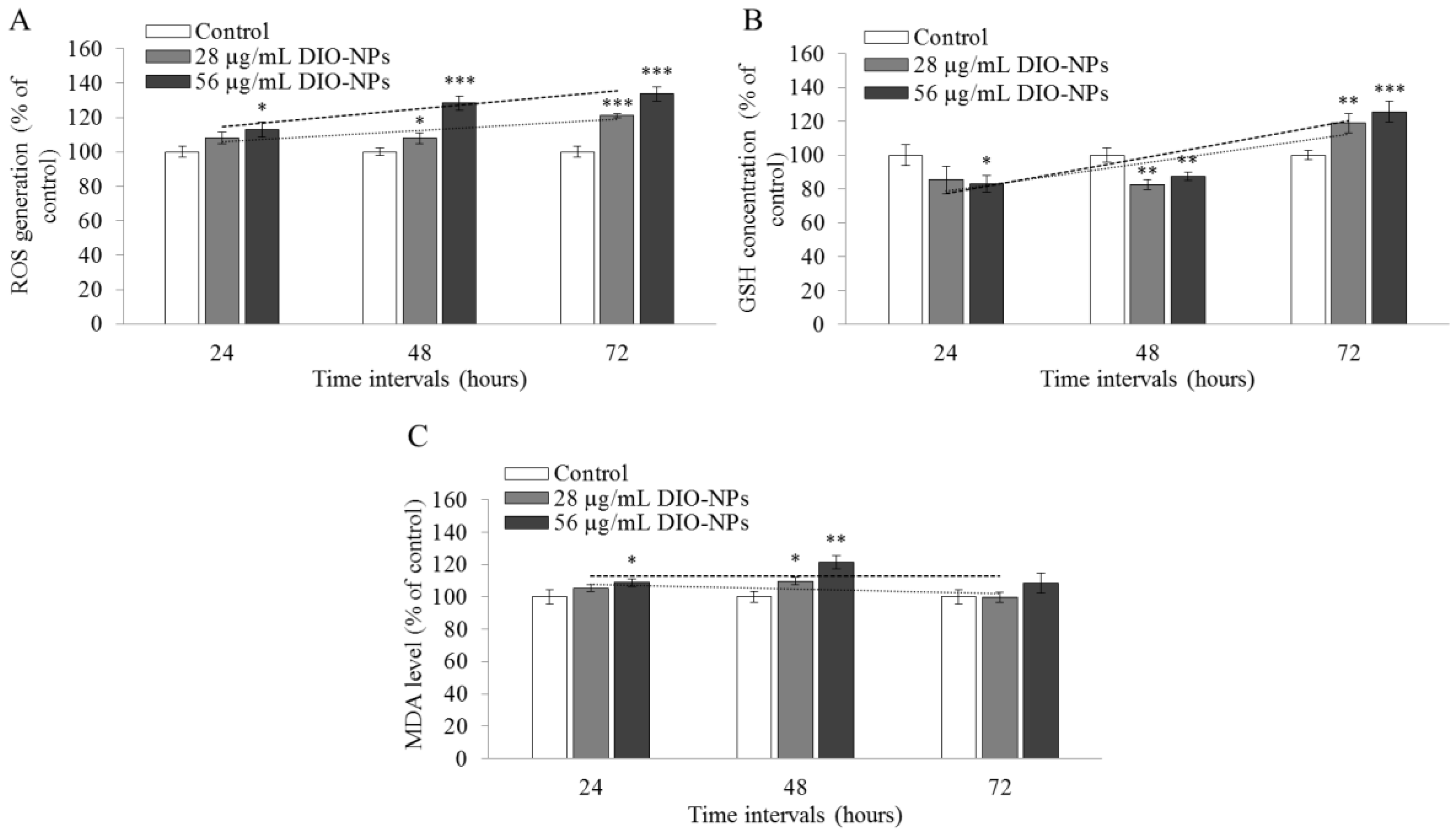
3.9. Evaluation of Oxidative Stress
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gupta, A.K.; Gupta, M. Synthesis and surface engineering of iron oxide nanoparticles for biomedical applications. Biomaterials 2005, 26, 3995–4021. [Google Scholar] [CrossRef] [PubMed]
- Ciobanu, C.S.; Iconaru, S.L.; Gyorgy, E.; Radu, M.; Costache, M.; Dinischiotu, A.; le Coustumer, P.; Lafdi, K.; Predoi, D. Biomedical properties and preparation of iron oxide-dextran nanostructures by MAPLE technique. Chem. Cent. J. 2012, 6, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahdavi, M.; Ahmad, M.B.; Haron, M.J.; Namvar, F.; Nadi, B.; Rahman, M.Z.; Amin, J. Synthesis, surface modification and characterisation of biocompatible magnetic iron oxide nanoparticles for biomedical applications. Molecules 2013, 18, 7533–7548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, J.K.; Park, J.M. Iron oxide-based superparamagnetic polymeric nanomaterials: Design, preparation, and biomedical application. Prog. Polym. Sci. 2011, 36, 168–189. [Google Scholar] [CrossRef] [Green Version]
- Salgueiriño-Maceira, V.; Correa-Duarte, M.A. Increasing the complexity of magnetic core/shell structured nanocomposites for biological applications. Adv. Mater. 2007, 19, 4131–4144. [Google Scholar] [CrossRef]
- Easo, S.L.; Mohanan, P.V. Dextran stabilized iron oxide nanoparticles: Synthesis, characterization and in vitro studies. Carbohydr. Polym. 2013, 92, 726–732. [Google Scholar] [CrossRef] [PubMed]
- Arbab, A.S.; Bashaw, L.A.; Miller, B.R.; Jordan, E.K.; Lewis, B.K.; Kalish, H.; Frank, J.A. Characterization of biophysical and metabolic properties of cells labeled with superparamagnetic iron oxide nanoparticles and transfection agent for cellular MR imaging. Radiology 2003, 229, 838–846. [Google Scholar] [CrossRef] [PubMed]
- Reimer, P.; Weissleder, R. Development and experimental application of receptor-specific MR contrast media. Radiology 1996, 36, 153–163. [Google Scholar] [CrossRef]
- Kilcoyne, S.H.; Gorisek, A. Magnetic properties of iron dextran. J. Magn. Magn. Mater. 1998, 177–181, 1457–1458. [Google Scholar] [CrossRef]
- Somsook, E.; Hinsin, D.; Buakhrong, P.; Teanchai, R.; Mophan, N.; Pohmakotr, M.; Shiowatana, J. Interactions between iron(III) and sucrose, dextran, or starch in complexes. J. Carbohydr. Polym. 2005, 61, 281–286. [Google Scholar] [CrossRef]
- Scrimshaw, N.S. Iron deficiency. Sci. Am. 1991, 265, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Gyurcsik, B.; Nagy, L. Carbohydrates as ligands: Coordination equilibria and structure of the metal complexes. Coord. Chem. Rev. 2000, 203, 81–149. [Google Scholar] [CrossRef]
- Hudson, J.Q.; Comstock, T.J. Considerations for optimal ironuse for anemia due to chronic kidney disease. Clin. Ther. 2001, 23, 1637–1671. [Google Scholar] [CrossRef]
- Kane, R.C. Intravenous iron replacement with sodium ferric gluconate complex in sucrose for iron deficiency anemia in adults. Curr. Ther. Res. Clin. Exp. 2003, 64, 263–268. [Google Scholar] [CrossRef]
- León-Félix, L.; Chaker, J.; Parise, M.; Coaquira, J.A.H.; de Los Santos Valladares, L.; Bustamante, A.; Garg, V.K.; Oliveira, A.C.; Morais, P.C. Synthesis and characterization of uncoated and gold-coated magnetite nanoparticles. Hyperfine Interact. 2014, 224, 179–188. [Google Scholar] [CrossRef]
- Ramos Guivar, J.A.; Bustamante, A.; Flores, J.; Mejía Santillan, M.; Osorio, A.M.; Martínez, A.I.; de Los Santos Valladares, L.; Barnes, C.H.W. Mössbauer study of intermediate superparamagnetic relaxation of maghemite (γ-Fe2O3) nanoparticles. Hyperfine Interact. 2014, 224, 89–97. [Google Scholar] [CrossRef]
- De Los Santos Valladares, L.; León Félix, L.; Espinoza Suarez, S.M.; Bustamante Dominguez, A.G.; Mitrelias, T.; Holmes, S.; Moreno, N.O.; Albino Aguiar, J.; Barnes, C.H.W. Preparation and crystallization of hollow α-Fe2O3 microspheres following the gas-bubble template method. Mater. Chem. Phys. 2016, 169, 21–27. [Google Scholar] [CrossRef]
- Zhang, Y.; Kohler, N.; Zhang, M. Surface modification of superparamagnetic magnetite nanoparticles and their intracellular uptake. Biomaterials 2002, 23, 1553–1561. [Google Scholar] [CrossRef]
- Lacava, L.M.; Lacava, Z.G.; da Silva, M.F.; Silva, O.; Chaves, S.B.; Azevedo, R.B.; Pelegrini, F.; Gansau, C.; Buske, N.; Sabolovic, D.; et al. Magnetic resonance of a dextran-coated magnetic fluid intravenously administered in mice. Biophys. J. 2001, 80, 2483–2486. [Google Scholar] [CrossRef]
- Silva, V.A.J.; Andrade, P.L.; Bustamante, A.; de Los Santos Valladares, L.; Mejía, M.; Souza, I.A.; Cavalcanti, K.P.S.; Silva, M.P.C.; Albino Aguiar, J. Magnetic and Mössbauer studies of fucan-coated magnetite nanoparticles for application on antitumoral activity. Hyperfine Interact. 2014, 224, 227–238. [Google Scholar] [CrossRef]
- Silva, V.A.J.; Andrade, P.L.; Silva, M.P.C.; Bustamante, A.; de Los Santos Valladares, L.; Albino Aguiar, J. Synthesis and characterization of Fe3O4 nanoparticles coated with fucan polysaccharides. J. Magn. Magn. Mater. 2013, 343, 138–143. [Google Scholar] [CrossRef]
- Andrade, P.L.; Silva, V.A.J.; Maciel, J.C.; Santillan, M.M.; Moreno, N.O.; de Los Santos Valladares, L.; Bustamante, A.; Pereira, S.M.B.; Silva, M.P.C.; Albino Aguiar, J. Preparation and characterization of cobalt ferrite nanoparticles coated with fucan and oleic acid. Hyperfine Interact. 2014, 224, 217–225. [Google Scholar] [CrossRef]
- Bautista, M.; Bomati-Miguel, O.; del Puerto Morales, M.; Serna, C.J.; Veintemillas-Verdaguer, S. Surface characterisation of dextran coated iron oxide nanoparticles prepared by laser pyrolysis and coprecipitation. J. Magn. Magn. Mater. 2005, 293, 20–27. [Google Scholar] [CrossRef]
- Iconaru, S.L.; Prodan, A.M.; Motelica-Heino, M.; Sizaret, S.; Predoi, D. Synthesis and characterization of polysaccharide-maghemite composite nanoparticles and their antibacterial properties. Nanoscale Res. Lett. 2012, 7, 576–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boutry, S.; Burtea, C.; Mahieu, I.; Murariu, O.; Laurent, S.; Vander Elst, L.; Muller, R.N. How to quantify iron in an aqueous or biological matrix: A technical note. Contrast Media Mol. Imaging 2009, 4, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Wan, C.P.; Myung, E.; Lau, B.H.S. An automated microfluorometric assay for monitoring oxidative burst activity of phagocytes. J. Immunol. Methods 1993, 159, 131–138. [Google Scholar] [CrossRef]
- Dinischiotu, A.; Stanca, L.; Gradinaru, D.; Petrache, S.N.; Radu, M.; Serban, A.I. Lipid peroxidation due to in vitro and in vivo exposure of biological samples to nanoparticles. In Oxidative Stress and Nanotechnology; Armstrong, D., Bharali, D.J., Eds.; Springer: New York, NY, USA, 2013; Volume 1028, pp. 155–164. [Google Scholar]
- Lutterotti, L. Total pattern fitting for the combined size-strainstress-texture determination in thin film diffraction. Nucl. Instrum. Meth. B 2010, 268, 334–340. [Google Scholar] [CrossRef]
- Kim, W.; Suh, C.-Y.; Cho, S.-W.; Roh, K.-M.; Kwon, H.; Song, K.; Shon, I.-J. A new method for the identification and quantification of magnetite-maghemite mixture using conventional X-ray diffraction technique. Talanta 2012, 94, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Dutta, P.; Manivannan, A.; Seehra, M.S. Magnetic properties of nearly defect-free maghemite nanocrystals. Phys. Rev. B 2004, 70, 174428. [Google Scholar] [CrossRef]
- Ramos Guivar, J.A.; Sanches, E.A.; Bruns, F.; Sadrollahi, E.; Morales, M.A.; López, E.O.; Litterst, F.J. Vacancy ordered γ-Fe2O3 nanoparticles functionalized with nanohydroxyapatite: XRD, FTIR, TEM, XPS and Mössbauer studies. Appl. Surf. Sci. 2016, 389, 721–734. [Google Scholar] [CrossRef]
- Jorgensen, J.E.; Mosegaard, L.; Thomsen, L.E.; Jensen, T.R.; Hanson, J.C. Formation of γ-Fe2O3 nanoparticles and vacancy ordering: An in situ X-ray powder diffraction study. J. Solid State Chem. 2007, 180, 180–185. [Google Scholar] [CrossRef]
- Musić, S.; Orehovec, Z.; Popović, S.; Czakó-Nagy, I. Structural properties of precipitates formed by hydrolisis of Fe3+ ions in Fe2(SO4)3 solutions. J. Mater. Sci. 1994, 29, 1991–1998. [Google Scholar] [CrossRef]
- Izumi, F.; Yamamoto, A.; Khasanova, N.R.; Kumazawa, S.; Hu, W.Z.; Kamiyama, T. Novel techniques of neutron powder diffraction and their applications to superconducting oxides. Physica C 2000, 335, 239–244. [Google Scholar] [CrossRef]
- David, W.I.F. Powder Diffraction: Least-Squares and Beyond. J. Res. Natl. Inst. Stand. Technol. 2004, 109, 107–123. [Google Scholar] [CrossRef] [PubMed]
- Toby, B.H. R factors in Rietveld analysis: How good is good enough? Powder Diffr. 2006, 21, 67–70. [Google Scholar] [CrossRef]
- Barrado, E.; Prieto, F.; Medina, J.; López, F.A. Characterisation of solid residues obtained on removal of Cr from waste water. J. Alloys Compd. 2002, 335, 203–209. [Google Scholar] [CrossRef]
- De Vidales, J.L.M.; López-Delgado, A.; Vila, E.; López, F.A. The effect of the starting solution on the physico-chemical properties of zinc ferrite synthesized at low temperature. J. Alloys Compd. 1999, 287, 276–283. [Google Scholar] [CrossRef]
- Predoi, D. A study on iron oxide nanoparticles coated with dextrin obtained by coprecipitation. Dig. J. Nanomater. Biostruct. 2007, 2, 169–173. [Google Scholar]
- Karnchanatat, A.; Puthong, S.; Sihanonth, P.; Piapukiew, J.; Sangvanich, P. Antioxidation and antiproliferation properties of polysaccharide-protein complex extracted from Phaeogyroporusportentosus (Berk. & Broome) McNabb. Afr. J. Microbiol. Res. 2013, 7, 1668–1680. [Google Scholar]
- Hradil, J.; Pisarev, A.; Babič, M.; Horák, D. Dextran-modified iron oxide nanoparticles. China Part. 2007, 5, 162–168. [Google Scholar] [CrossRef]
- Hair, M.L. Hydroxyl groups on silica surface. J. Non-Cryst. Solids 1975, 19, 299–309. [Google Scholar] [CrossRef]
- Farmer, V.C. The Infrared Spectra of Minerals; Mineralogical Society of Great Britain and Ireland: Twickenham, UK, 1974; pp. 305–330. [Google Scholar]
- Shafi, K.V.P.M.; Gedanken, A.; Prozorov, R.; Balogh, J. Sonochemical preparation and size-dependent properties of nanostructured CoFe2O4 particles. Chem. Mater. 1998, 10, 3445–3450. [Google Scholar] [CrossRef]
- Vijayakumar, R.; Koltypin, Y.; Felner, I.; Gedanken, A. Sonochemical synthesis and characterization of pure nanometersized Fe3O4 particles. Mater. Sci. Eng. A 2000, 286, 101–105. [Google Scholar] [CrossRef]
- Jiang, L.; Zhou, X.; Wei, G.; Lu, X.; Wei, W.; Qiu, J. Preparation and characterization of poly(glycidylmethacrylate)-grafted magnetic nanoparticles: Effects of the precursor concentration on polyol synthesis of Fe3O4 and [PMDETA]0/[CuBr2]0 ratios on SI-AGET ATRP. Appl. Surf. Sci. 2015, 357, 1619–1624. [Google Scholar] [CrossRef]
- Si, S.; Li, C.; Wang, X.; Yu, D.; Peng, Q.; Li, Y. Magnetic monodisperse Fe3O4 nanoparticles. Cryst. Growth Des. 2005, 5, 391–393. [Google Scholar] [CrossRef]
- Laurent, S.; Forge, D.; Port, M.; Roch, A.; Robic, C.; Vander Elst, L.; Muller, R.N. Magnetic iron oxide nanoparticles: Synthesis, stabilization, vectorization, physico-chemical characterizations and biological applications. Chem. Rev. 2008, 108, 2064–2110. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.; Stellacci, F. Effect of surface properties on nanoparticle–cell interactions. Small 2010, 6, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Laurent, S.; Tawil, N.; Yahia, L.; Mahmoudi, L. Protein-Nanoparticles Interactions. The Bio-Nano Interface; Springer: Berlin/Heidelberg, Germany, 2013; p. 15. [Google Scholar]
- Chen, C.-C.V.; Ku, M.-C.; Jayaseema, D.M.; Lai, J.-S.; Hueng, D.-Y.; Chang, C. Simple SPION incubation as an efficient intracellular labeling method for tracking neural progenitor cells using MRI. PLoS ONE 2013, 8, e56125. [Google Scholar] [CrossRef] [PubMed]
- Shang, L.; Nienhaus, K.; Nienhaus, G.U. Engineered nanoparticles interacting with cells: Size matters. J. Nanobiotechnol. 2014, 12, 1477–3155. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Huang, S.; Yu, K.J.; Clyne, A.M. Dextran and polymer polyethylene glycol (PEG) coating reduce both 5 and 30 nm iron oxide nanoparticle cytotoxicity in 2D and 3D cell culture. Int. J. Mol. Sci. 2012, 13, 5554–5570. [Google Scholar] [CrossRef] [PubMed]
- Thorek, D.L.; Tsourkas, A. Size, charge and concentration dependent uptake of iron oxide particles by non-phagocytic cells. Biomaterials 2008, 29, 3583–3590. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, S.M.; Singh, N.; Jenkins, G.; Williams, A.; Orbaek, A.; Barron, C. Dextran coated ultrafine superparamagnetic iron oxide nanoparticles: Compatibility with common fluorometric and colorimetric dyes. Anal. Chem. 2011, 83, 3778–3785. [Google Scholar] [CrossRef] [PubMed]
- Berry, C.C.; Wells, S.; Charles, S.; Curtis, A.S. Dextran and albumin derivatised iron oxide nanoparticles: Influence on fibroblasts in vitro. Biomaterials 2003, 24, 4551–4557. [Google Scholar] [CrossRef]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Time | Hydrodynamic Diameter (nm) | Zeta Potential (mV) |
|---|---|---|
| 0 h | 7.50 ± 0.50 | −30.37 ± 1.52 |
| 24 h | 8.18 ± 0.38 | −11.47 ± 1.24 |
| 48 h | 8.52 ± 1.10 | −9.92 ± 0.59 |
| 72 h | 7.73 ± 0.47 | −7.51 ± 1.01 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balas, M.; Ciobanu, C.S.; Burtea, C.; Stan, M.S.; Bezirtzoglou, E.; Predoi, D.; Dinischiotu, A. Synthesis, Characterization, and Toxicity Evaluation of Dextran-Coated Iron Oxide Nanoparticles. Metals 2017, 7, 63. https://doi.org/10.3390/met7020063
Balas M, Ciobanu CS, Burtea C, Stan MS, Bezirtzoglou E, Predoi D, Dinischiotu A. Synthesis, Characterization, and Toxicity Evaluation of Dextran-Coated Iron Oxide Nanoparticles. Metals. 2017; 7(2):63. https://doi.org/10.3390/met7020063
Chicago/Turabian StyleBalas, Mihaela, Carmen Steluta Ciobanu, Carmen Burtea, Miruna Silvia Stan, Eugenia Bezirtzoglou, Daniela Predoi, and Anca Dinischiotu. 2017. "Synthesis, Characterization, and Toxicity Evaluation of Dextran-Coated Iron Oxide Nanoparticles" Metals 7, no. 2: 63. https://doi.org/10.3390/met7020063