
Decontamination of Uranium-Contaminated Soil Sand Using Supercritical CO2 with a TBP–HNO3 Complex

Abstract
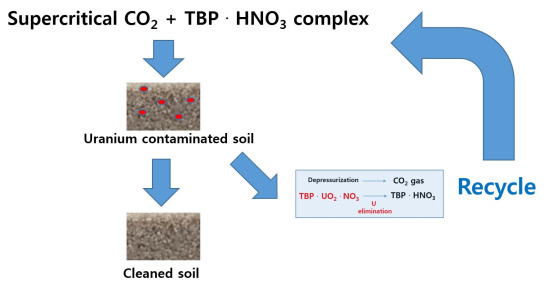
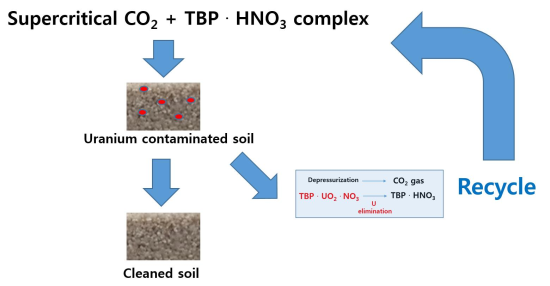
:
1. Introduction
2. Experiments
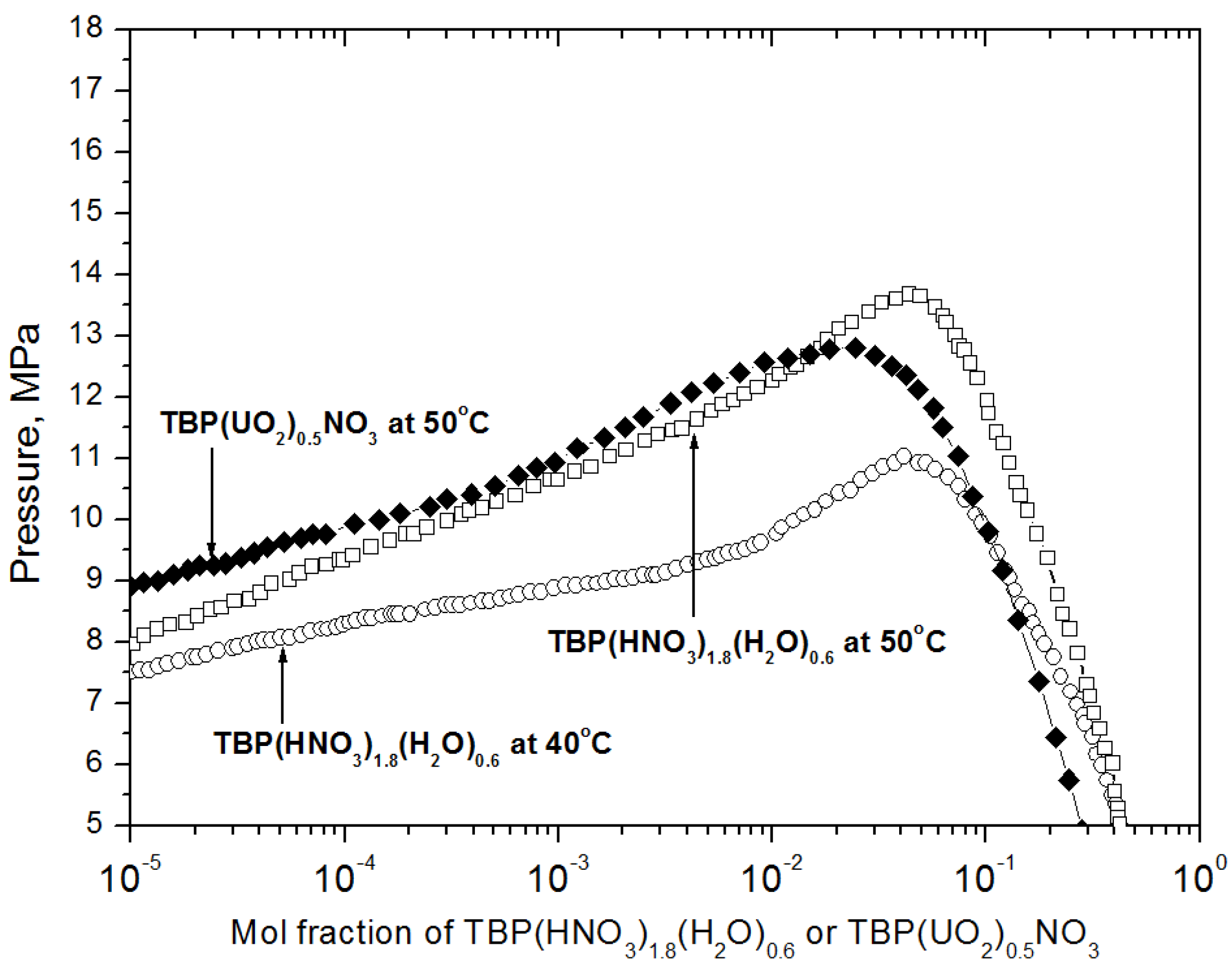
2.1. Preparation of the Reagent for Extraction and the Extraction Conditions
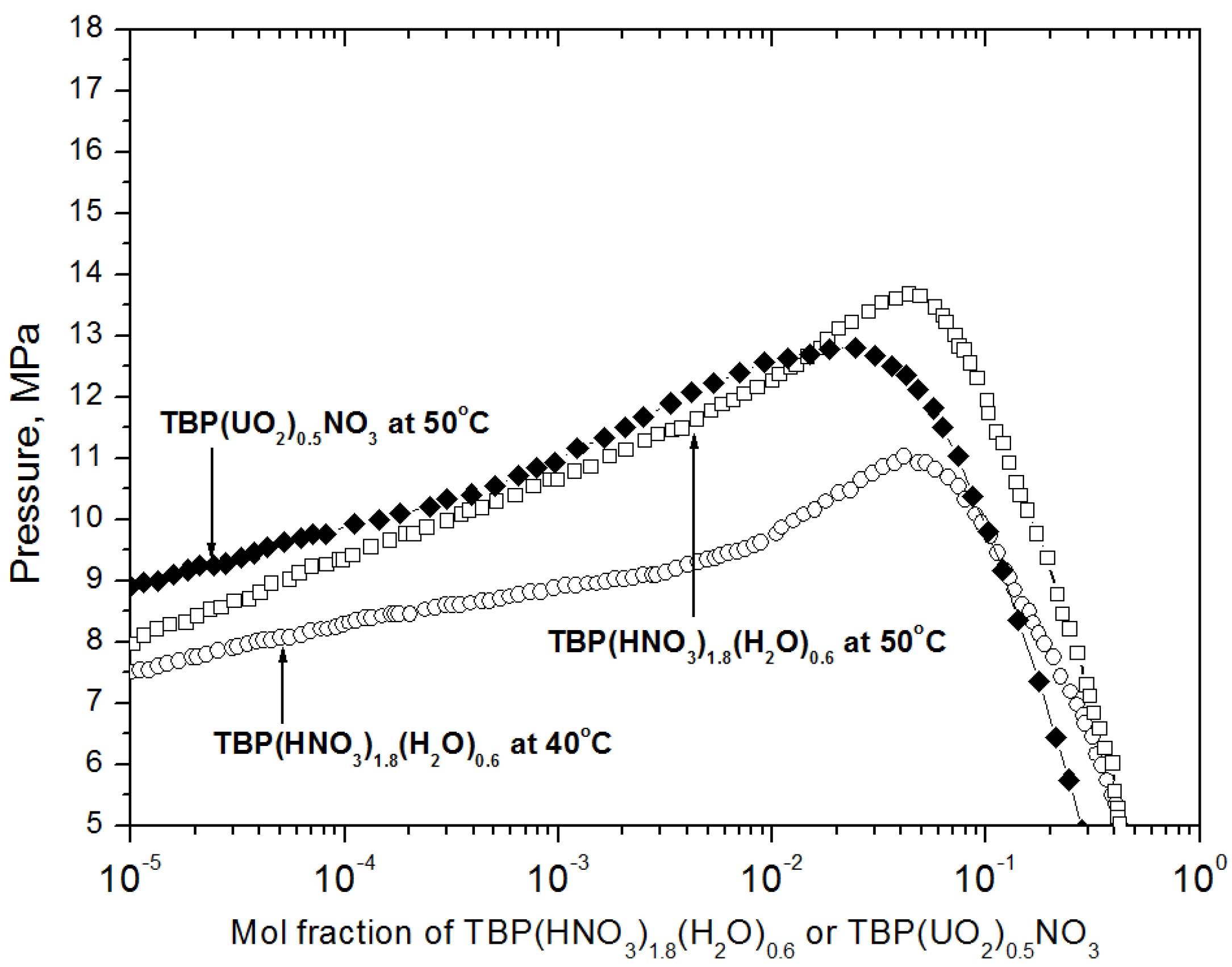
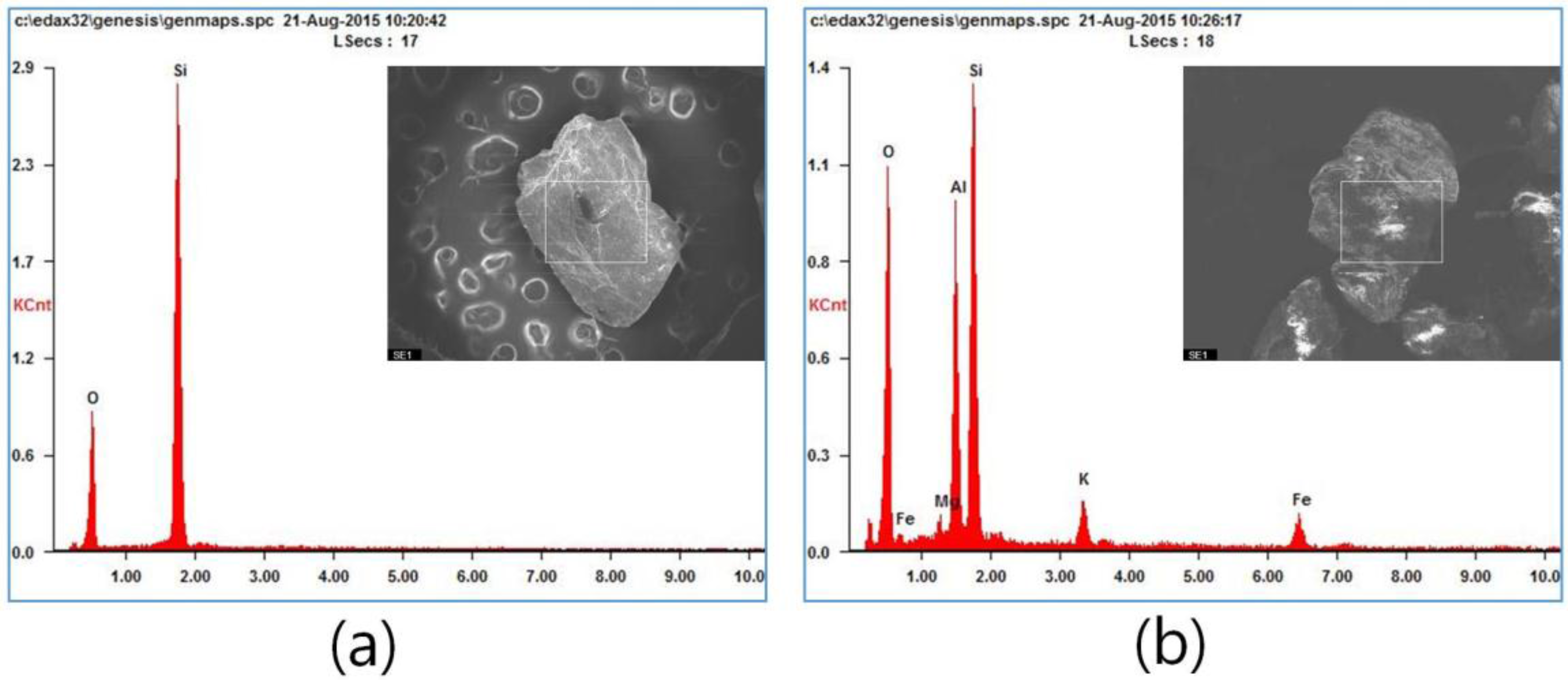
2.2. Specimen Preparation

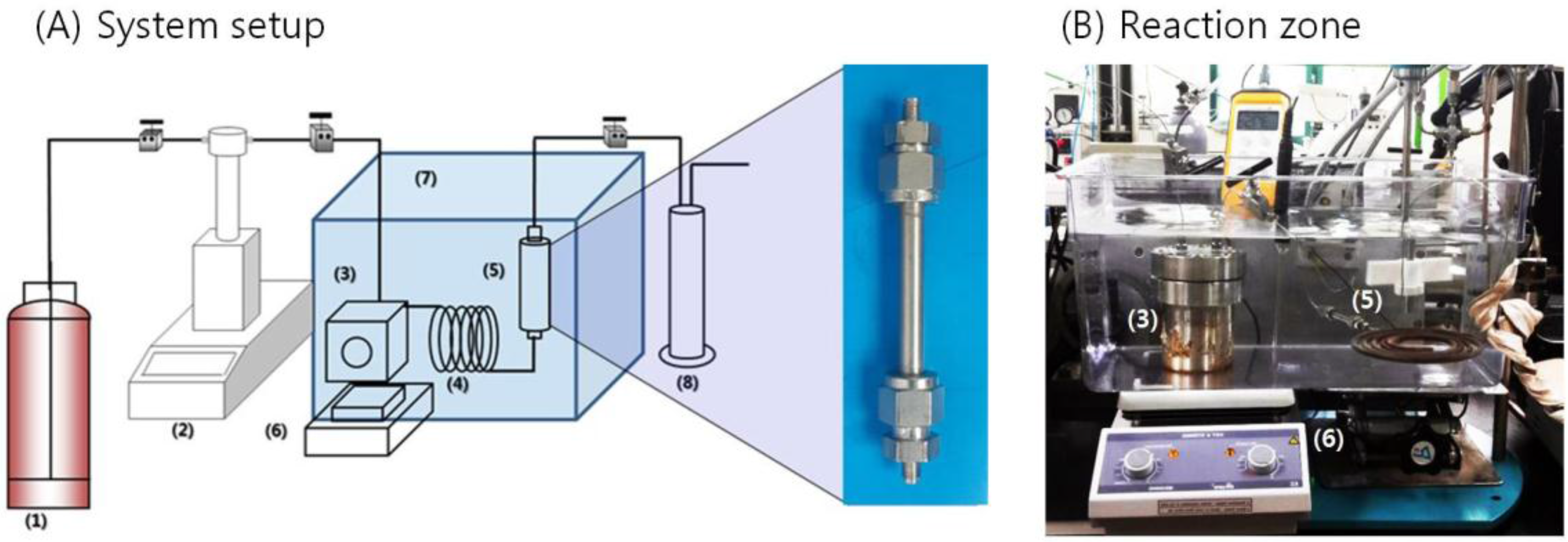
2.3. Experimental Process
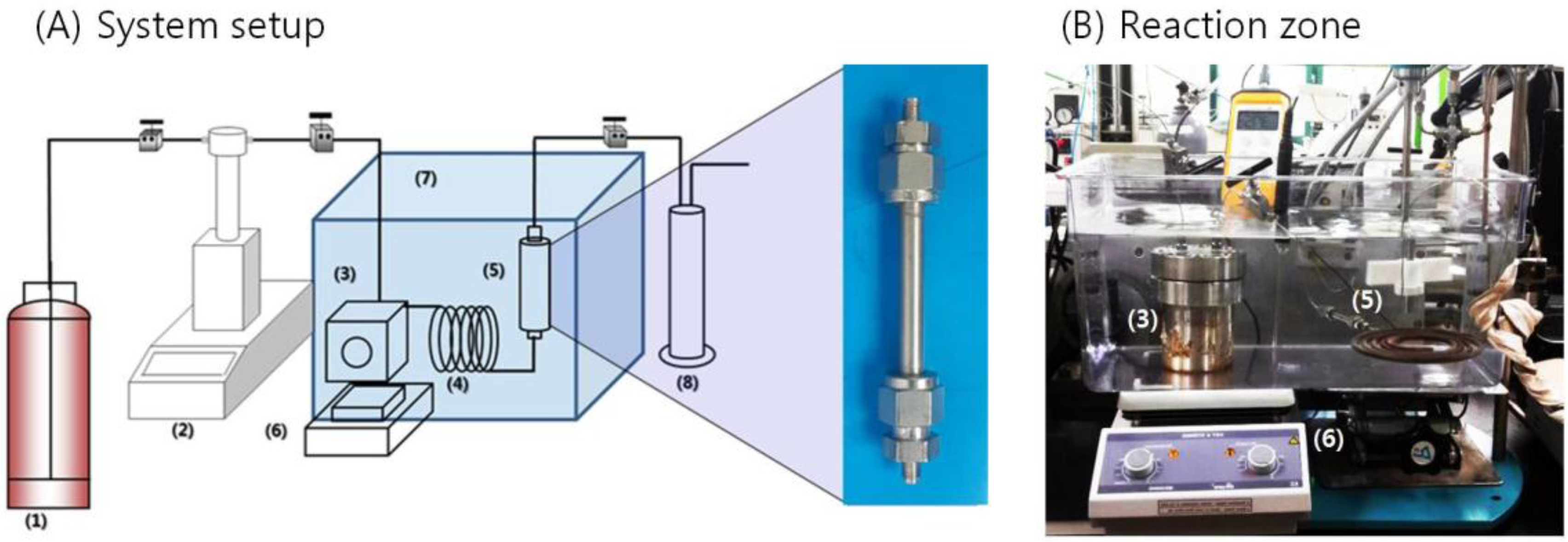

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Specimen | Description |
|---|---|---|
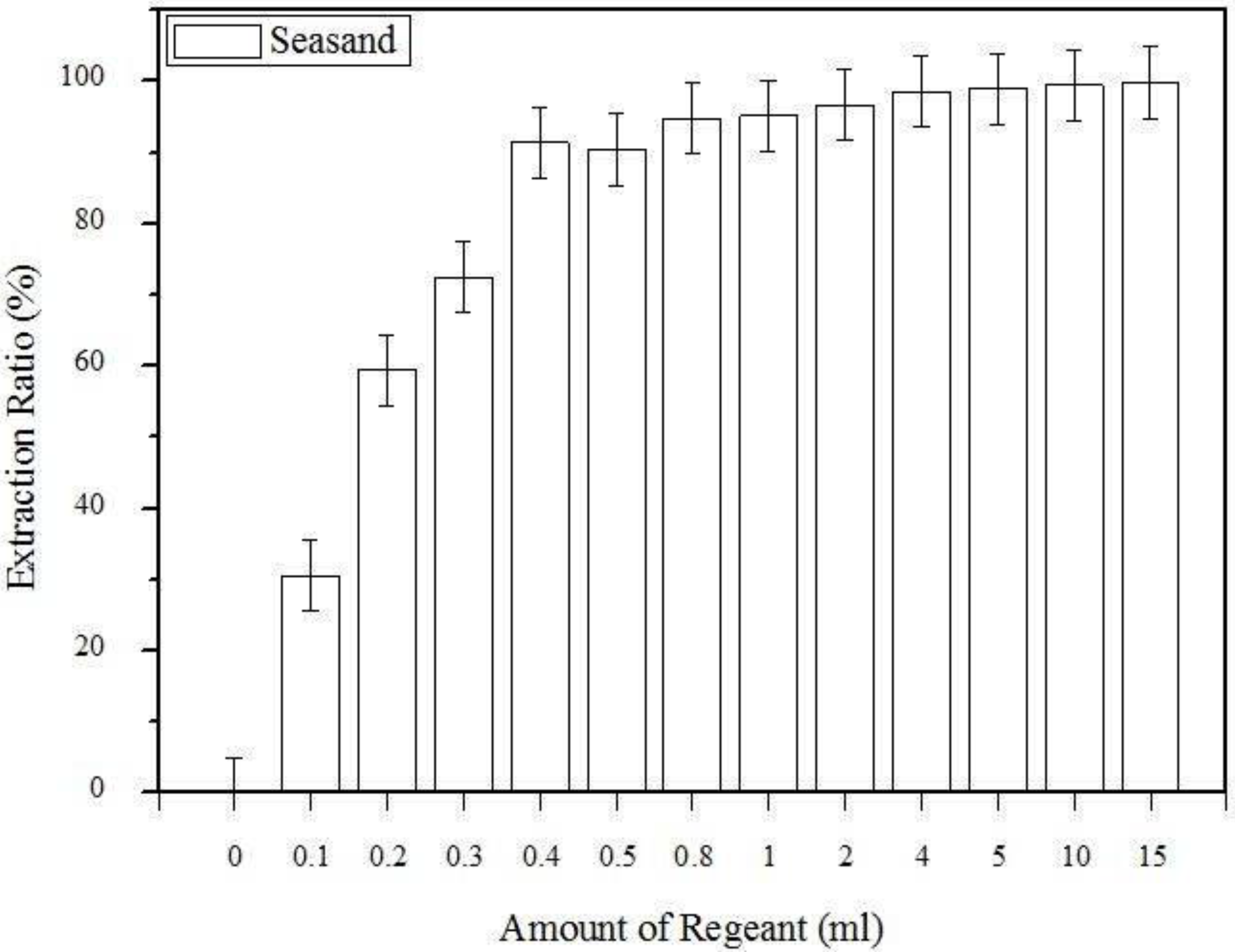
| Amount of the reagent used | Sea sand | 0.1–15 mL |
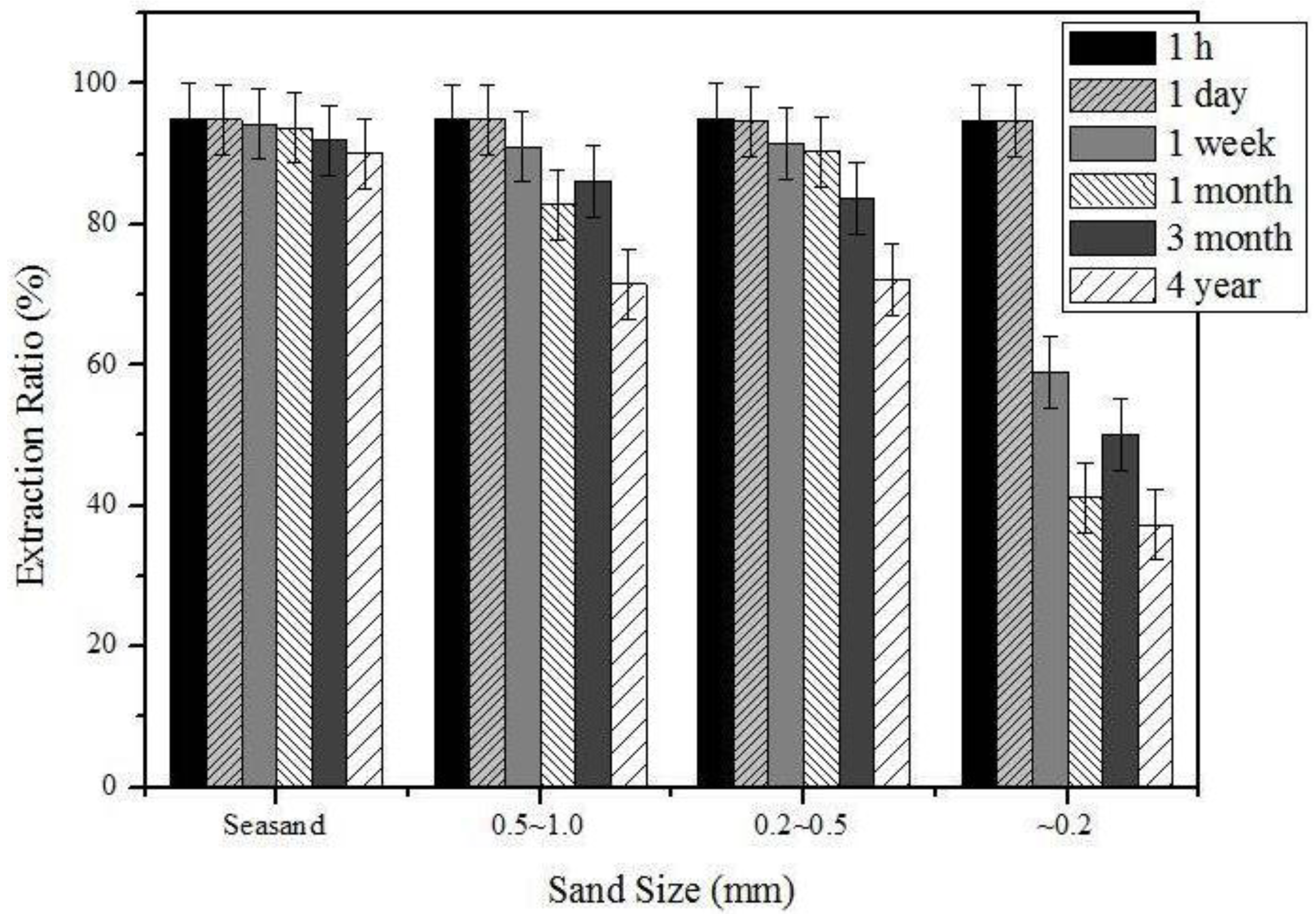
| Sand size (or type) | Sea sand | 1.0 mm (reagent amount: 0.5 mL) |
| Soil sand | Coarse (0.5–1.0 mm), medium (0.2–0.5 mm), fine (less than 0.2 mm) (reagent amount: 0.5 mL) | |
| Elapsed time after sample preparation | Sea sand | 1 h, 1 day, 1 week, 1 month, 4 years (reagent amount: 0.5 mL) |
2.4. Analysis of the Extracted Fraction of Uranium
3. Results and Discussion
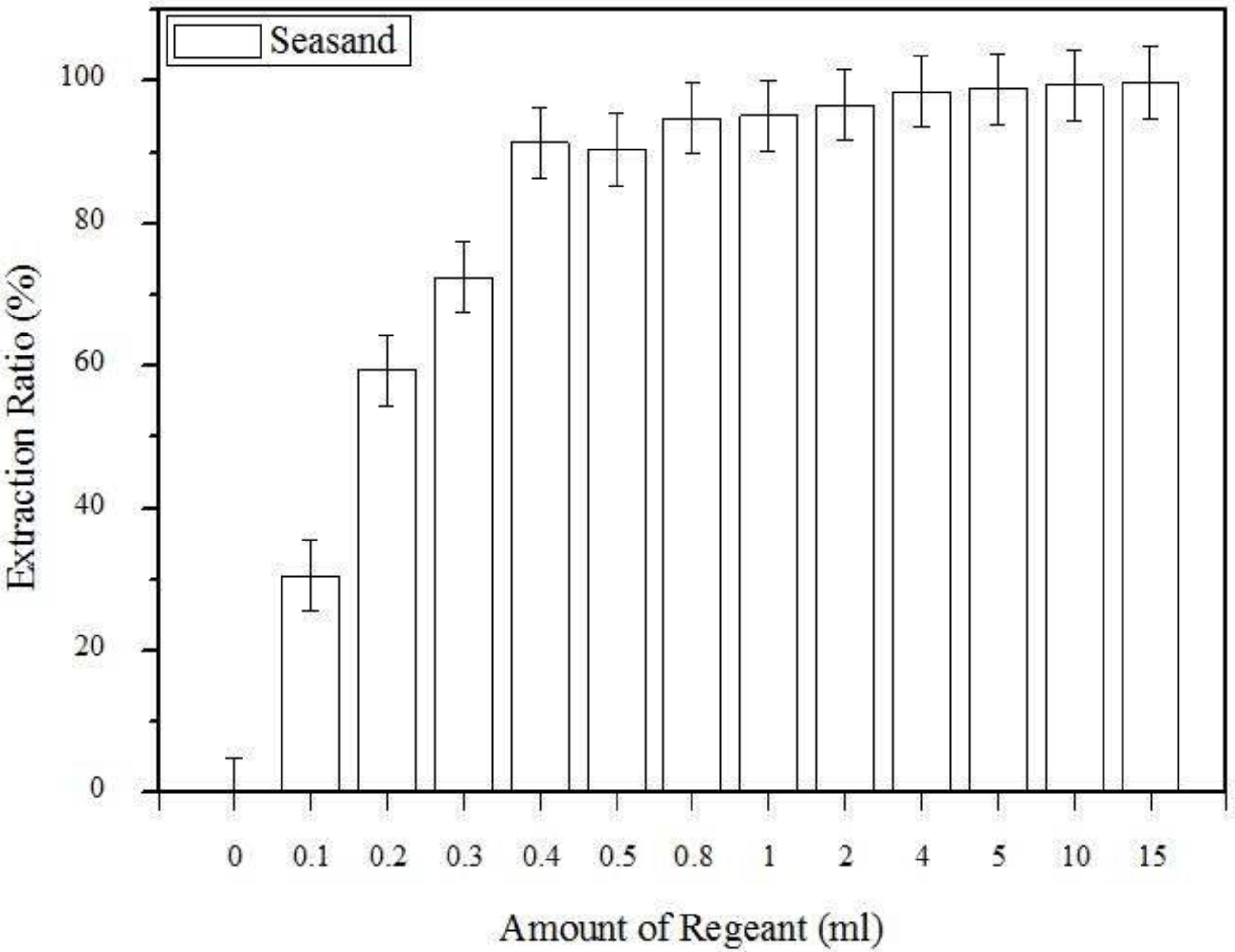
3.1. The Effect of the Amount of Extraction Regent Used
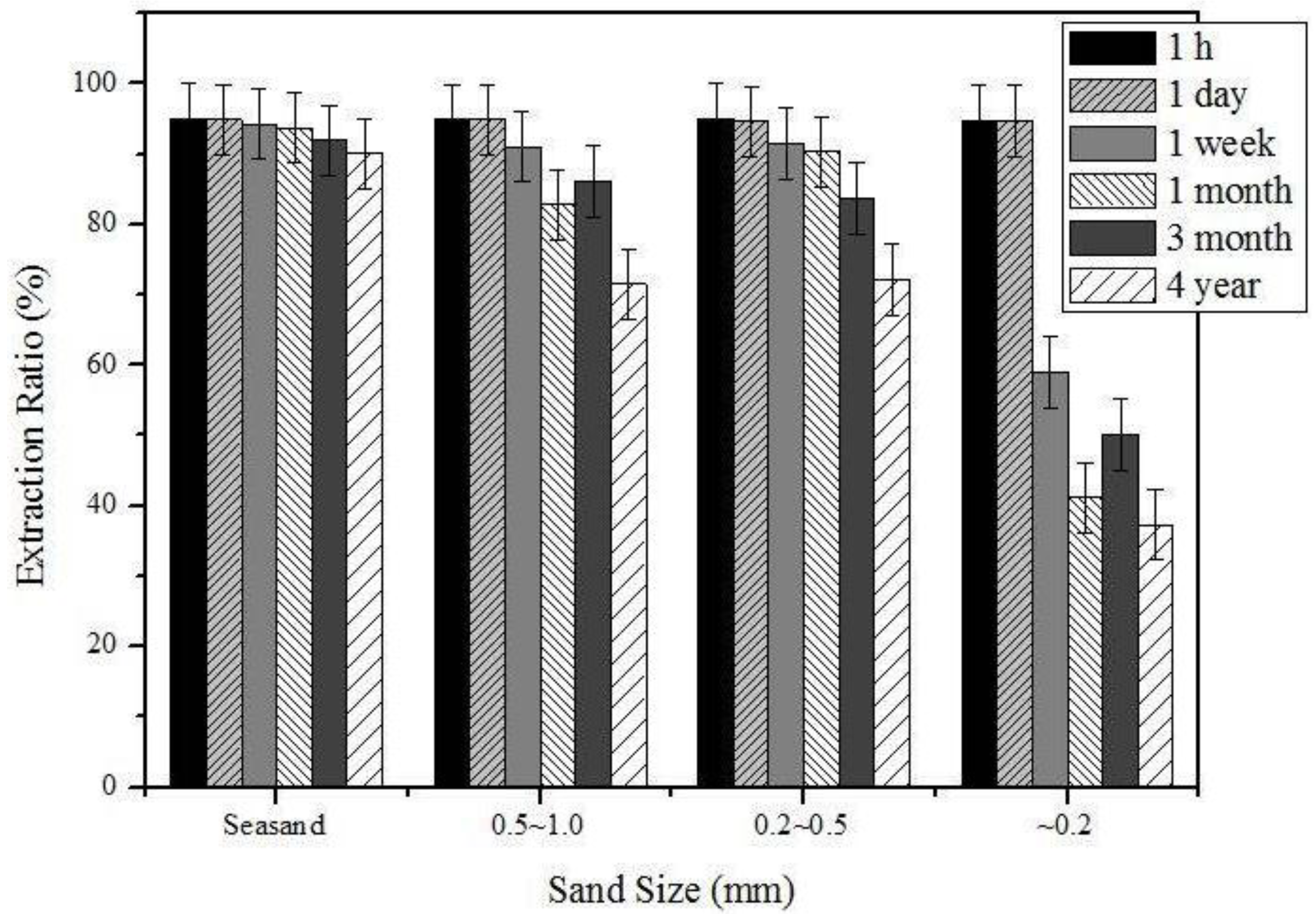
3.2. The Effects of the Sand Type and Elapsed Time after Sample Preparation


4. Conclusions
Acknowledgments
Author Contributions
Conflicts of interest
References
- Steinhauser, G.; Brandl, A.; Johnson, T.E. Comparison of the Chernobyl and Fukushima Nuclear Accidents: A review of the environmental impacts. Sci. Total Environ. 2014, 470–471, 800–817. [Google Scholar] [CrossRef] [PubMed]
- Calmon, P.; Gonze, M.A.; Mourlon, C. Modeling the Early-Phase Redistribution of Radiocesium Fallouts in an Evergreen Coniferous Forest after Chernobyl and Fukushima Accidents. Sci. Total Environ. 2015, 529, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Certini, G.; Scalenghe, R.; Woods, W.I. The Impact of Warfare on the Soil Environment. Earth Sci. Rev. 2013, 127, 1–15. [Google Scholar] [CrossRef]
- Park, K.; Lee, J.; Sung, J. Metal Extraction from the Artificially Contaminated Soil Using Supercritical CO2 with Mixed Ligands. Chemosphere 2013, 91, 616–622. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Jinhyun, S.; Koh, M.; Kim, H.; Kim, H. Decontamination of Radioactive Contaminants Using Liquid and Supercritical CO2. In Radioactive Waste; Rahman, R.A., Ed.; InTech: Rijeka, Croatia, 2012; pp. 219–238. [Google Scholar]
- Benedict, M.; Pigford, T.H.; Levi, H.W. Nuclear Chemical Engineering, 2nd ed.; McGraw-Hill Book Company: New York, NY, USA, 1981. [Google Scholar]
- Lin, Y.; Smart, N.G.; Wai, C.M. Supercritical Fluid Extraction of Uranium and Thorium from Nitric Acid Solutions with Organophosphorus Reagents. Environ. Sci. Technol. 1995, 29, 2706–2708. [Google Scholar] [CrossRef] [PubMed]
- McHardy, J.; Sawan, S.P. Supercritical Fluid Cleaning: Fundamentals, Technology and Applications; Noyes: Westwood, NJ, USA, 1998. [Google Scholar]
- Park, K.H.; Kim, H.W.; Kim, H.D.; Koh, M.S.; Ryu, J.D.; Kim, Y.E.; Lee, B.S.; Park, H.T. Application of CO2 Technology in Nuclear Decontamination. Han’guk Pyomyon Konghak Hoechi 2001, 34, 62–67. [Google Scholar]
- Carrott, M.J.; Waller, B.E.; Smart, N.G.; Wai, C.M. High solubility of UO2(NO3)2·2TBP complex in supercritical CO2. Chem. Commun. 1998. [Google Scholar] [CrossRef]
- Shimada, T.; Ogumo, S.; Ishihara, N.; Kosaka, Y.; Mori, Y. A Study on the Technique of Spent Fuel Reprocessing with Supercritical Fluid Direct Extraction Method (super-DIREX method). J. Nucl. Sci. Technol. 2002, 39, 757–760. [Google Scholar] [CrossRef]
- Erkey, C. Supercritical Carbon Dioxide Extraction of Metals from Aqueous Solutions: A Review. J. Supercrit. Fluids 2000, 17, 259–287. [Google Scholar] [CrossRef]
- Enokida, Y.; Yamamoto, I. Vapor-liquid Equilibrium of UO2(NO3)2·2TBP and Supercritical Carbon Dioxide Mixture. J. Nucl. Sci. Technol. 2002, 9, 270–273. [Google Scholar] [CrossRef]
- Tamika, O.; Meguro, Y.; Enokida, Y.; Yamamoto, I.; Yoshida, Z. Dissolution Behavior of Uranium Oxides with Supercritical CO2 Using HNO3–TBP Complex as a Reactant. J. Nucl. Sci. Technol. 2001, 38, 1097–1102. [Google Scholar] [CrossRef]
- Meguro, Y.; Iso, S.; Yoshida, Z.; Tomioka, O.; Enokida, Y.; Yamamoto, I. Decontamination of uranium Oxides from Solid Wastes by Supercritical CO2 Fluid Leaching Method Using HNO3–TBP Complex as a Reactant. J. Supercrit. Fluids 2004, 31, 141–147. [Google Scholar] [CrossRef]
- Sawada, K.; Uruga, K.; Koyama, T.; Shimada, T.; Mori, Y.; Enokida, Y.; Yamanoto, I. Stoichiometric relation for Extraction of Uranium from UO2 Powder Using TBP Complex with HNO3 and H2O in Supercritical CO2. J. Nucl. Sci. Technol. 2005, 42, 301–304. [Google Scholar] [CrossRef]
- Buol, S.W.; Southard, R.J.; Graham, R.C.; McDaniel, P.A. Soil Genesis and Classification, 6th ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2011. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, K.; Jung, W.; Park, J. Decontamination of Uranium-Contaminated Soil Sand Using Supercritical CO2 with a TBP–HNO3 Complex. Metals 2015, 5, 1788-1798. https://doi.org/10.3390/met5041788
Park K, Jung W, Park J. Decontamination of Uranium-Contaminated Soil Sand Using Supercritical CO2 with a TBP–HNO3 Complex. Metals. 2015; 5(4):1788-1798. https://doi.org/10.3390/met5041788
Chicago/Turabian StylePark, Kwangheon, Wonyoung Jung, and Jihye Park. 2015. "Decontamination of Uranium-Contaminated Soil Sand Using Supercritical CO2 with a TBP–HNO3 Complex" Metals 5, no. 4: 1788-1798. https://doi.org/10.3390/met5041788