
The Promising Features of New Nano Liquid Metals—Liquid Sodium Containing Titanium Nanoparticles (LSnanop)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
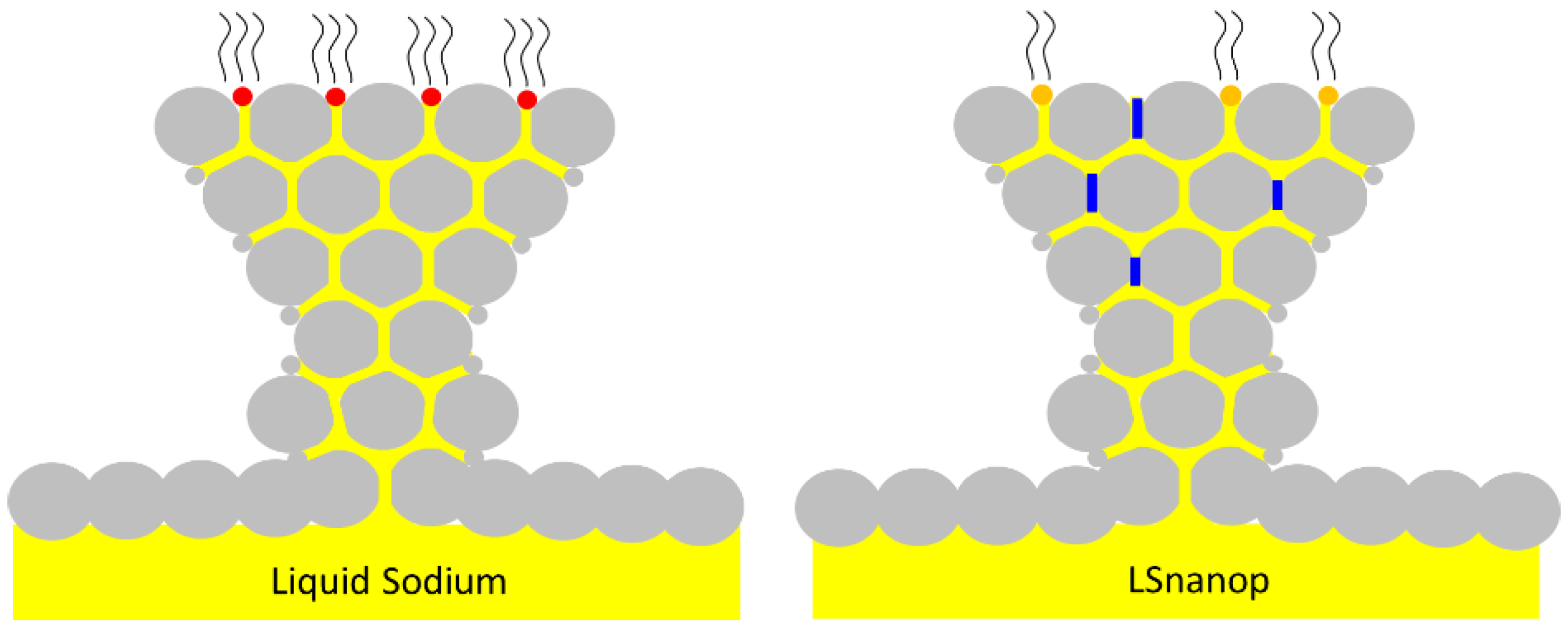
2. The Design of LSnanop

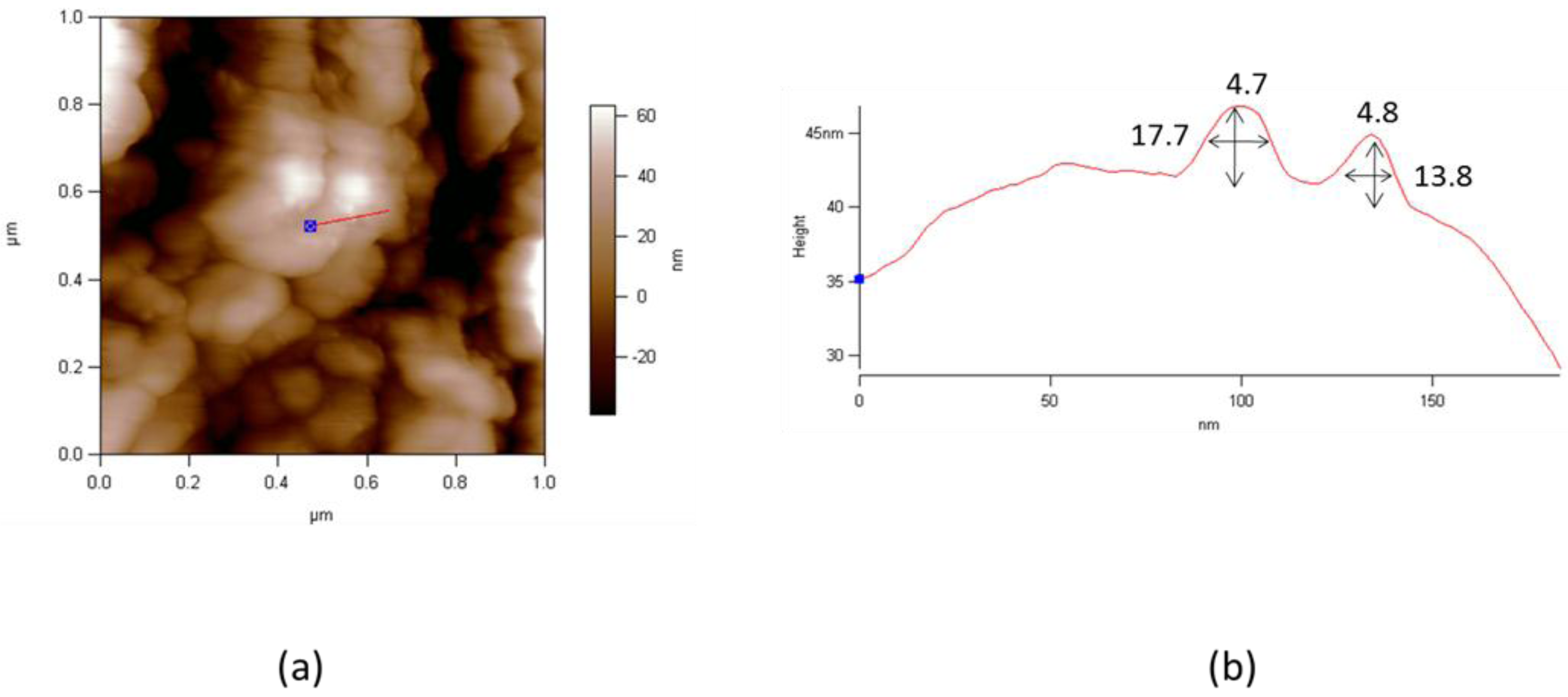
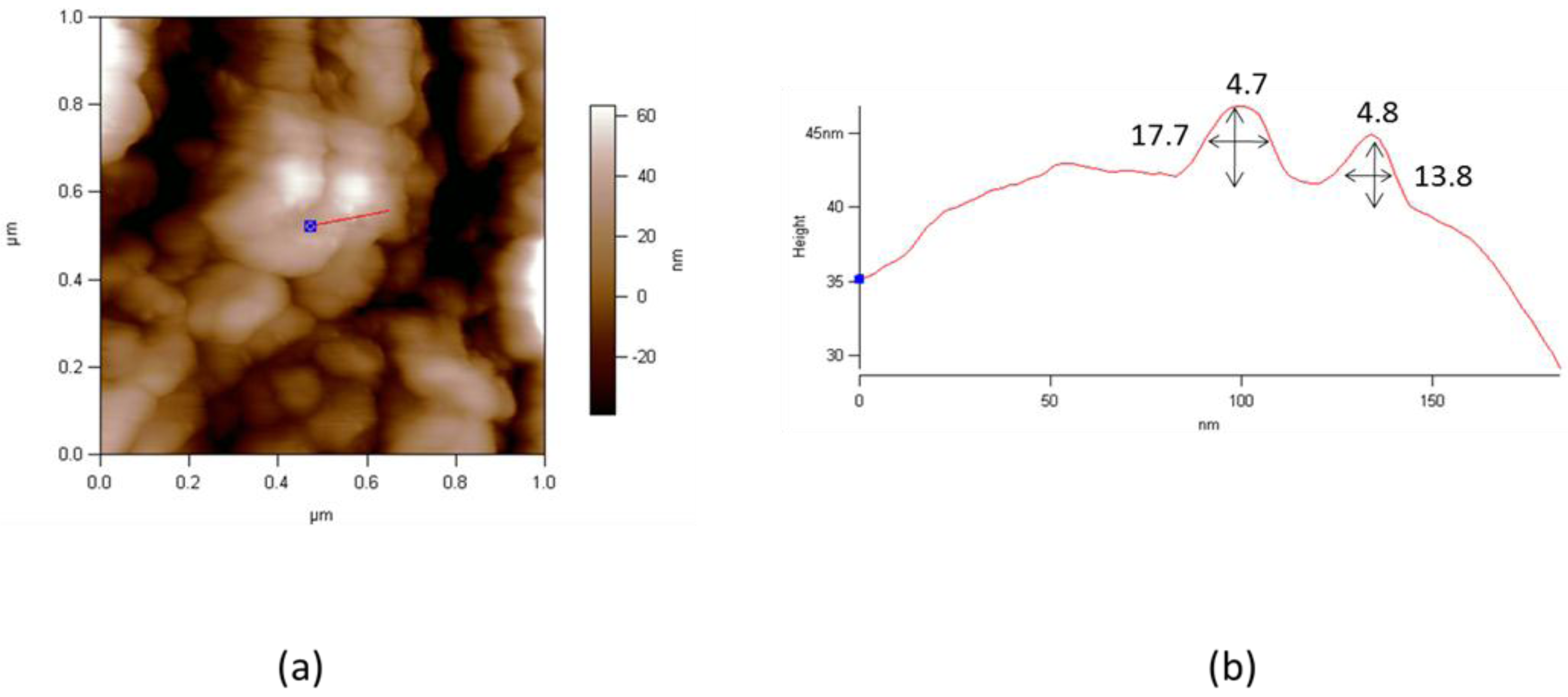
3. Preparation and Characterization of LSnanop

4. Fundamental View Point of LSnanop
5. Physicochemical Properties of LSnanop
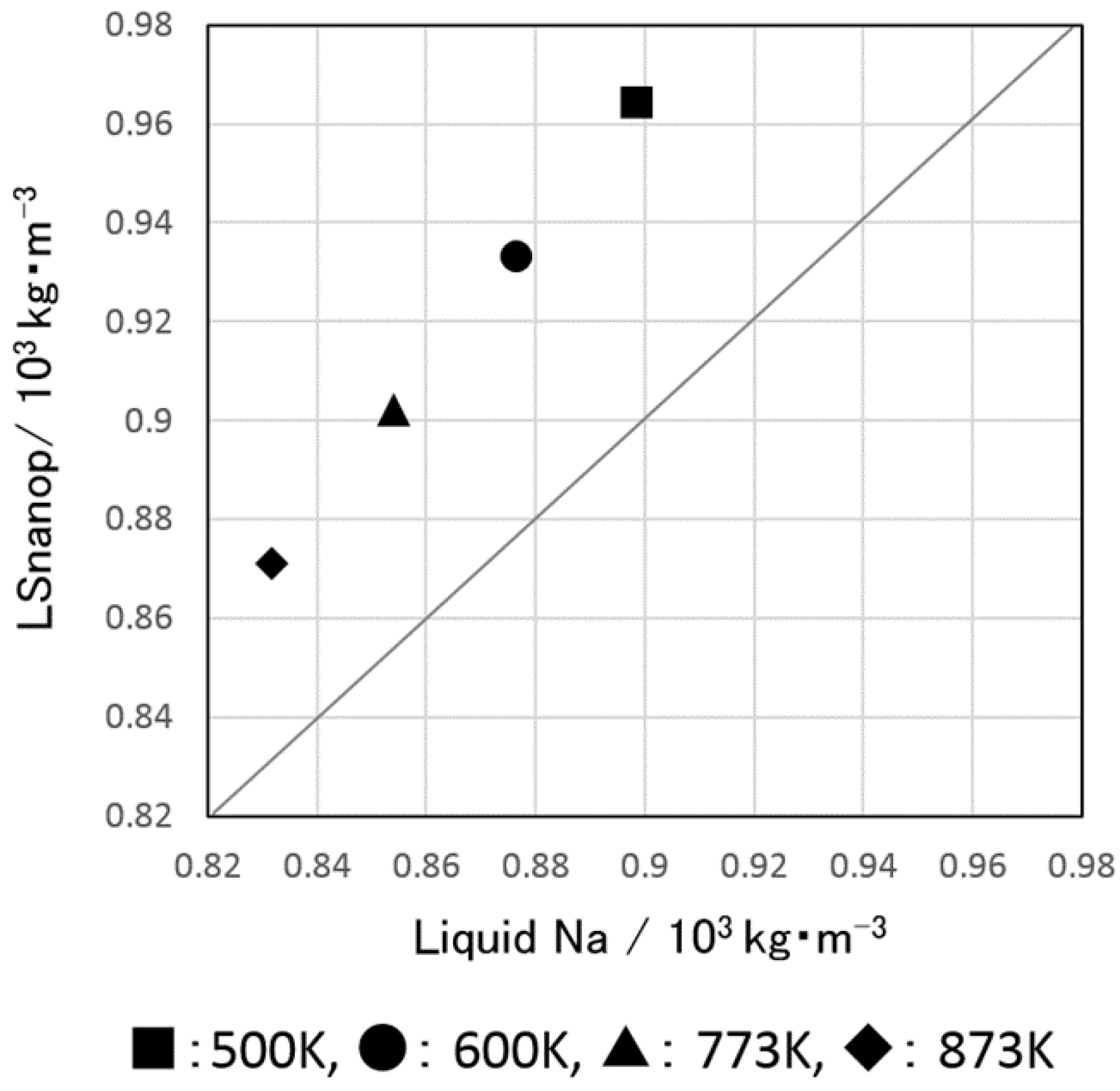
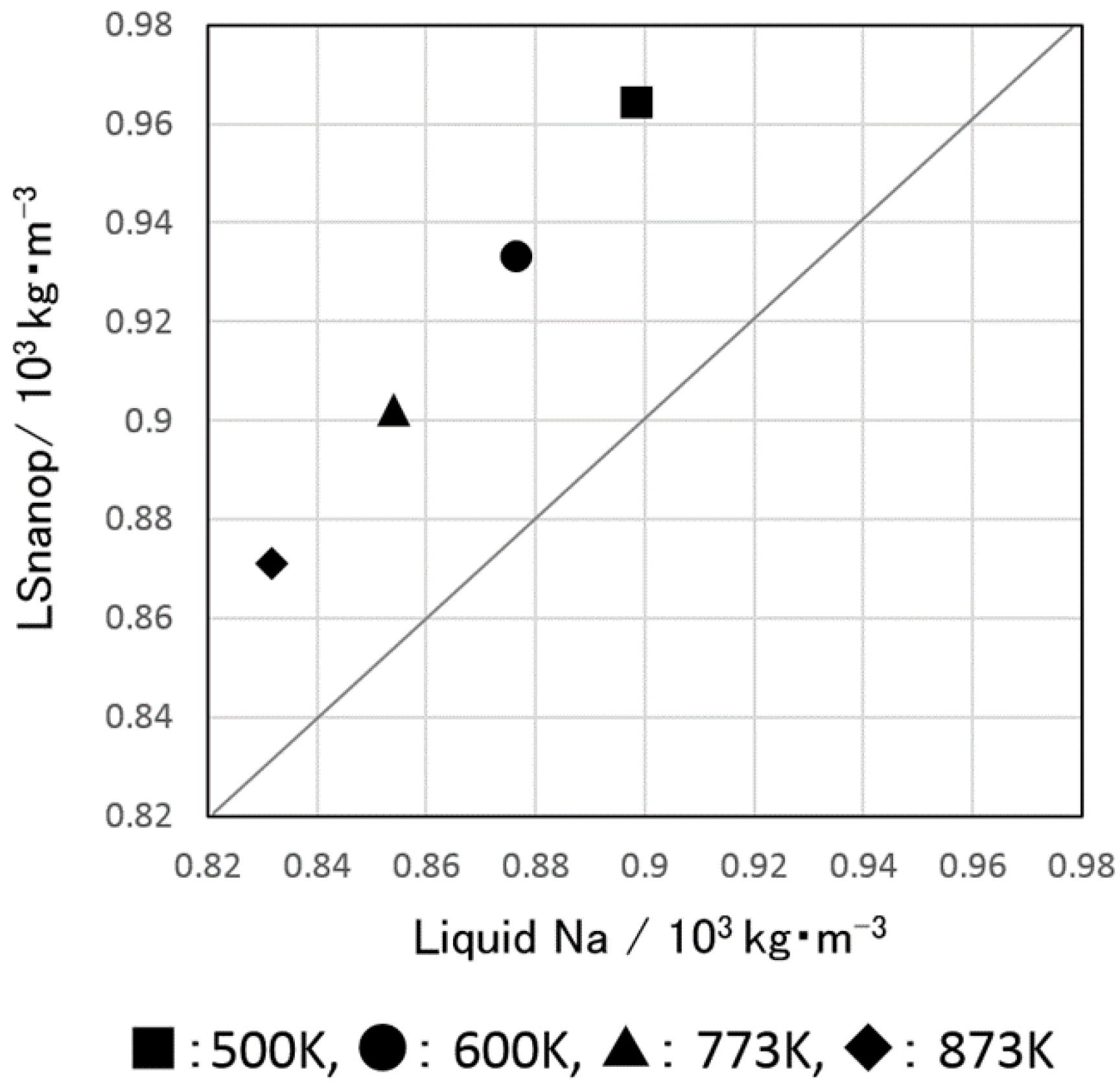
5.1. Density and Atomic Volume of LSnanop
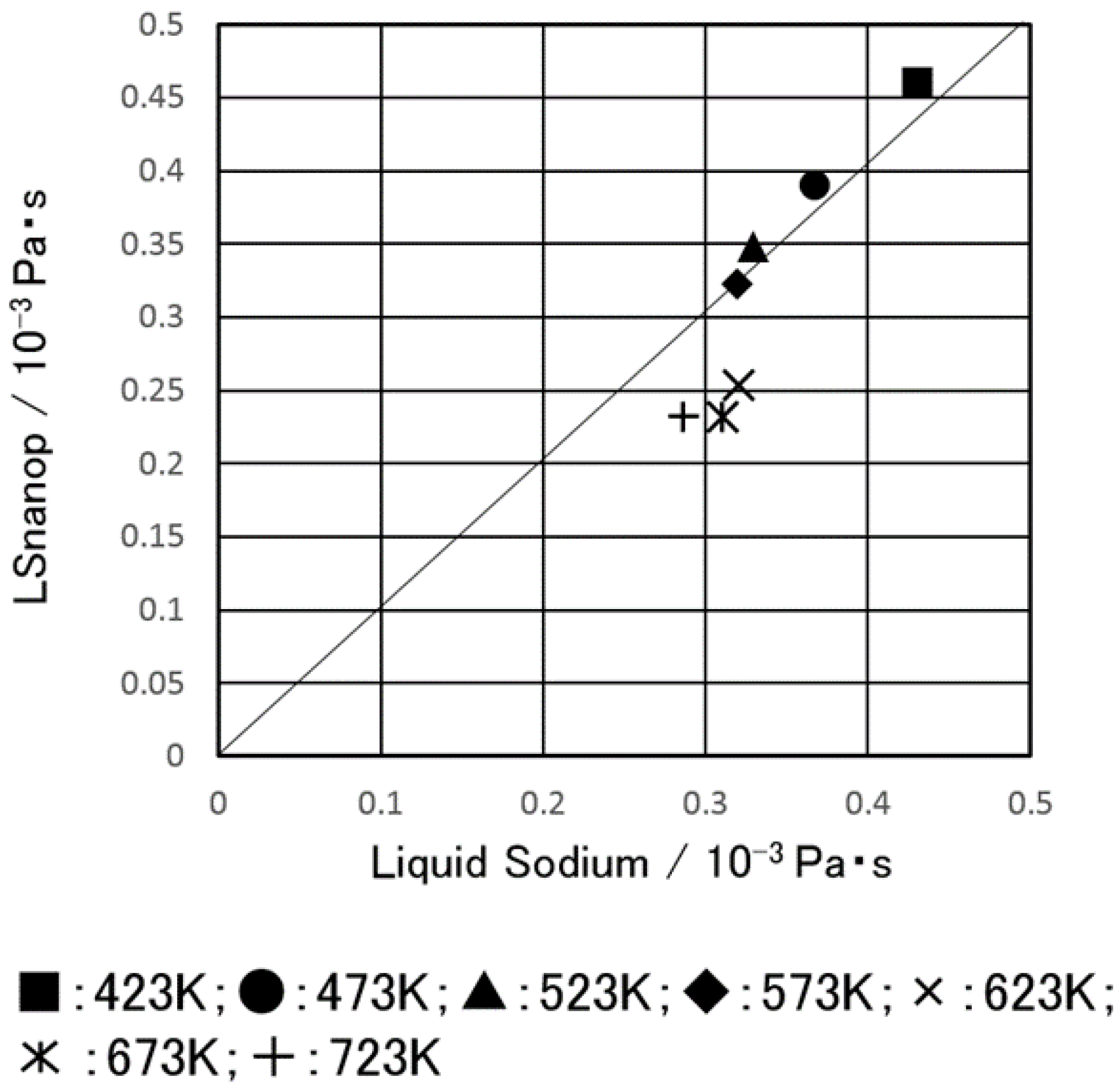
5.2. Shear Viscosity of LSnanop

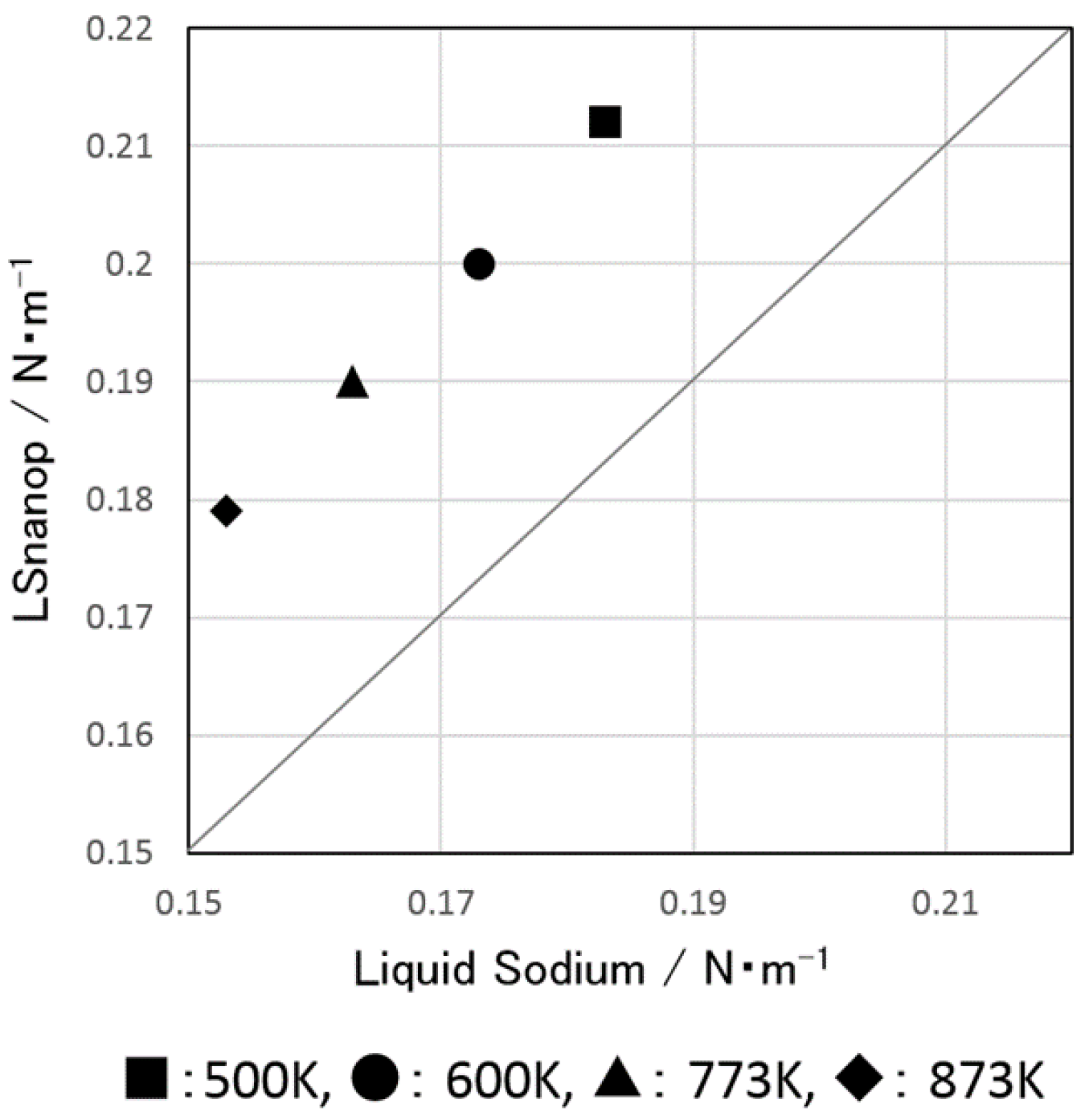
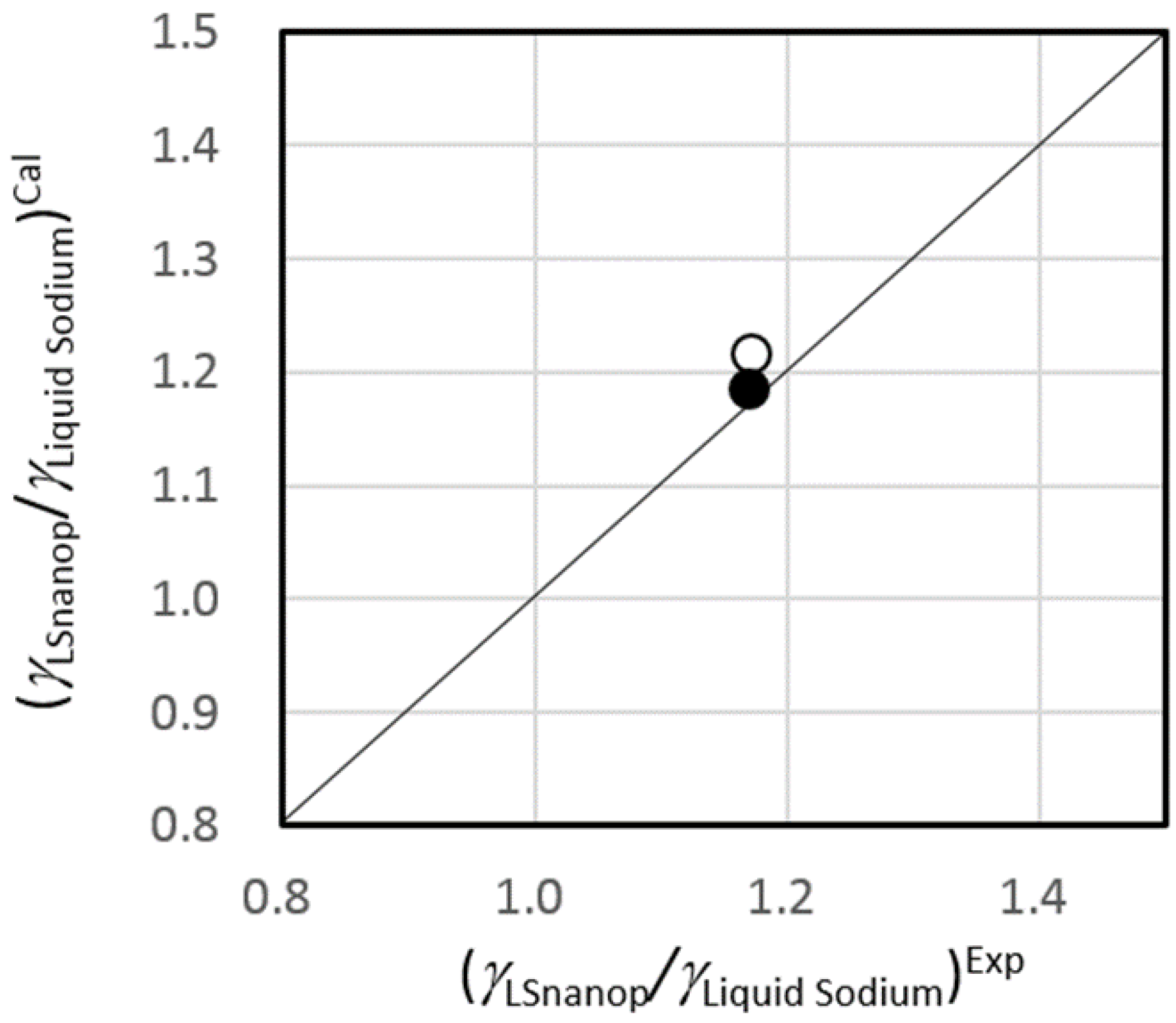
5.3. Surface Tension of LSnanop
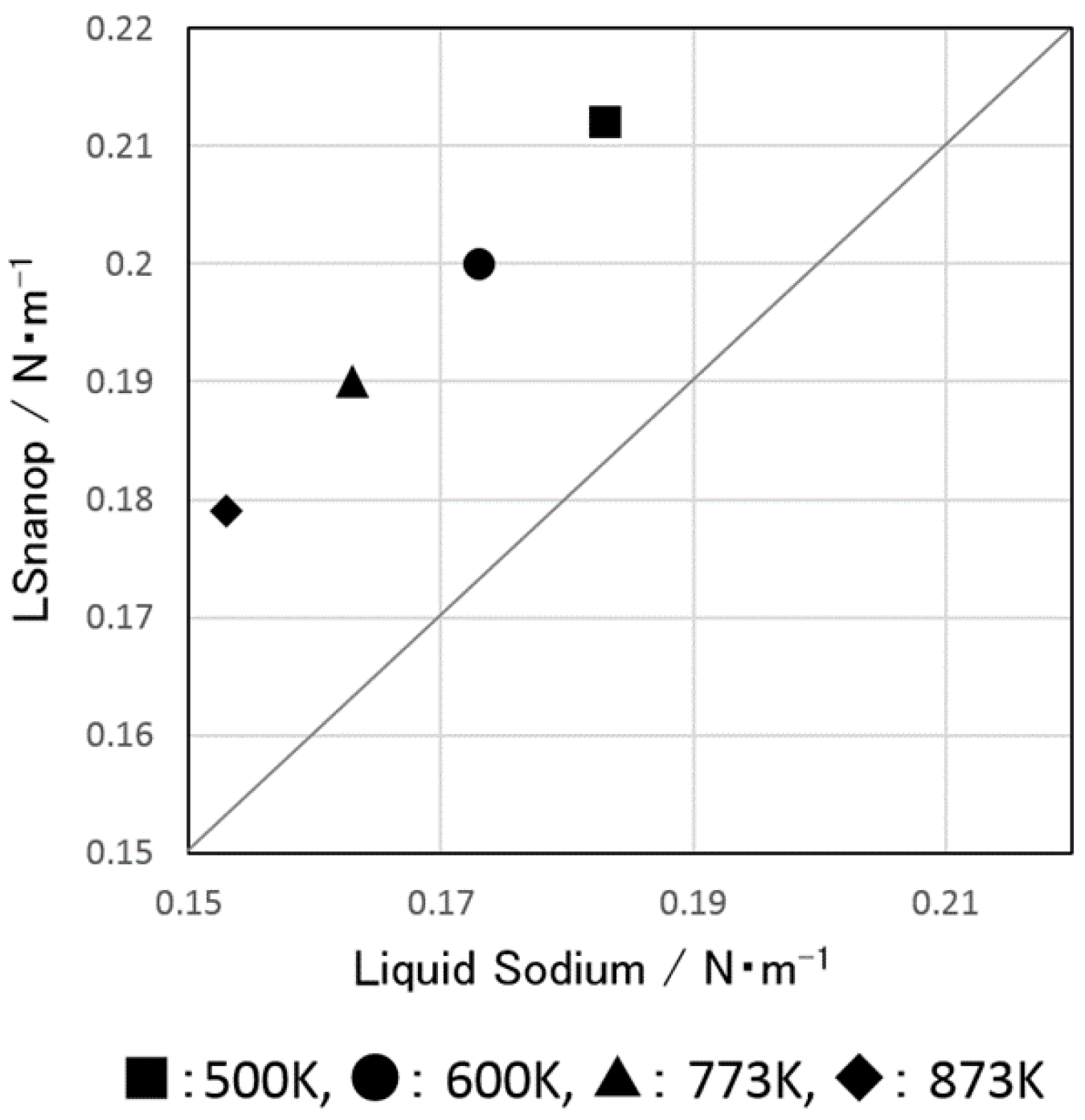
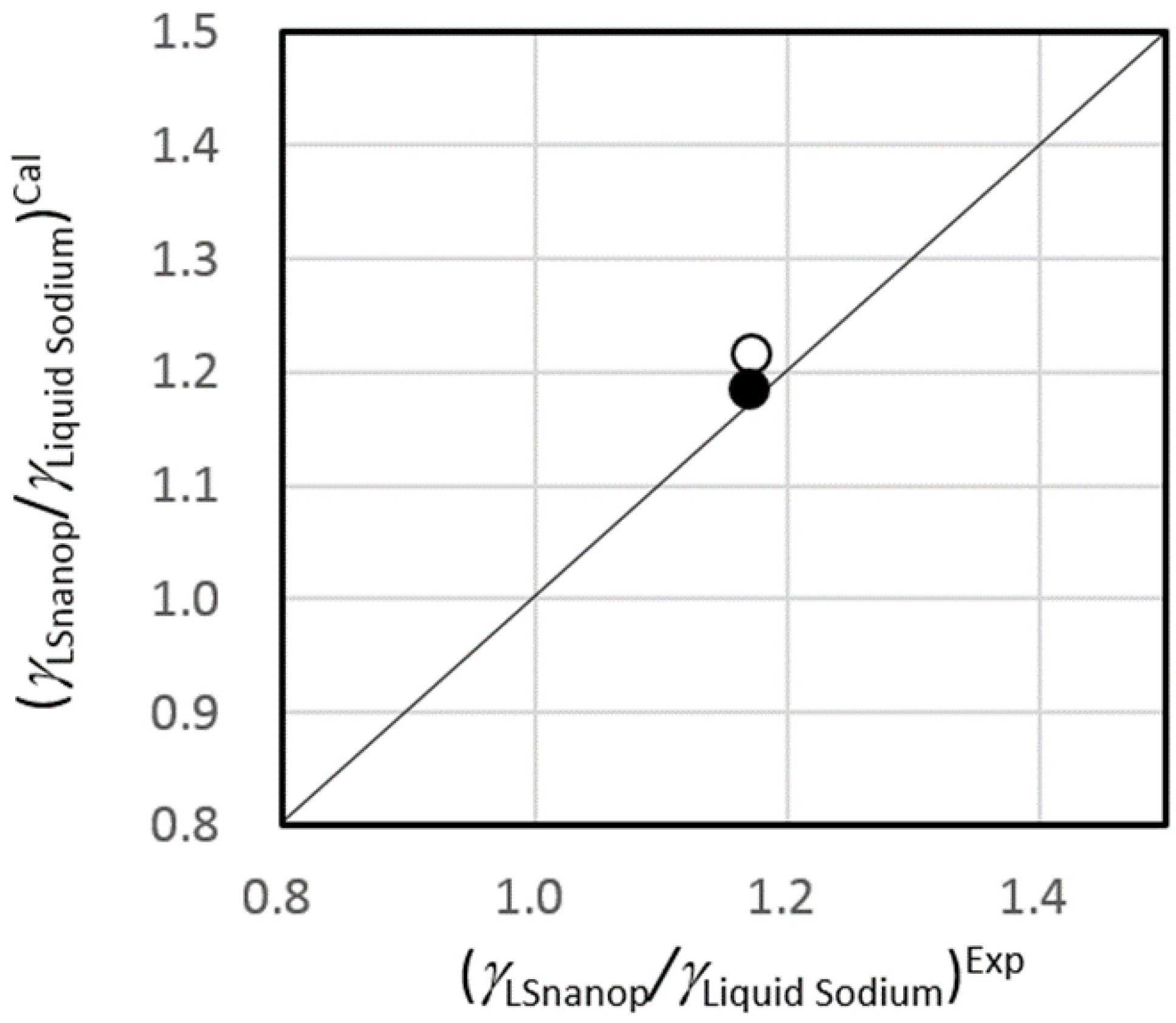
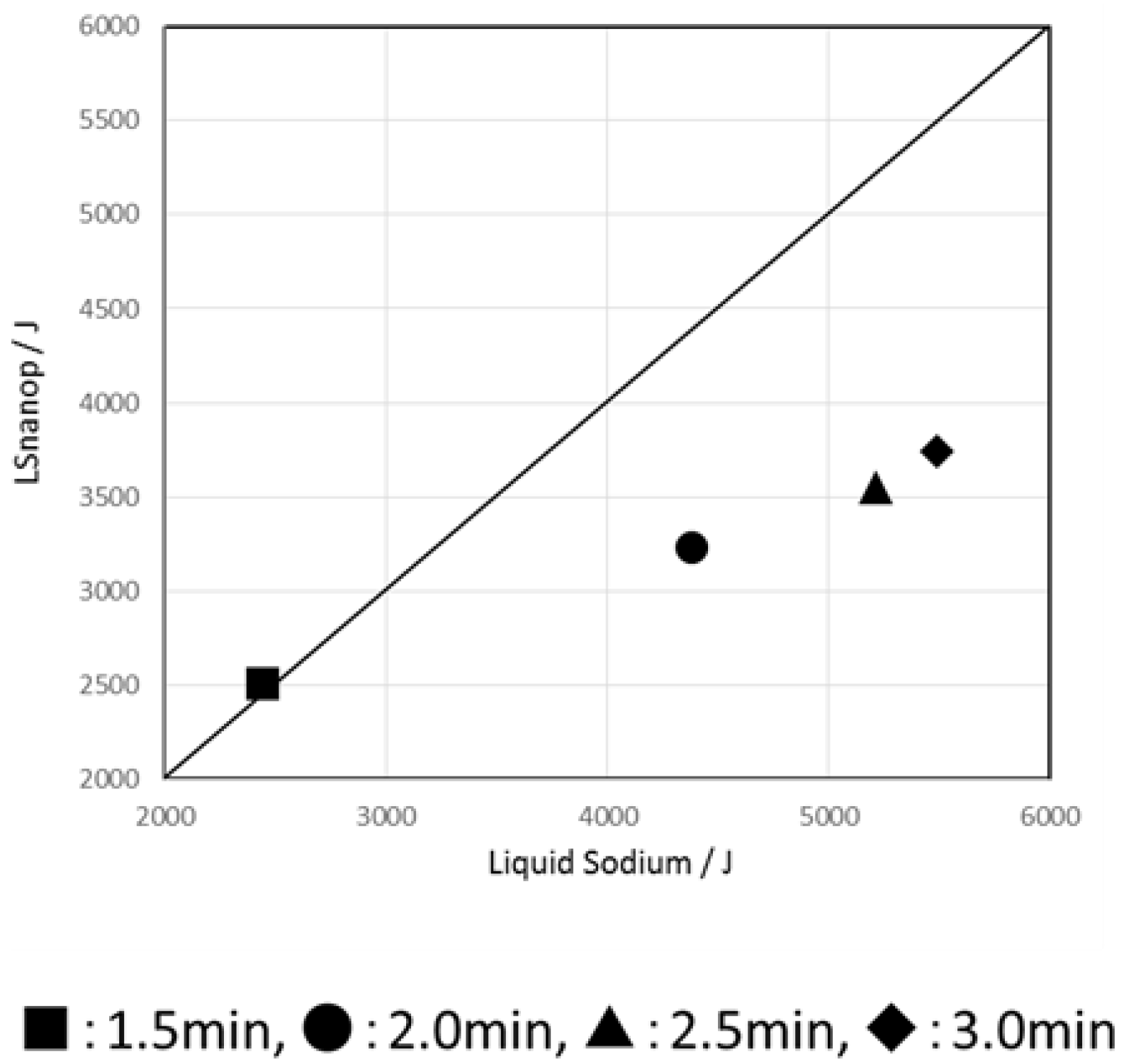
5.4. The Heat of Reaction to Water of LSnanop

6. Excess Cohesive Energy of LSnanop
6.1. The Determination of Excess Cohesive Energy of LSnanop from the Experimental Data of the Heat of Reaction to Water
6.2. Theoretical Analysis of Excess Cohesive Energy of LSnanop
6.2.1. The Analysis Based on the Born Model
6.2.2. The Screened Nanoparticles with Negative Charge in Liquid Sodium (SNP in Liquid Na)
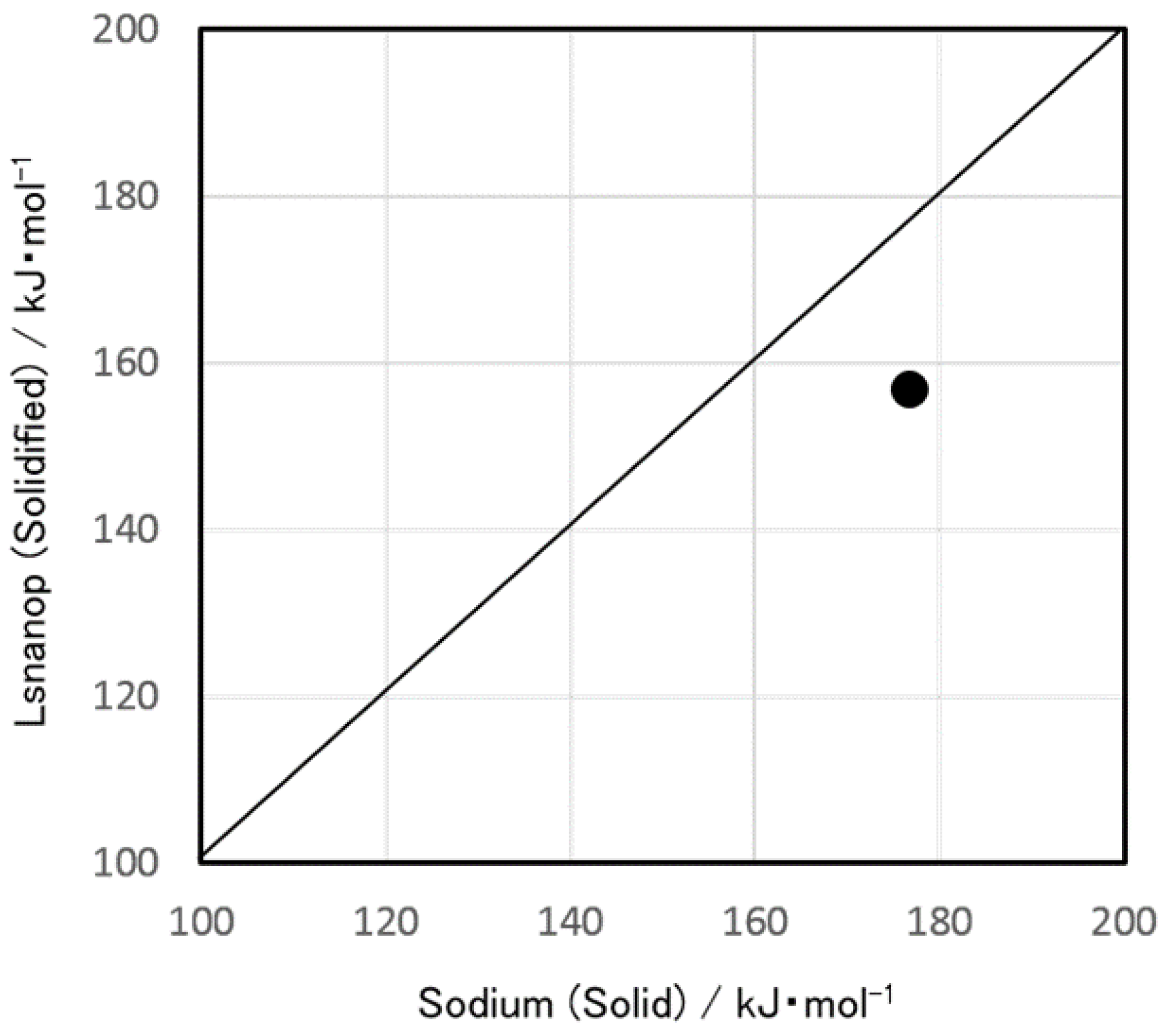
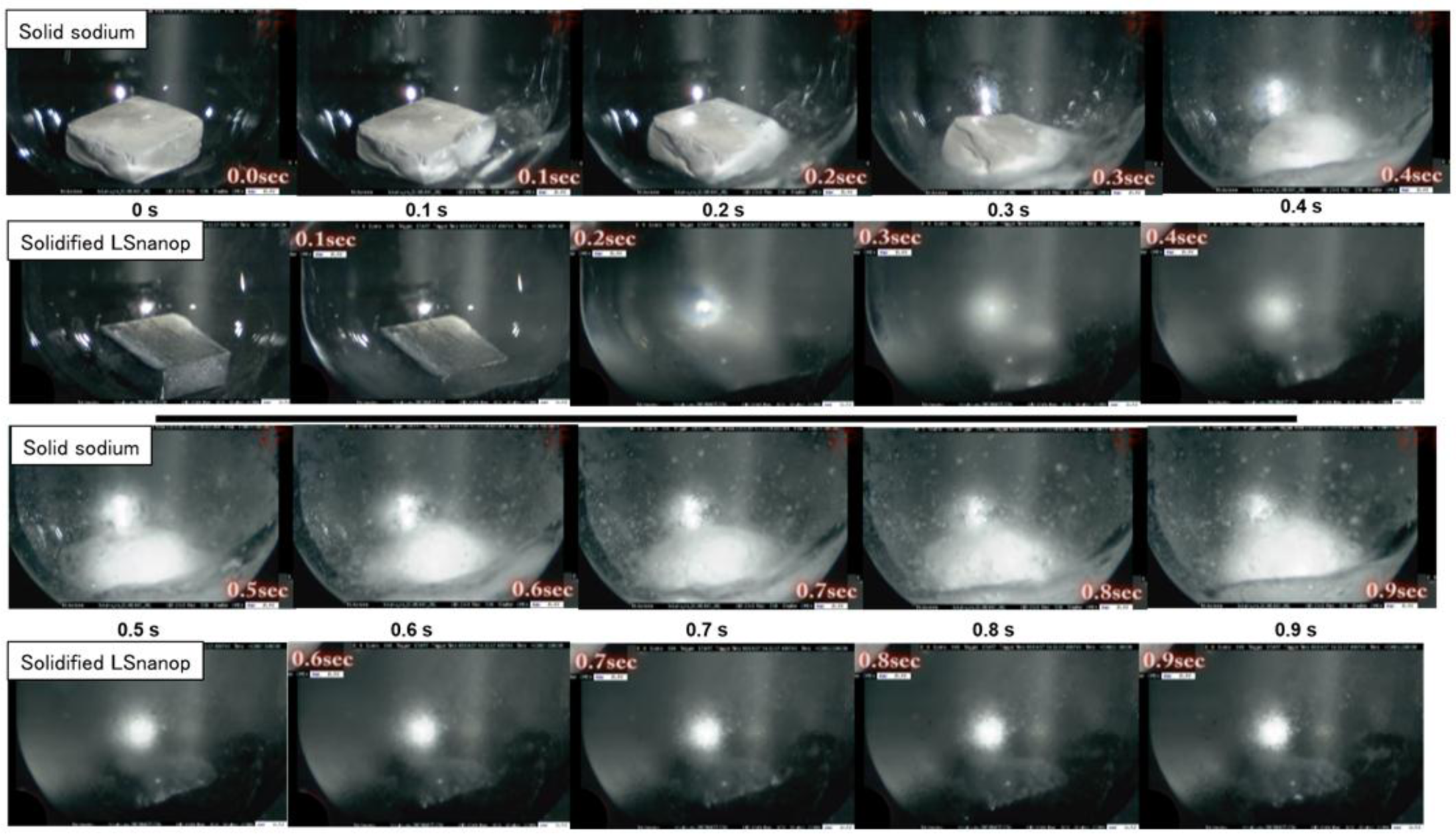
- Experimentally, a difference of heat of reaction to water was found between solidified LSnanop and solid Na. This experimental result is reliable, judging from the reproducibility of experiments and the preliminary error analysis of the thermal analysis.
- A similar decrease in the heat of reaction to water of LSnanop was also confirmed experimentally for the samples in a liquid state.
- The decrease of heat of reaction to water of LSnanop seems to be derived, not from the raise of the enthalpy of final state, but from the lowering of the enthalpy of initial state before this reaction.
- Based on Hess’ law, this difference is thought to be derived from the existence of excess cohesive energy for the LSnanop. Hess’ law is a rigorous law in thermodynamics based only on the fact that the enthalpy is a quantity of state.
- The experimentally estimated excess cohesive energy can be explained theoretically by the Born model and the “SNP in liquid Na” model. The transfer of electrons to Ti nanoparticles from Na atoms, which is the fundamental viewpoint of these models, is revealed by the ab initio DFT calculation [12] and ab initio MD simulation [9].
7. The Suppression of Reaction to Water and Oxygen of LSnanop
7.1. The Suppression of Reaction to Water

7.2. The Suppression of Reaction of LSnanop to Oxygen

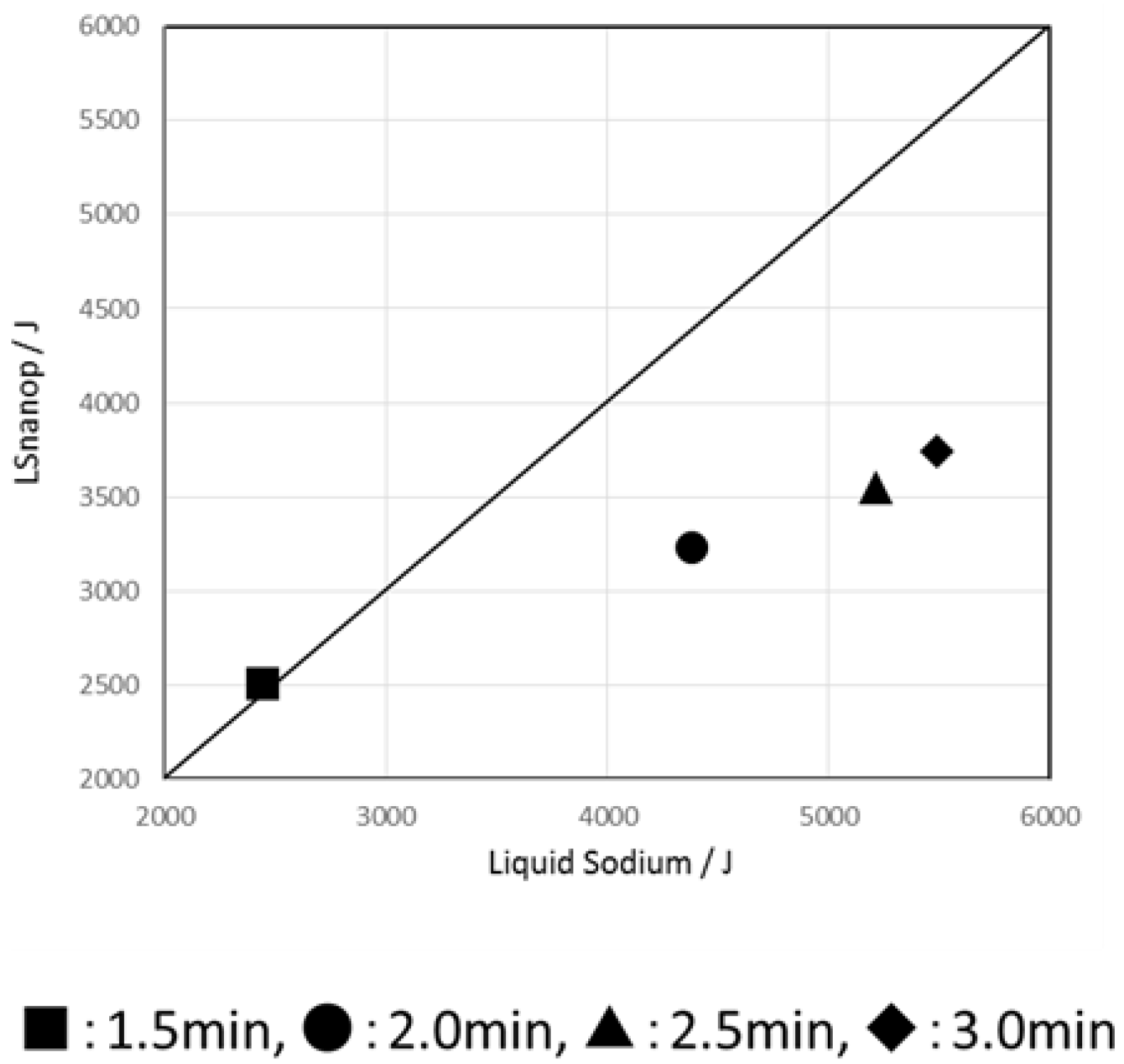

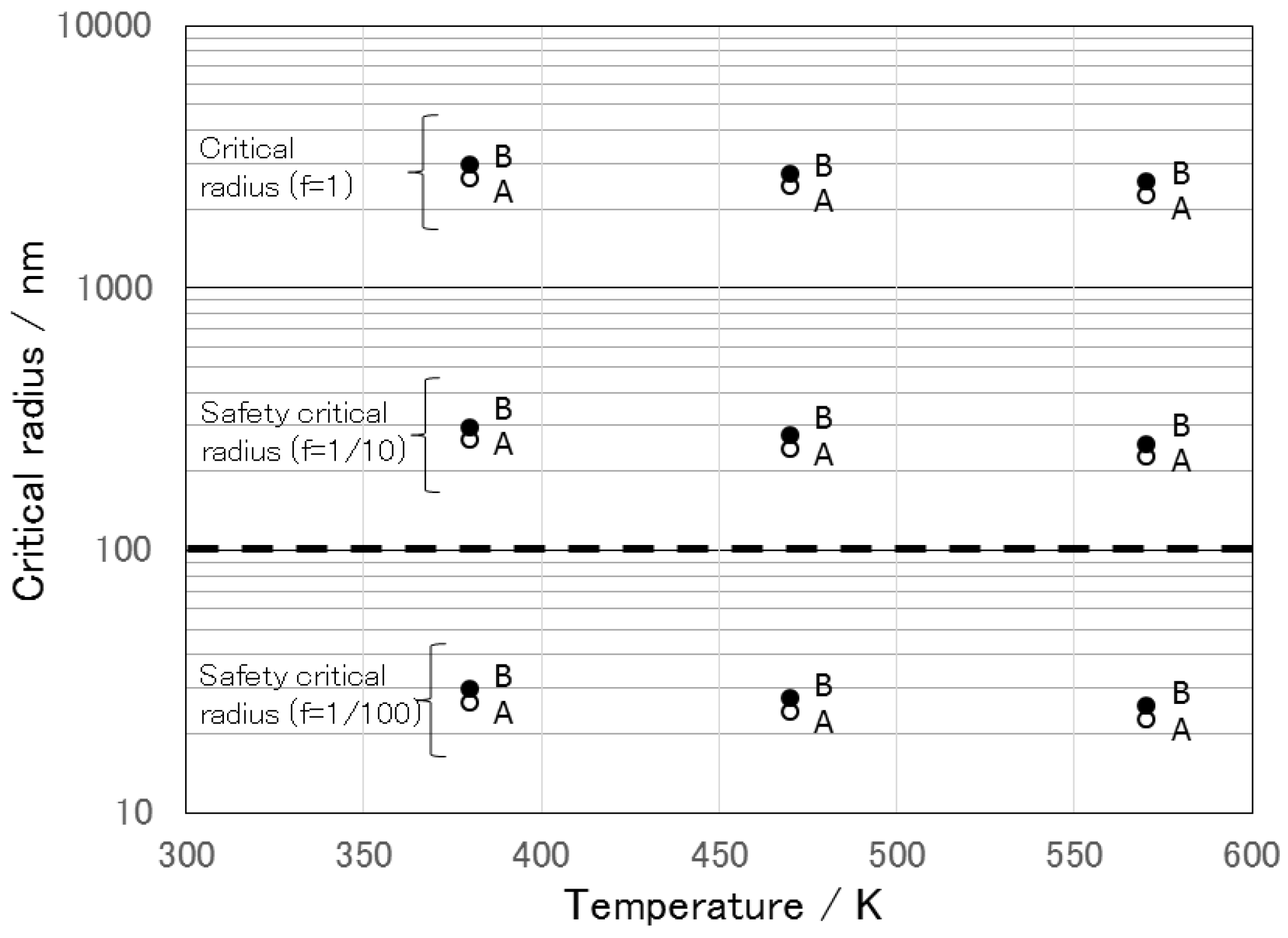
8. The Stability of LSnanop
9. Conclusions
Acknowledgments
Conflicts of Interest
References
- Faber, T.E. Introduction to the Theory of Liquid Metals; Cambridge Univ. Press: London, UK, 1972. [Google Scholar]
- Shimoji, M. Liquid Metals; Academic Press: London, UK, 1977. [Google Scholar]
- Cusack, N.E. The Physics of Structurally Disordered Matter: Introduction; Adam Hilger: Bristol, UK, 1987. [Google Scholar]
- Ziman, J.M. A Theory of the Electronic Properties of Liquid Metals I: The Monovalent Metals. Philos. Mag. 1961, 6, 1013–1034. [Google Scholar] [CrossRef]
- Das, S.K.; Choi, S.U.S.; Yu, W.; Pradeep, T. Nanofluids Science and Technology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008. [Google Scholar]
- Keeting, L.; Charles, S.W.; Popplewell, J. The prevention of diffusional growth of cobalt particles in mercury. J. Phys. F Metal Phys. 1984, 14, 3093–3100. [Google Scholar] [CrossRef]
- Abu-Aljarayesh, I.; Abu-Libdeh, A. Initial susceptibility of iron in mercury magnetic fluids. J. Magn. Magn. Mater. 1991, 96, 89–123. [Google Scholar] [CrossRef]
- Massart, J.R.; Rasolonjatovo, B.; Neveu, S.; Cabuil, V. Mercury-based cobalt magnetic fluids and cobalt nanoparticles. J. Magn. Magn. Mater. 2007, 308, 10–14. [Google Scholar] [CrossRef]
- Ara, K. Development of chemical reaction suppression technology of liquid sodium metal based on nanotechnology. In Annual Report of Japan Atomic Energy Agency by the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT); The Ministry of Education, Culture, Sports, Science and Technology of Japan: Tokyo, Japan, 2007. [Google Scholar]
- Ara, K.; Sugiyama, K.; Kitagawa, H.; Nagai, M.; Yoshioka, N. Study on chemical reactivity control of sodium by suspended nanoparticles I. J. Nucl. Sci. Technol. 2010, 47, 1165–1170. [Google Scholar] [CrossRef]
- Ara, K.; Sugiyama, K.; Kitagawa, H.; Nagai, M.; Yoshioka, N. Study on chemical reactivity control of sodium by suspended nanoparticles II. J. Nucl. Sci. Technol. 2010, 47, 1171–1181. [Google Scholar] [CrossRef]
- Saito, J.; Ara, K. A study of atomic interaction between suspended nanoparticles and sodium atoms in liquid sodium. Nucl. Eng. Des. 2010, 240, 2664–2673. [Google Scholar] [CrossRef]
- Saito, J.; Itami, T.; Ara, K. The preparation, physicochemical properties and the cohesive energy of liquid sodium containing titanium nanoparticles. J. Nanoparticle Res. 2012, 14, 1298–1315. [Google Scholar] [CrossRef]
- Nishimura, M.; Nagai, K.; Onojima, T.; Saito, J.; Ara, K.; Sugiyama, K. The sodium oxidation reaction and suppression effect of sodium with nanoparticles-growth behavior of dendritic oxide during oxidation. J. Nucl. Sci. Technol. 2012, 49, 71–77. [Google Scholar] [CrossRef]
- Kim, S.J.; Park, G.; Park, H.S.; Kim, M.H.; Baek, J. Dispersion behavior of 10 nm Ti nanoparticle in liquid Na: Theoretical approach. In Proceedings of the Ninth Korea-Japan Symposium on Nuclear Hydraulic and Safety, Buyeo, Korea, 16–19 November 2014.
- Darken, L.S.; Gurry, R.W. Physical Chemistry of Metals; McGrow-Hill Inc.: New York, NY, USA, 1953; p. 349. [Google Scholar]
- Goto, K. Magnetic Fluids in the Forefront of Physics No.23; Kyoritsu Pub.: Tokyo, Japan, 1989; p. 49. [Google Scholar]
- Bird, B.B.; Stewart, W.E.; Lightfoot, E.D. Transport Phenomena; John Wiley & Sons Inc.: New York, NY, USA, 1969; p. 513. [Google Scholar]
- Shimoji, M.; Itami, T. Atomic Transport in Liquid Metals; Trans Tech Pub.: Aedermannsdorf, Switzerland, 1986; p. 32. [Google Scholar]
- Fukunaga, K.; Ogata, K.; Nagai, M.; Oka, N.; Ara, K.; Saito, J.; Kitagawa, H.; Yamauchi, M. Influence factor of the nanoparticles generation by the flash vaporization method. In Proceeding of the 2008 Fall Meeting of Japan Institute of Metal, Kumamoto, Japan, 23–25 September 2008; p. 586.
- Japan Institute Metals. Metal Data Book, Revised ed.; Trans. JIM: Sendai, Japan, 1993. [Google Scholar]
- Waseda, Y. The Structure of Non-Crystalline Materials; McGrow Hill Inc.: New York, NY, USA, 1980; p. 58. [Google Scholar]
- Shpil’rain, E.E.; Yakimovich, K.A.; Fomin, V.A.; Skovorodjko, S.N.; Mozgovoi, A.G. Density and thermal expansion of liquid alkali metals. In Handbook of Thermodynamic and Transport Properties of Alkali Metals; Ohse, R.W., Ed.; Int. Union of Pure and Appl. Chemistry, Blackwell Scientific Pub.: Oxford, UK, 1985; p. 435. [Google Scholar]
- Shpil’rain, E.E.; Yakimovich, K.A.; Fomin, V.A.; Skovorodjko, S.N.; Mozgovoi, A.G. Dynamic and kinetic viscosity of liquid alkali Metals. In Handbook of Thermodynamic and Transport Properties of Alkali Metals; Ohse, R.W., Ed.; Int. Union of Pure and Appl. Chemistry, Blackwell Scientific Pub.: Oxford, UK, 1985; p. 753. [Google Scholar]
- Einstein, A. Eine neue Bestimmung der Moleküldimensionen. Ann. Phys. IV 1906, 19, 289–305. [Google Scholar] [CrossRef]
- Einstein, A. Berichtigung zu meimer Arbeit: “Eine neue Bestimmung der Moleküldimensionen”. Ann. Phys. 1911, 34, 591–592. [Google Scholar] [CrossRef]
- Allen, B.C. Surface Tension. In Handbook of Thermodynamic and Transport Properties of Alkali Metals; Ohse, R.W., Ed.; Int. Union of Pure and Appl. Chemistry, Blackwell Scientific Pub.: Oxford, UK, 1985; p. 691. [Google Scholar]
- Hudson, J.B. Surface Science Introduction; John Wiley Interscience: New York, NY, USA, 1998; p. 94. [Google Scholar]
- Emsley, J. The Elements, 3rd ed.; Oxford Univ. Press: New York, NY, USA, 1999; p. 216. [Google Scholar]
- Iida, T.; Guthrie, R.O. The Physical Properties of Liquid Metals; Clarendon Press: Oxford, UK, 1988; p. 132. [Google Scholar]
- Haynes, W.H.; Lide, D.R. CRC Handbook of Chemistry and Physics, 91st ed.; CRC Press: Boca Raton, FL, USA, 2010; pp. 5–85. [Google Scholar]
- Foust, O.J. Sodium-NaK Engineering Handbook Vol. I Sodium Chemistry and Physical Properties; Gordon and Breach, Science Pub.: New York, NY, USA, 1972. [Google Scholar]
- Thermodynamic Database Group. Japan Society of Calorimetry and Thermal Analysis. In Materials-Oriented Little Thermodynamic Database for PC (MALT2); (CD-ROM); Kagaku Gijutsu-Sha: Tokyo, Japan, 1992. [Google Scholar]
- Klotz, I.M.; Rosenberg, R.M. Chemical Thermodynamics Basic Theory and Methods, 6th ed.; John Wiley & Sons Inc.: New York, NY, USA, 2000; p. 42. [Google Scholar]
- Safran, S.A. Statistical Thermodynamics of Surfaces, Interface and Membrains; Perceus Books: New York, NY, USA, 1994. [Google Scholar]
- Thompson, R. The stability of metal particles and particle-plate interactions in liquid metals. In Material Behavior and Physical Chemistry in LIQUID METAL SYSTEMS; Borgstedt, H.U., Ed.; Plenum Press: New York, NY, USA, 1982. [Google Scholar]
- Hamaker, H.C. The London-van der Waals attraction between spherical particles. Physica 1937, 4, 1058–1072. [Google Scholar] [CrossRef]
- Lifshitz, E.W. The theory of molecular attraction forces between solid bodies. Sov. Phys. JETP Engl. Transl. 1956, 2, 73–83. [Google Scholar]
- Ferrante, J.; Smith, J.R. Theory of the bimetallic interface. Phys. Rev. 1985, 31, 3427–3434. [Google Scholar] [CrossRef]
- Andriotis, A.N. Bimetallic interface: A periodic planar jellium approach. Phys. Rev. 1993, 47, 6772–6775. [Google Scholar] [CrossRef]
- Kittel, C. Introduction to Solid State Physics, 2nd ed.; John Wiley & Sons Inc.: New York, NY, USA, 1967; p. 68. [Google Scholar]
- Lang, N.D.; Kohn, W. Theory of metallic surfaces: Charge density and surface energy. Phys. Rev. 1970, 1, 4555–4568. [Google Scholar] [CrossRef]
- Atkins, P.; de Paula, J. Physical Chemistry, 7th ed.; Oxford Univ. Press: New York, NY, USA, 2002; p. 255. [Google Scholar]
- Mott, N.F.; Jones, H. The Theory of the Properties of Metals and Alloys; Dover Pub. Inc.: New York, NY, USA, 1958; p. 124. [Google Scholar]
- Alonso, J.A.; March, N.H. Electrons in Metals and Alloys; Academic Press: Salt Lake City, UT, USA, 1889; p. 108. [Google Scholar]
- Raims, S. The Wave Mechanics of Electrons in Metals; North-Holland Pub.: Amsterdam, The Netherlands, 1967; p. 194. [Google Scholar]
- Sugano, S.; Koizumi, H. Microclusters; Springer-Verlag: Berlin/Heidelberg, Germany, 1998; p. 50. [Google Scholar]
- Dreirach, O.; Evans, R.; Güntherodt, H.J.; Kunzi, H.U. A simple muffin tin model for the electrical resistivity of liquid noble and transition metals and their alloys. J. Phys. F Metal Phys. 1972, 2, 709–725. [Google Scholar] [CrossRef]
- Itami, T. Condensed Matter-Liquid Transition Metals and Alloys in Condensed Matter-Disordered Solids; Srivastava, A.K., March, N.H., Eds.; World Scientific: Singapore, 1995; p. 206. [Google Scholar]
- Popplewell, J.; Charles, S.W.; Hoon, S.R. The long term stability of metallic ferromagnetic liquid. In Proceeding of the Second Conference on Advances in Magnetic Materials and Their Applications, London, UK, 1–3 September 1976.
- Nozaki, K.; Itami, T. Dual percolation transition of ionic conductor in the AgI-BN composite system. J. Phys. Condens. Matter 2004, 16, 7763–7767. [Google Scholar] [CrossRef]
- Nozaki, K.; Itami, T. The determination of the full set of characteristic values of percolation, percolation threshhold and critical exponents for the artificial composite with ionic conduction, Ag4RbI5-β-AgI. J. Phys. Condens. Matter 2006, 18, 2191–2198. [Google Scholar] [CrossRef]
- Nozaki, K.; Itami, T. The evolution of the ionic conduction of (AgI)x-(Ag2O)y-(B2O3)1-(x+y). J. Phys. Condens. Matter 2006, 18, 3617–3627. [Google Scholar] [CrossRef]
- Nozaki, K.; Itami, T. Ionic Conduction in Heterogeneous Systems Containing Superionic Conductors in Condensed Matter; Das, M.P., Ed.; Nova Scientific Pub.: New York, NY, USA, 2007; pp. 191–220. [Google Scholar]
- Bréchignac, C.; Cahuzac, P.H.; de Frutos, M. Asymmetric fission of Nan++ around the critical size of stability. Phys. Rev. Lett. 1990, 64, 2893–2896. [Google Scholar] [CrossRef] [PubMed]
- Saunders, W.A. Fisson and liquid-drop behavior of charged gold clusters. Phys. Rev. Lett. 1990, 64, 3046–3049. [Google Scholar] [CrossRef] [PubMed]
- Katakuse, I.; Ito, H.; Ichihara, T. Fission-like dissociation of doubly charged silver clusters. Int. J. Mass Spectrom. Ion Process. 1990, 97, 47–54. [Google Scholar] [CrossRef]
- Koizumi, H.; Sugano, S.; Ishii, Y. Shell correction study of fission of doubly charged silver clusters. Z. Phys. 1993, 28, 223–234. [Google Scholar] [CrossRef]
- Buffat, P.H.; Borel, J.P. Size effect on the melting temperature of gold particles. Phys. Rev. 1976, 13, 2287–2298. [Google Scholar] [CrossRef]
- Ohshima, K.; Harada, J. A X-ray diffraction study of soft surface vibrations of FCC fine metal particles. J. Phys. C Solid State Phys. 1984, 17, 1607–1616. [Google Scholar] [CrossRef]
- Derjaguin, B. Friction and adhesion, IV. The theory of adhesion of small particles. Kolloid Zeits 1934, 69, 155–164. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Itami, T.; Saito, J.-i.; Ara, K. The Promising Features of New Nano Liquid Metals—Liquid Sodium Containing Titanium Nanoparticles (LSnanop). Metals 2015, 5, 1212-1240. https://doi.org/10.3390/met5031212
Itami T, Saito J-i, Ara K. The Promising Features of New Nano Liquid Metals—Liquid Sodium Containing Titanium Nanoparticles (LSnanop). Metals. 2015; 5(3):1212-1240. https://doi.org/10.3390/met5031212
Chicago/Turabian StyleItami, Toshio, Jun-ichi Saito, and Kuniaki Ara. 2015. "The Promising Features of New Nano Liquid Metals—Liquid Sodium Containing Titanium Nanoparticles (LSnanop)" Metals 5, no. 3: 1212-1240. https://doi.org/10.3390/met5031212