
Pharmacogenomics Guided-Personalization of Warfarin and Tamoxifen


1
Schulich School of Medicine and Dentistry, Western University, London, ON N6A 5A5, Canada
2
Department of Medicine, Division of Clinical Pharmacology, Western University, London, ON N6A 5A5, Canada
3
Department of Physiology and Pharmacology, Western University, London, ON N6A 5A5, Canada
4
Department of Oncology, Western University, London, ON N6A 5A5, Canada
*
Author to whom correspondence should be addressed.
J. Pers. Med. 2017, 7(4), 20; https://doi.org/10.3390/jpm7040020
Submission received: 25 October 2017
/
Revised: 23 November 2017
/
Accepted: 7 December 2017
/
Published: 13 December 2017
(This article belongs to the Special Issue Cytochrome P450 Variation in Pharmacogenomics)
Abstract
:The use of pharmacogenomics to personalize drug therapy has been a long-sought goal for warfarin and tamoxifen. However, conflicting evidence has created reason for hesitation in recommending pharmacogenomics-guided care for both drugs. This review will provide a summary of the evidence to date on the association between cytochrome P450 enzymes and the clinical end points of warfarin and tamoxifen therapy. Further, highlighting the clinical experiences that we have gained over the past ten years of running a personalized medicine program, we will offer our perspectives on the utility and the limitations of pharmacogenomics-guided care for warfarin and tamoxifen therapy.
1. Introduction
Warfarin and tamoxifen are widely prescribed and clinically important drugs for the treatment of conditions that require anticoagulation and estrogen receptor (ER)-positive breast cancer, respectively. The clinical response and efficacy of both drugs are variable likely due in part to genetic differences in pharmacogenes that mediate their metabolism and clearance. While genomics-based clinical guidelines have been established for several drugs, whether pharmacogenomics can be used to personalize treatment for individual patients remains controversial for warfarin and tamoxifen. This review aims to discuss the evidence to date that seek to correlate pharmacogenomic testing and the clinical outcomes in the context of each drug. Further, we will provide clinical insights and future perspectives based on our experiences with the implementation of personalized medicine strategies for warfarin and tamoxifen within a large acute care hospital setting.
2. Personalizing Warfarin Therapy
Warfarin is an oral anticoagulant that is indicated for the treatment and prevention of thrombosis related complication such as stroke and pulmonary embolism among patients with atrial fibrillation, prosthetic heart valves and venous thrombosis. Warfarin is an inhibitor of vitamin K epoxide reductase complex subunit 1 (VKORC1), which prevents the cycling of vitamin K to its active metabolite. Reduction in levels of active vitamin K1 leads to a deficiency in many components for the coagulation cascade including factors II, VII, IX, and X. Vitamin K antagonism has proven to be difficult to manage due to marked interpatient variability of response originating from genetic, environmental and iatrogenic influences. Warfarin therapy requires frequent monitoring of the international normalized ratio (INR) of prothrombin time. Healthy individuals have an INR near 1 and the target range for the majority of warfarin-treated patients is 2–3. Out of range INR can be a warning sign of reduced medication compliance, dietary changes, and potential drug interactions that increase the risk of adverse events, where low INR fails to prevent thrombotic events and high INR results in increased risk of bleeding [1]. The adverse event rate for warfarin therapy is a major difficulty that is confronted by clinicians. For example, Budnitz et al. found that adverse drug events accounted for 2.5% of visits to American emergency departments, and that warfarin alone accounted for approximately 6% of these visits [2]. A follow up study in elderly adults over 65 years of age demonstrated that 3.6% of emergency department visits were due to adverse drug reactions, of which 17% were attributable to warfarin [3]. The dangerously high adverse event rate with warfarin usage has led to a number of studies, both in vitro and in vivo, examining the pathways and determinants that govern the observed variation in warfarin dose response, and development of more predictive dosing algorithms, including those that take into account patient-specific pharmacogenomic information.
2.1. Warfarin Metabolism by Cytochrome P450s
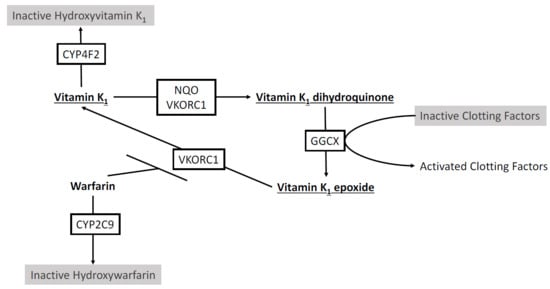
Warfarin is delivered as a racemic mixture whereby the S-enantiomer is a significantly more potent inhibitor of VKORC1, to the extent that S-warfarin is believed to be the clinically relevant compound [4]. The enantiomers are metabolized by different cytochrome P450 (CYP) enzymes [5] with S-warfarin metabolized by CYP2C9 to the inactive S-7-hydroxywarfarin in the liver (Figure 1) [6]. CYP2C9 is known to harbor common genetic variations. Specifically, CYP2C9 variant alleles *2 and *3, are not only common, but result in an enzyme with impaired activity and decreased warfarin turnover in vitro [7,8]. CYP2C9*2 and CYP2C9*3 have been shown to have an allele frequency of 12.5% and 8.5% in Caucasian populations [8], and genotyping for these alleles has demonstrated correlation with reduced dosage requirements and a greater likelihood of over anticoagulation [9,10,11,12,13]. CYP2C9 genotyping has been found to account for approximately 12% of the variation in warfarin dose requirement in Caucasian populations [12], leaving a large portion of the variability to be explained by other genetic factors and the environment.
Cytochrome P450 enzymes also play a direct role in the vitamin K cycle, distinct from their activity in warfarin metabolism. CYP4F2 has been shown to influence warfarin activity through its function as a vitamin K1 oxidase, resulting in the removal of vitamin K1 from the vitamin K cycle [14]. Warfarin also breaks the vitamin K cycle by impairing VKORC1 and preventing transformation to active vitamin K metabolite, therefore CYP4F2 activity increases the effect of warfarin anticoagulation (Figure 1). Caldwell et al. identified a CYP4F2 nonsynonymous single nucleotide polymorphism (SNP) rs2108622 that results if V433M coding mutation. This polymorphism (rs2108622) was then shown to be significantly associated with increased warfarin dose requirements in a Caucasian population [15]. CYP4F2 rs2108622 results in reduced CYP4F2 expression and activity effectively increasing the homeostatic pool of vitamin K1 that must be reduced for effective anticoagulation, potentially necessitating an increased dose of warfarin [14]. The rs2108622 variant of CYP4F2 has been predicted to account for between 1% and 2% of warfarin dose variation in Caucasian populations [12,15]. As well, CYP4F2 genotyping has demonstrated that the difference in warfarin dosing appears to occur during the induction phase of treatment [16,17]. Combining the two cytochrome P450 enzymes, CYP2C9 and CYP4F2, and their variants could predict approximately 15% of the variation in warfarin dose response in Caucasian populations.
VKORC1 is the target enzyme that is inhibited by warfarin and the rate limiting enzyme in the vitamin K cycle, allowing for warfarin to effectively prevent the production of vitamin K dependent coagulation factors (Figure 1) [18,19]. Elucidation of the gene VKORC1 as the target for warfarin response, through the in-depth genetic assessment of patients exhibiting warfarin resistance was a crucial discovery that led to the identification of common genetic variations that predict warfarin dose and sensitivity in the general population [20,21]. Indeed, the VKORC1 SNP rs9923231 (*2) has now been well established as an important determinant of VKORC1 response to warfarin therapy [22,23]. Genotyping of VKORC1 demonstrates that VKORC1*2 individuals are sensitized to the effects of warfarin, and therefore require a reduced dose [24], and genotyping studies have shown that VKORC1*2 can account for over 30% of the variation in warfarin dose [25,26]. Studies combining the impact of VKORC1, CYP2C9, and CYP4F2 variants have shown that VKORC1 and CYP2C9 have the most appreciable effect and account for 40% of variation in warfarin dose requirements in Caucasian populations. With up to 40% of warfarin variation in Caucasian patients accounted for through genetic polymorphisms, additional variation can be incorporated from clinical variables. Age, weight, body surface area, sex, smoking status, and indication for warfarin use have all been implicated to account for some of the variation in warfarin response [27,28]. In Caucasian populations, the combination of genetic and clinical factors predicted 57% of variation [28].
2.2. The Warfarin Clinical Trial Debate
The wealth of literature on the determinants of warfarin dose requirements has led to the development of a number of pharmacogenomics-guided dosing algorithms both for initiation and maintenance dosing of warfarin [28,29,30,31]. Efforts to validate pharmacogenomics based algorithms for warfarin dosing have shown that such algorithms can improve the accuracy of dose prediction (Table 1). However, converting theoretical benefit into validated clinical benefit has proven difficult, with randomized trials producing both positive and neutral results for pharmacogenomics based algorithms. Two large randomized controlled trials that were published in 2013 attempted to answer whether there was a benefit to utilizing pharmacogenetics in the dosing of warfarin [32,33]. The European Pharmacogenetics of Anticoagulant Therapy (EU-PACT) trial utilized a pharmacogenomic algorithm including clinical data, CYP2C9*2, CYP2C9*3, and VKORC1*2 genotype for the induction phase of warfarin therapy against a standard clinical dosing regimen. When comparing the primary outcome of time in therapeutic INR range and secondary outcomes of excessive anticoagulation, and time to stable INR, the EU-PACT trial demonstrated a benefit for use of pharmacogenetic algorithms during initiation of warfarin therapy [32]. However, the Clarification of Optimal Anticoagulation through Genetics (COAG) trial published in the same month also used a very similar algorithm when compared against a control group that utilized clinical data for dose prediction during induction therapy, and failed to find a statistically significant difference in the same primary outcome, time in therapeutic range [33]. A trial in Italian Caucasians also failed to detect a difference in time in therapeutic INR range when compared against a clinically guided dosing algorithm [34].
Discrepancies among clinical trials have led to many questions over the value of pharmacogenetic-guided dosing. The EU-PACT trial can be criticized for the control group lacking any standardized clinical algorithm for dosing decisions where the typical loading dose regimen may have resulted in unreasonable levels of over anticoagulation in the control arm. However, at the time of publication, the authors argue common practice did not utilize clinical algorithms and dosing with a standardized format was more translatable [32]. Meanwhile, the COAG trial has received criticism over the stratification of patients by age and indication for warfarin therapy, with implications that the atrial fibrillation and venous thromboembolism patients may respond differently to the anticoagulation effect of warfarin [35]. The EU-PACT trial used pharmacogenetic dosing from day 1 of the trial, while due to the constraints of the study design, in the COAG trial many patients received pharmacogenetics information after the first dose and some authors believe it is the earliest dosing that benefits from the input of pharmacogenetics [34,35]. Additionally, the indication for warfarin use has been implicated as a determinant factor [35] with a greater benefit predicted in atrial fibrillation [31,36]. While the EU-PACT and COAG trials differed in the proportion of patients for each indication, no significant effect was observed upon further analysis by the authors [35]. The largest difference between the EU-PACT and COAG trials is found within the patient population. The EU-PACT trial consisted of a largely homogenous population of European Caucasians, while the COAG trial was a mixed population of Caucasians and African Americans. Ethnic differences in the prevalence of CYP2C9 and VKORC1 genotypes have long been known, but these trials bring this to the center of the debate.
Compounding the difficulty in understanding the clinical value of pharmacogenetics in warfarin dosing is the recent publication of the Genetics Informatics Trial (GIFT) of warfarin to prevent deep vein thrombosis post-operatively in patients older than 65 [37]. GIFT is the largest trial to date, with 1650 patients enrolled across six centres, allowing for the use of clinical outcomes as an endpoint. GIFT found that genotype guided warfarin dosing improved the composite outcome of major bleeding, INR > 4, venous thromboembolism, or death, when compared to a clinically guided dosing algorithm. The scale of GIFT allowed for the power to detect rare events and showed a significant improvement of the primary outcome between the groups. The secondary outcome in GIFT, time in the therapeutic INR range, show a significant benefit in the genotype guided group. Interestingly, the largest benefit in the secondary outcome was observed in patients considered the high-risk subgroup, those where the genotype and clinical algorithms differed by greater than 1 mg/day. Some key differences between this and previous trials are the inclusion of CYP4F2*3 genotyping and the application of the algorithm for 11 days, twice as long as previous trials [37]. As well, GIFT enrolled patients with a single indication for warfarin therapy, thereby potentially influencing the statistical significance of the study and limiting its translation to other indications. The results of GIFT add to the debate for pharmacogenomics in warfarin dosing, while the real-world application and cost-effectiveness of these programs has yet to be tested.
2.3. Tailoring Pharmacogenomics-Based Warfarin Dosing Algorithms
Pharmacogenomic guided warfarin dosing algorithms continue to show promise to optimize warfarin dose selection. However, one key caveat that must be kept in mind is that much of the prospective clinical trials-based evidence has been primarily derived from Caucasian populations. Even among subjects who reside in similar parts of the world, variant allele frequencies can differ widely. For example, it has been common knowledge among clinicians that subjects of Asian descent require lower warfarin dosing on average, and Lee et al. provided evidence that 87% of their Chinese study population harbored the VKORC1*2 variant. However, within that same study, the Malay cohort carrier frequency was 65% and the Indian population had much lower rates of 12% [41]. A further review of the prevalence of VKORC1 variants in east Asian populations found that 90% of study subjects were variant carriers, while in the rest of Asia there exists a wide range from 14% to 80% in VKORC1 variant carrier frequency [42]. In some studies, such as the COAG trial, which enrolled a large number of African-American patients, the investigators failed to include additional SNPs in VKORC1 and CYP2C9 that are more prevalent and more strongly associated with warfarin dose adjustment in patients of African descent [43]. Indeed, a dosing algorithm using these two novel markers showed significant improvement on the amount of variation explained (27%) as compared to the International Warfarin Pharmacogenetics Consortium (IWPC) algorithm (16%), but overall, the power remains low and further exploration for additional genetic markers in this population is warranted [44]. The variation in allele frequencies between ethnicities impairs the translation of pharmacogenomics-based warfarin dosing algorithms, as accounting for all of the alleles may be too costly and not accounting for certain alleles may limit its utility. To effectively implement pharmacogenomics-guided dosing algorithms, clinicians may need to tailor algorithms for different ethnicities by accounting for alleles that are most prevalent within that ethnic population.
2.4. Insights from Clinical Implementation of Pharmacogenomics-Guided Warfarin Therapy
Since 2006, our research team has been involved in the implementation of pharmacogenomics-based personalized medicine for real-world patient care. In 2008, with the support of our hospital (London Health Sciences Centre, London Ontario Canada), we started a personalized medicine clinic that was focused on optimization of drug dosing or selection using patient-specific pharmacogenomic information. Over the past nine years we have obtained informed pharmacogenetics research consent from over 4000 patients, as part of our personalized medicine research program. We are able to incorporate clinical assessments, as well as relevant genomics-guided recommendations for drugs that are known to be affected by pharmacogenetic variation, such as, warfarin, tamoxifen, azathioprine, tacrolimus, clopidogrel, capecitabine, 5-fluorouracil, and irinotecan. Feedback from referring physicians indicates that they all value our personalized medicine-based approach to warfarin dosing. In the following sections, our perspective/opinion, based on our experience of utilizing warfarin pharmacogenomic information, both for inpatient care and in the ambulatory clinic setting is outlined.
When and how fast to genotype: In our experience, carrying out warfarin pharmacogenomic testing is valuable during the initiation of warfarin therapy, or when a stable dose of warfarin has not been attained. Carrying out genotyping for patients who are already at a stable dose and adequate INR time in therapeutic range is likely unnecessary. The majority of our pharmacogenomics-guided recommendations are provided within 24 h. However, we noted that for most of the patients who are just starting warfarin therapy, one or two days of a typical standard dose (e.g., 5 mg) does not significantly alter INR trajectory even among carriers of risk alleles in VKORC1 or CYP2C9.
Timely discharge from hospital: As our team gained more experience with inpatient consultation requests for warfarin dosing and management, we learned the capability to predict warfarin dose based on their pharmacogenomic makeup gives far greater confidence with regards to the dose of warfarin required at the time of discharge. This is related to the fact that it typically takes 7–10 days, from initiation of warfarin to reach target INR. Frequently, in a hospitalized setting, the treating physicians worry about an unexpectedly rapid or slow rise in INR, and tend to keep such patients in the hospital longer than necessary.
Warfarin pharmacogenomics can be used as rationale for initiating alternate treatments: The emergence of direct oral anticoagulants (DOACs) has meant that there is now an alternative to warfarin, particularly for the patients who require oral anticoagulation for nonvalvular atrial fibrillation or venous thromboembolism (VTE). For many patients, the cost of DOACs vs. warfarin, may be the key issue. In our experience, patients who are predicted to require low or very low warfarin dose based on their pharmacogenomic profile, tend to have more variable INRs and are less able to maintain adequate time in therapeutic range. Greater bleeding risk among variant carriers in CYP2C9 and VKORC1 was clearly demonstrated in the Effective Anticoagulation with Factor Xa Next Generation in Atrial Fibrillation-Thrombolysis in Myocardial Infarction 48 (ENGAGE AF-TIMI 48) clinical trial that compared edoxaban vs. warfarin [45]. Therefore, for patients in our clinic who are at greater bleeding risk during the first 90 days of therapy based on their CYP2C9 and VKORCI genetics and who qualify for either warfarin or DOAC therapy, we are more likely to initiate a DOAC or switch them to a DOAC.
Dedicated expertise and clinic for follow-up still needed: While pharmacogenomic assessment can provide a reasonable estimate of predicted warfarin dose, routine INR monitoring and further dose adjustment, regardless of predicted dose, is still required. Moreover, warfarin drug interactions are common, both from inhibitors of CYP2C9, those that affect gut vitamin K synthesis (e.g., antibiotics), and inducers of P450 enzymes (e.g., phenytoin, phenobarbital, carbamazepine, rifampin). In severely ill patients, caution is warranted in terms of over-reliance on pharmacogenomic-based warfarin dose as such patients tend to be quite ill, often with multi-organ dysfunction and poor oral intake. Therefore, a team or a clinic with expertise in warfarin dosing and management is highly recommended.
3. Personalizing Tamoxifen Therapy
Tamoxifen, a selective estrogen receptor modulator (SERM) that functions as an antagonist within the breast and reproductive organs, has been widely used to treat and prevent recurrence of ER-positive breast cancer since the 1970s [46,47,48]. While most commonly used as an adjuvant therapy, tamoxifen was the first drug approved by the US Food and Drug Administration (FDA) as a chemo-preventative agent for women at high risk for breast cancer. The benefits of long-term endocrine treatment for improved disease-free survival have been demonstrated in two large randomized controlled trials for patients with early-stage hormone receptor positive disease, shifting the paradigm of treatment strategy from 5 years to 10 years of extended tamoxifen therapy [49,50,51]. Despite a reduction in recurrence rates, there is high variability observed in response to tamoxifen. Many factors, including disease type, nodal involvement, drug adherence, concomitant medications, treatment with chemotherapy, and the presence of pharmacogenetic polymorphisms all likely contribute to variation in tamoxifen efficacy [47]. Currently, the goal of personalized tamoxifen therapy to improve breast cancer outcomes is primarily focused on understanding the contribution of pharmacogenomics to tamoxifen metabolism [52].
3.1. Tamoxifen Metabolism by Cytochrome P450s
Tamoxifen is a prodrug that undergoes extensive hepatic metabolism by cytochrome P450 enzymes to form primary metabolites 4-hydroxytamoxifen (4-OH-TAM) and N-desmethyl-tamoxifen (NDM-TAM), which are both further converted to 4-hydroxy-N-desmethyl-tamoxifen, better known as endoxifen [46]. It is well established that the rate limiting enzyme in tamoxifen to endoxifen bioactivation is CYP2D6, while other enzymes (CYP3A4, CYP2C9, CYP2C8, and CYP2C19) likely have a smaller contribution [46,47]. While 4-OH-TAM and endoxifen share similar affinities for ER binding, approximately 100-fold greater than tamoxifen itself, endoxifen is considered to play the lead efficacious role [53]. Circulating endoxifen concentration is on average six times greater than 4-OH-TAM, and has been shown in vitro to cause ERα degradation in a concentration dependent manner [53,54]. Endoxifen concentrations are highly variable among patients, ranging from <5 nM to >100 nM, with CYP2D6 being thought to be the main source driving this variability [55].
CYP2D6, primarily expressed in the liver, is responsible for the metabolism of approximately 25% of prescription drugs currently on the market [46]. To date, there are over 100 identified single nucleotide polymorphisms (SNPs) within CYP2D6, making it the most polymorphic CYP enzyme [56]. In addition, patients can carry multiple copies of CYP2D6. The presence of decreased (e.g., *9, *10, *17, *41) or null (e.g., *3, *4, *5, *6, *7, *8) function variants can be translated to a phenotypic classification of CYP2D6 metabolic activity as either intermediate metabolizer (IM) or poor metabolizer (PM), respectively, while patients without these variants are normal metabolizers (NM) or ultrarapid metabolizers (UM) if multiple copies of CYP2D6 are present [46,47]. In an effort to standardize terms for clinical pharmacogenomic results, the Clinical Pharmacogenetics Implementation Consortium has redefined CYP2D6 extensive metabolizers (EM) as normal metabolizers, and as such, will be referred to as NMs within this review [57]. It is well established that CYP2D6 metabolic activity correlates with endoxifen concentration with PMs having significantly lower systemic exposure [58,59,60,61]. However, there is marked interpatient variation in endoxifen concentration that is observed within CYP2D6 phenotype groups. It is likely that some of this variation can be explained by the lack of standardization for determining CYP2D6 activity. The number of variants interrogated, their differential effect on CYP2D6 activity, and how they are combined to define phenotype can create a source of discordance. For example, using simple phenotyping, patients with *1/*41 and *41/*4 genotypes would both be categorized as IMs, while the effect of each genotype on CYP2D6 activity is considerably different. To better describe metabolic activity, many studies have used more defined phenotypic groups (e.g., NM/IM and IM/PM) or have opted to implement a CYP2D6 activity score (AS) in place of phenotype, where the AS is considered the sum of the values assigned to each CYP2D6 allele (e.g., *1/*41 and *41/*4 would have AS of 1.5 and 0.5, respectively) [62].
The extent of variation in CYP2D6 activity due to the presence of variant alleles differs among ethnicities [63,64]. The CYP2D6 PM phenotype is more commonly observed in Caucasians due to the higher prevalence of the non-functional alleles CYP2D6*4 and *5. Gene duplications leading to genotype defined UMs and the reduced function *41 allele are most prevalent in Middle Eastern populations [63]. The high frequency (approximately 41%) of the reduced function *10 allele among Asians suggests that CYP2D6 mediated drug metabolism may be slower in this population [64]. Approximately, 35% of the allele variation observed in African and African American populations is comprised of reduced function alleles, primarily *17 and *29, respectively, with African Americans having a higher frequency of non-functional alleles [63,64]. The likelihood of yet unidentified functionally consequential variants poses a further challenge when comparing results between studies from patients of different ethnic backgrounds.
In addition to genotype, CYP2D6 activity can be affected by the co-administration of interacting medications. Selective serotonin reuptake inhibitors (SSRIs) are commonly prescribed for depression, but are also frequently used to alleviate hot flash symptoms, one of the most common side effects of tamoxifen therapy [46,65]. SSRIs, can be classified as mild, moderate, and strong inhibitors based on their demonstrated ability to inhibit the metabolic activity of CYP2D6. When taken concomitantly with tamoxifen, strong inhibitors, such as paroxetine, fluoxetine, and buproprion, can result in a dramatic reduction in the ability to form endoxifen [66] and may impact tamoxifen efficacy [67]. The use of moderate (duloxetine, sertraline) and mild (venlafaxine, desvenlafaxine, citalopram, and escitalopram) inhibitors are thought to have less of an impact on tamoxifen metabolism, however, the individual effect may vary significantly suggesting that if a SSRI is required the lowest dose of a mild inhibitor should be the preferred option to minimize the impact on CYP2D6 activity [65].
3.2. CYP2D6 and Tamoxifen Clinical Outcomes
The importance of CYP2D6 to tamoxifen metabolism and subsequent endoxifen formation has provided logical rationale for the hypothesis that CYP2D6 genotype correlates with tamoxifen efficacy. If CYP2D6 genotype correlated strongly with outcomes, then up-front genotyping tests could be offered to predict the risk of recurrence. For higher risk patients with reduced or absent CYP2D6 activity, alternate dosing or treatment strategies could be proactively offered to personalize endocrine therapy potentially improving survival outcomes. However, studies spanning the past two decades fail to provide conclusive evidence for recommending CYP2D6 genotyping as a predictive marker of tamoxifen efficacy. While some studies have demonstrated a significant correlation, many have failed to reproduce these results (summarized in Table 2).
Early studies observed significant correlation between null and reduced CYP2D6 activity alleles and worse disease outcomes, including higher rates of recurrence [68,69,70]. Similarly, Lammers et al. found that CYP2D6 PM phenotype was associated with shorter overall survival (OS) in metastatic breast cancer patients prescribed 40 mg daily tamoxifen. This study also showed that CYP2D6 inhibitor use was an independent predictor of OS [71]. Schroth et al. conducted a large retrospective study with a median follow up period of 6.3 years, including United States (US) and German cohorts of post-menopausal women diagnosed with early breast cancer and demonstrated that patients with reduced or non-function CYP2D6 alleles had worse disease-free survival (DFS) [72]. Further, CYP2D6 PM phenotype was found to be associated with a higher risk of disease events only in the tamoxifen arm of the Austrian Breast and Colorectal Cancer Study Group trial (ABCSG) 8. This increased risk was not observed in patients who switched to the aromatase inhibitor anastrozole after two years of tamoxifen [73]. Disease outcomes were also correlated with CYP2D6 activity when phenotype was categorized based on activity score rather than metabolizer status [74,75]. In addition, several studies have shown that homozygosity for CYP2D6 *10, which is more prevalent in Asian populations, was associated with worse DFS and recurrence-free survival (RFS) [76,77]. Kiyotani et al. conducted a study with 282 Japanese breast cancer patients receiving tamoxifen monotherapy, and showed that the presence of two variant alleles was associated with worse RFS [78]. Recently, Saladores et al. found that poor CYP2D6 activity correlated with shorter distant relapse free survival, irrespective of ethnicity [75]. CYP2D6 PM male breast cancer patients have also been shown to have a higher risk of recurrence, which remained significant when adjusted for nodal status and tumor size [79].
Retrospective data from two large double-blind trials, the Arimidex, Tamoxifen, Alone, or in Combination (ATAC) and the Breast International Group (BIG) 1–98 trial, sparked controversy by failing to validate an association between CYP2D6 genotype and tamoxifen efficacy [80,81]. A large population based case-cohort study in the United Kingdom (UK) also failed to observe an association between the common CYP2D6*4 variant and breast cancer specific survival, however, they did note that the null CYP2D6*6 allele may affect survival in patients taking tamoxifen [82]. Other studies investigating the role of *4 and reduced function variants in patients from various ethnicities, including a recent study by Hertz et al., were unable to validate a predictive role for CYP2D6 [83,84,85,86]. Interestingly, Kiyotani et al. noted that although no association between CYP2D6 genotype and RFS was observed in patients receiving tamoxifen-combination therapy, a significant association was shown in patients on tamoxifen monotherapy [87]. They suggest that the difference in tamoxifen regimen could explain some of the contradictions in the literature, as many studies failing to validate a role for CYP2D6 were comprised of patients on combination therapy.
Study heterogeneity resulting from varying inclusion criteria, length of treatment, concomitant medications, adherence data, measured outcomes, and DNA sources combined with non-standardized genotype classification has impeded the ability to conclusively determine the association between CYP2D6 phenotype and tamoxifen efficacy. To address heterogeneity related to use of DNA extracted from tumor infiltrated tissue, Ahern et al. conduced a quantitative bias analysis based on observed concordance rates of CYP2D6 genotypes to examine whether call errors could bias the estimates of association. They determined that genotyping errors have a negligible effect on measured outcomes, suggesting that DNA source is unlikely to be a major contributor to study discrepancies [85]. Recently, several meta-analyses have been conducted to ascertain the benefit of CYP2D6 genotyping [88,89,90,91,92]. Results from the International Tamoxifen Pharmacogenomics Consortium meta-analysis from studies conducted globally, suggested that CYP2D6 might indeed impact tamoxifen benefit [91]. While most of the analyses conducted to date have demonstrated that CYP2D6 variant phenotypes appear to be associated with reduced survival outcomes, the associations are based upon small, heterogeneous studies with large differences in comparator groups. As such, CYP2D6 is likely important for tamoxifen efficacy, but there remains insufficient robust evidence to support the recommendation of CYP2D6 genotyping for personalizing tamoxifen therapy. As most studies to date have been retrospective in nature, large, well-designed prospective studies with more homogenous populations are required to fully elucidate the predictive value of CYP2D6.
3.3. Tamoxifen Metabolism by Other CYP P450 Enzymes
Tamoxifen metabolism is affected by additional CYP enzymes, including CYP3A4, CYP2C9, and CYP2C19, however, the overall impact of polymorphisms within these genes appears minor when compared to CYP2D6 [93,94,95]. CYP3A4 activity displays high inter-individual variability and is susceptible to drug interactions [96]. Concomitant administration of rifampicin, a CYP3A4 inducer, surprisingly resulted in the dramatic reduction in plasma endoxifen concentration [97,98]. Additionally, carriers of CYP3A4*22, thought to be a reduced function variant, have been shown to have significantly increased tamoxifen and metabolite exposure. This effect is more pronounced in CYP2D6 PMs suggesting that it may provide some functional compensation in the absence of CYP2D6 metabolic activity [60,99]. Although several studies have failed to find an association between CYP2C9 variants and tamoxifen metabolism [60,93,100], two studies have observed a significant effect of CYP2C9 activity on endoxifen concentration [61,95]. The magnitude of this effect was small and likely does not have significant impact on clinical outcomes [95]. A putative role for CYP2C19 has been controversial with some studies demonstrating higher endoxifen concentration and improved outcomes in CYP2C19*17 (increased activity variant) carriers [69,94,101,102], while other studies failed to validate these findings [100,103]. A recent retrospective analysis of the International Tamoxifen Pharmacogenomics Consortium dataset of over 2000 patients observed no effect of CYP2C19 variants (*2, reduced function and *17) on tamoxifen outcomes, an effect that remained after accounting for CYP2D6 genotype, concluding that CYP2C19 genotype should likely not be considered when personalizing tamoxifen therapy [104]. The role of phase II enzymes (SULTs and UGTs) and transporters (ABCB1, ABCC2) have been also been investigated and are reviewed in [93,105].
SULTs and UGTs play a key role in the metabolism of tamoxifen by catalyzing the inactivation and subsequent elimination of tamoxifen and its metabolites. Similar to CYP2D6, the genes that encode for SULTs and UGTs are polymorphic with some variants impacting tamoxifen metabolism. SULT1A1 is thought to be the primary SULT enzyme that is responsible for the sulfation of endoxifen and 4-OH-TAM [105]. SULT1A1*2 is associated with reduced activity, but does not appear to correlate with endoxifen concentrations. However, carriers of SULT1A2 variants, including SULT1A2*2 and SULT1A2*3 have been associated with higher plasma endoxifen and 4-OH-TAM concentrations [93,106]. Variants within UGT1A4, UGT2B15, and UGT2B7 have been shown to affect glucuronidation activity [93]. A study by Romero-Lorca et al. investigated the effect of variants on the concentration of endoxifen and 4-OH-TAM glucuronidated metabolites. Patients with variants UGT1A448Val, UGT2B7268Tyr, or with wildtype genotypes for UGT2B17nodel and UGT2B15523Lys exhibited increased concentrations of active endoxifen and 4-OH-TAM [107]. A recent follow up study by the same group observed a trend for CYP2D6 PM patients to have higher active metabolite concentrations if they were carriers of the favorable genotypes they had previously identified, suggesting that genetic variation in SULTs and UGTs may contribute to improved algorithms for predicting tamoxifen outcomes [108]. Moreover, a trend for a higher risk of recurrence was noted for patients carrying a combination of SULT1A1*2/*2:UGT2B15*1/*2 or SULT1A1*2/*2:UGT2B15*2/*2 variant alleles [83]. At present, more studies are needed to better understand the role of SULT and UGT polymorphisms on tamoxifen metabolism and efficacy.
3.4. Therapeutic Drug Monitoring of Tamoxifen
Amid conflicting results regarding CYP2D6 activity as a biomarker for tamoxifen efficacy, more studies are now investigating the potential for endoxifen concentration to predict outcomes. As discussed above, many factors can influence the systemic exposure of endoxifen, therefore, determining the CYP2D6 genotype is likely not enough to predict the concentration of active metabolite. Fox et al., reported that greater than 50% of low endoxifen could not be explained by CYP2D6 genotype or use of inhibitory medications [109]. Studies in vitro, as well as in a murine tumor growth inhibition (TGI) model, have demonstrated that the effect of endoxifen ER antagonism is concentration-dependent [61,110]. Based on pharmacokinetic-pharmacodynamic modeling, optimal TGI was predicted for patients attaining endoxifen concentrations >40 nM, while <15 nM endoxifen was predicted to achieve sub-optimal (83%) TGI [110]. A putative threshold level of endoxifen has been demonstrated by two large retrospective studies. Madlensky et al., observed that patients within the lowest quintile of endoxifen concentration (<15 nM) had higher rates of recurrence when compared to the upper four quintiles [55]. Similarly, Saladores et al., reported that patients with <14 nM endoxifen had significantly short distant relapse-free survival as compared to patients with levels >35 nM [75].
Several dose escalation studies have been designed to elevate endoxifen in CYP2D6 PMs and have consistently shown that an increased daily tamoxifen dose of 30–40 mg can significantly increase levels to be similar to NM averages, without any noted increase in adverse events [111,112,113,114,115]. Additionally, Fox et al., performed dose escalations in patients based on baseline endoxifen concentration rather than CYP2D6 genotype. Patients with <30 nM endoxifen after eight weeks of tamoxifen on the standard dose (20 mg/day) underwent dose increases by 10 mg/day increments until target endoxifen (>30 nM) or a maximum of 60 mg/day was reached. Endoxifen levels of >15 nM were achieved in 96% of the patient cohort after dose escalation when compared to 76% at baseline, with 76% of patients attaining >30 nM endoxifen [109]. This study demonstrates the potential utility of personalizing tamoxifen therapy to improve outcomes through therapeutic drug monitoring (TDM) of endoxifen. Patients with sub-therapeutic endoxifen exposure or poor adherence can be quickly identified within the first 4–8 weeks after tamoxifen initiation, providing early rationale for clinicians to continue with tamoxifen therapy or switch to an alternate treatment plan. The use of TDM may be limited in some clinical settings, therefore the development of algorithms to predict endoxifen concentration based on clinical, environmental and genetic factors are warranted. An early algorithm developed by our team based on a patient cohort of approximately 200 patients, achieved a predictive accuracy of 89%, with a cross validation estimated accuracy of 85% [60]. Larger prospective trials are needed to further develop highly predictive models and confirm the utility of TDM for personalizing tamoxifen therapy. The undertaking of such lengthy prospective trials may in fact be unrealistic due to the large body of conflicting evidence to date. However, the possibility of replacing tamoxifen with endoxifen therapy is on the horizon, as the first phase I trial results have demonstrated acceptable toxicity with promising efficacy in patients with endocrine-refractory, metastatic breast cancer [116].
3.5. Clinical Perspectives Based on Experience from a Personalized Tamoxifen Clinic
Clinical application of CYP2D6 genotyping as a way of predicting response to tamoxifen therapy has not been widely accepted. Over the past seven years, as a part of our personalized medicine research program, we have carried out CYP2D6 genotyping, and TDM measuring both tamoxifen and endoxifen plasma concentrations using a LC-MS/MS system in approximately 800 patients. Shown below are key observations from our clinic.
CYP2D6 genotyping alone is inadequate: We have observed that nearly 5% of patients, primarily of Caucasian descent, are CYP2D6 PMs. However, only a portion (60%) of those predicted to be PMs have endoxifen concentrations below 15 nM. Additionally, nearly 20% of patients predicted to be CYP2D6 IMs were observed to have endoxifen concentrations of less than 15 nM. Current clinical standard does not require or suggest tamoxifen dosing be changed based on CYP2D6 genotype. However, we observed that a portion of patients are likely receiving sub-optimal benefit from tamoxifen and patients with low endoxifen may benefit from higher daily tamoxifen doses.
Endoxifen measurement provides reassurance for patients regarding adequacy of tamoxifen metabolism: In our clinic, we often hear from our patients that they experience little to no side effects from tamoxifen, and are thus very concerned that they may not be attaining benefit from tamoxifen therapy. Since our team not only carries out CYP2D6 genotyping, but also measures endoxifen plasma concentration, we are able to demonstrate to our patients that in fact, presence or absence of side effects cannot be used as a predictor of adequate tamoxifen metabolism.
Tamoxifen and endoxifen measurement and patient compliance: Tamoxifen’s half-life is nearly seven days, thus major changes in tamoxifen and endoxifen concentrations are reflective of the overall tamoxifen compliance in the previous month prior to endoxifen measurement. Thus, when we observe markedly lower than expected tamoxifen and endoxifen concentrations, we are able to discuss the importance of medication compliance. Interestingly, our patients who are provided with their own endoxifen metabolism data, demonstrate a greater willingness to continue tamoxifen. This holds true even when patients experience significant side effects such as hot flashes, as they feel more reassured regarding the adequacy of their tamoxifen metabolism, and the likelihood of long-term clinical benefit.
Endoxifen measurement is useful in quantifying the extent of tamoxifen drug interactions: One of most common and clinically impactful roles of our tamoxifen clinic relates to our ability to quantify tamoxifen drug interactions. A significant proportion of our tamoxifen patients are on antidepressants. Low doses of antidepressants are often prescribed by oncologists to reduce hot flash symptoms. Most antidepressants, particularly those in the SSRI class, act as inhibitors of CYP2D6 activity. SSRIs, such as paroxetine, fluoxetine, and bupropion are well known potent inhibitors of CYP2D6. In fact, the product monograph of such SSRIs cautions an interaction with tamoxifen that may lead to reduced endoxifen formation. By measuring a patient’s endoxifen concentration, we can demonstrate the net effect of such interactions. For patients with a low endoxifen concentration who are taking potent CYP2D6 inhibitors, we are able to document a two-fold increase in endoxifen concentration when such patients discontinue the SSRI or are switched to SSRIs with low to moderate CYP2D6 inhibitory effect [60]. We note that patients who are CYP2D6 NMs or UMs tend to exhibit endoxifen concentration above 15 nM, even when taking potent CYP2D6 inhibitors. Such information can be useful in some cases where the patient is unwilling to switch to an alternative antidepressant. Conversely, we have been able to document the deleterious effect of CYP metabolism inducing drugs, such as phenytoin and rifampin, to a reduction in both tamoxifen and endoxifen plasma concentrations [98,117].
4. Conclusions
Genetic variation in CYP enzymes is increasingly recognized as clinically important. Although the relationship between molecular basis of genetic variation in CYP enzymes and their expression and function is well established, the application using such information to guide relevant drug therapy in real-world patients has taken longer than expected. Much of the delay has been related to the lack of randomized clinical trials data, as well as the cost-effectiveness of pharmacogenomics-based approach. While current evidence may not support the implementation of pharmacogenomics testing that may be impractical or unfeasible in certain clinical settings, the use of available genetic information should be utilized to optimize drug therapy. Clinical guidelines have been established for warfarin dosing in patients with known genetic information [118], and guidelines for tamoxifen are in currently in development.
For warfarin, it is now becoming clear that preemptive genotyping can aid in better dosing decisions, and the most recent large scale randomized trials data suggest genotype guided approach results in better outcome. For medications such as tamoxifen, we are still awaiting decisive large scale clinical outcomes data, in terms of optimal genotype or endoxifen-based dosing. Given that tamoxifen is usually only a part of overall breast cancer treatment strategy (surgery, chemotherapy, radiation therapy), it will likely require a very large sample size and a long follow-up period to provide such conclusive evidence. What is becoming clear is that endoxifen is the major active metabolite, and the observed endoxifen level could be viewed as a surrogate marker of adequate tamoxifen dosing, and for mitigation of potentially deleterious drug interactions. The recently completed Phase I study using endoxifen suggest that there is the potential for endoxifen therapy in the future, potentially bypassing the current concerns relating to CYP2D6 and tamoxifen [116].
Acknowledgments
This work was supported by the Wolfe Medical Research Chair in Pharmacogenomics and the Canadian Institutes of Health Research, Drug Safety and Effectiveness Network (DSEN-PREVENT, FRN-117588) and Ontario Research Fund-Research Excellence Round 8.
Author Contributions
All authors contributed to the writing and editing of the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Nelson, W.W.; Wang, L.; Baser, O.; Damaraju, C.V.; Schein, J.R. Out-of-range INR values and outcomes among new warfarin patients with non-valvular atrial fibrillation. Int. J. Clin. Pharm. 2015, 37, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Budnitz, D.S.; Pollock, D.A.; Weidenbach, K.N.; Mendelsohn, A.B.; Schroeder, T.J.; Annest, J.L. National surveillance of emergency department visits for outpatient adverse drug events. JAMA 2006, 296, 1858–1866. [Google Scholar] [CrossRef] [PubMed]
- Budnitz, D.S.; Shehab, N.; Kegler, S.R.; Richards, C.L. Medication use leading to emergency department visits for adverse drug events in older adults. Ann. Intern. Med. 2007, 147, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Choonara, I.A.; Cholerton, S.; Haynes, B.P.; Breckenridge, A.M.; Park, B.K. Stereoselective interaction between the R enantiomer of warfarin and cimetidine. Br. J. Clin. Pharmacol. 1986, 21, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Kaminsky, L.S.; Zhang, Z.Y. Human P450 metabolism of warfarin. Pharmacol. Ther. 1997, 73, 67–74. [Google Scholar] [CrossRef]
- Rettie, A.E.; Korzekwa, K.R.; Kunze, K.L.; Lawrence, R.F.; Eddy, A.C.; Aoyama, T.; Gelboin, H.V.; Gonzalez, F.J.; Trager, W.F. Hydroxylation of warfarin by human cDNA-expressed cytochrome P-450: A role for P-4502C9 in the etiology of (S)-warfarin-drug interactions. Chem. Res. Toxicol. 1992, 5, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Furuya, H.; Fernandez-Salguero, P.; Gregory, W.; Taber, H.; Steward, A.; Gonzalez, F.J.; Idle, J.R. Genetic polymorphism of CYP2C9 and its effect on warfarin maintenance dose requirement in patients undergoing anticoagulation therapy. Pharmacogenet. Genom. 1995, 5, 389–392. [Google Scholar] [CrossRef]
- Stubbins, M.J.; Harries, L.W.; Smith, G.; Tarbit, M.H.; Wolf, C.R. Genetic analysis of the human cytochrome P450 CYP2C9 locus. Pharmacogenet. Genom. 1996, 6, 429–439. [Google Scholar] [CrossRef]
- Aithal, G.P.; Day, C.P.; Kesteven, P.J.; Daly, A.K. Association of polymorphisms in the cytochrome P450 CYP2C9 with warfarin dose requirement and risk of bleeding complications. Lancet 1999, 353, 717–719. [Google Scholar] [CrossRef]
- Higashi, M.K.; Veenstra, D.L.; Kondo, L.M.; Wittkowsky, A.K.; Srinouanprachanh, S.L.; Farin, F.M.; Rettie, A.E. Association between CYP2C9 genetic variants and anticoagulation-related outcomes during warfarin therapy. JAMA 2002, 287, 1690–1698. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.M.; Johnson, J.A.; Langaee, T.Y.; Feng, H.; Stanaway, I.B.; Schwarz, U.I.; Ritchie, M.D.; Stein, C.M.; Roden, D.M.; Smith, J.D.; et al. A genome-wide scan for common genetic variants with a large influence on warfarin maintenance dose. Blood 2008, 112, 1022–1027. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, F.; McGinnis, R.; Bourgeois, S.; Barnes, C.; Eriksson, N.; Soranzo, N.; Whittaker, P.; Ranganath, V.; Kumanduri, V.; McLaren, W.; et al. A genome-wide association study confirms VKORC1, CYP2C9, and CYP4F2 as principal genetic determinants of warfarin dose. PLoS Genet. 2009, 5, e1000433. [Google Scholar] [CrossRef] [PubMed]
- Lindh, J.D.; Holm, L.; Andersson, M.L.; Rane, A. Influence of CYP2C9 genotype on warfarin dose requirements—A systematic review and meta-analysis. Eur. J. Clin. Pharmacol. 2009, 65, 365–375. [Google Scholar] [CrossRef] [PubMed]
- McDonald, M.G.; Rieder, M.J.; Nakano, M.; Hsia, C.K.; Rettie, A.E. CYP4F2 is a vitamin K1 oxidase: An explanation for altered warfarin dose in carriers of the V433M variant. Mol. Pharmacol. 2009, 75, 1337–1346. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, M.D.; Awad, T.; Johnson, J.A.; Gage, B.F.; Falkowski, M.; Gardina, P.; Hubbard, J.; Turpaz, Y.; Langaee, T.Y.; Eby, C.; et al. CYP4F2 genetic variant alters required warfarin dose. Blood 2008, 111, 4106–4112. [Google Scholar] [CrossRef] [PubMed]
- Bejarano-Achache, I.; Levy, L.; Mlynarsky, L.; Bialer, M.; Muszkat, M.; Caraco, Y. Effects of CYP4F2 polymorphism on response to warfarin during induction phase: A prospective, open-label, observational cohort study. Clin. Ther. 2012, 34, 811–823. [Google Scholar] [CrossRef] [PubMed]
- Tatarunas, V.; Lesauskaite, V.; Veikutiene, A.; Grybauskas, P.; Jakuska, P.; Jankauskiene, L.; Bartuseviciute, R.; Benetis, R. The effect of CYP2C9, VKORC1 and CYP4F2 polymorphism and of clinical factors on warfarin dosage during initiation and long-term treatment after heart valve surgery. J. Thromb. Thrombolysis 2014, 37, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Suttie, J.W. The biochemical basis of warfarin therapy. Adv. Exp. Med. Biol. 1987, 214, 3–16. [Google Scholar] [PubMed]
- Zimmermann, A.; Matschiner, J.T. Biochemical basis of hereditary resistance to warfarin in the rat. Biochem. Pharmacol. 1974, 23, 1033–1040. [Google Scholar] [CrossRef]
- Li, T.; Chang, C.Y.; Jin, D.Y.; Lin, P.J.; Khvorova, A.; Stafford, D.W. Identification of the gene for vitamin K epoxide reductase. Nature 2004, 427, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Rost, S.; Fregin, A.; Ivaskevicius, V.; Conzelmann, E.; Hortnagel, K.; Pelz, H.J.; Lappegard, K.; Seifried, E.; Scharrer, I.; Tuddenham, E.G.; et al. Mutations in VKORC1 cause warfarin resistance and multiple coagulation factor deficiency type 2. Nature 2004, 427, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Rieder, M.J.; Reiner, A.P.; Gage, B.F.; Nickerson, D.A.; Eby, C.S.; McLeod, H.L.; Blough, D.K.; Thummel, K.E.; Veenstra, D.L.; Rettie, A.E. Effect of VKORC1 haplotypes on transcriptional regulation and warfarin dose. N. Engl. J. Med. 2005, 352, 2285–2293. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, M.D.; Berg, R.L.; Zhang, K.Q.; Glurich, I.; Schmelzer, J.R.; Yale, S.H.; Vidaillet, H.J.; Burmester, J.K. Evaluation of genetic factors for warfarin dose prediction. Clin. Med. Res. 2007, 5, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.Y.; Chen, J.J.; Lee, M.T.; Wung, J.C.; Chen, Y.F.; Charng, M.J.; Lu, M.J.; Hung, C.R.; Wei, C.Y.; Chen, C.H.; et al. A novel functional VKORC1 promoter polymorphism is associated with inter-individual and inter-ethnic differences in warfarin sensitivity. Hum. Mol. Genet. 2005, 14, 1745–1751. [Google Scholar] [CrossRef] [PubMed]
- Bodin, L.; Verstuyft, C.; Tregouet, D.A.; Robert, A.; Dubert, L.; Funck-Brentano, C.; Jaillon, P.; Beaune, P.; Laurent-Puig, P.; Becquemont, L.; et al. Cytochrome P450 2C9 (CYP2C9) and vitamin K epoxide reductase (VKORC1) genotypes as determinants of acenocoumarol sensitivity. Blood 2005, 106, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Limdi, N.A.; Wadelius, M.; Cavallari, L.; Eriksson, N.; Crawford, D.C.; Lee, M.T.; Chen, C.H.; Motsinger-Reif, A.; Sagreiya, H.; Liu, N.; et al. Warfarin pharmacogenetics: A single VKORC1 polymorphism is predictive of dose across 3 racial groups. Blood 2010, 115, 3827–3834. [Google Scholar] [CrossRef] [PubMed]
- Hillman, M.A.; Wilke, R.A.; Caldwell, M.D.; Berg, R.L.; Glurich, I.; Burmester, J.K. Relative impact of covariates in prescribing warfarin according to CYP2C9 genotype. Pharmacogenetics 2004, 14, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Gage, B.F.; Eby, C.; Johnson, J.A.; Deych, E.; Rieder, M.J.; Ridker, P.M.; Milligan, P.E.; Grice, G.; Lenzini, P.; Rettie, A.E.; et al. Use of pharmacogenetic and clinical factors to predict the therapeutic dose of warfarin. Clin. Pharmacol. Ther. 2008, 84, 326–331. [Google Scholar] [CrossRef] [PubMed]
- International Warfarin Pharmacogenetics Consortium; Klein, T.E.; Altman, R.B.; Eriksson, N.; Gage, B.F.; Kimmel, S.E.; Lee, M.T.; Limdi, N.A.; Page, D.; Roden, D.M.; et al. Estimation of the warfarin dose with clinical and pharmacogenetic data. N. Engl. J. Med. 2009, 360, 753–764. [Google Scholar] [PubMed]
- Wu, A.H.; Wang, P.; Smith, A.; Haller, C.; Drake, K.; Linder, M.; Valdes, R., Jr. Dosing algorithm for warfarin using CYP2C9 and VKORC1 genotyping from a multi-ethnic population: Comparison with other equations. Pharmacogenomics 2008, 9, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Gong, I.Y.; Tirona, R.G.; Schwarz, U.I.; Crown, N.; Dresser, G.K.; Larue, S.; Langlois, N.; Lazo-Langner, A.; Zou, G.; Roden, D.M.; et al. Prospective evaluation of a pharmacogenetics-guided warfarin loading and maintenance dose regimen for initiation of therapy. Blood 2011, 118, 3163–3171. [Google Scholar] [CrossRef] [PubMed]
- Pirmohamed, M.; Burnside, G.; Eriksson, N.; Jorgensen, A.L.; Toh, C.H.; Nicholson, T.; Kesteven, P.; Christersson, C.; Wahlstrom, B.; Stafberg, C.; et al. A randomized trial of genotype-guided dosing of warfarin. N. Engl. J. Med. 2013, 369, 2294–2303. [Google Scholar] [CrossRef] [PubMed]
- Kimmel, S.E.; French, B.; Kasner, S.E.; Johnson, J.A.; Anderson, J.L.; Gage, B.F.; Rosenberg, Y.D.; Eby, C.S.; Madigan, R.A.; McBane, R.B.; et al. A pharmacogenetic versus a clinical algorithm for warfarin dosing. N. Engl. J. Med. 2013, 369, 2283–2293. [Google Scholar] [CrossRef] [PubMed]
- Pengo, V.; Zambon, C.F.; Fogar, P.; Padoan, A.; Nante, G.; Pelloso, M.; Moz, S.; Frigo, A.C.; Groppa, F.; Bozzato, D.; et al. A randomized trial of pharmacogenetic warfarin dosing in naive patients with non-valvular atrial fibrillation. PLoS ONE 2015, 10, e0145318. [Google Scholar] [CrossRef] [PubMed]
- Kimmel, S.E.; French, B.; Geller, N.L.; Investigators, C. Genotype-guided dosing of vitamin K antagonists. N. Engl. J. Med. 2014, 370, 1763–1764. [Google Scholar] [PubMed]
- Gong, I.Y.; Schwarz, U.I.; Crown, N.; Dresser, G.K.; Lazo-Langner, A.; Zou, G.; Roden, D.M.; Stein, C.M.; Rodger, M.; Wells, P.S.; et al. Clinical and genetic determinants of warfarin pharmacokinetics and pharmacodynamics during treatment initiation. PLoS ONE 2011, 6, e27808. [Google Scholar] [CrossRef] [PubMed]
- Gage, B.F.; Bass, A.R.; Lin, H.; Woller, S.C.; Stevens, S.M.; Al-Hammadi, N.; Li, J.; Rodriguez, T., Jr.; Miller, J.P.; McMillin, G.A.; et al. Effect of genotype-guided warfarin dosing on clinical events and anticoagulation control among patients undergoing hip or knee arthroplasty: The GIFT randomized clinical trial. JAMA 2017, 318, 1115–1124. [Google Scholar] [CrossRef] [PubMed]
- Caraco, Y.; Blotnick, S.; Muszkat, M. CYP2C9 genotype-guided warfarin prescribing enhances the efficacy and safety of anticoagulation: A prospective randomized controlled study. Clin. Pharmacol. Ther. 2008, 83, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Verhoef, T.I.; Ragia, G.; de Boer, A.; Barallon, R.; Kolovou, G.; Kolovou, V.; Konstantinides, S.; Le Cessie, S.; Maltezos, E.; van der Meer, F.J.; et al. A randomized trial of genotype-guided dosing of acenocoumarol and phenprocoumon. N. Engl. J. Med. 2013, 369, 2304–2312. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.L.; Horne, B.D.; Stevens, S.M.; Grove, A.S.; Barton, S.; Nicholas, Z.P.; Kahn, S.F.; May, H.T.; Samuelson, K.M.; Muhlestein, J.B.; et al. Randomized trial of genotype-guided versus standard warfarin dosing in patients initiating oral anticoagulation. Circulation 2007, 116, 2563–2570. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Ng, S.S.; Oldenburg, J.; Chong, P.Y.; Rost, S.; Guo, J.Y.; Yap, H.L.; Rankin, S.C.; Khor, H.B.; Yeo, T.C.; et al. Interethnic variability of warfarin maintenance requirement is explained by VKORC1 genotype in an Asian population. Clin. Pharmacol. Ther. 2006, 79, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Gaikwad, T.; Ghosh, K.; Shetty, S. VKORC1 and CYP2C9 genotype distribution in Asian countries. Thromb. Res. 2014, 134, 537–544. [Google Scholar] [CrossRef] [PubMed]
- Perera, M.A.; Gamazon, E.; Cavallari, L.H.; Patel, S.R.; Poindexter, S.; Kittles, R.A.; Nicolae, D.; Cox, N.J. The missing association: Sequencing-based discovery of novel SNPs in VKORC1 and CYP2C9 that affect warfarin dose in African Americans. Clin. Pharmacol. Ther. 2011, 89, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, W.; Gamazon, E.R.; Aquino-Michaels, K.; Patel, S.; O’Brien, T.J.; Harralson, A.F.; Kittles, R.A.; Barbour, A.; Tuck, M.; McIntosh, S.D.; et al. Ethnicity-specific pharmacogenetics: The case of warfarin in African Americans. Pharmacogenom. J. 2014, 14, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Mega, J.L.; Walker, J.R.; Ruff, C.T.; Vandell, A.G.; Nordio, F.; Deenadayalu, N.; Murphy, S.A.; Lee, J.; Mercuri, M.F.; Giugliano, R.P.; et al. Genetics and the clinical response to warfarin and edoxaban: Findings from the randomised, double-blind ENGAGE AF-TIMI 48 trial. Lancet 2015, 385, 2280–2287. [Google Scholar] [CrossRef]
- Higgins, M.J.; Stearns, V. CYP2D6 polymorphisms and tamoxifen metabolism: Clinical relevance. Curr. Oncol. Rep. 2010, 12, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Zembutsu, H. Pharmacogenomics toward personalized tamoxifen therapy for breast cancer. Pharmacogenomics 2015, 16, 287–296. [Google Scholar] [CrossRef] [PubMed]
- Jameera Begam, A.; Jubie, S.; Nanjan, M.J. Estrogen receptor agonists/antagonists in breast cancer therapy: A critical review. Bioorg. Chem. 2017, 71, 257–274. [Google Scholar] [CrossRef] [PubMed]
- Bhave, M.A.; Henry, N.L. Extended Endocrine Therapy: Is 5 Years Enough? Curr. Oncol. Rep. 2017, 19, 16. [Google Scholar] [CrossRef] [PubMed]
- Davies, C.; Pan, H.; Godwin, J.; Gray, R.; Arriagada, R.; Raina, V.; Abraham, M.; Medeiros Alencar, V.H.; Badran, A.; Bonfill, X.; et al. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet 2013, 381, 805–816. [Google Scholar] [CrossRef]
- Gray, R.G.; Rea, D.; Handley, K.; Bowden, S.J.; Perry, P.; Earl, H.M.; Poole, C.J.; Bates, T.; Chetiyawardana, S.; Dewar, J.A.; et al. aTTom: Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years in 6953 women with early breast cancer. J. Clin. Oncol. 2013, 31. [Google Scholar] [CrossRef]
- Del Re, M.; Citi, V.; Crucitta, S.; Rofi, E.; Belcari, F.; van Schaik, R.H.; Danesi, R. Pharmacogenetics of CYP2D6 and tamoxifen therapy: Light at the end of the tunnel? Pharmacol. Res. 2016, 107, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Hawse, J.R.; Subramaniam, M.; Cicek, M.; Wu, X.; Gingery, A.; Grygo, S.B.; Sun, Z.; Pitel, K.S.; Lingle, W.L.; Goetz, M.P.; et al. Endoxifen’s molecular mechanisms of action are concentration dependent and different than that of other anti-estrogens. PLoS ONE 2013, 8, e54613. [Google Scholar] [CrossRef] [PubMed]
- Lim, Y.C.; Desta, Z.; Flockhart, D.A.; Skaar, T.C. Endoxifen (4-hydroxy-N-desmethyl-tamoxifen) has anti-estrogenic effects in breast cancer cells with potency similar to 4-hydroxy-tamoxifen. Cancer Chemother. Pharmacol. 2005, 55, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Madlensky, L.; Natarajan, L.; Tchu, S.; Pu, M.; Mortimer, J.; Flatt, S.W.; Nikoloff, D.M.; Hillman, G.; Fontecha, M.R.; Lawrence, H.J.; et al. Tamoxifen metabolite concentrations, CYP2D6 genotype, and breast cancer outcomes. Clin. Pharmacol. Ther. 2011, 89, 718–725. [Google Scholar] [CrossRef] [PubMed]
- Gaedigk, A. Complexities of CYP2D6 gene analysis and interpretation. Int. Rev. Psychiatry 2013, 25, 534–553. [Google Scholar] [CrossRef] [PubMed]
- Caudle, K.E.; Dunnenberger, H.M.; Freimuth, R.R.; Peterson, J.F.; Burlison, J.D.; Whirl-Carrillo, M.; Scott, S.A.; Rehm, H.L.; Williams, M.S.; Klein, T.E.; et al. Standardizing terms for clinical pharmacogenetic test results: Consensus terms from the Clinical Pharmacogenetics Implementation Consortium (CPIC). Genet. Med. 2017, 19, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Desta, Z.; Stearns, V.; Ward, B.; Ho, H.; Lee, K.H.; Skaar, T.; Storniolo, A.M.; Li, L.; Araba, A.; et al. CYP2D6 genotype, antidepressant use, and tamoxifen metabolism during adjuvant breast cancer treatment. J. Natl. Cancer Inst. 2005, 97, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Borges, S.; Desta, Z.; Li, L.; Skaar, T.C.; Ward, B.A.; Nguyen, A.; Jin, Y.; Storniolo, A.M.; Nikoloff, D.M.; Wu, L.; et al. Quantitative effect of CYP2D6 genotype and inhibitors on tamoxifen metabolism: Implication for optimization of breast cancer treatment. Clin. Pharmacol. Ther. 2006, 80, 61–74. [Google Scholar] [CrossRef] [PubMed]
- Teft, W.A.; Gong, I.Y.; Dingle, B.; Potvin, K.; Younus, J.; Vandenberg, T.A.; Brackstone, M.; Perera, F.E.; Choi, Y.H.; Zou, G.; et al. CYP3A4 and seasonal variation in vitamin D status in addition to CYP2D6 contribute to therapeutic endoxifen level during tamoxifen therapy. Breast Cancer Res. Treat. 2013, 139, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Murdter, T.E.; Schroth, W.; Bacchus-Gerybadze, L.; Winter, S.; Heinkele, G.; Simon, W.; Fasching, P.A.; Fehm, T.; German, T.; Group, A.I.C.; et al. Activity levels of tamoxifen metabolites at the estrogen receptor and the impact of genetic polymorphisms of phase I and II enzymes on their concentration levels in plasma. Clin. Pharmacol. Ther. 2011, 89, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Gaedigk, A.; Simon, S.D.; Pearce, R.E.; Bradford, L.D.; Kennedy, M.J.; Leeder, J.S. The CYP2D6 activity score: Translating genotype information into a qualitative measure of phenotype. Clin. Pharmacol. Ther. 2008, 83, 234–242. [Google Scholar] [CrossRef] [PubMed]
- LLerena, A.; Naranjo, M.E.; Rodrigues-Soares, F.; Penas, L.E.M.; Farinas, H.; Tarazona-Santos, E. Interethnic variability of CYP2D6 alleles and of predicted and measured metabolic phenotypes across world populations. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1569–1583. [Google Scholar] [CrossRef] [PubMed]
- Bradford, L.D. CYP2D6 allele frequency in European Caucasians, Asians, Africans and their descendants. Pharmacogenomics 2002, 3, 229–243. [Google Scholar] [CrossRef] [PubMed]
- Sideras, K.; Ingle, J.N.; Ames, M.M.; Loprinzi, C.L.; Mrazek, D.P.; Black, J.L.; Weinshilboum, R.M.; Hawse, J.R.; Spelsberg, T.C.; Goetz, M.P. Coprescription of tamoxifen and medications that inhibit CYP2D6. J. Clin. Oncol. 2010, 28, 2768–2776. [Google Scholar] [CrossRef] [PubMed]
- Binkhorst, L.; Mathijssen, R.H.; van Herk-Sukel, M.P.; Bannink, M.; Jager, A.; Wiemer, E.A.; van Gelder, T. Unjustified prescribing of CYP2D6 inhibiting SSRIs in women treated with tamoxifen. Breast Cancer Res. Treat. 2013, 139, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.M.; Juurlink, D.N.; Gomes, T.; Duong-Hua, M.; Pritchard, K.I.; Austin, P.C.; Paszat, L.F. Selective serotonin reuptake inhibitors and breast cancer mortality in women receiving tamoxifen: A population based cohort study. BMJ 2010, 340, c693. [Google Scholar] [CrossRef] [PubMed]
- Goetz, M.P.; Rae, J.M.; Suman, V.J.; Safgren, S.L.; Ames, M.M.; Visscher, D.W.; Reynolds, C.; Couch, F.J.; Lingle, W.L.; Flockhart, D.A.; et al. Pharmacogenetics of tamoxifen biotransformation is associated with clinical outcomes of efficacy and hot flashes. J. Clin. Oncol. 2005, 23, 9312–9318. [Google Scholar] [CrossRef] [PubMed]
- Schroth, W.; Antoniadou, L.; Fritz, P.; Schwab, M.; Muerdter, T.; Zanger, U.M.; Simon, W.; Eichelbaum, M.; Brauch, H. Breast cancer treatment outcome with adjuvant tamoxifen relative to patient CYP2D6 and CYP2C19 genotypes. J. Clin. Oncol. 2007, 25, 5187–5193. [Google Scholar] [CrossRef] [PubMed]
- Ramon y Cajal, T.; Altes, A.; Pare, L.; del Rio, E.; Alonso, C.; Barnadas, A.; Baiget, M. Impact of CYP2D6 polymorphisms in tamoxifen adjuvant breast cancer treatment. Breast Cancer Res. Treat. 2010, 119, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Lammers, L.A.; Mathijssen, R.H.; van Gelder, T.; Bijl, M.J.; de Graan, A.J.; Seynaeve, C.; van Fessem, M.A.; Berns, E.M.; Vulto, A.G.; van Schaik, R.H. The impact of CYP2D6-predicted phenotype on tamoxifen treatment outcome in patients with metastatic breast cancer. Br. J. Cancer 2010, 103, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Schroth, W.; Goetz, M.P.; Hamann, U.; Fasching, P.A.; Schmidt, M.; Winter, S.; Fritz, P.; Simon, W.; Suman, V.J.; Ames, M.M.; et al. Association between CYP2D6 polymorphisms and outcomes among women with early stage breast cancer treated with tamoxifen. JAMA 2009, 302, 1429–1436. [Google Scholar] [CrossRef] [PubMed]
- Goetz, M.P.; Suman, V.J.; Hoskin, T.L.; Gnant, M.; Filipits, M.; Safgren, S.L.; Kuffel, M.; Jakesz, R.; Rudas, M.; Greil, R.; et al. CYP2D6 metabolism and patient outcome in the Austrian Breast and Colorectal Cancer Study Group trial (ABCSG) 8. Clin. Cancer Res. 2013, 19, 500–507. [Google Scholar] [CrossRef] [PubMed]
- Damodaran, S.E.; Pradhan, S.C.; Umamaheswaran, G.; Kadambari, D.; Reddy, K.S.; Adithan, C. Genetic polymorphisms of CYP2D6 increase the risk for recurrence of breast cancer in patients receiving tamoxifen as an adjuvant therapy. Cancer Chemother. Pharmacol. 2012, 70, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Saladores, P.; Murdter, T.; Eccles, D.; Chowbay, B.; Zgheib, N.K.; Winter, S.; Ganchev, B.; Eccles, B.; Gerty, S.; Tfayli, A.; et al. Tamoxifen metabolism predicts drug concentrations and outcome in premenopausal patients with early breast cancer. Pharmacogenom. J. 2015, 15, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Sun, Y.; Yao, L.; Shi, L.; Wu, Y.; Ouyang, T.; Li, J.; Wang, T.; Fan, Z.; Fan, T.; et al. Association between CYP2D6 *10 genotype and survival of breast cancer patients receiving tamoxifen treatment. Ann. Oncol. 2008, 19, 1423–1429. [Google Scholar] [CrossRef] [PubMed]
- Kiyotani, K.; Mushiroda, T.; Sasa, M.; Bando, Y.; Sumitomo, I.; Hosono, N.; Kubo, M.; Nakamura, Y.; Zembutsu, H. Impact of CYP2D6*10 on recurrence-free survival in breast cancer patients receiving adjuvant tamoxifen therapy. Cancer Sci. 2008, 99, 995–999. [Google Scholar] [CrossRef] [PubMed]
- Kiyotani, K.; Mushiroda, T.; Imamura, C.K.; Hosono, N.; Tsunoda, T.; Kubo, M.; Tanigawara, Y.; Flockhart, D.A.; Desta, Z.; Skaar, T.C.; et al. Significant effect of polymorphisms in CYP2D6 and ABCC2 on clinical outcomes of adjuvant tamoxifen therapy for breast cancer patients. J. Clin. Oncol. 2010, 28, 1287–1293. [Google Scholar] [CrossRef] [PubMed]
- Abreu, M.H.; Gomes, M.; Menezes, F.; Afonso, N.; Abreu, P.H.; Medeiros, R.; Pereira, D.; Lopes, C. CYP2D6*4 polymorphism: A new marker of response to hormonotherapy in male breast cancer? Breast 2015, 24, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Rae, J.M.; Drury, S.; Hayes, D.F.; Stearns, V.; Thibert, J.N.; Haynes, B.P.; Salter, J.; Sestak, I.; Cuzick, J.; Dowsett, M.; et al. CYP2D6 and UGT2B7 genotype and risk of recurrence in tamoxifen-treated breast cancer patients. J. Natl. Cancer Inst. 2012, 104, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Regan, M.M.; Leyland-Jones, B.; Bouzyk, M.; Pagani, O.; Tang, W.; Kammler, R.; Dell’orto, P.; Biasi, M.O.; Thurlimann, B.; Lyng, M.B.; et al. CYP2D6 genotype and tamoxifen response in postmenopausal women with endocrine-responsive breast cancer: The breast international group 1–98 trial. J. Natl. Cancer Inst. 2012, 104, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Abraham, J.E.; Maranian, M.J.; Driver, K.E.; Platte, R.; Kalmyrzaev, B.; Baynes, C.; Luccarini, C.; Shah, M.; Ingle, S.; Greenberg, D.; et al. CYP2D6 gene variants: Association with breast cancer specific survival in a cohort of breast cancer patients from the United Kingdom treated with adjuvant tamoxifen. Breast Cancer Res. 2010, 12, R64. [Google Scholar] [CrossRef] [PubMed]
- Nowell, S.A.; Ahn, J.; Rae, J.M.; Scheys, J.O.; Trovato, A.; Sweeney, C.; MacLeod, S.L.; Kadlubar, F.F.; Ambrosone, C.B. Association of genetic variation in tamoxifen-metabolizing enzymes with overall survival and recurrence of disease in breast cancer patients. Breast Cancer Res. Treat. 2005, 91, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Park, I.H.; Ro, J.; Park, S.; Lim, H.S.; Lee, K.S.; Kang, H.S.; Jung, S.Y.; Lee, S. Lack of any association between functionally significant CYP2D6 polymorphisms and clinical outcomes in early breast cancer patients receiving adjuvant tamoxifen treatment. Breast Cancer Res. Treat. 2012, 131, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Ahern, T.P.; Hertz, D.L.; Damkier, P.; Ejlertsen, B.; Hamilton-Dutoit, S.J.; Rae, J.M.; Regan, M.M.; Thompson, A.M.; Lash, T.L.; Cronin-Fenton, D.P. Cytochrome P-450 2D6 (CYP2D6) Genotype and Breast Cancer Recurrence in Tamoxifen-Treated Patients: Evaluating the Importance of Loss of Heterozygosity. Am. J. Epidemiol. 2017, 185, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Hertz, D.L.; Kidwell, K.M.; Hilsenbeck, S.G.; Oesterreich, S.; Osborne, C.K.; Philips, S.; Chenault, C.; Hartmaier, R.J.; Skaar, T.C.; Sikora, M.J.; et al. CYP2D6 genotype is not associated with survival in breast cancer patients treated with tamoxifen: Results from a population-based study. Breast Cancer Res. Treat. 2017, 166, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Kiyotani, K.; Mushiroda, T.; Hosono, N.; Tsunoda, T.; Kubo, M.; Aki, F.; Okazaki, Y.; Hirata, K.; Takatsuka, Y.; Okazaki, M.; et al. Lessons for pharmacogenomics studies: Association study between CYP2D6 genotype and tamoxifen response. Pharmacogenet. Genom. 2010, 20, 565–568. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Liu, Y.; Liu, Z.; You, J.; Chen, Z.; Wang, J.; Peng, Q.; Xie, L.; Li, R.; Li, S.; et al. CYP2D6 polymorphisms influence tamoxifen treatment outcomes in breast cancer patients: A meta-analysis. Cancer Chemother. Pharmacol. 2013, 72, 287–303. [Google Scholar] [CrossRef] [PubMed]
- Lum, D.W.; Perel, P.; Hingorani, A.D.; Holmes, M.V. CYP2D6 genotype and tamoxifen response for breast cancer: A systematic review and meta-analysis. PLoS ONE 2013, 8, e76648. [Google Scholar] [CrossRef] [PubMed]
- Jung, J.A.; Lim, H.S. Association between CYP2D6 genotypes and the clinical outcomes of adjuvant tamoxifen for breast cancer: A meta-analysis. Pharmacogenomics 2014, 15, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Province, M.A.; Goetz, M.P.; Brauch, H.; Flockhart, D.A.; Hebert, J.M.; Whaley, R.; Suman, V.J.; Schroth, W.; Winter, S.; Zembutsu, H.; et al. CYP2D6 genotype and adjuvant tamoxifen: Meta-analysis of heterogeneous study populations. Clin. Pharmacol. Ther. 2014, 95, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Hwang, G.S.; Bhat, R.; Crutchley, R.D.; Trivedi, M.V. Impact of CYP2D6 polymorphisms on endoxifen concentrations and breast cancer outcomes. Pharmacogenom. J. 2017. [Google Scholar] [CrossRef] [PubMed]
- Binkhorst, L.; Mathijssen, R.H.; Jager, A.; van Gelder, T. Individualization of tamoxifen therapy: Much more than just CYP2D6 genotyping. Cancer Treat. Rev. 2015, 41, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Powers, J.L.; Buys, S.S.; Fletcher, D.; Melis, R.; Johnson-Davis, K.L.; Lyon, E.; Malmberg, E.M.; McMillin, G.A. Multigene and drug interaction approach for tamoxifen metabolite patterns reveals possible involvement of CYP2C9, CYP2C19, and ABCB1. J. Clin. Pharmacol. 2016, 56, 1570–1581. [Google Scholar] [CrossRef] [PubMed]
- Marcath, L.A.; Deal, A.M.; Van Wieren, E.; Danko, W.; Walko, C.M.; Ibrahim, J.G.; Weck, K.E.; Jones, D.R.; Desta, Z.; McLeod, H.L.; et al. Comprehensive assessment of cytochromes P450 and transporter genetics with endoxifen concentration during tamoxifen treatment. Pharmacogenet. Genom. 2017, 27, 402–409. [Google Scholar] [CrossRef] [PubMed]
- Lamba, J.K.; Lin, Y.S.; Schuetz, E.G.; Thummel, K.E. Genetic contribution to variable human CYP3A-mediated metabolism. Adv. Drug Deliv. Rev. 2002, 54, 1271–1294. [Google Scholar] [CrossRef]
- Binkhorst, L.; van Gelder, T.; Loos, W.J.; de Jongh, F.E.; Hamberg, P.; Moghaddam-Helmantel, I.M.; de Jonge, E.; Jager, A.; Seynaeve, C.; van Schaik, R.H.; et al. Effects of CYP induction by rifampicin on tamoxifen exposure. Clin. Pharmacol. Ther. 2012, 92, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Henderson, S.L.; Teft, W.A.; Kim, R.B. Profound reduction in tamoxifen active metabolite endoxifen in a breast cancer patient treated with rifampin prior to initiation of an anti-TNFα biologic for ulcerative colitis: A case report. BMC Cancer 2016, 16, 304. [Google Scholar] [CrossRef] [PubMed]
- Antunes, M.V.; de Oliveira, V.; Raymundo, S.; Staudt, D.E.; Gossling, G.; Biazus, J.V.; Cavalheiro, J.A.; Rosa, D.D.; Mathy, G.; Wallemacq, P.; et al. CYP3A4*22 is related to increased plasma levels of 4-hydroxytamoxifen and partially compensates for reduced CYP2D6 activation of tamoxifen. Pharmacogenomics 2015, 16, 601–617. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.S.; Chen, X.A.; Singh, O.; Yap, Y.S.; Ng, R.C.; Wong, N.S.; Wong, M.; Lee, E.J.; Chowbay, B. Impact of CYP2D6, CYP3A5, CYP2C9 and CYP2C19 polymorphisms on tamoxifen pharmacokinetics in Asian breast cancer patients. Br. J. Clin. Pharmacol. 2011, 71, 737–750. [Google Scholar] [CrossRef] [PubMed]
- van Schaik, R.H.; Kok, M.; Sweep, F.C.; van Vliet, M.; van Fessem, M.; Meijer-van Gelder, M.E.; Seynaeve, C.; Lindemans, J.; Wesseling, J.; Van’t Veer, L.J.; et al. The CYP2C19*2 genotype predicts tamoxifen treatment outcome in advanced breast cancer patients. Pharmacogenomics 2011, 12, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; He, J.; He, G.H.; He, J.C.; Xu, F.; Xu, G.L. Association of CYP2C19 polymorphisms with survival of breast cancer patients using tamoxifen: Results of a meta-analysis. Asian Pac. J. Cancer Prev. 2014, 15, 8331–8335. [Google Scholar] [CrossRef] [PubMed]
- Moyer, A.M.; Suman, V.J.; Weinshilboum, R.M.; Avula, R.; Black, J.L.; Safgren, S.L.; Kuffel, M.J.; Ames, M.M.; Ingle, J.N.; Goetz, M.P. SULT1A1, CYP2C19 and disease-free survival in early breast cancer patients receiving tamoxifen. Pharmacogenomics 2011, 12, 1535–1543. [Google Scholar] [CrossRef] [PubMed]
- Damkier, P.; Kjaersgaard, A.; Barker, K.A.; Cronin-Fenton, D.; Crawford, A.; Hellberg, Y.; Janssen, E.A.M.; Langefeld, C.; Ahern, T.P.; Lash, T.L. CYP2C19*2 and CYP2C19*17 variants and effect of tamoxifen on breast cancer recurrence: Analysis of the International Tamoxifen Pharmacogenomics Consortium dataset. Sci. Rep. 2017, 7, 7727. [Google Scholar] [CrossRef] [PubMed]
- Kiyotani, K.; Mushiroda, T.; Nakamura, Y.; Zembutsu, H. Pharmacogenomics of tamoxifen: Roles of drug metabolizing enzymes and transporters. Drug Metab. Pharmacokinet. 2012, 27, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Santander, A.; Gaibar, M.; Novillo, A.; Romero-Lorca, A.; Rubio, M.; Chicharro, L.M.; Tejerina, A.; Bandres, F. Relationship between genotypes Sult1a2 and Cyp2d6 and tamoxifen metabolism in breast cancer patients. PLoS ONE 2013, 8, e70183. [Google Scholar] [CrossRef] [PubMed]
- Romero-Lorca, A.; Novillo, A.; Gaibar, M.; Bandres, F.; Fernandez-Santander, A. Impacts of the Glucuronidase Genotypes UGT1A4, UGT2B7, UGT2B15 and UGT2B17 on Tamoxifen Metabolism in Breast Cancer Patients. PLoS ONE 2015, 10, e0132269. [Google Scholar] [CrossRef] [PubMed]
- Novillo, A.; Romero-Lorca, A.; Gaibar, M.; Rubio, M.; Fernandez-Santander, A. Tamoxifen metabolism in breast cancer treatment: Taking the focus off the CYP2D6 gene. Pharmacogenom. J. 2017, 17, 109–111. [Google Scholar] [CrossRef] [PubMed]
- Fox, P.; Balleine, R.L.; Lee, C.; Gao, B.; Balakrishnar, B.; Menzies, A.M.; Yeap, S.H.; Ali, S.S.; Gebski, V.; Provan, P.; et al. Dose Escalation of Tamoxifen in Patients with Low Endoxifen Level: Evidence for Therapeutic Drug Monitoring-The TADE Study. Clin. Cancer Res. 2016, 22, 3164–3171. [Google Scholar] [CrossRef] [PubMed]
- Gong, I.Y.; Teft, W.A.; Ly, J.; Chen, Y.H.; Alicke, B.; Kim, R.B.; Choo, E.F. Determination of clinically therapeutic endoxifen concentrations based on efficacy from human MCF7 breast cancer xenografts. Breast Cancer Res. Treat. 2013, 139, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Irvin, W.J., Jr.; Walko, C.M.; Weck, K.E.; Ibrahim, J.G.; Chiu, W.K.; Dees, E.C.; Moore, S.G.; Olajide, O.A.; Graham, M.L.; Canale, S.T.; et al. Genotype-guided tamoxifen dosing increases active metabolite exposure in women with reduced CYP2D6 metabolism: A multicenter study. J. Clin. Oncol. 2011, 29, 3232–3239. [Google Scholar] [CrossRef] [PubMed]
- Kiyotani, K.; Mushiroda, T.; Imamura, C.K.; Tanigawara, Y.; Hosono, N.; Kubo, M.; Sasa, M.; Nakamura, Y.; Zembutsu, H. Dose-adjustment study of tamoxifen based on CYP2D6 genotypes in Japanese breast cancer patients. Breast Cancer Res. Treat. 2012, 131, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Martinez de Duenas, E.; Ochoa Aranda, E.; Blancas Lopez-Barajas, I.; Ferrer Magdalena, T.; Bandres Moya, F.; Chicharro Garcia, L.M.; Gomez Capilla, J.A.; Zafra Ceres, M.; de Haro, T.; Romero Llorens, R.; et al. Adjusting the dose of tamoxifen in patients with early breast cancer and CYP2D6 poor metabolizer phenotype. Breast 2014, 23, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Dezentje, V.O.; Opdam, F.L.; Gelderblom, H.; Hartigh den, J.; Van der Straaten, T.; Vree, R.; Maartense, E.; Smorenburg, C.H.; Putter, H.; Dieudonne, A.S.; et al. CYP2D6 genotype- and endoxifen-guided tamoxifen dose escalation increases endoxifen serum concentrations without increasing side effects. Breast Cancer Res. Treat. 2015, 153, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Hertz, D.L.; Rae, J.M. Individualized tamoxifen dose escalation: Confirmation of feasibility, question of utility. Clin. Cancer Res. 2016, 22, 3121–3123. [Google Scholar] [CrossRef] [PubMed]
- Goetz, M.P.; Suman, V.J.; Reid, J.M.; Northfelt, D.W.; Mahr, M.A.; Ralya, A.T.; Kuffel, M.; Buhrow, S.A.; Safgren, S.L.; McGovern, R.M.; et al. First-in-human phase I study of the tamoxifen metabolite Z-endoxifen in women with endocrine-refractory metastatic breast cancer. J. Clin. Oncol. 2017, 35, 3391–3400. [Google Scholar] [CrossRef] [PubMed]
- Gryn, S.E.; Teft, W.A.; Kim, R.B. Profound reduction in the tamoxifen active metabolite endoxifen in a patient on phenytoin for epilepsy compared with a CYP2D6 genotype matched cohort. Pharmacogenet. Genom. 2014, 24, 367–369. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.A.; Caudle, K.E.; Gong, L.; Whirl-Carrillo, M.; Stein, C.M.; Scott, S.A.; Lee, M.T.; Gage, B.F.; Kimmel, S.E.; Perera, M.A.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for pharmacogenetics-guided warfarin dosing: 2017 update. Clin. Pharmacol. Ther. 2017, 102, 397–404. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic of the effect of warfarin and involvement of CYP2C9, CYP4F2, and vitamin K epoxide reductase complex subunit 1 (VKORC1) on the vitamin K cycle. VKORC1 and NAD(P)H:Quinone oxidoreductase (NQO) reduce vitamin K1 to active vitamin K1 dihydroquinone. Gamma-glutamyl carboxylase (GGCX) catalyzes carboxylation of glutamate residues activating clotting factors II, VII, IX, and X in a vitamin K1 dihydroquinone dependent manner. VKORC1 reduces vitamin K1 epoxide to vitamin K1, restarting the cycle. Warfarin impairs VKORC1 and the reduction of vitamin K1. Warfarin is metabolized by CYP2C9. Vitamin K1 can be removed from the cycle by CYP4F2 by hydroxylation.
Figure 1.
Schematic of the effect of warfarin and involvement of CYP2C9, CYP4F2, and vitamin K epoxide reductase complex subunit 1 (VKORC1) on the vitamin K cycle. VKORC1 and NAD(P)H:Quinone oxidoreductase (NQO) reduce vitamin K1 to active vitamin K1 dihydroquinone. Gamma-glutamyl carboxylase (GGCX) catalyzes carboxylation of glutamate residues activating clotting factors II, VII, IX, and X in a vitamin K1 dihydroquinone dependent manner. VKORC1 reduces vitamin K1 epoxide to vitamin K1, restarting the cycle. Warfarin impairs VKORC1 and the reduction of vitamin K1. Warfarin is metabolized by CYP2C9. Vitamin K1 can be removed from the cycle by CYP4F2 by hydroxylation.

{kind=link}
{kind=link}
Table 1.
Association of pharmacogenomics with warfarin outcomes.
| Studies | Design | N | Population | Alleles | Outcomes | P |
|---|---|---|---|---|---|---|
| Positive association | ||||||
| Primohamed et al., 2013 [32] | RCT, genotype guided vs. standard dose | 455 | 98% White 1% Black 1% Asian | CYP2C9*2 CYP2C9*3 VKORC1*2 | Improved time within therapeutic INR (67.4% vs. 60.3%); reduction in INR > 4, reduced time to therapeutic INR | <0.001 |
| Gage et al., 2017 [37] | RCT, genotype guided vs. clinical | 1597 | 91% White 6% Black 2% Asian 1% Other | CYP2C9*2 CYP2C9*3 VKORC1*2 CYP4F2*3 | Reduced composite measure of major bleeding, INR > 4, death, and VTE (10.8% vs. 14.7%). In hip and knee arthroplasty patients | <0.02 |
| Caraco et al., 2008 [38] | RCT, Genotype vs. clinical | 191 | Unavailable | CYP2C9*2 CYP2C9*3 | Reduction in time to first therapeutic INR (2.73 days earlier) and reduction in time to stable INR (18.1 days earlier) | <0.001 |
| Gage et al., 2008 [28] | Validation of dosing algorithm | 292 | 93% Caucasian 15% Black 2% Hispanic | CYP2C9*2 CYP2C9*3 VKORC1*2 | Pharmacogenomic dose prediction more accurate than clinical dose prediction (53% vs. 17% of explained variability, respectively) | <0.0001 |
| IWPC, 2009 [29] | Validation of dosing algorithm | 1009 | 55% White 30% Asian 10% Black 5% Other | CYP2C9*2 CYP2C9*3 VKORC1*2 # | Pharmacogenomic dose prediction more accurate than clinical dose prediction (accurately identified 49.4% vs. 33.3% of patients requiring ≤21 mg warfarin per week, respectively) | <0.001 |
| Gong et al., 2011 [31] | Validation of dosing algorithm | 167 | 95% White 2% Black 2%Asian 1% Other | CYP2C9*2 CYP2C9*3 VKORC1*2 CYP4F2*3 | Demonstrated the safe effective prediction of dose limiting variation | N/A |
| Negative association | ||||||
| Kimmel et al., 2013 [33] | RCT, Genotype guided vs. clinical | 1015 | 66%White 27% Black 7% Hispanic | CYP2C9*2 CYP2C9*3 VKORC1*2 | No difference in time in therapeutic INR (45.2% vs. 45.4%) No difference in anticoagulation control or dose prediction | 0.91 |
| Verhoef et al., 2013 [39] | RCT, Genotype guided vs. clinical | 1597 | 98% White | CYP2C9*2 CYP2C9*3 VKORC1*2 | No difference in time in therapeutic INR range (61.6% vs. 60.2%) | 0.47 |
| Pengo et al., 2015 [34] | RCT, Genotype guided vs. standard | 180 | 100% White | CYP2C9*2 CYP2C9*3 VKORC1*2 CYP4F2*3 | No difference in out of range INRs (45.6% vs. 43.6%) or time in therapeutic INR range (51.9% vs. 53.3%) | 0.79 0.71 |
| Anderson et al., 2007 [40] | RCT, Genotype guided vs. standard | 200 | 94% White | CYP2C9*2 CYP2C9*3 VKORC1*2 | No difference in time in therapeutic INR range (30.7% vs. 33.1%) | 0.47 |
RCT, randomized control trial; INR, International normalization ratio; VTE, venous thromboembolism; #, VKORC1*2 or one of six other linked SNPs, N/A, not available.
Table 2.
Association of CYP2D6 pharmacogenomics with tamoxifen outcomes.
| Studies | N | Alleles | DNA Source | Conclusions | Outcome | HR (95% CI) | P |
|---|---|---|---|---|---|---|---|
| Positive association | |||||||
| Goetz et al., 2005 [68] | 190 | *4 | PE-tissue, buccal swabs | *4/*4 patients had worse RFS and DFS | RFS DFS | 2.71 (1.15–6.41) 2.44 (1.22–4.90) | 0.023 0.012 |
| Schroth et al., 2007 [69] | 206 | *4, *5, *10, *41, CNV | normal breast tissue | Decreased function alleles (*4, *5, *10 and *41) were associated with higher rates of recurrence and shorter relapse free periods | RFS EFS | 2.24 (1.16–4.33) 1.89 (1.10–3.25) | 0.02 0.02 |
| Ramón et al., 2010 [70] | 91 | 33 alleles | blood | Patients with *4/*4, *4/*41, *1/*5 or *2/*5 genotypes had shorter DFS | 0.016 | ||
| Lammers et al., 2010 [71] | 99 | *3, *4, *5, *6, *10, *41 | blood | PMs had worse overall survival compared to NMs | OS | 2.09 (1.06–4.12) | 0.034 |
| Schroth et al., 2009 [72] | 1325 | *3,*4, *5, *10, *41 | blood, fresh frozen or PE-tissue | Decreased activity (NM/IM; PM) had worse EFS and DFS | EFS DFS | 1.35 (1.08–1.68) 1.31 (1.06–1.61) | 0.007 0.02 |
| Goetz et al., 2013 [73] | 453 | *3, *4, *6, *10, *41 | PE- tissue | PM/PM patients had higher risk of disease event compared to NM/NM patients | OR | 2.45 (1.05–5.73 | 0.04 |
| Damodaran et al., 2012 [74] | 132 | *1, *2, *4, *5, *10 | blood | CYP2D6 activity scores <0.5 had worse RFS compared to activity scores >1 | RFS | 7.29 (2.92–18.2) | <0.001 |
| Saladores et al., 2015 [75] | 587 | *3, *4, *5, *6, *9, *10, *41, CNV | blood | Improved DRFS was associated with increased CYP2D6 activity score | DRFS | 0.62 (0.43–0.9) | 0.013 |
| Xu et al., 2008 [76] | 152 | *10 | blood, fresh frozen or PE-tissue | *10/*10 was associated with worse DFS | DFS | 4.7 (1.1–20.0) | 0.04 |
| Kiyotani et al., 2008 [77] | 67 | *4, *5, *6, *10, *14, *18, *21, *41 | blood | *10/*10 genotype had worse RFS | RFS | 10.04 (1.17–86.3) | 0.036 |
| Kiyotani et al., 2010 [78] | 282 | *4, *5, *6, *10, *14B, *18, *21, *36, *41, CNV | blood | Presence of two variant alleles was associated with worse RFS compared to patients with no variants | RFS | 9.52 (2.79–32.45) | <0.0001 |
| Negative association | |||||||
| Rae et al., 2012 [80] | 588 | *2, *3, *4, *6, *10, *41 | PE-tissue | PMs did not have reduced recurrence rates compared to NMs | RFS | 0.99 (0.48–2.08) | 0.99 |
| Regan et al., 2012 [81] | 973 | *2, *3, *4, *5, *6, *7, *10, *17, *41 | PE-tissue | IMs and PMs treated with tamoxifen monotherapy were not associated with BCFI | BCFI | 0.86 (0.6–1.24) | 0.35 |
| Abraham et al., 2010 [82] | 3155 | *4, *5, *6, *9, *10, *41, CNV | blood | PM/IM patients did not have reduced survival outcomes compared to NMs | BCSS | 0.93 (0.55–1.57) | 0.78 |
| Nowell et al., 2005 [83] | 160 | *3, *4, *6 | PE-tissue | *4/*4, *1/*4 were not associated with reduced DFS compared to *1/*1 | DFS | 0.67 (0.33–1.35) | 0.19 |
| Park et al., 2012 [84] | 716 | *2, *5, *10, *41 | blood | Homozygous variant carriers did not have reduced RFS | RFS | 1.14 (0.68–1.92) | 0.61 |
| Hertz et al., 2017 [86] | 476 | *2, *3, *4, *6, *10, *41, CNV | Fresh frozen tumors | CYP2D6 activity score was not associated with RFS | RFS | 1.16 (0.84–1.62) | 0.37 |
| Kiyotani et al., 2010 [87] | 167 | *1, *4, *5, *10, *21, *36, *41 | blood | No association between genotype and RFS in patients on tamoxifen-combined therapy | RFS | 0.64 (0.20–1.99) | 0.44 |
HR, Hazard ratio; CI, confidence interval; PE, paraffin-embedded; RFS, recurrence free survival; DFS, disease free survival; CNV, copy number variation; EFS, event free survival; NM, CYP2D6 normal metabolizer; OS, overall survival; IM, CYP2D6 intermediate metabolizer; PM, CYP2D6 poor metabolizer; DRFS, distant relapse free survival; BCFI, breast cancer-free interval; BCSS, breast cancer specific survival.
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Wigle, T.J.; Jansen, L.E.; Teft, W.A.; Kim, R.B. Pharmacogenomics Guided-Personalization of Warfarin and Tamoxifen. J. Pers. Med. 2017, 7, 20. https://doi.org/10.3390/jpm7040020
AMA Style
Wigle TJ, Jansen LE, Teft WA, Kim RB. Pharmacogenomics Guided-Personalization of Warfarin and Tamoxifen. Journal of Personalized Medicine. 2017; 7(4):20. https://doi.org/10.3390/jpm7040020
Chicago/Turabian StyleWigle, Theodore J., Laura E. Jansen, Wendy A. Teft, and Richard B. Kim. 2017. "Pharmacogenomics Guided-Personalization of Warfarin and Tamoxifen" Journal of Personalized Medicine 7, no. 4: 20. https://doi.org/10.3390/jpm7040020
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.