Sialidosis: A Review of Morphology and Molecular Biology of a Rare Pediatric Disorder



1
Department of Laboratory Medicine and Pathology (5B4.09), University of Alberta, 8440 112 St NW, Edmonton, AB T6G 2B7, Canada
2
Department of Pediatrics, Stollery Children’s Hospital, University of Alberta Hospital, Edmonton, AB T6G 2B7, Canada
*
Author to whom correspondence should be addressed.
Diagnostics 2018, 8(2), 29; https://doi.org/10.3390/diagnostics8020029
Submission received: 23 February 2018
/
Revised: 22 April 2018
/
Accepted: 22 April 2018
/
Published: 25 April 2018
(This article belongs to the Special Issue Diagnosis and Management of Pediatric Diseases)
Abstract
:Sialidosis (MIM 256550) is a rare, autosomal recessive inherited disorder, caused by α-N-acetyl neuraminidase deficiency resulting from a mutation in the neuraminidase gene (NEU1), located on 6p21.33. This genetic alteration leads to abnormal intracellular accumulation as well as urinary excretion of sialyloligosaccharides. A definitive diagnosis is made after the identification of a mutation in the NEU1 gene. So far, 40 mutations of NEU1 have been reported. An association exists between the impact of the individual mutations and the severity of clinical presentation of sialidosis. According to the clinical symptoms, sialidosis has been divided into two subtypes with different ages of onset and severity, including sialidosis type I (normomorphic or mild form) and sialidosis type II (dysmorphic or severe form). Sialidosis II is further subdivided into (i) congenital; (ii) infantile; and (iii) juvenile. Despite being uncommon, sialidosis has enormous clinical relevance due to its debilitating character. A complete understanding of the underlying pathology remains a challenge, which in turn limits the development of effective therapeutic strategies. Furthermore, in the last few years, some atypical cases of sialidosis have been reported as well. We herein attempt to combine and discuss the underlying molecular biology, the clinical features, and the morphological patterns of sialidosis type I and II.
1. Introduction
Sialidosis, an autosomal recessive disorder, occurs due to a structural defect in the neuraminidase gene and is characterized by abnormal tissue accumulation as well as urinary excretion of sialylated oligosaccharides and glycolipids [1].
The human neuraminidase gene is located at chromosome band 6p21.3, where the HLA locus is also reported to be located [2]. Until 1977, deficiency of Neuraminidase 1 (NEU1) was thought to be associated with classical mucolipidosis I, a severe and rapidly progressive lysosomal storage disease with onset at birth or shortly after birth [3,4]. In 1977, the term sialidosis was first used to describe the syndrome of two siblings having a visual impairment and mild neurological manifestations that slowly developed in their adolescence. Enzymatic assays in cultured fibroblasts and leukocytes from these siblings exhibited an isolated deficiency of NEU1 [3,4]. Later, Sialidosis was classified into two types: Sialidosis Type I (normomorphic) and Sialidosis Type II (dysmorphic) [5]. In this study, we review the underlying genetics and molecular mechanisms of sialidosis and their correlation with clinical and morphological findings.
2. Background
Sialidase (neuraminidase, EC 3.2.1.18) catalyzes the hydrolysis of terminal sialic acid residues of glycoconjugates. Sialidase has been extensively studied in viruses and bacteria. In these microorganisms, its function is to destroy the sialic acid-containing receptors at the surface of host cells to mobilize bacterial nutrients. In mammals, three types of sialidases have been reported, including the lysosomal, plasma membrane, and cytosolic localized enzymes [1].
In human lysosomes, the degradation of complex macromolecular substrates requires the synergistic action of multiple hydrolases that act synergistically to carry out the degradation process of complex macromolecular substrates efficiently. One such efficient catalytic team is formed by three hydrolases which are ubiquitous, but differentially expressed: the serine carboxypeptidase, protective protein/cathepsin A (PPCA), the sialidase, Neuraminidase-1 (NEU1), and the glycosidase β-Galactosidase (β-GAL) [6]. The different expression of three enzymes can be explained by the fact that deficiency of each leads to three distinct lysosomal storage disorders (LSDs): galactosialidosis (GS) or PPCA deficiency with a secondary combined deficiency of NEU1 and β-GAL, sialidosis or NEU1 deficiency, and GM1-gangliosidosis (GM1) or β-GAL deficiency. Each disease is inherited as an autosomal recessive trait and is distinguished by variable clinical phenotypes, ranging from congenital forms to infantile/juvenile forms. All three present as a systemic disease, involving visceral organs, bone, cartilage, muscle and the nervous system [6].
Catalytic activation of NEU1 is PPCA dependent: The function of NEU1 is to initiate the catabolism glycoproteins and glycolipids, by cleaving their terminal sialic acids. For this, NEU1 depends on its interaction with the auxiliary protein protective protein/cathepsin A (PPCA). PPCA is essential for the stability of NEU1 and acts as a molecular chaperone for the subcellular localization and compartmentalization [7]. It can be assumed that NEU1 mutations that affect its interaction with PPCA may also lead to disease, even if the residues forming the active site of the enzyme remain intact [8]. In fact, researchers using the crystal structures of bacterial sialidases as templates have investigated numerous NEU1 amino acid substitutions related to different clinical phenotypes. Most of those substitutions appear to be located at the core surface of the molecule, demonstrating that they may affect the interaction of NEU1 with its chaperone PPCA. Three pathogenic mutations, F260Y, L270F, and A298V, gathered at the surface of the bacterial sialidases. These enzymes were correctly synthesized yet degraded instantly since the resulting proteins failed to associate with PPCA [9]. A putative region of interaction between NEU1 and PPCA was noticed after a careful study of the hydrodynamic properties of these two proteins. This region appeared to be crucial for NEU1 binding to the precursor form of PPCA. Therefore, NEU1 mutations affecting amino acids within this domain may affect the stability of the enzyme and subsequent PPCA-mediated transportation to lysosomes [7,8,9]. It is crucial to remember that a primary defect of PPCA results in the lysosomal disorder galactosialidosis [2]. The similarity in the clinical symptoms of these two disorders can be attributed to the fact that the absence of functional PPCA results in a near-complete secondary deficiency of NEU1 [10].
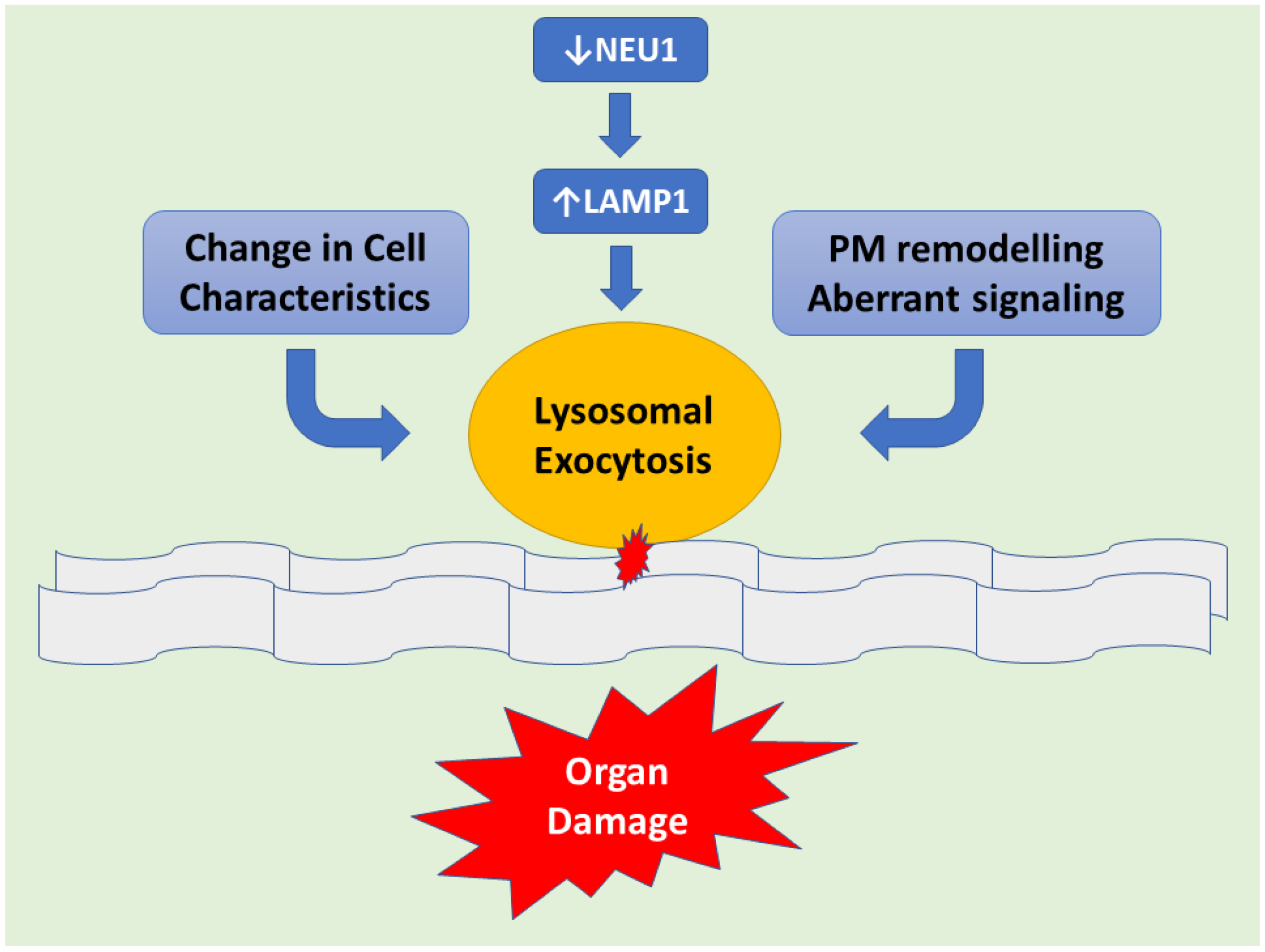
Mechanisms of Pathogenesis in Sialidosis explained with the help of the Mouse Model: The Neu1−/− knockout (KO) mouse model is helpful in understanding the underlying molecular mechanism(s) of sialidosis. Neu1−/− mice demonstrated NEU1 as a negative regulator of lysosomal exocytosis. It was observed that in hematopoietic cells, NEU1 negatively regulates lysosomal exocytosis by processing the sialic acids on the lysosomal membrane protein 1 (LAMP1). LAMP1 is an integral membrane protein and plays a useful role in the docking of lysosomes at the plasma membrane (PM). Deficiency or impaired NEU1 activity causes defective processing of the sialic acids on LAMP1, causing accumulation of LAMP1 in an over-sialylated state with a prolonged half-life. This accumulation of over-sialylated LAMP1 increases the number of LAMP1-marked lysosomes that dock at the PM, poised to engage in lysosomal exocytosis upon an influx of calcium. LAMP1’s essential function in the docking of lysosomes to the PM has been further supported by the fact that silencing LAMP1 in NEU1-deficient cells normalize the number of lysosomes docked at the PM and thus decreased the extent of lysosomal exocytosis. Hence, NEU1 loss of function ultimately results in the excessive extracellular release of lysosomal luminal contents from deficient cells of several tissues and organs (Figure 1). Excessive lysosomal exocytosis has now been associated with various pathological manifestations which are characteristic of sialidosis, including its role in neurodegeneration and links with Alzheimer’s disease, hearing loss, muscle atrophy and splenomegaly [11]. Moreover, phagocytosis in macrophages, which is regulated by NEU1, as well as NEU1-dependent regulation of insulin signaling are the other pathways that have been considered as additional pathological mechanisms involved in the NEU1 loss of function of the organs [12].
3. Morphological and Clinical Aspects of Sialidosis and NEU1 Mutation(s)
3.1. Method of Study Selection, Criteria, and Data Extraction
Initially, an electronic search was done on Google Scholar, Scopus, and PUBMED. The search was conducted between the time of January 1980 to January 2018. The words used were sialidosis I, sialidosis II, congenital sialidosis, infantile sialidosis, juvenile sialidosis and combinations of these words. In the next step, reference lists of the publications were checked to identify any additional studies. The search was limited to studies published in English in the open literature in peer-reviewed journals.
Cases of sialidosis I typically present later in life (second to third decade); therefore, sialidosis I was discussed briefly in this study. However, for sialidosis II, all cases found were included in the paper since this condition is present in the pediatric group of patients.
Sialidosis (MIM #256550) is known as an autosomal recessive inherited disease. However numerous times, genetic alterations have been found in NEU1 of unrelated sialidosis patients. So far, more than 40 mutations within the NEU1 gene have been identified in patients with sialidosis types I and II. Age of onset and severity of the clinical manifestations are paralleled with NEU1 mutations and the level of residual neuraminidase activity, demonstrating the existence of significant genotype-phenotype correlation in sialidosis.
NEU1 protein variants have been categorized into three groups based on biochemical properties. In the first group, the mutant enzyme stays catalytically inactive and does not localize to the lysosomes; whereas in the second group, the mutant protein localizes to the lysosomes yet is enzymatically inactive. Finally, in the third group, the mutant protein has residual activity and localizes to the lysosomes. An association appears to exist between individual mutations and the clinical severity of sialidosis [7]. In the least severe form of sialidosis, the modification is thought to cause a decrease in sialidase activity. However, mutant sialidase has residual activity as well as localizing to the lysosomes. Hence, leading to sialidosis I.
3.2. Sialidosis I
Also known as cherry-red spot myoclonus syndrome, type I sialidosis is the less severe, non-neuropathic subtype of this disease. Patients typically exhibit symptoms in the second or third decade of life. Symptoms may include gait abnormalities, decreased visual acuity, or both. Patients usually have no physical defects. Their intelligence level may range from normal to slightly impaired [13]. Myoclonus is an essential feature of sialidosis I, which over the course of the disease tends to get disabling. Precipitating factors may include light touch, sound stimuli, voluntary movements, passive joint movements, voluntary movements, and dysarthria. Action myoclonus, intentional tremors, cerebellar ataxia, and hyperreflexia are the other commonly found symptoms [14]. Muscle strength may remain normal. However, hypotonia can be seen. In some cases, the patients may become wheelchair-bound as the disease progresses [7]. Laboratory tests for sialidosis include a thin-layer chromatography test that is a useful screening test to find an abnormal urinary oligosaccharide pattern. Peripheral blood smear or bone marrow smear may show the presence of storage granules in lymphocytes. Deficiency of the lysosomal sialidase activity (neuraminidase) can be demonstrated in cultured skin fibroblasts obtained from a skin biopsy and is an important diagnostic step. Importantly, in enzymatic studies, sialidosis is differentiated from galactosialidosis by analyzing the enzymatic activity of β-galactosidase which should be normal. The final diagnosis is made after whole genome sequencing. As mentioned earlier, symptoms and their extent of severity are closely associated with the type of NEU1 mutations involved and subsequently the levels of residual enzyme activity [15].
Pathologically, cytoplasmic accumulation of sialyloligosaccharides has been observed in many neurons in the central nervous systems (CNS) of sialidosis patients. Moreover, neuroradiological imaging studies frequently reveal diffuse brain atrophy in the advanced stage of sialidosis type I, particularly of the cerebellar area [16]. However, initial neuroradiological investigations can be unremarkable in sialidosis 1. Studies also suggest that the major clinical effects seen are caused by changes at a level above the brainstem [17,18,19,20]. Further investigation in this area will help in understanding the underlying pathological mechanism which will consequently lead to the availability of better therapeutic approaches.
Atypical Cases of Sialidosis I
In the last decade, one crucial observation regarding sialidosis I is the presence of isolated instances of action myoclonus. Myoclonus, which is considered an essential feature of sialidosis I, has been seen in patients in the absence of other classic symptoms (macular cherry-red spot and sialyloligosacchariduria). After whole genome sequencing, mutations in NEU1 were identified. This aspect elucidates the fact that mutations affecting NEU1 activity can exist in the absence of other clinical signs that are characteristic of sialidosis [21]. This aspect warrants further studies in this area to investigate if there are more cases of sialidosis I than initially predicted.
3.3. Sialidosis II
Based on the age at onset of the symptoms, type II sialidosis is further divided into three subtypes: (i) congenital or hydropic (in utero); (ii) infantile (0–12 months); and (iii) juvenile (2–20 years) [7,22].
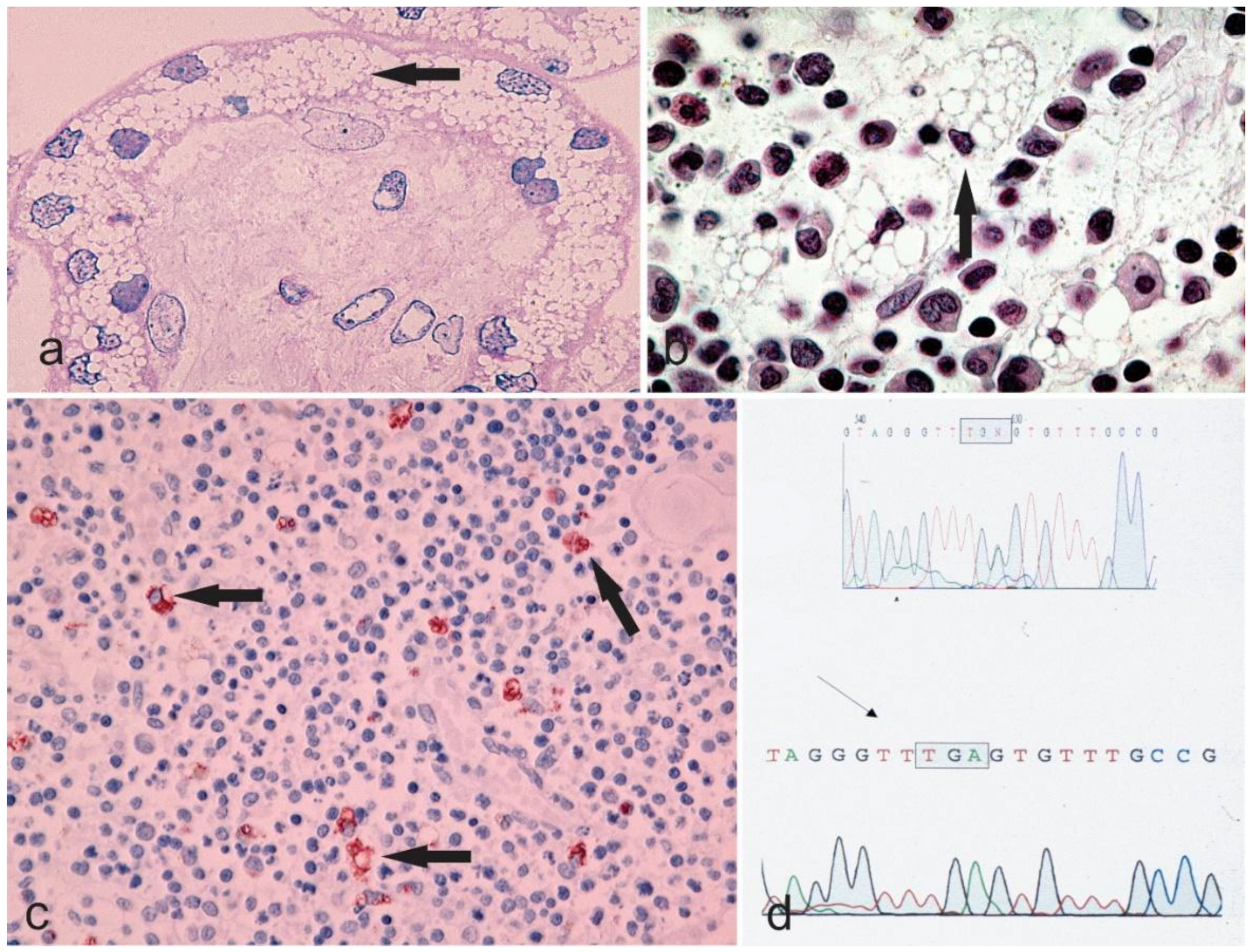
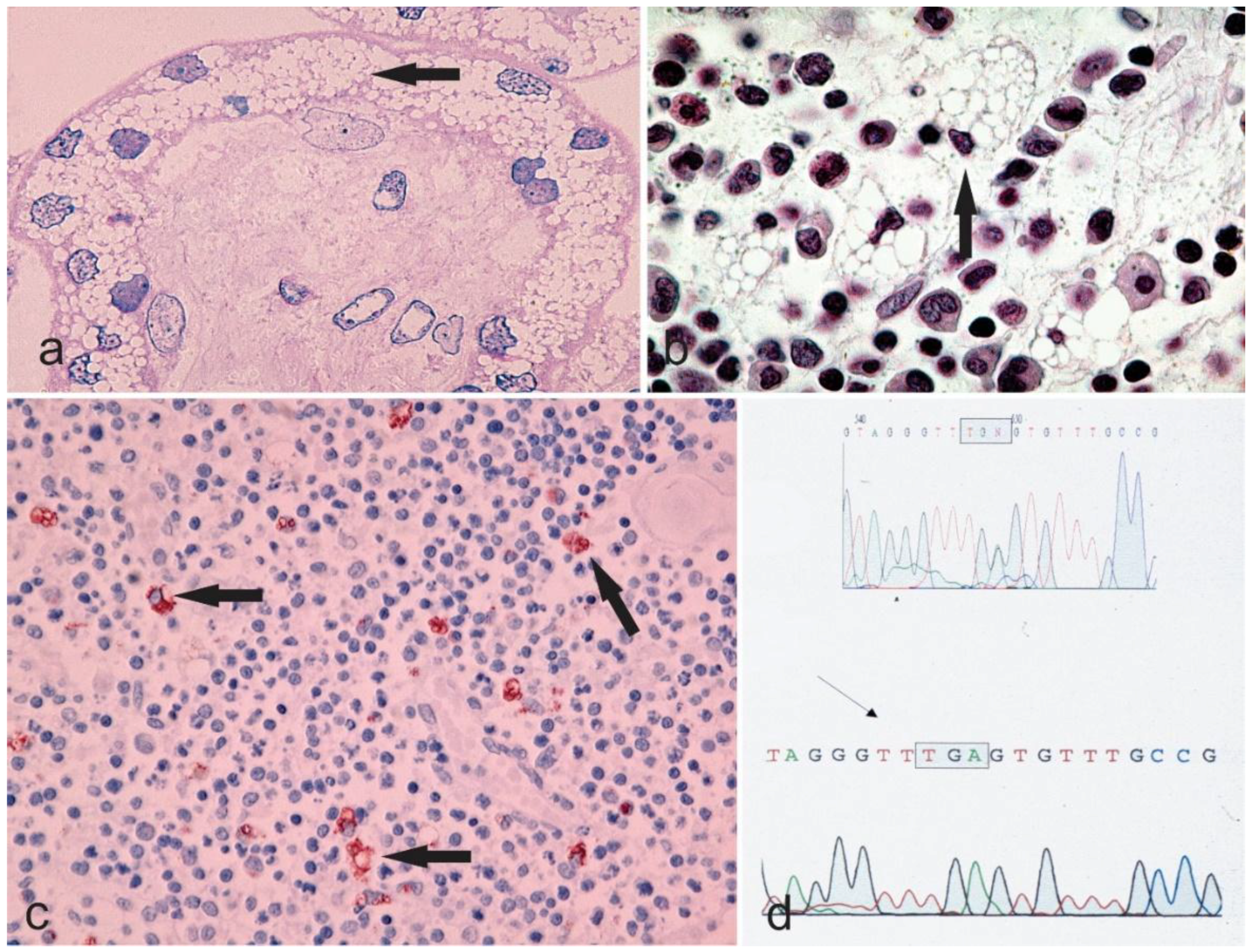
The congenital or hydropic subtype: Profoundly severe mutational alterations may lead to a complete absence of lysosomal neuraminidase and are lethal during fetal development or at birth. The congenital type of sialidosis, which is the severe form of the disease, is thought to be the result of such mutations. It manifests itself prenatally and is characterized by ascites and hydrops fetalis, hepatomegaly and stillbirths or death at a very early age [7]. Table 1 summarizes the cases of congenital sialidosis reported from 1979 until now (cases presented before 1979 can be found in a previous review [5]). It can be observed that hydrops, ascites, and edema are the distinguishing features of the severe, congenital group of the disease, followed by coarse features, dysostosis multiplex, and hepatosplenomegaly. Renal involvement, cardiac anomalies, ophthalmic finding, myoclonus, inguinal hernia, telangiectasias, petechiae, bluish to purpuric macules and hydrocephalus are the clinical features that may infrequently manifest [9,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46]. Studies reporting histopathological features of congenital sialidosis are limited. In one study, light microscopy of fetal tissues (after pregnancy was terminated at 20 weeks) exhibited vacuolation in the liver, bone marrow, kidney, and brain. In addition, an abnormal pattern of vacuolations was also present in the endocrine organs such as the thyroid gland, adrenal gland, hypophysis, testes as well as in the thymus. Moreover, vacuolation of the placenta demonstrated that in congenital sialidosis abnormal storage takes place during the early fetal period [36]. Figure 2 illustrates vacuolation of placenta, spleen, and thymus of a patient with congenital sialidosis.
Infantile/juvenile subtype: It has been suggested that NEU1 mutations in which mutant enzymes localize to the lysosomes yet stay enzymatically inactive lead to the infantile/juvenile subtype of sialidosis II, which is characterized by the development of progressive mucopolysaccharidosis-like phenotype; Patients presenting with coarse facies, visceromegaly, dysostosis multiplex, vertebral deformities, mental retardation [7,47,48,49,50,51,52,53,54,55]. Table 2 summarizes the clinical features of the infantile/juvenile subtype. Skeletal abnormalities, mainly dysostosis multiplex appear to be a consistent feature of infantile/juvenile phenotypes, along with coarse facies, hepatosplenomegaly, and severe mental retardation. Ocular manifestations, including cherry red spots, cataracts, nystagmus, strabismus, and corneal clouding are common as well while hypotonia, renal involvement, and cardiac anomalies are relatively infrequent findings. It is noticeable that earlier onset of disease has a fulminant course. While in late-onset, the patient may survive for more extended periods (only four patients survived for more than two decades). Hearing loss and ataxia may present in the late onset of infantile/juvenile form and tend to worsen with time. One case of sialidosis II was reported in the last decade, in which the patient developed myoclonic seizures at the age of 17, followed by dysphagia and dysphonia. There was a marked delay in motor and cognitive functions since childhood which worsened over time. However, the patient was able to achieve an elementary school education. At the time of diagnosis, the patient was 30 years old and bedridden, with an advanced degree of mental deficiency. It is notable that the patient was able to achieve education demonstrates that her cognitive delay was not as severe in childhood but progressed with age [55]. This detail seems essential and may aid in future developing therapeutic interventions in this area. Also, it is noteworthy that myoclonus, which is considered a characteristic of sialidosis I, is found in both Congenital as well as Infantile/Juvenile subtypes. (Table 1 and Table 2).
NEU1 knockout model Neu1−/− mice developed a systemic and neurodegenerative condition that is comparable to the early onset of sialidosis II [10] thus helping in understanding the clinical manifestation of this disease. Some typical clinical features that have been studied and explained are as follows. Hepatosplenomegaly is a fairly standard feature in sialidosis II. Enlargement of the spleen observed in Neu1−/−mice is consistent with this finding. Histological studies of spleens in mutant mice showed a time-dependent expansion of total splenic cell counts along with an increased number of erythroid precursors and megakaryocytes. Along with these changes in the spleen, an increased number of hematopoietic progenitors in the peripheral blood and an overall lower number of these cells in the bone marrow (BM) was found. Thus, indicating extramedullary hematopoiesis (EMH) as the possible cause of splenic hypertrophy in Neu1−/−. Furthermore, immunohistochemical studies of mutant livers showed a high number of erythroblasts, suggesting extramedullary hematopoiesis in this organ as well. Pathological studies of livers revealed initial ballooning followed by progressive, age-dependent filling of sinusoidal cells and hepatocytes with vacuoles [10]. Skeletal abnormalities frequently occur in sialidosis II. According to researchers, a possible hypothesis explaining the underlying pathogenesis is the impaired function of osteoclasts resulting in inefficient bone remodeling and consequently leading to bone deformities [10]. Hearing loss is another feature of sialidosis II that appears late in the disease. Studies of the (KO)model showed both conductive and neurosensory defects contribute to hearing loss in Neu1−/− mice. It has been suggested that the absence of NEU1 and the potential exacerbation of lysosomal exocytosis in the inner ear are responsible for the occurrence of hearing the loss in Neu1−/− mice. Histopathological studies exhibited thickened, cerumen occlusion in the external auditory canal, while in the middle ear infiltration of connective tissue with signs of chronic inflammation was noticed. Also, in many cells of the extensive cochlea vacuolization was seen [56]. In the brain, Neu1-null mice show extensive vacuolization of the epithelial cells of the choroid plexus and the endothelial cells of the ependymal layer 31, 33. Microglia and perivascular macrophages were among the most affected cells. This feature was prominently noticeable in the dentate gyrus and the hippocampus, but these cells are also scattered throughout the cortex and in the cerebellum, causing a widespread microgliosis [10]. Additionally, in the hippocampal region of the Neu1-null mice, recent studies have suggested a link between NEU1 deficiency-exacerbated lysosomal exocytosis and the spontaneous occurrence of Alzheimer’s disease (AD)-like amyloidogenic process. Essentially accumulation in endo-lysosomes of an over sialylated amyloid precursor protein (APP), which is a newly identified substrate of NEU1, initiates the process. Next Endo-lysosomal APP is proteolytically cleaved to generate amyloid β-peptide isoforms (Aβ), which is ultimately released extracellularly by excessive lysosomal exocytosis. The finding that intracranial injection of NEU1 in the AD mouse model reduces the numbers of amyloid plaques and the levels of amyloid peptides is exceptionally important, as it demonstrates that NEU1 can be explored as a therapeutic approach for AD [57].
4. Therapeutic Interventions for Sialidosis
Due to the rarity of the disease, establishing optimum therapeutic measures remains a challenge although many promising methods are being proposed and being investigated. One attempt of enzyme replacement therapy (ERT) in Neu1−/− mice using a recombinant NEU1 enzyme purified from overexpressing insect cells was made, which helped significantly in increasing levels of the NEU1 protein, and this treatment achieved subsequent correction of the underlying pathology in most of the systemic organs. However, the recombinant protein turned out to be highly immunogenic in the mutant mice and thus causes a severe immune response. Consequently, the therapeutic use of ERT became restricted [58,59]. Another study investigated the efficacy of the immuno-suppressant (Celastrol) along with a proteasomal inhibitor (MG132) as a therapeutic option for sialidosis. Researchers found that MG132 enhances enzyme activity and its localization in cells expressing defective sialidase provided promising results [60]. Additionally, chaperone-mediated gene therapy using a new mouse model has been suggested. The new mouse model ubiquitously expresses a NEU1 variant, which has a V54M amino acid substitution found in an adult patient with type I sialidosis. Mutant mice exhibited signs of lysosomal disease after one year of age, with low residual NEU1 activity detected in most organs and cell types. Injection of aged mutant mice with AAV-PPCA caused improvement of symptoms in the disease phenotype, hence suggesting that some NEU1 mutations associated with type I sialidosis may respond to PPCA-chaperone-mediated gene therapy [61]. Researchers believe that the treatment may be useful for other NEU1 mutations, particularly those in patients with type I sialidosis [8].
5. Conclusions and Future Perspectives
Sialidosis is an autosomal recessive disease resulting due to a mutation in the neuraminidase (NEU1) gene. Missense mutations appear to be the most commonly occurring. Among these mutations, a significant molecular heterogeneity is present, with a mixed variety of clinical phenotypes presenting either as sialidosis I or sialidosis II with different levels of severity. The onset of disease, the severity of symptoms, and prognosis differ in sialidosis I and sialidosis II. Bonten et al. demonstrated that this association between the clinical phenotype and severity of the disease is the result of the residual activity of the mutant enzymes. Patients with type II disease have catalytic inactive neuraminidase, while patients with the mild type I disease have active catalytic neuraminidase. In the type I disease, there is an absence of any obvious physical defects, and life expectancy remains unaffected. However, progressive visual loss and myoclonus, which may tend to get worse with time, can be disabling. In type II, the congenital subtype takes a fulminant course with very low life expectancy. Ascites and edema are the prominent features of this subgroup of the disease. In the infantile/juvenile sub type, characteristic features include hepatosplenomegaly, dysostosis multiplex, coarse facies, cherry-red spot, myoclonus, and severe mental retardation. Some clinical manifestations such as ataxia and hearing loss may become progressively severe with age. In the last decade, atypical cases of sialidosis 1 have been reported, and this aspect warrants further studies in this area to investigate if there are more cases of sialidosis I than initially predicted. Furthermore, the possibility suggested by previous researchers that, in addition to neuraminidase mutations, environmental factors including diet, prophylactic therapies, or other genetic factors may have some effect on the penetrance and severity of the disease and/or phenotype of the disease seems more plausible [7]. Perhaps in the future, a more detailed prenatal history inquiring about the diet, medication, and different types of environmental exposure of parents may help the researchers understand this aspect of the disease. Therapeutic options for sialidosis remain limited. Researchers have emphasized exploring treatment options for sialidosis I [8]. In sialidosis I, symptoms are relatively mild and appear late. Although intelligence level remains normal, myoclonus can be debilitating. Therefore, effective therapy may improve quality of life. In sialidosis type II patients, an early onset, systemic involvement, and a fulminant course make it more challenging to provide treatment. Perhaps it would be worthwhile to explore the suggestion of taking an in utero approach for therapy in the future. Alternatively, carrier detection in affected families, prenatal molecular diagnosis, and improved genetic counseling seem to be a suitable practical approaches. Finally, NEU1 appears to have a crucial link with the CNS. Studies provide substantial evidence to demonstrate the NEU1 enzyme has a correlation with Alzheimer’s disease. Further understanding of NEU1 and its link with the CNS may provide useful information regarding genetics, pathogenesis, and treatment of not only sialidosis, but also Alzheimer’s disease.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pshezhetsky, A.V.; Richard, C.; Michaud, L.; Igdoura, S.; Wang, S.; Elsliger, M.A.; Qu, J.; Leclerc, D.; Gravel, R.; Dallaire, L.; et al. Cloning, expression and chromosomal mapping of human lysosomal sialidase and characterization of mutations in sialidosis. Nat. Genet. 1997, 15, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Bonten, E.; van der Spoel, A.; Fornerod, M.; Grosveld, G.; d’Azzo, A. Characterization of human lysosomal neuraminidase defines the molecular basis of the metabolic storage disorder sialidosis. Genes Dev. 1996, 10, 3156–3169. [Google Scholar] [CrossRef] [PubMed]
- Cantz, M.; Gehler, J.; Spranger, J. Mucolipidosis I: Increased sialic acid content and deficiency of an alpha-n-acetylneuraminidase in cultured fibroblasts. Biochem. Biophys. Res. Commun. 1977, 74, 732–738. [Google Scholar] [CrossRef]
- Sphranger, J.; Gehler, J.; Cantz, M. Mucolipidosis i—A sialidosis. Am. J. Med. Genet. 1977, 1, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Lowden, J.A.; O’Brien, J.S. Sialidosis: A review of human neuraminidase deficiency. Am. J. Hum. Genet. 1979, 31, 1–18. [Google Scholar] [PubMed]
- Bonten, E.J.; Annunziata, I.; d’Azzo, A. Lysosomal multienzyme complex: Pros and cons of working together. Cell. Mol. Life Sci. 2014, 71, 2017–2032. [Google Scholar] [CrossRef] [PubMed]
- Bonten, E.J.; Arts, W.F.; Beck, M.; Covanis, A.; Donati, M.A.; Parini, R.; Zammarchi, E.; d’Azzo, A. Novel mutations in lysosomal neuraminidase identify functional domains and determine clinical severity in sialidosis. Hum. Mol. Genet. 2000, 9, 2715–2725. [Google Scholar] [CrossRef] [PubMed]
- D’Azzo, A.; Machado, E.; Annunziata, I. Pathogenesis, emerging therapeutic targets and treatment in sialidosis. Expert Opin. Orphan Drugs 2015, 3, 491–504. [Google Scholar] [CrossRef] [PubMed]
- Lukong, K.E.; Elsliger, M.A.; Chang, Y.; Richard, C.; Thomas, G.; Carey, W.; Tylki-Szymanska, A.; Czartoryska, B.; Buchholz, T.; Criado, G.R.; et al. Characterization of the sialidase molecular defects in sialidosis patients suggests the structural organization of the lysosomal multienzyme complex. Hum. Mol. Genet. 2000, 9, 1075–1085. [Google Scholar] [CrossRef] [PubMed]
- De Geest, N.; Bonten, E.; Mann, L.; de Sousa-Hitzler, J.; Hahn, C.; d’Azzo, A. Systemic and neurologic abnormalities distinguish the lysosomal disorders sialidosis and galactosialidosis in mice. Hum. Mol. Genet. 2002, 11, 1455–1464. [Google Scholar] [CrossRef] [PubMed]
- Yogalingam, G.; Bonten, E.J.; van de Vlekkert, D.; Hu, H.; Moshiach, S.; Connell, S.A.; d’Azzo, A. Neuraminidase 1 is a negative regulator of lysosomal exocytosis. Dev. Cell 2008, 15, 74–86. [Google Scholar] [CrossRef] [PubMed]
- Dridi, L.; Seyrantepe, V.; Fougerat, A.; Pan, X.; Bonneil, E.; Thibault, P.; Moreau, A.; Mitchell, G.A.; Heveker, N.; Cairo, C.W.; et al. Positive regulation of insulin signaling by neuraminidase 1. Diabetes 2013, 62, 2338–2346. [Google Scholar] [CrossRef] [PubMed]
- Sobral, I.; Cachulo Mda, L.; Figueira, J.; Silva, R. Sialidosis type i: Ophthalmological findings. BMJ Case Rep. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Franceschetti, S.; Canafoglia, L. Sialidoses. Epileptic Disord. 2016, 18, 89–93. [Google Scholar] [PubMed]
- Takahashi, Y.; Nakamura, Y.; Yamaguchi, S.; Orii, T. Urinary oligosaccharide excretion and severity of galactosialidosis and sialidosis. Clin. Chim. Acta 1991, 203, 199–210. [Google Scholar] [CrossRef]
- Sekijima, Y.; Nakamura, K.; Kishida, D.; Narita, A.; Adachi, K.; Ohno, K.; Nanba, E.; Ikeda, S. Clinical and serial mri findings of a sialidosis type i patient with a novel missense mutation in the neu1 gene. Intern. Med. 2013, 52, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.Z.; Lai, S.C.; Lu, C.S.; Weng, Y.H.; Chuang, W.L.; Chen, R.S. Abnormal cortical excitability with preserved brainstem and spinal reflexes in sialidosis type i. Clin. Neurophysiol. 2008, 119, 1042–1050. [Google Scholar] [CrossRef] [PubMed]
- Palmeri, S.; Villanova, M.; Malandrini, A.; van Diggelen, O.P.; Huijmans, J.G.; Ceuterick, C.; Rufa, A.; DeFalco, D.; Ciacci, G.; Martin, J.J.; et al. Type i sialidosis: A clinical, biochemical and neuroradiological study. Eur. Neurol. 2000, 43, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.H.; Tipton, R.E.; Ch’ien, L.T.; Reynolds, L.W.; Miller, C.S. Sialidase (alpha-n-acetyl neuraminidase) deficiency: The enzyme defect in an adult with macular cherry-red spots and myoclonus without dementia. Clin. Genet. 1978, 13, 369–379. [Google Scholar] [CrossRef] [PubMed]
- Franceschetti, S.; Uziel, G.; Di Donato, S.; Caimi, L.; Avanzini, G. Cherry-red spot myoclonus syndrome and alpha-neuraminidase deficiency: Neurophysiological, pharmacological and biochemical study in an adult. J. Neurol. Neurosurg. Psychiatry 1980, 43, 934–940. [Google Scholar] [CrossRef] [PubMed]
- Canafoglia, L.; Robbiano, A.; Pareyson, D.; Panzica, F.; Nanetti, L.; Giovagnoli, A.R.; Venerando, A.; Gellera, C.; Franceschetti, S.; Zara, F. Expanding sialidosis spectrum by genome-wide screening: Neu1 mutations in adult-onset myoclonus. Neurology 2014, 82, 2003–2006. [Google Scholar] [CrossRef] [PubMed]
- Caciotti, A.; Di Rocco, M.; Filocamo, M.; Grossi, S.; Traverso, F.; d’Azzo, A.; Cavicchi, C.; Messeri, A.; Guerrini, R.; Zammarchi, E.; et al. Type ii sialidosis: Review of the clinical spectrum and identification of a new splicing defect with chitotriosidase assessment in two patients. J. Neurol. 2009, 256, 1911–1915. [Google Scholar] [CrossRef] [PubMed]
- Kelly, T.E.; Bartoshesky, L.; Harris, D.J.; McCauley, R.G.; Feingold, M.; Schott, G. Mucolipidosis i (acid neuraminidase deficiency). Three cases and delineation of the variability of the phenotype. Am. J. Dis. Child. 1981, 135, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Riches, W.G.; Smuckler, E.A. A severe infantile mucolipidosis. Clinical, biochemical, and pathologic features. Arch. Pathol. Lab. Med. 1983, 107, 147–152. [Google Scholar] [PubMed]
- Gillan, J.E.; Lowden, J.A.; Gaskin, K.; Cutz, E. Congenital ascites as a presenting sign of lysosomal storage disease. J. Pediatr. 1984, 104, 225–231. [Google Scholar] [CrossRef]
- Beck, M.; Bender, S.W.; Reiter, H.L.; Otto, W.; Bassler, R.; Dancygier, H.; Gehler, J. Neuraminidase deficiency presenting as non-immune hydrops fetalis. Eur. J. Pediatr. 1984, 143, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Guibaud, P.; Cottin, X.; Maire, I.; Boyer, S.; Guibaud, S.; Coicaud, C.; Bellon-Azzouzi, C.; Duvernois, J.P. fetal ascites as a manifestation of infantile sialidosis. Significance of a study of oligosaccharides in amniotic fluid. J. Genet. Hum. 1985, 33, 317–324. [Google Scholar] [PubMed]
- Johnson, W.G.; Thomas, G.H.; Miranda, A.F.; Driscoll, J.M.; Wigger, J.H.; Yeh, M.N.; Schwartz, R.C.; Cohen, C.S.; Berdon, W.E.; Koenigsberger, M.R. Congenital sialidosis: Biochemical studies: Clinical spectrum in four sibs; two successful prenatal diagnoses (abstract). Am. J. Hum. Genet. 1980, 32, A43. [Google Scholar]
- Yamano, T.; Shimada, M.; Matsuzaki, K.; Matsumoto, Y.; Yoshihara, W.; Okada, S.; Inui, K.; Yutaka, T.; Yabuuchi, H. Pathological study on a severe sialidosis (alpha-neuraminidase deficiency). Acta Neuropathol. 1986, 71, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Tabardel, Y.; Soyeur, D.; Vivario, E.; Senterre, J. primary neuraminidase deficiency with prenatal disclosure. Arch. Fr. Pediatr. 1989, 46, 737–740. [Google Scholar] [PubMed]
- Ries, M.; Deeg, K.H.; Wolfel, D.; Ibel, H.; Maier, B.; Buheitel, G. Colour doppler imaging of intracranial vasculopathy in severe infantile sialidosis. Pediatr. Radiol. 1992, 22, 179–181. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Takahashi, Y.; Yamaguchi, S.; Omiya, S.; Orii, T.; Yara, A.; Gushiken, M. Severe infantile sialidosis—The characteristics of oligosaccharides isolated from the urine and the abdominal ascites. Tohoku J. Exp. Med. 1992, 166, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.; Fahnenstich, H.; Haverkamp, F.; Platz, H.; Hansmann, M.; Bartmann, P. sialidosis and galactosialidosis as the cause of non-immunologic hydrops fetalis. Z Geburtshilfe Neonatol. 1997, 201, 177–180. [Google Scholar] [PubMed]
- Ovali, F.; Samanci, N.; Guray, A.; Akdogan, Z.; Akdeniz, C.; Dagoglu, T.; Petorak, I. Congenital sialidosis. Turk. J. Pediatr. 1998, 40, 447–451. [Google Scholar] [PubMed]
- Sergi, C.; Beedgen, B.; Kopitz, J.; Zilow, E.; Zoubaa, S.; Otto, H.F.; Cantz, M.; Linderkamp, O. Refractory congenital ascites as a manifestation of neonatal sialidosis: Clinical, biochemical and morphological studies in a newborn syrian male infant. Am. J. Perinatol. 1999, 16, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Sergi, C.; Penzel, R.; Uhl, J.; Zoubaa, S.; Dietrich, H.; Decker, N.; Rieger, P.; Kopitz, J.; Otto, H.F.; Kiessling, M.; et al. Prenatal diagnosis and fetal pathology in a turkish family harboring a novel nonsense mutation in the lysosomal alpha-n-acetyl-neuraminidase (sialidase) gene. Hum. Genet. 2001, 109, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, T.; Molitor, G.; Lukong, K.E.; Praun, M.; Genzel-Boroviczeny, O.; Freund, M.; Pshezhetsky, A.V.; Schulze, A. Clinical presentation of congenital sialidosis in a patient with a neuraminidase gene frameshift mutation. Eur. J. Pediatr. 2001, 160, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Uhl, J.; Penzel, R.; Sergi, C.; Kopitz, J.; Otto, H.F.; Cantz, M. Identification of a ctl4/neu1 fusion transcript in a sialidosis patient. FEBS Lett. 2002, 521, 19–23. [Google Scholar] [CrossRef]
- Donati, M.A.; Caciotti, A.; Bardelli, T.; Dani, C.; d’Azzo, A.; Morrone, A.; Zammarchi, E. Congenital sialidosis hydrops fetalis neuraminidase hydrocephalus sialidosis congenita idrope fetale neuraminidase idrocefalo. Ital. J. Pediatr. 2003, 29, 404–410. [Google Scholar]
- Penzel, R.; Uhl, J.; Kopitz, J.; Beck, M.; Otto, H.F.; Cantz, M. Splice donor site mutation in the lysosomal neuraminidase gene causing exon skipping and complete loss of enzyme activity in a sialidosis patient. FEBS Lett. 2001, 501, 135–138. [Google Scholar] [CrossRef]
- Itoh, K.; Naganawa, Y.; Matsuzawa, F.; Aikawa, S.; Doi, H.; Sasagasako, N.; Yamada, T.; Kira, J.; Kobayashi, T.; Pshezhetsky, A.V.; et al. Novel missense mutations in the human lysosomal sialidase gene in sialidosis patients and prediction of structural alterations of mutant enzymes. J. Hum. Genet. 2002, 47, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez Criado, G.; Pshezhetsky, A.V.; Rodriguez Becerra, A.; Gomez de Terreros, I. Clinical variability of type ii sialidosis by c808t mutation. Am. J. Med. Genet. A 2003, 116A, 368–371. [Google Scholar] [CrossRef] [PubMed]
- Pattison, S.; Pankarican, M.; Rupar, C.A.; Graham, F.L.; Igdoura, S.A. Five novel mutations in the lysosomal sialidase gene (neu1) in type ii sialidosis patients and assessment of their impact on enzyme activity and intracellular targeting using adenovirus-mediated expression. Hum. Mutat. 2004, 23, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Loren, D.J.; Campos, Y.; d’Azzo, A.; Wyble, L.; Grange, D.K.; Gilbert-Barness, E.; White, F.V.; Hamvas, A. Sialidosis presenting as severe nonimmune fetal hydrops is associated with two novel mutations in lysosomal alpha-neuraminidase. J. Perinatol. 2005, 25, 491–494. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Son, S.K.; Park, J.H.; Song, J.S.; Cheon, C.K. Neu1 mutation in a korean infant with type 2 sialidosis presenting as isolated fetal ascites. Pediatr. Neonatol. 2015, 56, 68–69. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.H.; Kim, Y.M.; Kim, J.H.; Kim, G.H.; Lee, B.S.; Kim, C.J.; Yoo, H.J.; Yoo, H.W. Histological, biochemical, and genetic characterization of early-onset fulminating sialidosis type 2 in a korean neonate with hydrops fetalis. Brain Dev. 2014, 36, 171–175. [Google Scholar] [CrossRef] [PubMed]
- Winter, R.M.; Swallow, D.M.; Baraitser, M.; Purkiss, P. Sialidosis type 2 (acid neuraminidase deficiency): Clinical and biochemical features of a further case. Clin. Genet. 1980, 18, 203–210. [Google Scholar] [CrossRef] [PubMed]
- King, M.; Cockburn, F.; MacPhee, G.B.; Logan, R.W. Infantile type 2 sialidosis in a pakistani family—A clinical and biochemical study. J. Inherit. Metab. Dis. 1984, 7, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Oohira, T.; Nagata, N.; Akaboshi, I.; Matsuda, I.; Naito, S. The infantile form of sialidosis type ii associated with congenital adrenal hyperplasia: Possible linkage between hla and the neuraminidase deficiency gene. Hum. Genet. 1985, 70, 341–343. [Google Scholar] [CrossRef] [PubMed]
- Young, I.D.; Young, E.P.; Mossman, J.; Fielder, A.R.; Moore, J.R. Neuraminidase deficiency: Case report and review of the phenotype. J. Med. Genet. 1987, 24, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Bakker, H.D.; Abeling, N.G.G.M.; Staalman, C.R.; van Gennip, A.H. Thirty-years follow-up of a patient with sialidosis. J. Inherit. Metab. Dis. 1998, 21, 116. [Google Scholar]
- Schiff, M.; Maire, I.; Bertrand, Y.; Cochat, P.; Guffon, N. Long-term follow-up of metachronous marrow-kidney transplantation in severe type ii sialidosis: What does success mean? Nephrol. Dial. Transplant. 2005, 20, 2563–2565. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez Gonzalez, G.; Jimenez Lopez, I. anesthetic management of a boy with sialidosis. Rev. Esp. Anestesiol. Reanim. 2006, 53, 253–256. [Google Scholar] [PubMed]
- Ranganath, P.; Sharma, V.; Danda, S.; Nandineni, M.R.; Dalal, A.B. Novel mutations in the neuraminidase-1 (neu1) gene in two patients of sialidosis in india. Indian J. Med. Res. 2012, 136, 1048–1050. [Google Scholar] [PubMed]
- Vieira de Rezende Pinto, W.B.; Sgobbi de Souza, P.V.; Pedroso, J.L.; Barsottini, O.G. Variable phenotype and severity of sialidosis expressed in two siblings presenting with ataxia and macular cherry-red spots. J. Clin. Neurosci. 2013, 20, 1327–1328. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Steigelman, K.A.; Bonten, E.; Hu, H.; He, W.; Ren, T.; Zuo, J.; d’Azzo, A. Vacuolization and alterations of lysosomal membrane proteins in cochlear marginal cells contribute to hearing loss in neuraminidase 1-deficient mice. Biochim. Biophys. Acta 2010, 1802, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Annunziata, I.; Patterson, A.; Helton, D.; Hu, H.; Moshiach, S.; Gomero, E.; Nixon, R.; d’Azzo, A. Lysosomal NEU1 deficiency affects amyloid precursor protein levels and amyloid-β secretion via deregulated lysosomal exocytosis. Nat. Commun. 2013, 4, 2734. [Google Scholar] [CrossRef] [PubMed]
- Bonten, E.J.; Wang, D.; Toy, J.N.; Mann, L.; Mignardot, A.; Yogalingam, G.; D’Azzo, A. Targeting macrophages with baculovirus-produced lysosomal enzymes: Implications for enzyme replacement therapy of the glycoprotein storage disorder galactosialidosis. FASEB J. 2004, 18, 971–973. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Bonten, E.J.; Yogalingam, G.; Mann, L.; d’Azzo, A. Short-term, high dose enzyme replacement therapy in sialidosis mice. Mol. Genet. Metab. 2005, 85, 181–189. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, E.M.; Igdoura, S.A. The therapeutic potential of pharmacological chaperones and proteosomal inhibitors, celastrol and mg132 in the treatment of sialidosis. Mol. Genet. Metab. 2012, 107, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Bonten, E.J.; Yogalingam, G.; Hu, H.; Gomero, E.; van de Vlekkert, D.; d’Azzo, A. Chaperone-mediated gene therapy with recombinant aav-ppca in a new mouse model of type i sialidosis. Biochim. Biophys. Acta 2013, 1832, 1784–1792. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic representation demonstrating downstream of NEU1 deficiency leads to LAMP1 accumulation causing an increased number of lysosomes at the plasmatic membrane (PM) resulting in exacerbated lysosomal exocytosis. Lysosomal-associated membrane protein 1 (LAMP1), aka lysosome-associated membrane glycoprotein 1 or CD107a, is a protein that in humans is determined by the LAMP1 gene. This abnormal release of lysosomal content causes extracellular PM remodeling. Hence changes in cell characteristics take place with subsequent organ pathogenesis.
Figure 1.
Schematic representation demonstrating downstream of NEU1 deficiency leads to LAMP1 accumulation causing an increased number of lysosomes at the plasmatic membrane (PM) resulting in exacerbated lysosomal exocytosis. Lysosomal-associated membrane protein 1 (LAMP1), aka lysosome-associated membrane glycoprotein 1 or CD107a, is a protein that in humans is determined by the LAMP1 gene. This abnormal release of lysosomal content causes extracellular PM remodeling. Hence changes in cell characteristics take place with subsequent organ pathogenesis.
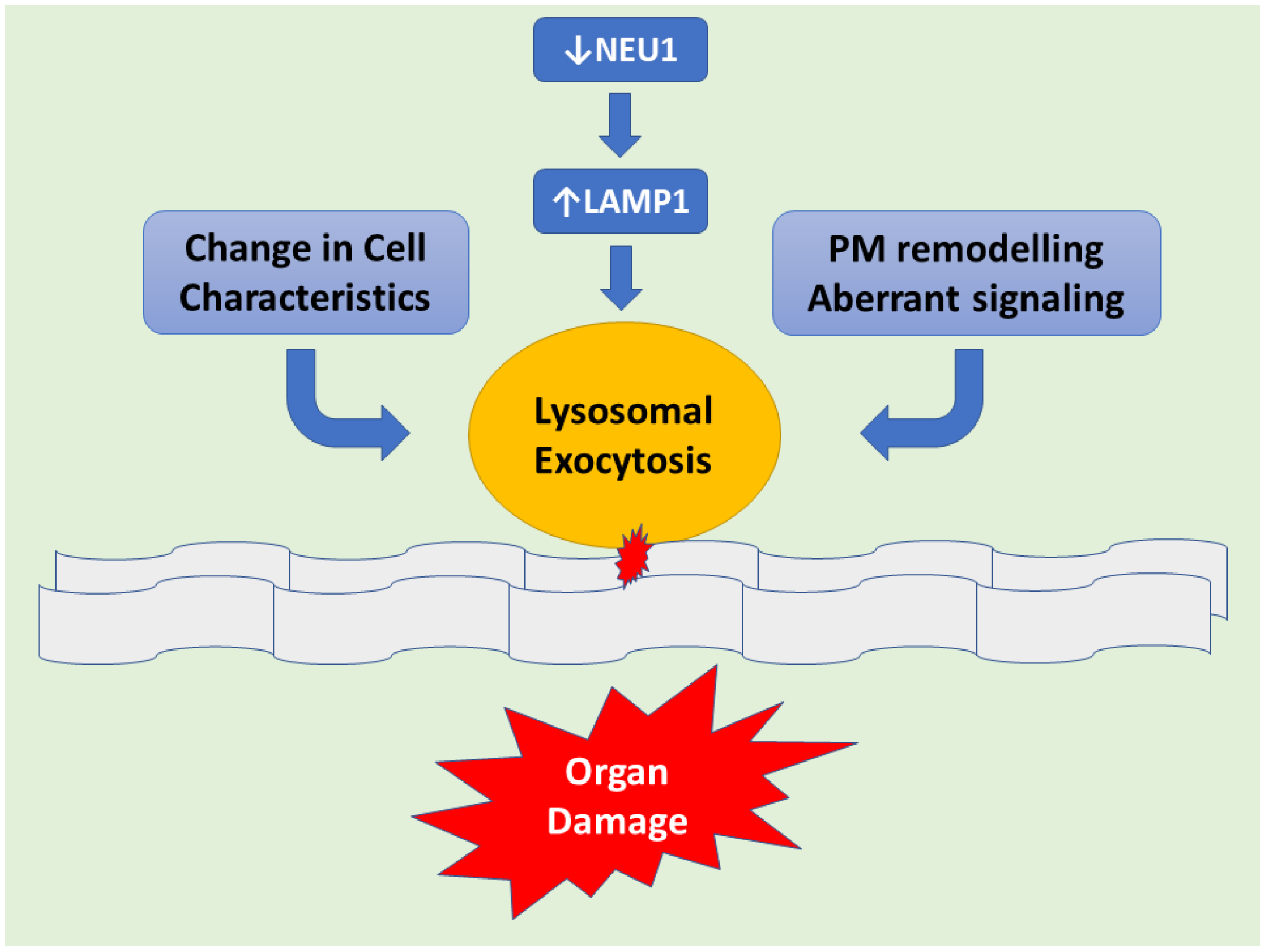
Figure 2.
Microphotographs a–c showing the vacuolar degeneration of syncytium-trophoblast of a placenta (Hematoxylin & Eosin staining, 400×), bone marrow (Hematoxylin & Eosin staining, 630×, the arrow points to a cell with margination of the nucleus due to an engulfment of the cytoplasm with undigested material), and thymus (Anti-CD68 immunostaining, 200× with the arrows highlighting the macrophages) from a sialidosis pregnancy. The chromatogram in d shows the genetic alteration of the sialidosis gene.
Figure 2.
Microphotographs a–c showing the vacuolar degeneration of syncytium-trophoblast of a placenta (Hematoxylin & Eosin staining, 400×), bone marrow (Hematoxylin & Eosin staining, 630×, the arrow points to a cell with margination of the nucleus due to an engulfment of the cytoplasm with undigested material), and thymus (Anti-CD68 immunostaining, 200× with the arrows highlighting the macrophages) from a sialidosis pregnancy. The chromatogram in d shows the genetic alteration of the sialidosis gene.

{kind=link}
{kind=link}
Table 1.
Clinical and Morphological findings of patients suffering from sialidosis II, congenital subtype form. (Cases from 1980 until present are included). M = male; F = female; n/a = not available; n/r = not reported; no = not detected.
Table 1.
Clinical and Morphological findings of patients suffering from sialidosis II, congenital subtype form. (Cases from 1980 until present are included). M = male; F = female; n/a = not available; n/r = not reported; no = not detected.
| References (Name of the First Author) | Gender | General Presentation | Nervous System | Ophthalmologic Findings | Skeleton | Respiratory Distress/Infections | Renal Involvement | Cardiac Involvement | Others | Course of Disease |
|---|---|---|---|---|---|---|---|---|---|---|
| Kelly, T.E. [23] | F | Ascites, edema, hepatosplenomegaly course features | n/r | n/r | Dysostosis multiplex | n/r | n/r | Cardiac anomalies | n/r | Exitus at 26 months |
| Riches, W.G. [24] | F | Hydrops, ascites, hepatosplenomegaly | n/r | n/r | n/r | n/r | n/r | n/r | n/r | Exitus at 4 months |
| Gillan, J.E. [25] | M | Hydrops, ascites, edema, hepatosplenomegaly course features | n/r | n/r | n/r | n/r | n/r | n/r | n/r | Exitus at 3 days |
| Beck, M. [26] | F | Hydrops, ascites, edema, hepato-splenomegaly, coarse features | n/r | n/r | n/r | present | present | n/r | n/r | Exitus at 6 months |
| Guibaud, P. [27] | F | Hydrops, ascites, edema, hepato-splenomegaly, coarse features | n/r | Corneal clouding | Dysostosis multiplex | n/r | n/r | n/r | n/r | n/r |
| Johnson, W.G. [28] | M M F F | Hydrops, Ascites, edema | Seizures | n/r | n/r | n/r | n/r | n/r | Telangiectasia | stillborn, 1 month, 3 months, alive 3 months |
| Yamano, T. [29] | M | Hydrops, ascites, edema, hepato-splenomegaly | n/a | no | no | n/a | n/a | n/a | n/a | Exitus at 56 days |
| Tabardel, Y. [30] | n/r | Hydrops, ascites, coarse features, hepatosplenomegaly | n/r | n/r | n/r | n/r | n/r | Cardiac anomalies | Petechiae | n/r |
| Ries, M. [31] | n/a | hydrops, ascites | n/r | Cherry red spots | n/r | n/r | n/r | n/r | n/r | Exitus at 28 days |
| Lukong, K.E. [9] | F | hydrops | n/r | n/r | n/r | n/r | n/r | n/r | n/r | Exitus at 82 days |
| Nakamura, Y. [32] | F | Ascites, coarse features, hepato-splenomegaly, inguinal hernia | Psychomotor retardation | n/r | Dysostosis multiplex | n/r | n/r | Cardiac anomalies | n/a | At the age of 2 months, patient was alive |
| Schmidt, M. [33] | F | Hydrops, ascites, edema, hepato-splenomegaly | Seizures | n/r | n/r | n/r | present | n/r | n/r | Exitus at 5 months |
| Ovali, F. [34] | M | Hydrops, ascites, coarse features, hepato-splenomegaly, inguinal hernia | n/r | n/r | n/r | n/r | present | n/r | n/r | Exitus at 27 days |
| Sergi, C. [35] | M | Hydrops, ascites, coarse features, hepato-splenomegaly, inguinal hernia | n/r | Corneal clouding | n/r | n/r | n/r | n/r | n/r | Exitus at 28 days |
| Sergi, C. [36] | M | Hydrops, edema ascites, hepato-splenomegaly | n/r | n/r | n/r | n/r | present | n/rn/r | n/r | Exitus at 2 months |
| Buchholz, T. [37] | M | Hydrops, edema ascites, hepato-splenomegaly | n/a | n/a | n/a | present | n/a | Cardiac anomalies | Telangiectasia Hypotonia | Exitus at 82 days |
| Uhl, J. [38] | M M | Hydrops, edema, ascites in both patients | n/a | n/a | n/a | n/a | n/a | n/r | Polydactyly in patient 1 | n/a |
| Donati, M.A. [39] | F | Hydrops, ascites, edema, coarse features, hepato-splenomegaly, Inguinal hernia | Psychomotor retardation, Hydrocephalus | yellow/rretina | Dysostosis multiplex | present | present | Cardiac anomalies | Telangiectasia Hypotonia, Petechiae | Exitus at 19 months |
| Penzel, R. [40] | F | Hydrops, ascites, edema | Seizures | n/r | n/r | n/r | n/r | n/r | n/r | n/r |
| Itoh, K. [41] | M | Hydrops, ascites, edema, hepato-splenomegaly | n/r | n/r | n/r | n/r | n/r | n/r | n/r | Exitus at 27 days |
| Rodriguez Criado, G. [42] | M | Hydrops, coarse features, hepatosplenomegaly | Psychomotor retardation | n/r | Dysostosis multiplex | n/r | n/r | Cardiac anomalies | Hypotonia | Exitus at 20 months |
| Pattison, S. [43] | n/r | n/r | n/r | n/r | n/r | n/r | n/r | n/r | n/r | Exitus at Patient 1; 3 months Patient 2; 2 months |
| Loren, D.J. [44] | n/r | Hydrops, ascites, edema, hepato-splenomegaly | n/r | n/r | no | n/r | n/r | n/r | n/r | Alive at the age of three months |
| Caciotti, A. [22] | F | Coarse features, hepatosplenomegaly | Psychomotor retardation | n/r | Dysostosis multiplex | n/r | present | Cardiac anomalies | Telangiectasia Hypotonia, petechiae | Exitus at 1 year |
| Bonten, E.J. [7] | F | Hydrops, hepato-splenomegaly | Psychomotor retardation, Hydrocephalus | No corneal opacity | Dysostosis multiplex, Joint contractures | n/r | n/r | cardiomyopathy | Cardiac anomalies | Exitus at 18 months |
| Lee, Y.J. [45] | F | Hydrops, ascites, coarse features, hepatosplenomegaly | n/r | Bilateral congenital cataracts with foveal hypoplasia | n/r | n/r | n/r | n/r | Telangiectasia Hypotonia, bluish to purpuric macules mild thrombocytopenia | Exitus a t9 months |
| Lee, B.H. [46] | F | Hydrops, ascites, coarse features, edema, hepato-splenomegaly | n/r | n/r | n/r | Present | n/r | Cardiomegaly with huge patent ductus arteriosus (PDA), Ventriculomegaly | Hypotonia | Exitus a t3 months |
Table 2.
Clinical and Morphological findings of patients suffering from sialidosis II, subtype infantile/Juvenile form (Cases from 1980 until the date of submission are included). M = male; F = female; n/a = not available n/r = not reported; no = not detected; ECG = electrocardiogram; ** Authors reported the case “suspected as sialidosis II” after other congenital errors of metabolism investigated during her childhood, such as mucopolysaccharidosis, were excluded. No formal genomic testing is reported in the study.
Table 2.
Clinical and Morphological findings of patients suffering from sialidosis II, subtype infantile/Juvenile form (Cases from 1980 until the date of submission are included). M = male; F = female; n/a = not available n/r = not reported; no = not detected; ECG = electrocardiogram; ** Authors reported the case “suspected as sialidosis II” after other congenital errors of metabolism investigated during her childhood, such as mucopolysaccharidosis, were excluded. No formal genomic testing is reported in the study.
| References (Name of the First Author) | Gender | General Presentation | Nervous System | Ophthalmologic Findings | Skeleton | Respiratory Distress/Infections | Renal Involvement | Cardiac Involvement | Others | Course of Disease |
|---|---|---|---|---|---|---|---|---|---|---|
| Winter, R.M. [47] | M <1 year | Coarse feature | Psychomotor delay, Seizures | Visual loss | Dysostosis Multiplex | n/r | n/r | n/r | Hearing loss, Inguinal hernia | 22 years |
| Kelly, T.E. [23] | F <1 year | Coarse feature, Hepatosplenomegaly | Psychomotor delay, seizures/myoclonic jerks | Cherry red spot | Dysostosis Multiplex | present | n/r | present | Umbilical hernia | 5 and half years |
| Kelly, T.E. [23] | F Birth | Coarse feature, Hepatosplenomegaly | Psychomotor delay | Cataract | Dysostosis Multiplex | n/r | n/r | present | Hearing loss, Umbilical hernia, Hypotonia | 24 months |
| King, M. [48] | M 5 months | Coarse feature, Hepatosplenomegaly | Psychomotor delay Ataxia | Cherry red spot, corneal Clouding, Cataract, | Dysostosis Multiplex | n/r/ | n/r | n/r | Hearing loss | 13 years |
| King, M. [48] | F N/A | Coarse feature, Hepatosplenomegaly | Psychomotor delay | Cherry red spot, Cataract, | Dysostosis Multiplex | n/r | n/r | n/r | Hearing loss | 12 years |
| Oohira, T. [49] | F <1 year | Coarse feature, Hepatosplenomegaly | Psychomotor delay, Ataxia, myoclonic jerks | Cherry red spots | Dysostosis Multiplex | n/r | n/r | n/r | Hypotonia | 5 years |
| Young, I.D. [50] | M 18 months | Coarse feature | Psychomotor delay, Ataxia, myoclonic jerks | Cherry red spot, Nystagmus, Optic atrophy | Dysostosis Multiplex | nr | n/r | n/r | Hearing Loss, Hypotonia | 12 years |
| Bakker, H.D. [51] | F 6 months | Coarse feature | Psychomotor delay | Strabismus, Nystagmus | n/r | n/r | n/r | n/r | Hearing Loss, Hypotonia | 30 years |
| Rodriguez Criado, G. [42] | M <1 year | Coarse feature, Hepatosplenomegaly | Psychomotor delay, Myoclonic movements Ataxia | n/r | Dysostosis Multiplex | n/r | n/r | present | Hearing Loss, Hypotonia | 13 years |
| Rodriguez Criado, G. [42] | M 16 months | Coarse feature, Hepatosplenomegaly | Psychomotor delay | n/r | Dysostosis Multiplex | n/r | n/r | absent | Hearing Loss, Hypotonia | 11 years |
| Pattison, S. [43] | n/r | Coarse feature, Hepatosplenomegaly | n/r | n/r | Dysostosis Multiplex | n/r | n/r | n/r | n/r | 3 years |
| Pattison, S. [43] | n/r | Coarse feature, Hepatosplenomegaly | n/r | n/r | Dysostosis Multiplex | n/r | n/r | n/r | n/r | 3 years |
| Schiff, M. [52] | F <1 year | Coarse feature, Hepatosplenomegaly | Psychomotor delay | n/r | Dysostosis Multiplex | n/r | present | n/r | n/r | 11 years |
| Gonzalez Gonzalez G [53] | n/r | n/r | Myoclonic epilepsy | n/r | Dysostosis Multiplex | n/r | n/r | present | n/r | 14 years |
| Caciotti, A. [22] | M 1 year | Coarse feature | Psychomotor delay Seizures | Cherry red spot, cataract, | Dysostosis Multiplex | n/r | n/r | n/r | Hearing Loss | 9 years |
| Bonten, E.J. [7] | M birth | Coarse feature, Hepatosplenomegaly | Developmental delay, Orbital hypoplasia | normal | Craniosynostosis | n/r | n/r | n/r | n/r | Progressing at 4 months |
| Bonten, E.J. [7] | F 12 years | n/r | Psychomotor delay, Seizures Ataxia, Dysmetria Spasticity | Cherry red spots | Dysostosis Multiplex, microcephaly | n/r | n/r | ECG specific alterations of repolarization | Hearing Lossl | Progressing at 28 years. |
| Ranganath, P. [54] | F 18 months | Coarse facies, hepatomegaly | n/a | Mild corneal haziness, bilateral fundal Cherry red spots | Macrocephaly | n/r | n/r | Cardiac anomalies | Protuberant tongue, gum hypertrophy, generalized hypertrichosis, large Mongolian spots on the back, umbilical hernia | n/r |
| de Rezende Pintoi ** [55] | F 30 years | a high forehead and low-set ears | Advanced degree of mental deficiency.lower limb spasticity, and facial and limb myoclonic jerks | Bilateral macular cherry-red spots | n/r | n/r | n/r | n/r | A marked delayed in motor and cognitive functions present since childhood. Cognitive and motor skills had worsened over 10 years | n/r |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Khan, A.; Sergi, C. Sialidosis: A Review of Morphology and Molecular Biology of a Rare Pediatric Disorder. Diagnostics 2018, 8, 29. https://doi.org/10.3390/diagnostics8020029
AMA Style
Khan A, Sergi C. Sialidosis: A Review of Morphology and Molecular Biology of a Rare Pediatric Disorder. Diagnostics. 2018; 8(2):29. https://doi.org/10.3390/diagnostics8020029
Chicago/Turabian StyleKhan, Aiza, and Consolato Sergi. 2018. "Sialidosis: A Review of Morphology and Molecular Biology of a Rare Pediatric Disorder" Diagnostics 8, no. 2: 29. https://doi.org/10.3390/diagnostics8020029
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.