3.1. Oxidoreductase Activity of Pyrrhotite
In a prebiotic scenario, the synthesis of pyruvic acid is of great interest. Many studies have focused on the role of this keto acid in the synthesis of other organic compounds with metabolic relevance, such as acetic acid, methylsuccinic acid and other cyclized compounds [
23], oxalacetic acid, acetoacetic acid, fumaric acid, and succinic acid [
24]. In the first steps toward the formation of the protometabolic system prior to the origin of living biochemistry, the redox reactions catalyzed by transition metals, such as those in the iron-sulfur minerals, were necessary, as is exhibited by the conserved organometallic enzymes of the most primitive metabolic pathways for carbon fixation and energy generation. This supports the iron-sulfur world hypothesis, wherein the formation of pyrite from pyrrhotite may have provided enough reducing power to catalyze these fundamental redox reactions [
20,
21]. However, the pyrrhotite-pyrite transformation coupled with redox reaction is not a sufficient condition for the building of the iron-sulfur world. As Schoonen
et al. pointed out [
25], the pyrrhotite-pyrite couple is not capable of reducing CO
2, although it is termodinamically favorable, due to the energetically unfavorable electron transfer from the pyrrhotite valence band to the LUMO (lowest unoccupied molecular orbital) of CO
2, especially at higher temperatures. In consequence, to evaluate the pyrrhotite system in the context of iron-sulfur world hypothesis, we studied its redox properties and design an experimental system that overcome the limitations of the FeS/FeS
2 couple.
In our experiments, the initial goal was to mimic the oxidoreductase activity of the fermentative enzyme lactate dehydrogenase (LDH) and to connect the pyruvate-lactate redox reaction to the FeS/pyrite system (1). This reaction is essential for explaining the behavior of lactate-pyruvate in sulfur rich mediums.
In a biochemical system, the LDH enzyme catalyzes the redox interconversion between pyruvate and lactate. In bacteria, there are two types of LDH: NADH-dependent and NADH-independent. In the first group, the pyruvate resulting from the glycolysis is reduced to lactate, regenerating the nicotinamide adenine dinucleotide (NAD
+) from the reduced nicotinamide adenine dinucleotide (NADH) produced in the earlier step. In the second group, which mostly consists of anaerobic bacteria, the LDH does not need the cofactor NADH, and the lactate is used as a carbon source in the subsequent steps of metabolism [
26]. One of these non-NADH dependent LDH is the lactate cyctochrome c reductase, which uses a Fe(III)-porphyrin complex as an electron acceptor to yield pyruvic acid and Fe(II)-porphyrin.
To test the activity of the FeS/H
2S system as a redox catalyst, we performed two different sets of experiments. First, we used pyruvic acid as a reagent to see whether this system can reduce it to lactic acid under different pH conditions. All of the reactions were carried out at 130 °C for 5 h in an anaerobic atmosphere.
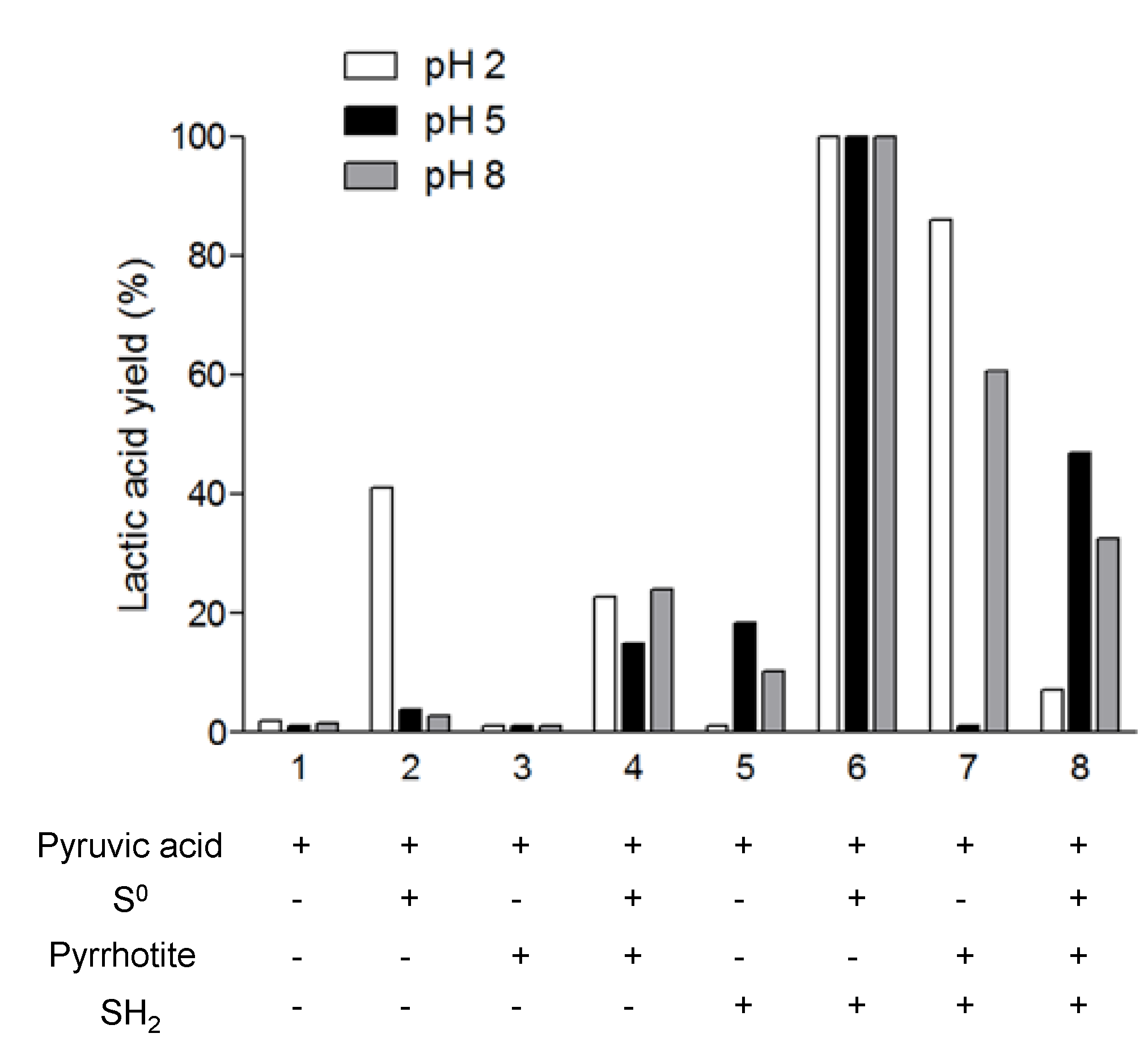
Figure 4 shows the percentage of lactic acid synthesis related to the initial amount of pyruvic acid present in the mixture.
If we look at the third column in
Figure 4, we note that pyrrhotite
per se is not able to promote the synthesis of lactic acid. However, when it is mixed with H
2S at either an acidic or basic pH, there is a large reduction in pyruvic acid, greater than 50%. These results differ from those of Wang
et al. [
16], who found that the
in situ precipitation of FeS reduced the pyruvic acid and that its mixture with H
2S decreased the yield of the reaction.
Figure 4.
Lactic acid synthesis by pyruvic acid reduction coupled to iron sulfide/sulfur system. Role of pH and sulfur oxidation state.
Figure 4.
Lactic acid synthesis by pyruvic acid reduction coupled to iron sulfide/sulfur system. Role of pH and sulfur oxidation state.
The highest rate of lactic acid production (100%) is obtained by mixing elemental sulfur (Sº) and H
2S. If we compare these results with mixtures 2 and 4, where these reagents were used separately, we can see that the activity of the Sº and the H
2S might be coupled, thereby enhancing the reaction. It is interesting to note that in mixture 2, Sº can reduce the pyruvic acid mainly at an acidic pH. Wang
et al. [
16] determined that Sº, at temperatures higher than 113 °C and at neutral pH, acts as an oxidant. Nevertheless, according to our results, Sº is not able to oxidize lactic acid (
Figure 5), but it is a reducing agent at lower pH.
We performed four reactions using pyrrhotite to clarify its role in the pyruvic acid reduction. The reactions correspond to conditions 3, 4, 7, and 8. Comparing them, we note that the highest yields of lactic synthesis are obtained in pyrrhotite/H2S. In this case, the amount of lactic synthesis is higher than in reaction 5, where only H2S was added. If the reaction medium contains pyrrhotite with Sº and H2S, the rate of pyruvic reduction is decreased compared with reaction 6. However, lactic acid synthesis in reaction 4, pyrrhotite + Sº, is enhanced at neutral and basic pH compared with reaction 2. Overall, we observe that in the presence of pyrrhotite at an acidic pH, the addition of Sº to the mixture decreases the rate of pyruvic acid reduction, though the rate is enhanced at neutral and basic pH.
Taken together, our results demonstrate that the pyrrhotite is able to reduce pyruvic acid into lactic acid only in the presence of Sº and/or H
2S. This result is important for explaining the reaction product in
Section 3.2.
Figure 5.
Pyruvic acid synthesis by lactic acid oxidation coupled with iron sulfide/sulfur system. Role of pH and sulfur oxidation state.
Figure 5.
Pyruvic acid synthesis by lactic acid oxidation coupled with iron sulfide/sulfur system. Role of pH and sulfur oxidation state.
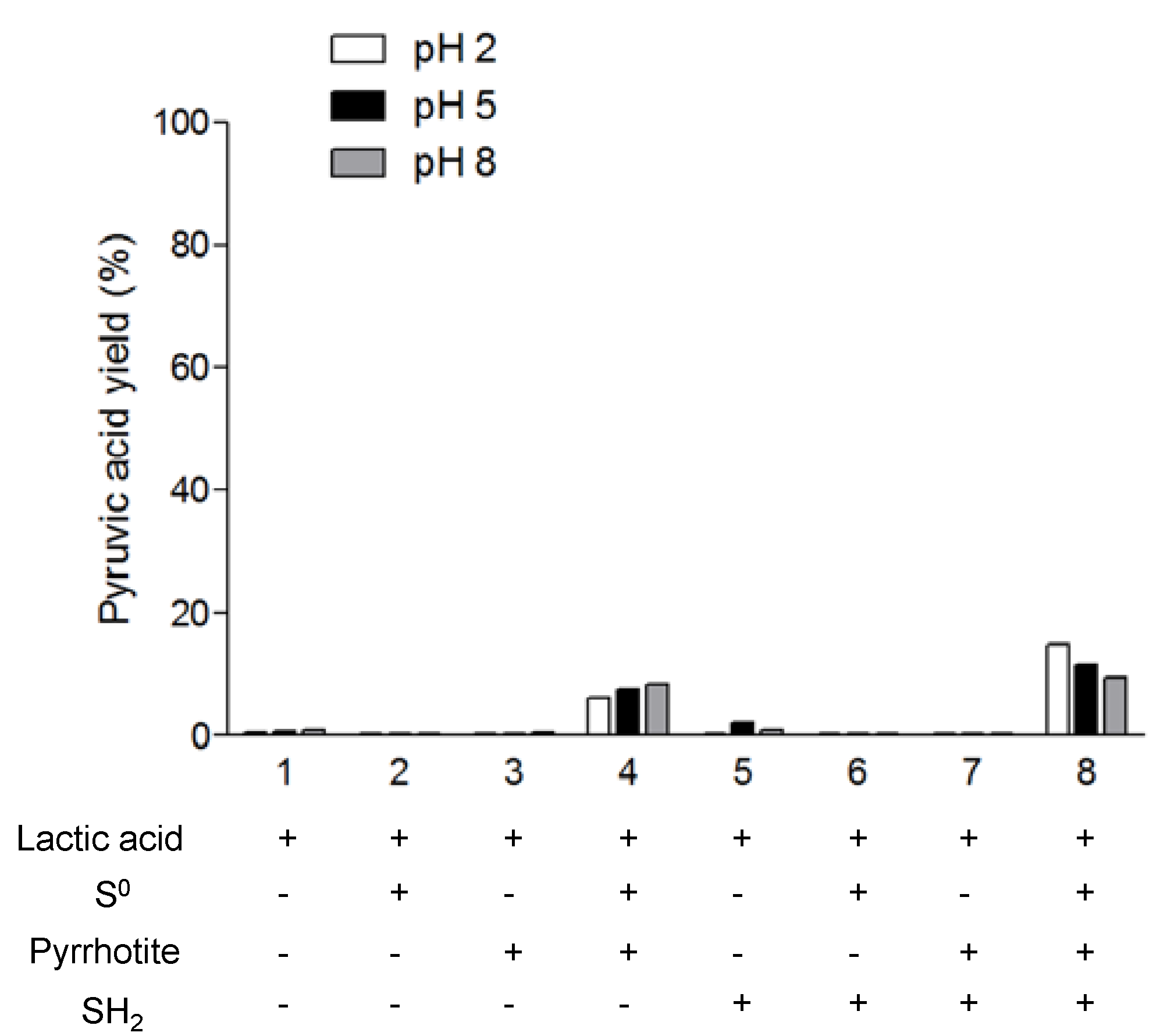
In the second set of experiments, we tested the reverse reaction, that is, the oxidation of lactic acid into pyruvic acid in sulfur rich systems.
Figure 5 shows the percentage of pyruvic acid synthesis related to the initial amount of lactic acid. First, we note that the oxidation rates are lower than the reduction rates. In addition, the oxidation takes place only in the presence of pyrrhotite and Sº (reaction 4 and 8). There is no detectable pyruvic acid synthesis at reaction 7 (pyrrhotite + H
2S); however, in the presence of elemental sulfur (reaction 8), the presence of H
2S enhances the lactic acid oxidation compared with reaction 4 (pyrrhotite + Sº). In this case, our results are in agreement with those of Wang
et al. [
16] and support the idea of a coupled reaction system between FeS/Sº/H
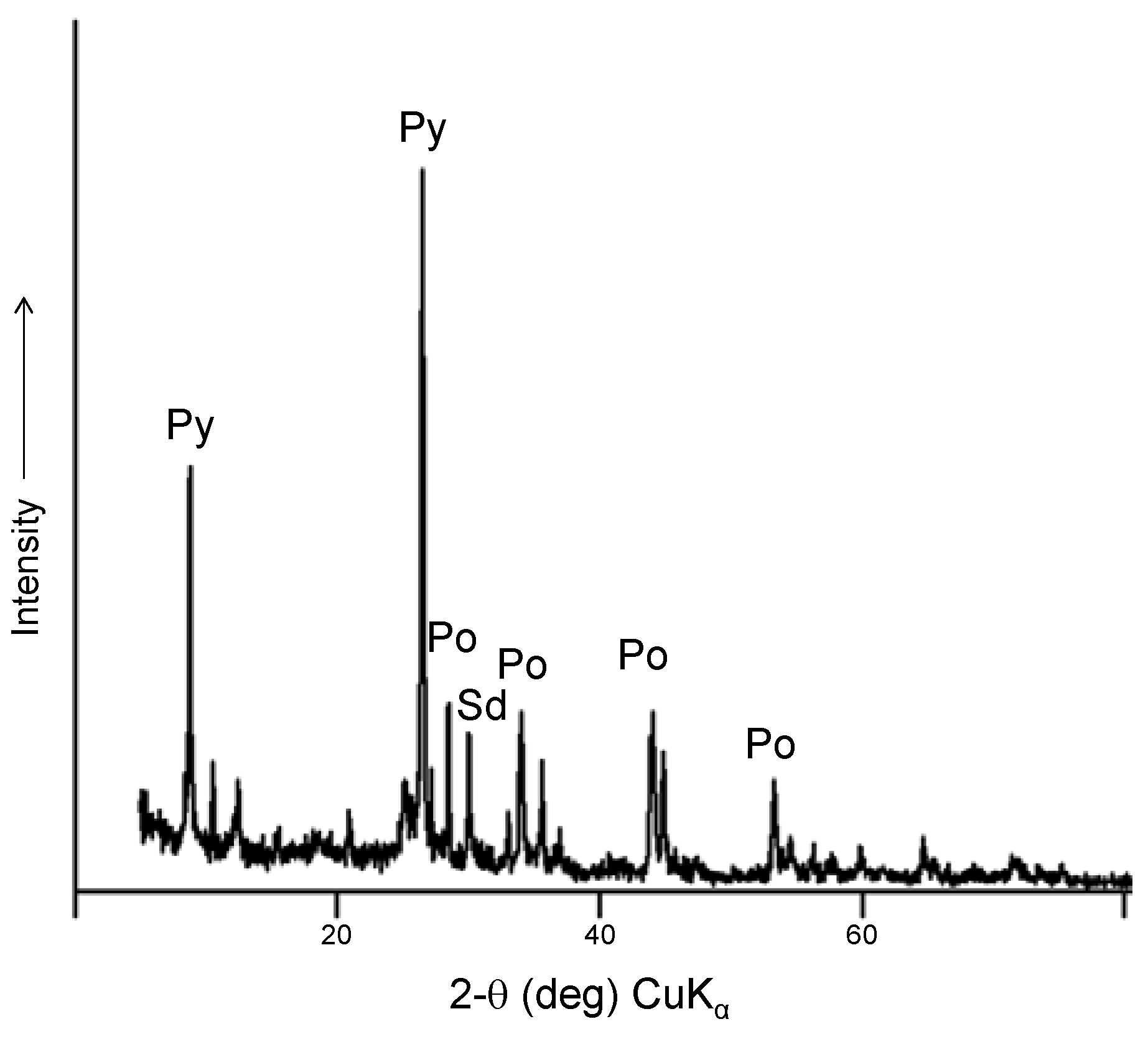
2S, mainly at acidic pH, and with a significant formation of pyrite, which is identified by XRD analysis of the solid material after the reaction (
Figure 6). According to the results, the presence of H
2S decreases the rate of oxidation at basic pH compared to acidic pH., The hypothesis used to explain this pH dependence is that the protonated state of the lactic acid affects the reaction. However, in our experiments, when we combined Sº with pyrrhotite, the effect of pH was the opposite of that in reaction 8; thus, it seems more reasonable to conclude that H
2S is the factor causing the pH-dependent behavior of the reaction. In fact, at pH 2 and 5, the free sulfide is in the form of H
2S, whereas at pH 8, it is in SH
− form. If we consider that there is a coupled reaction between FeS/Sº/H
2S, the form of all the components under different pH conditions will affect the overall reaction. Additionally, the lactic acid could form stable complexes with iron, e.g., Fe(Lac)
+ and Fe(Lac)
2. The formation of iron-lactate complexes is an important factor in the explanation of the differences in the reactions. Lactic acid promotes iron mobilization from the mineral; simulations performed using Geochemist’s Workbench showed that the formation of Fe(III) lactate complexes is associated with pyrrhotite transformation in pyrite in the presence of Sº. This could explain the lack of oxidation to pyruvate.
Considering the role of elemental sulfur in the oxidation of lactic acid, it is interesting to highlight that in the presence of pyrrhotite, it promotes the oxidation at low pH, which is in contrast with the results obtained for the reduction, where elemental sulfur decreases the yield of lactic synthesis. Thus, these results suggest that at 130 °C the Sº acts as an oxidant when the pH is acidic and as a reductant at neutral and basic pH.
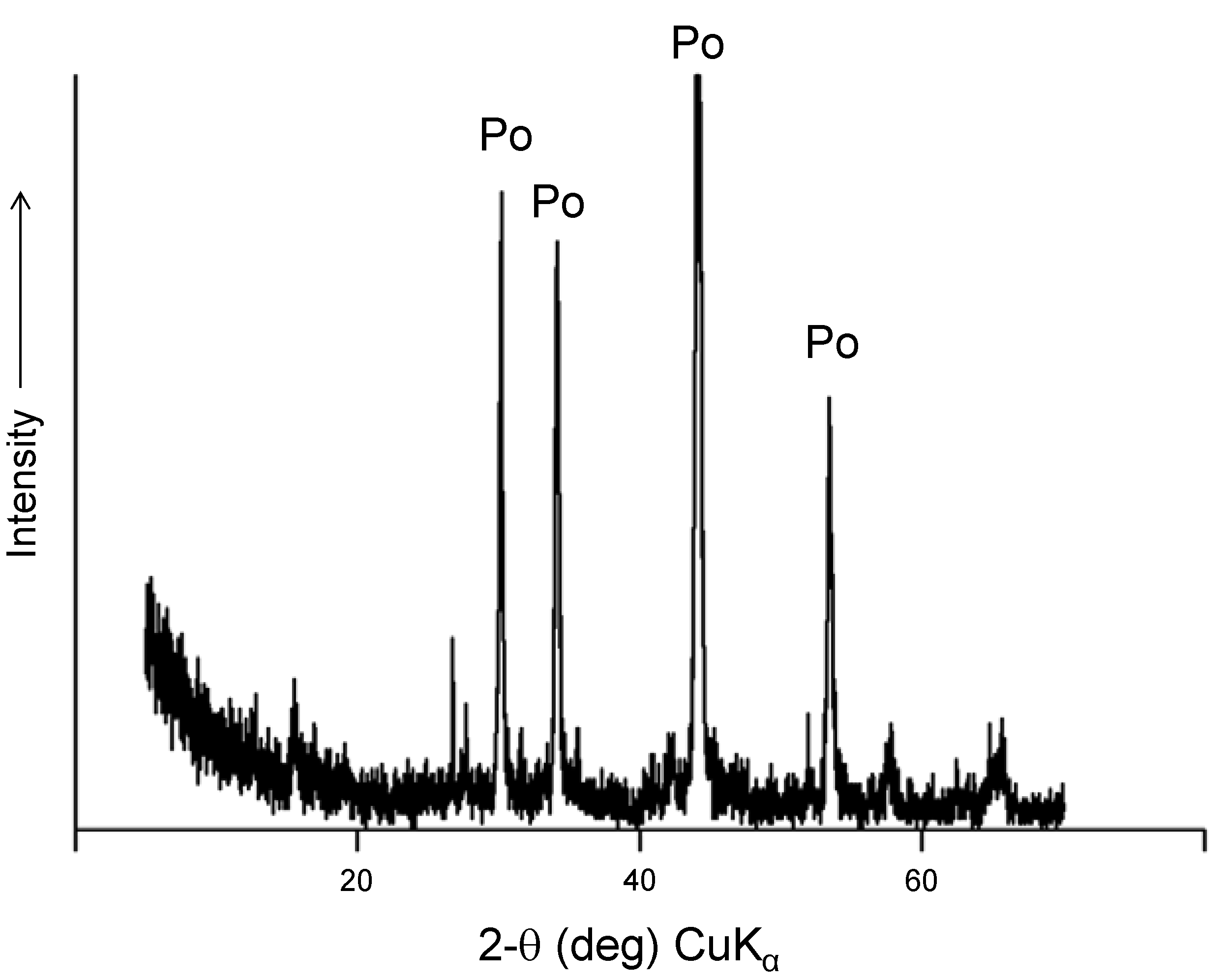
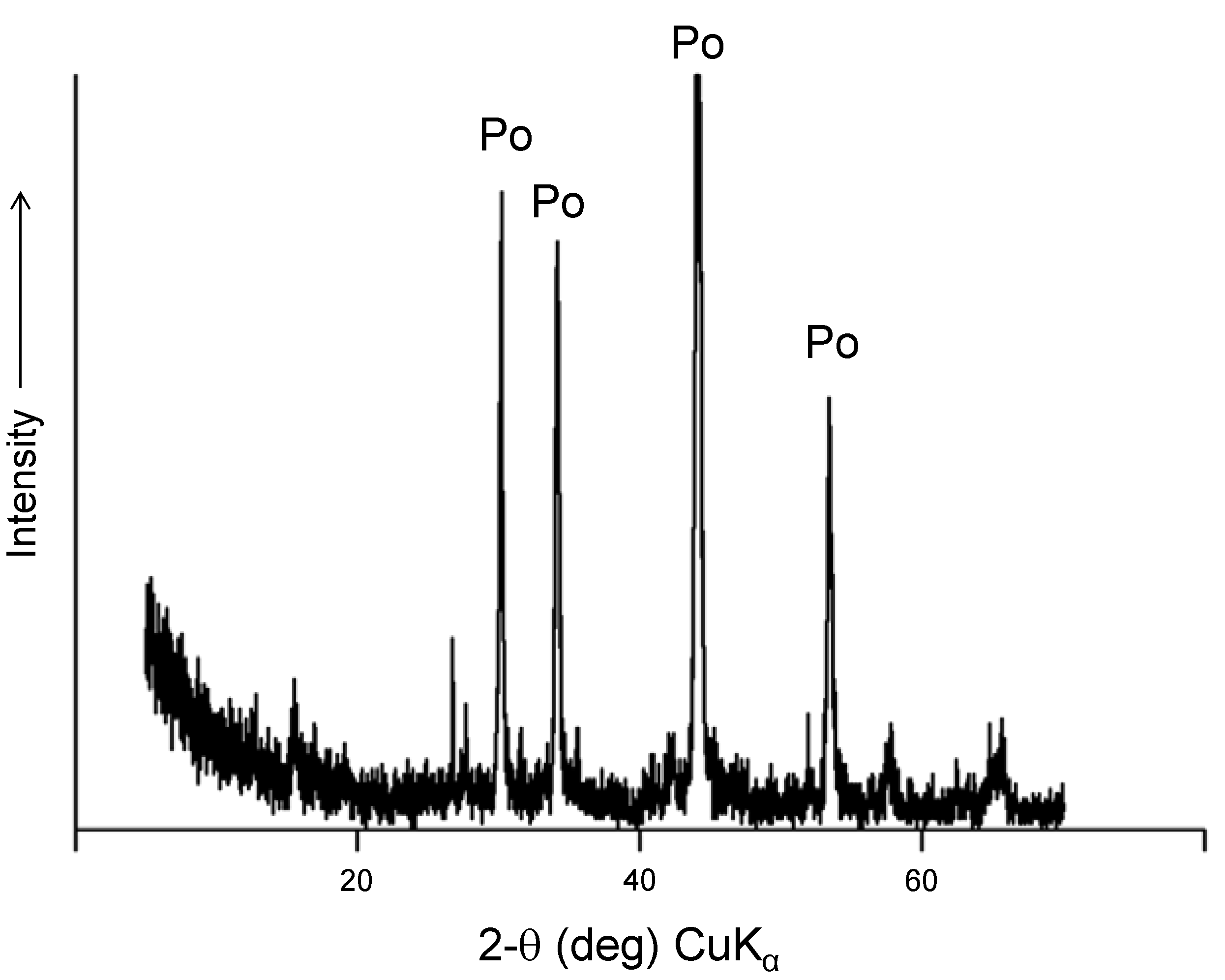
Figure 6.
Powder X-ray diffraction of pyrrhotite ore after experiments. The identified phases include pyrite and siderite.
Figure 6.
Powder X-ray diffraction of pyrrhotite ore after experiments. The identified phases include pyrite and siderite.
Overall, our results demonstrate that the pyrrhotite/Sº/H
2S system has the ability to mimic the oxidoreductase activity and that it drives lactate-pyruvate redox chemistry in low temperature hydrothermal systems. The smaller yield of lactic synthesis in the presence of pyrrhotite, Sº and H
2S in comparison with the same mixture without pyrrhotite (
Figure 4) might be explained by the fact that in the presence of pyrrhotite the coupling reaction system is promoted at the same time as the reduction, and the oxidation is pH-dependent.
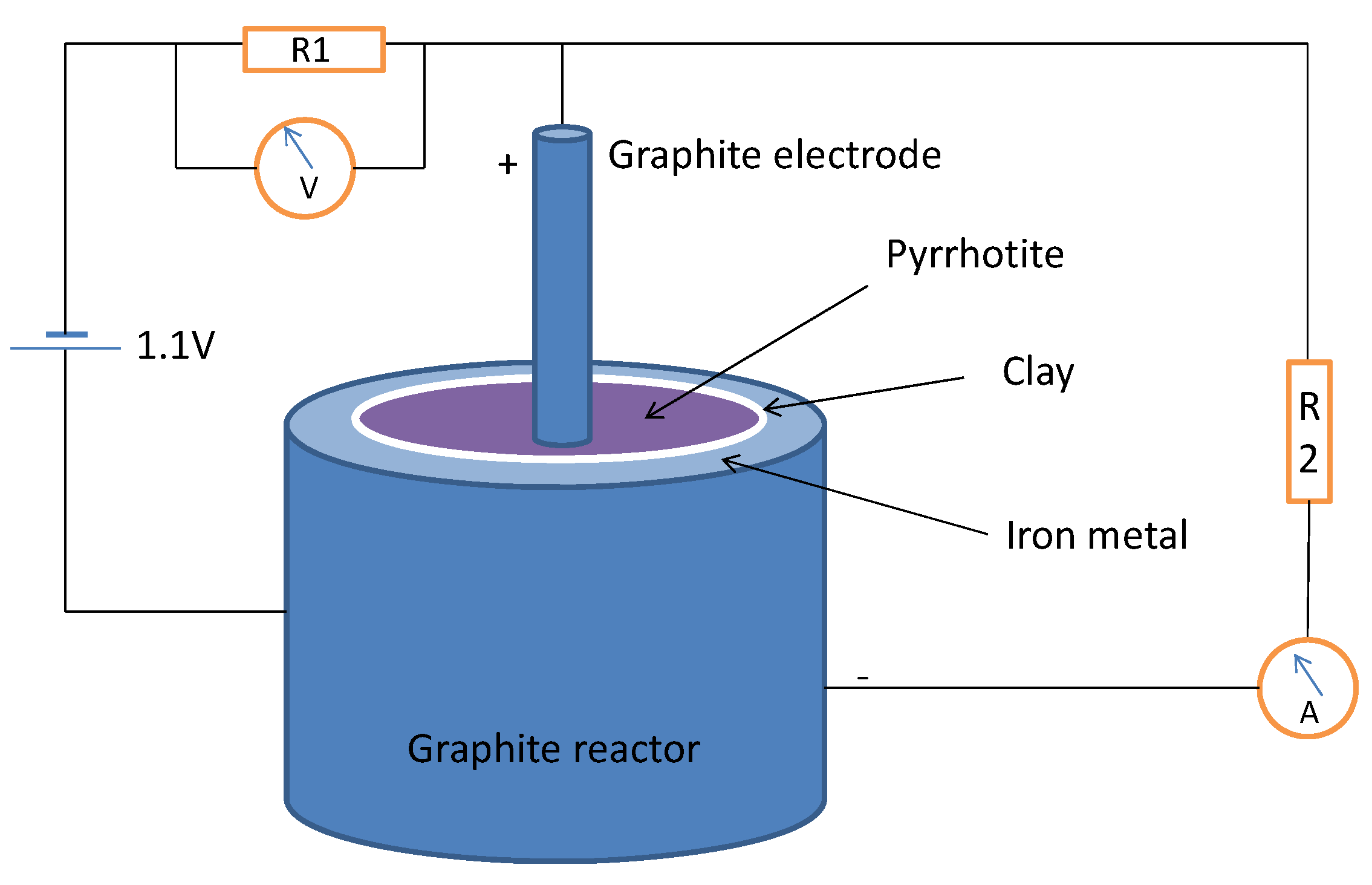
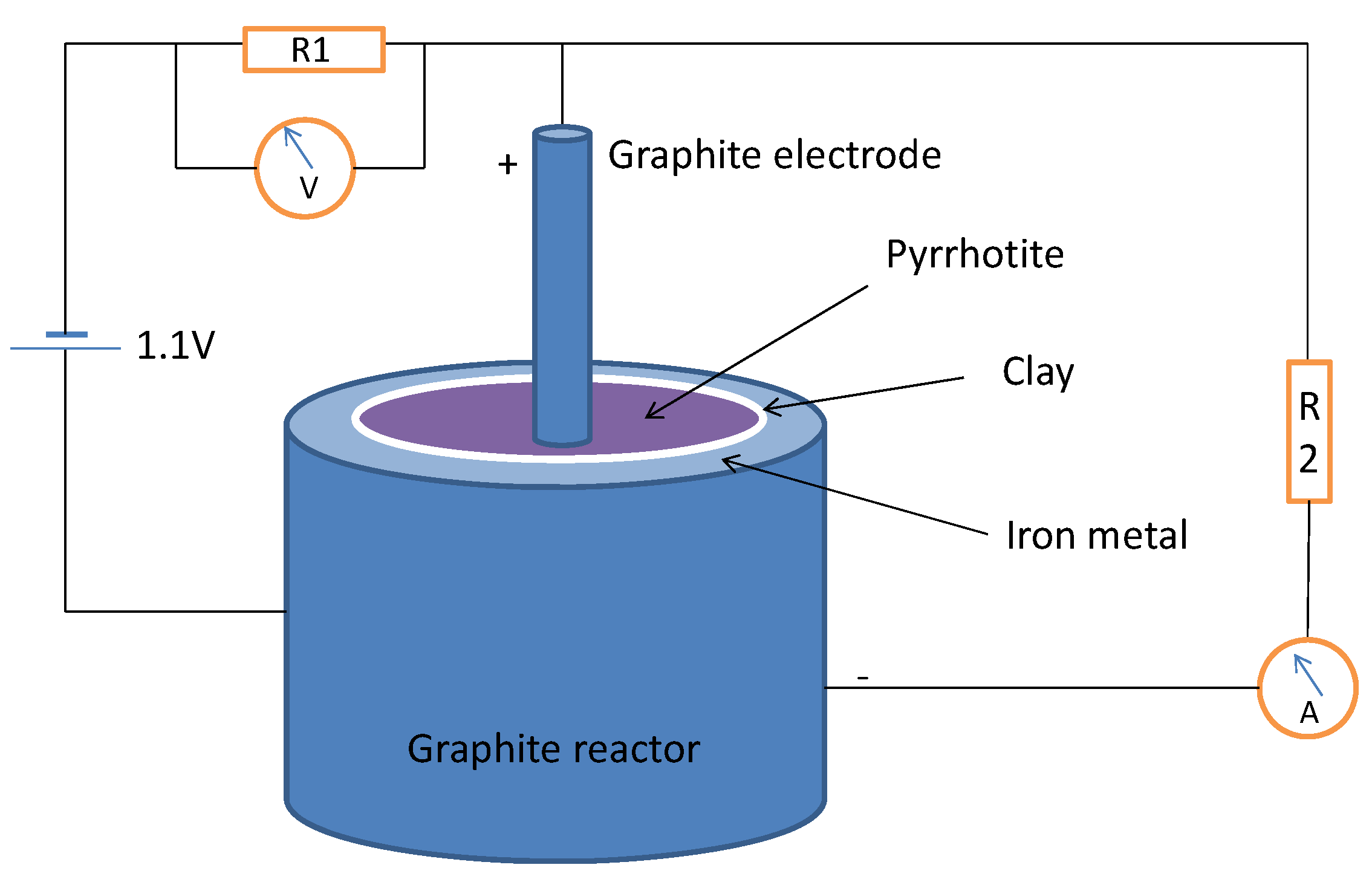
3.2. Pyrrhotite/Iron Metal in Thioester Reductive Carboxylation: a Ferredoxin Mimic?
Metallic iron is an extremely rare mineral in the modern Earth’s crust, mainly due to its instability in oxic conditions, but it may have been abundant in the early Earth before the rise of an oxygen-rich atmosphere. Iron is a good source of electrons, through its oxidation to Fe
2+ (standard reduction potential, Eº = −440 mV). We constructed an electrochemical cell using a powder iron metal paste in water-HSO
4− at pH 5 as the anode, separated by a clay barrier from the cathode, which was formed by pyrrhotite containing 1 mmol of ethyl thioacetate and wetted with a NH
4+ HCO
3− 50 mM solution (pH 9) containing 0.15 mmol of Na
2S; we used the cell to test whether iron metal could be an additional source of reducing power and a supply of mobile iron under mild conditions. The system was connected to a power supply at 1.1 V under a nitrogen atmosphere in the circuit depicted in
Figure 3; the current began at 17.4 µA, rose to a maximum of 23 µA and 1.87 V after 6 h and then decayed to a constant current of 10.5 µA. The organic solutes were analyzed after three days of standing in anoxic conditions at room temperature; the analysis shows a significant quantity of lactic acid (
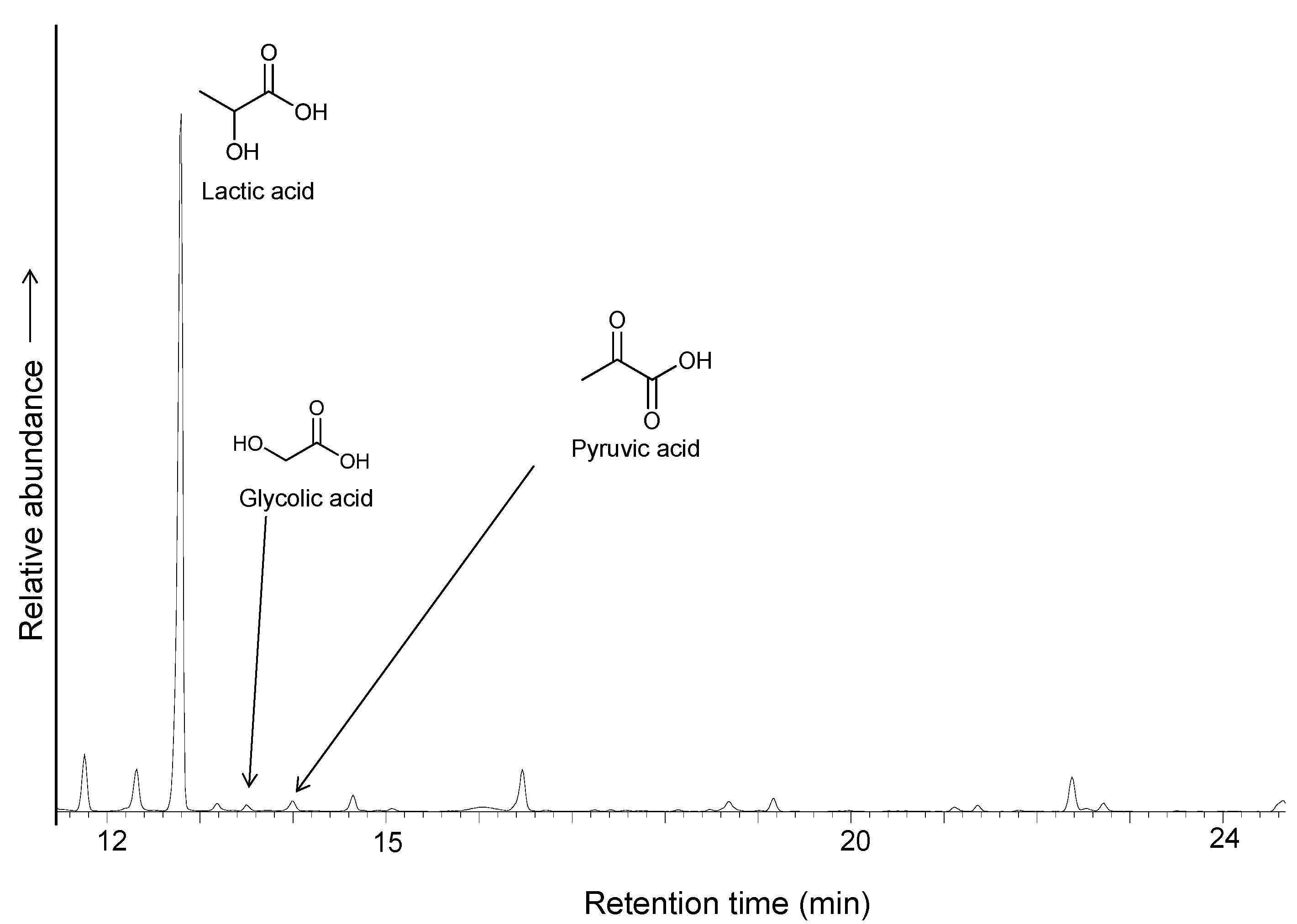
Figure 7) with an estimated yield of 6.5% of the added ethyl thioacetate. Pyruvic acid was also detected, as well as glycolic acid and glycine. Aliphatic amines and unidentified organic compounds, including sulfur-containing molecules, were formed.
Figure 7.
GC/MS chromatogram of the TMS derivatives of identified products obtained by carboxylation of ethylthioacetate coupled to the iron/pyrrhotite/sulfide electrochemical system.
Figure 7.
GC/MS chromatogram of the TMS derivatives of identified products obtained by carboxylation of ethylthioacetate coupled to the iron/pyrrhotite/sulfide electrochemical system.
No carbon chains greater than C3 were detected. The X-ray diffraction of pyrrhotite conducted after the experiment shows peaks of remaining pyrrhotite, pyrite, siderite and unidentified iron oxides, possibly wustite, ferrihydrite, and “green rust” (iron hydroxycarbonate). A control experiment performed without an external voltage source showed a significantly lower yield of lactic acid (less than 1%). An additional control experiment without thioester does not show formation of C2 or C3 organic compounds, suggesting that lactate/pyruvate are formed after carboxylation of thioacetate ester and that the formation of organic compounds directly from carbon dioxide is not possible under these conditions.
The voltage source generates an electrochemical gradient and can supply additional electrons to the pyrrhotite-S
2−/pyrite redox couple, which favors CO
2 reduction and is not an efficient electron acceptor due to its large overpotential (−2.22 V
vs. SCE). The presence of iron-sulfur clusters can significantly reduce the CO
2 reduction overpotential, promoting carbon fixation [
20]. Under our chosen conditions, the anodic oxidation of iron metal could supply Fe
2+ cations to the system, with subsequent formation of FeS
(aq), which constitutes the building blocks of crystalline forms of ferrous sulfides [
27]. The soluble iron-sulfur complexes that constitute the aqueous form of FeS could have similar properties to the iron-sulfur clusters found in the metalloprotein ferredoxin, which may mediate the electron transfer reaction for the reductive carboxylation of thioacetic acid [
28]. The formation of these protoferredoxins [
8] could be favored by the formation of organic sulfur species, as ethanethiol, derived from ethyl thioacetate, which could form complexes with the formula [Fe
4S
4(SC
2H
5)
4]
3−. Under these conditions, the external source and the iron could constitute the electron donors necessary to keep the iron-sulfur complexes in a reduced state and to overcome the reduction potential of the pyruvate synthesis from CO
2 and thioester (Eº = −500 mV). Hence, our experiment can be considered to be a biomimetic pathway to the biosynthesis of pyruvate from acetyl-CoA, promoted by PFOR and reduced ferredoxin [
17]. The strongly favored presence of lactic acid, which is the major organic product of the experiment, can be explained by the formation of stable Fe-lactic acid complexes, which stabilize both the lactic acid and the iron in solution.
The role of iron-sulfur clusters in the reductive carboxylation were demonstrated in the seminal work of Nakajima
et al. [
29], in which an alpha-keto acid is formed under mild conditions as an intermediate in the synthesis of phenylalanine from n-octyl-phenylthioacetate, catalyzed by a synthetic iron-sulfur cluster that models ferredoxin. Although the Nakajima reaction helped to conceptualize the possibility of iron-sulfur clusters as non-enzymatic catalysts and its potential role during life origin, the reaction is not geochemically extrapolable, as it has been performed in non-aqueous conditions and with hydrosulfite as electron donor (Eº = −0.660V). The electron donor role, as suggested in the Iron-Sulfur world theory, could be assumed by the pyrite formation from pyrrhotite in a geochemical environment [
30]. In fact, the control experiment without an external voltage source shows a significantly lower yield in the formation of lactic acid, suggesting that a low potential electron donor could be necessary for the process. To test this possibility, we performed an experiment using the same electrochemical cell design but without using an external voltage source and adding 1 mmol of hydroquinone (Eº = −0.699V). Hydroquinone can act as analog of the biological ubiquinol and can perform electron transfer reactions on the surface of minerals [
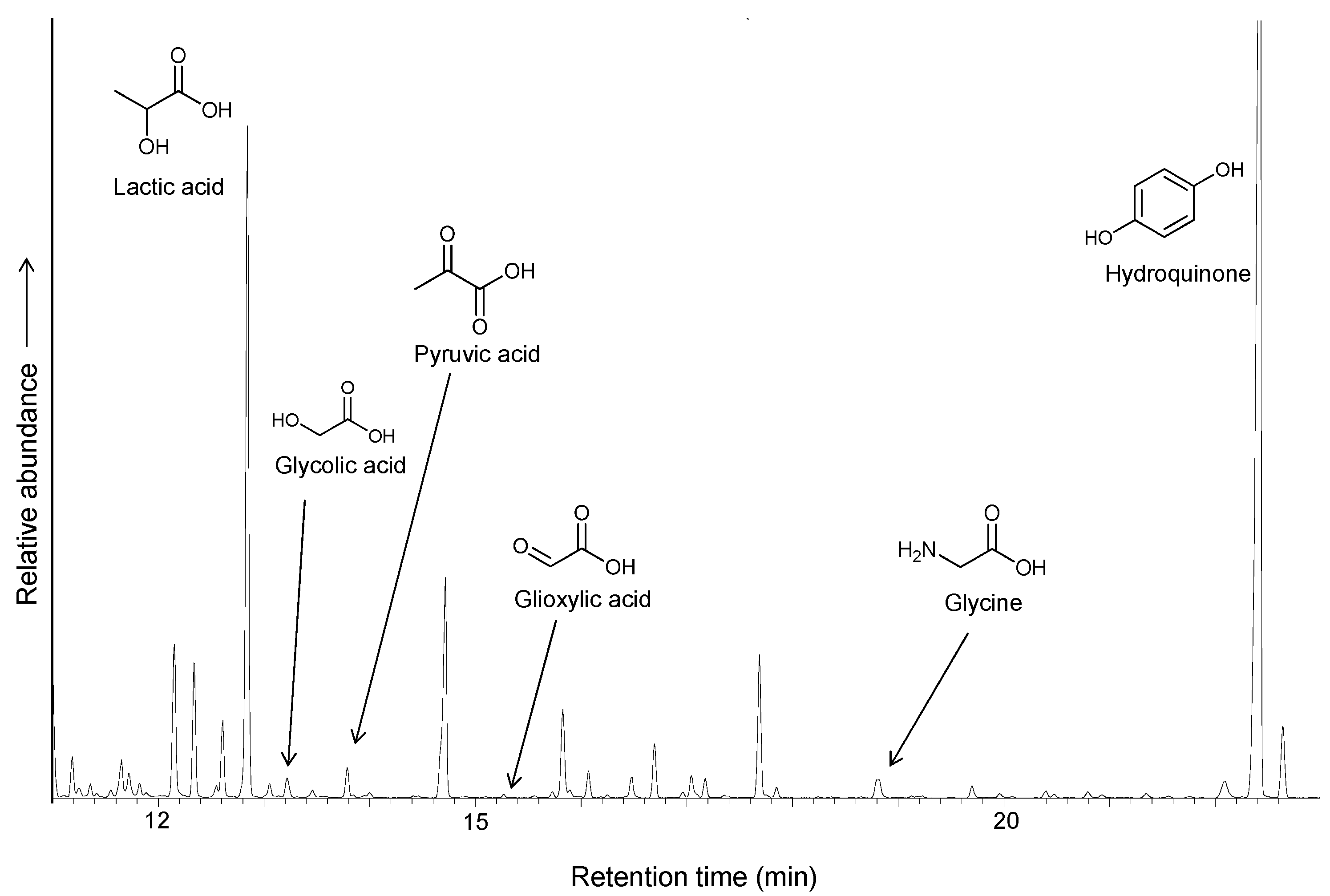
31]. The model biochemical reaction that motivates the selection of hydroquinone as an electron donor is the formation of pyruvate by direct carboxylation of acetic acid, promoted by (quinone) pyruvate dehydrogenase. The presence of hydroquinone promotes the synthesis of lactic acid, increasing the yield to 10.5% and suggesting that electrons can be transferred through iron sulfur clusters or surfaces, similar to the ubiquinol/iron-sulfur system in biochemistry (
Figure 8).
Our experiments show that iron metal is essential to the reaction, possibly as an electron source and soluble iron source. However, what is the role of pyrrhotite? A control experiment performed using silica sand instead of pyrrhotite does not show the formation of C
3 derivatives, which indicates that pyrrhotite plays an essential role in the formation of pyruvate/lactate. Although the synthesis of pyruvate, promoted by FeS, has been reported by G.D. Cody
et al. [
32], the high CO pressure experimental conditions suggest a different mechanism. Under our mild conditions, two facts could help to explain the role of pyrrhotite. (i) The pyrrhotite structure is complex; it is a non-stoichiometric, iron defective sulfide, with surface vacancies and distortions [
33]. The surface chemistry of pyrrhotite, although not widely studied, should be different than of troilite (its stoichiometric equivalent) and the synthetic FeS phases usually used in prebiotic chemistry. The electronic properties of pyrrhotite particles could allow them to be used as electron transfer conduits to the species in solution or to newly formed membraneous precipitates, in a electrochemical induced pathway similar to the photoelectrochemical formation of alpha-ketoglutaric acid from pyruvate, catalyzed by sphalerite particles [
34]. (ii) The formation of pyrite in the presence of hydrogen sulfide, which, in synergy with the formation of soluble protoferredoxin clusters, could supply reducing power in the form of molecular hydrogen. This pyrite-pulled reaction connects the prebiotic chemistry with the biochemistry; this is an equivalent mechanism to that in which ferredoxin is maintained in a reduced state by hydrogen in chemolithotrophs [
12]. Although the system shows considerable chemical complexity, and further work is necessary to explain the products obtained, the common key feature is the disequilibrium. We established an electrochemical reactor, with dissipation of a potential and pH gradient, together with the formation of new mineral precipitates, following the theory postulated by M. Russell [
35,
36]. The main difference is that the Russell theory proposed an exhalative submarine model that involves the mixture of two fluids at different temperature and pH: The Hadean ocean water and the hydrothermal fluid. The formation of membranous iron sulfide precipitate in the fluid interface promoted an environment where accumulation of clusters and organics and the dissipation of electrochemical gradients through membrane, promotes the protometabolic reactions. We propose in this experiment a fluid-rock interaction model in which iron metal and iron sulfur minerals promoted the formation of new species and the adequate environment for the carbon fixation, by formation of new iron solid species and soluble clusters. The presence of previously formed organic precursors (such as thioesters or alkyl thiols, in the reported experiment) is necessary to the increase of organic complexity in the mild conditions tested.
Figure 8.
GC/MS chromatogram of the TMS derivatives of products obtained by carboxylation of ethylthioacetate coupled to the iron/pyrrhotite/sulfide electrochemical system, with hydroquinone as an additional electron donor.
Figure 8.
GC/MS chromatogram of the TMS derivatives of products obtained by carboxylation of ethylthioacetate coupled to the iron/pyrrhotite/sulfide electrochemical system, with hydroquinone as an additional electron donor.
An interesting feature of the reaction is the formation of glycolic acid and glycine. The reductive amination of alpha-keto acids in the presence of ammonium to yield amino acids has been reported under mild conditions in the presence of Fe
2+ [
37]. In our results, we did not identify any amino acids larger than glycine. The glycine could be synthesized from glyoxylic acid by transamination using other amino acids as nitrogen donors [
38] and by reductive amination in the presence of strong reducing agents [
39]. The possible presence of glyoxylic acid as an intermediate under our conditions connects the pyrrhotite/electrochemical reduction with the synthesis of lactate, which is promoted by the highly reducing photo-generated conduction band electrons in the zinc sulfide mineral sphalerite [
40]. This photo-electrochemical synthesis leads to the direct formation of lactate by a reaction between carbon dioxide and glyoxylic acid. It is possible that the electronic properties of pyrrhotite favor a similar pathway in the electrochemical cell (
Figure 9), although oxalic acid was not identified and there was no evidence of thioacetate independent carbon fixation. It is interesting to note the lack of organic acids greater than C
3, which suggests that there are no further reactions involving pyruvic acid or that reduction to lactic acid or degradation (for example, formation of oxaloacetate and degradation to pyruvate) are kinetically favored.
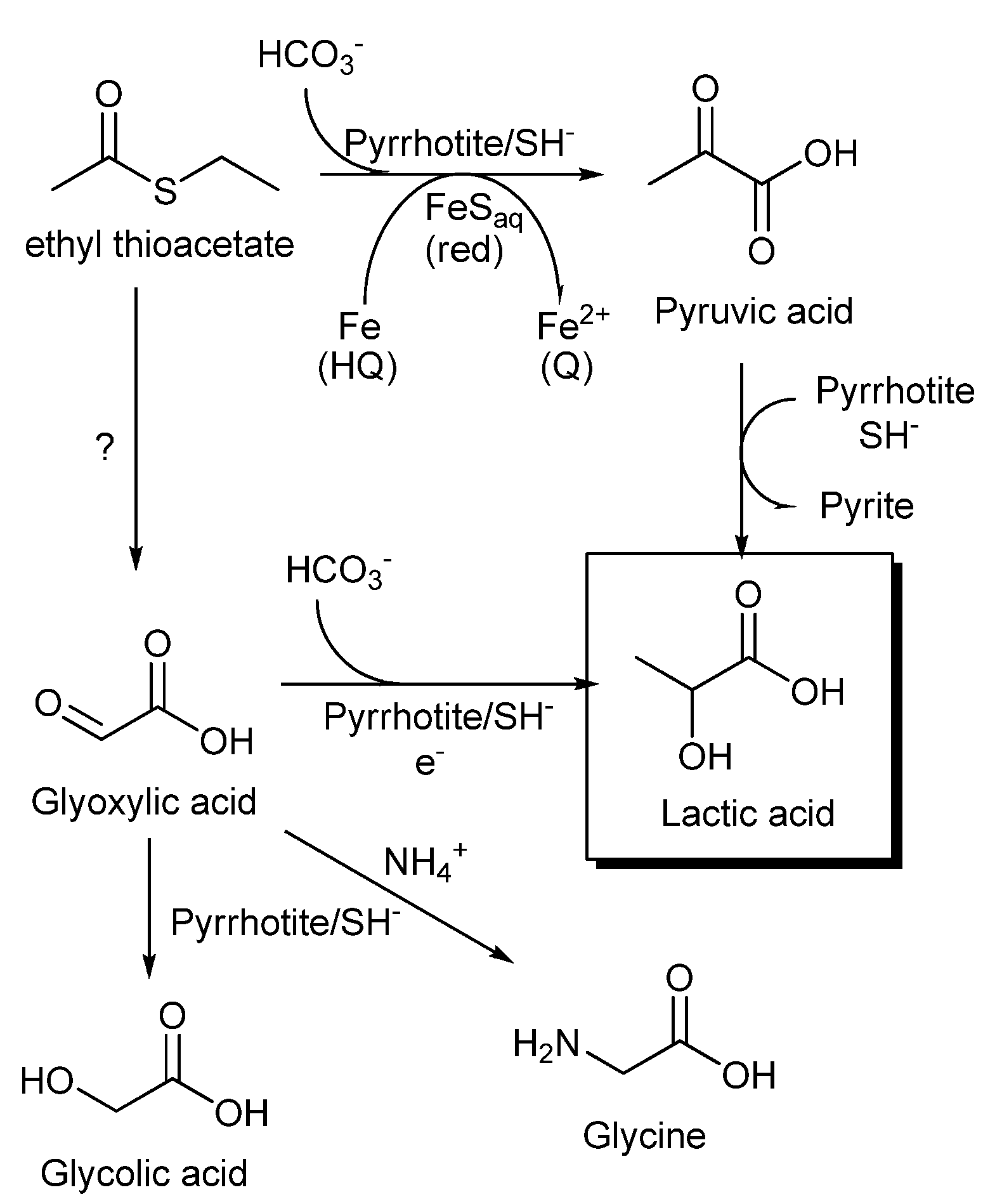
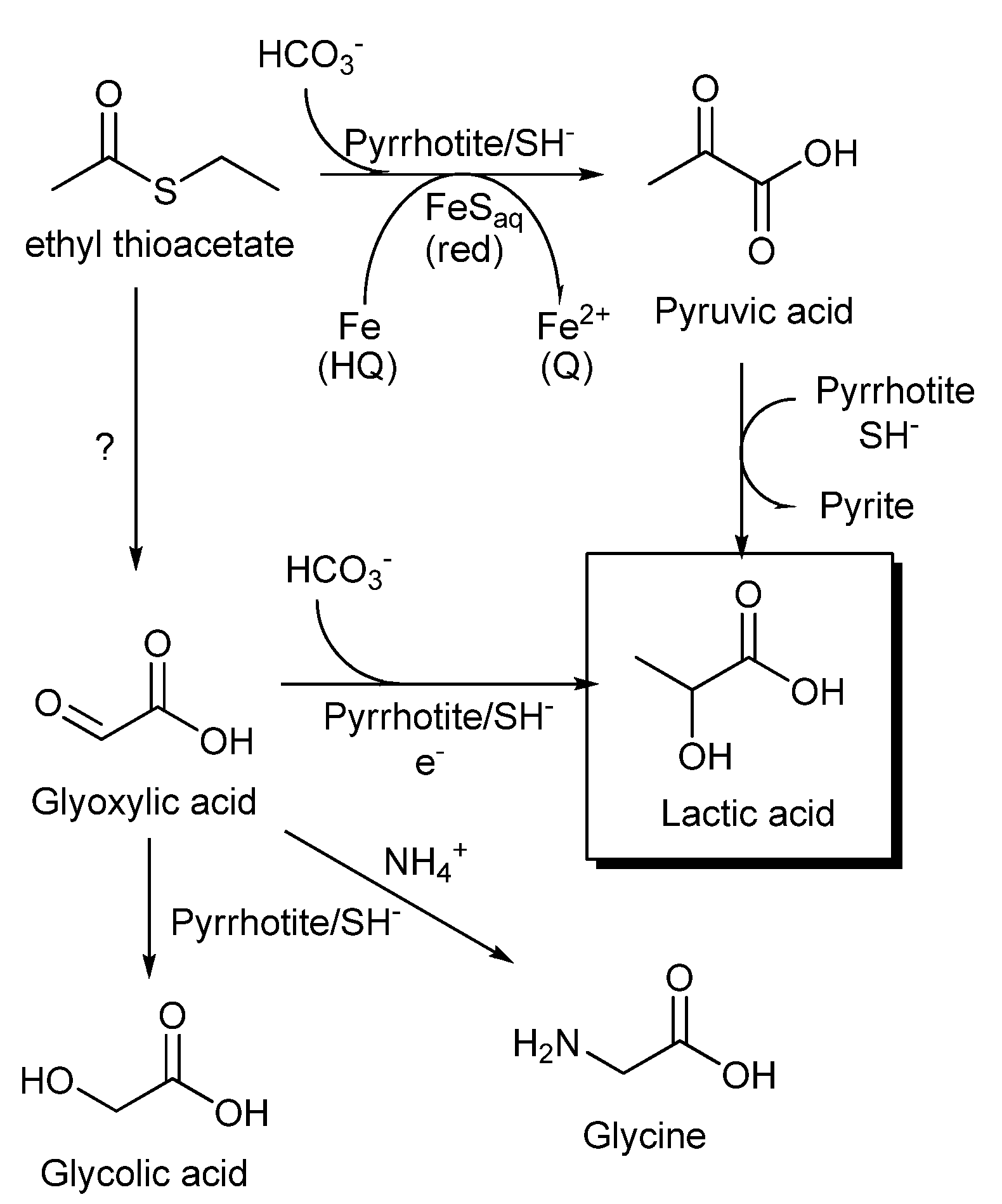
Figure 9.
Proposed routes to the synthesis of lactic acid by carboxylation of an alkyl thioacetate ester. The synthesis of pyruvic acid could be regarded as a prebiotic analog of reductive carboxylation of acetyl CoA promoted by PFOR, with iron-sulfur clusters of ferredoxin as electron donors.
Figure 9.
Proposed routes to the synthesis of lactic acid by carboxylation of an alkyl thioacetate ester. The synthesis of pyruvic acid could be regarded as a prebiotic analog of reductive carboxylation of acetyl CoA promoted by PFOR, with iron-sulfur clusters of ferredoxin as electron donors.
In summary (
Figure 9), the formation of lactic acid under our experimental conditions can be explained through pyruvate synthesis by the reductive carboxylation of thioacetate ester, followed by a reduction to lactic acid (see
Section 3.1). Additionally, the presence of glycolic acid and glycine could suggest alternative pathways, including the glyoxylate intermediary. Further experiments are necessary to explore the potential of electrochemical gradients in combination with sulfide minerals and soluble iron sulfide complexes in the origin of a biochemical system. The identification of unidentified amino- and thio- derivatives found in our experiments could help to explain the complex mechanism implicated in the biomimetic reductive carboxylation of a thioester. Aside from establishing the precise mechanism that this system uses for the formation of C
3 acids from C
2 precursors and carbon dioxide, our experiments show that iron sulfides in a non-equilibrium environment, characterized by the presence of electrochemical gradients and soluble and solid iron species in diverse oxidation states, could support biologically useful reactions. This supports the idea that transition metal sulfides play a role in the development of protometabolism.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}