Towards Greener Lixiviants in Value Recovery from Mine Wastes: Efficacy of Organic Acids for the Dissolution of Copper and Arsenic from Legacy Mine Tailings

Abstract
:1. Introduction
2. Methodology
2.1. Site Description
2.2. Mine Tailing Collection Procedure
2.3. Physical and Chemical Characterisation of the Mine Tailings
2.4. Hydrometallurgical Extraction
3. Results and Discussion
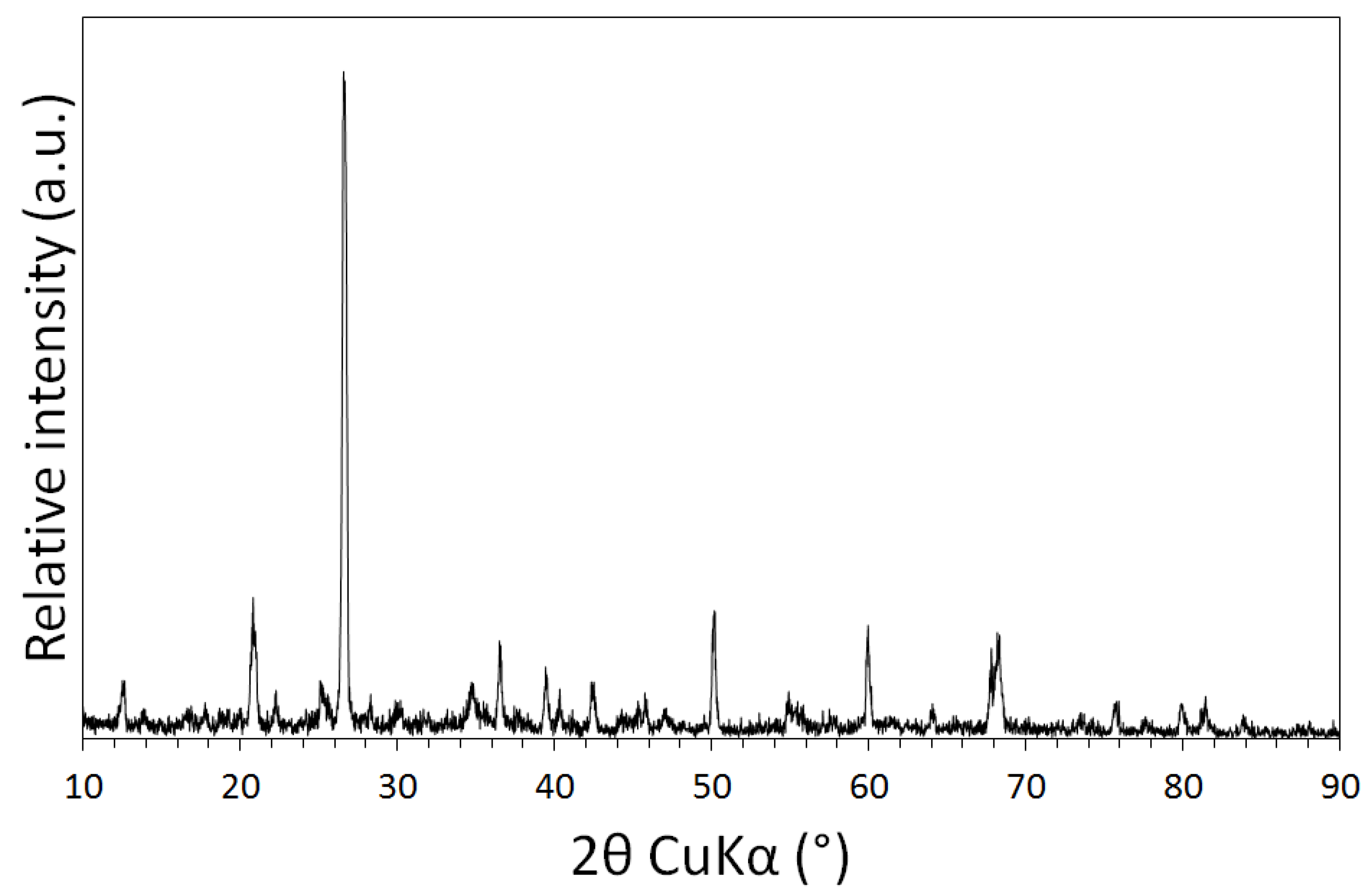
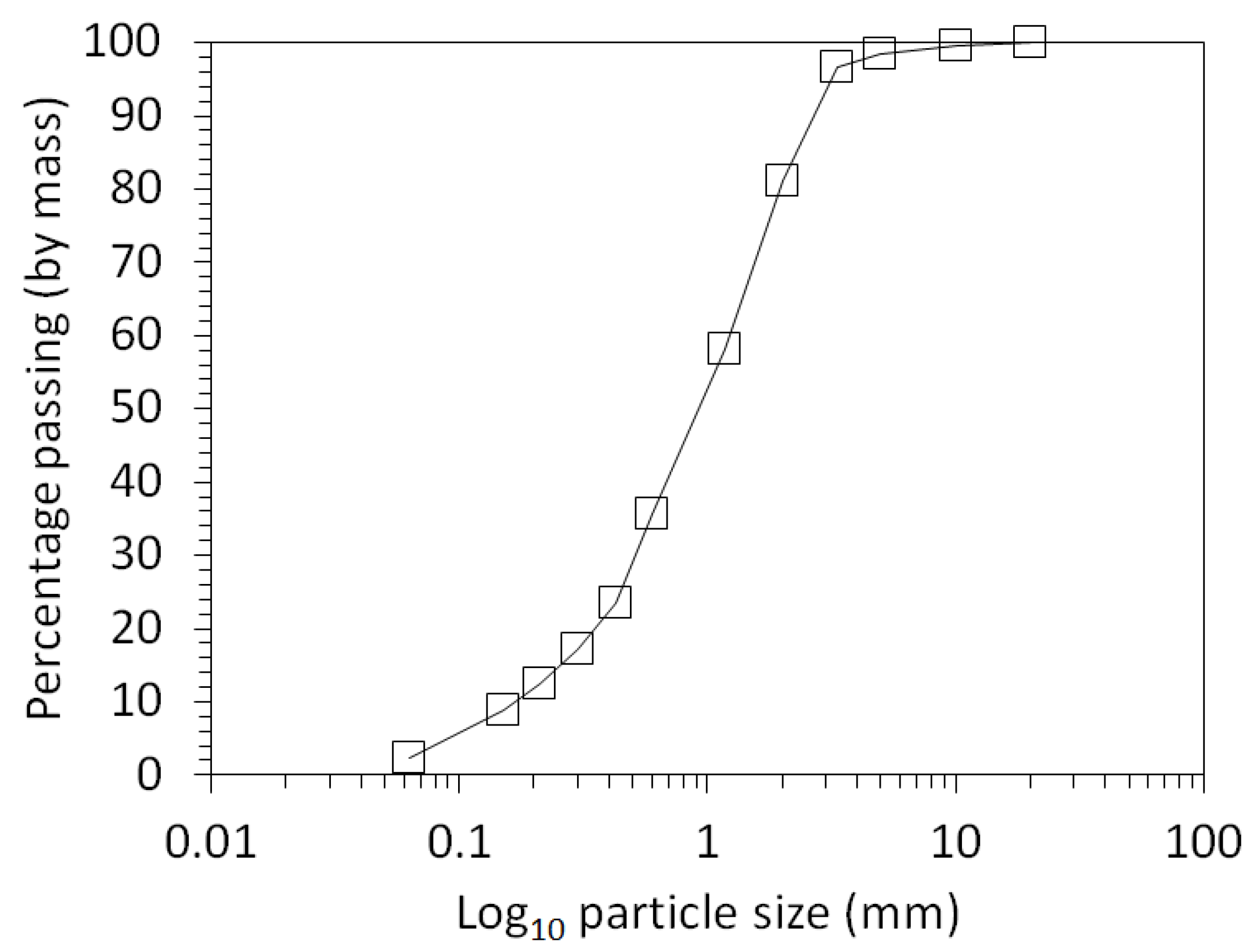
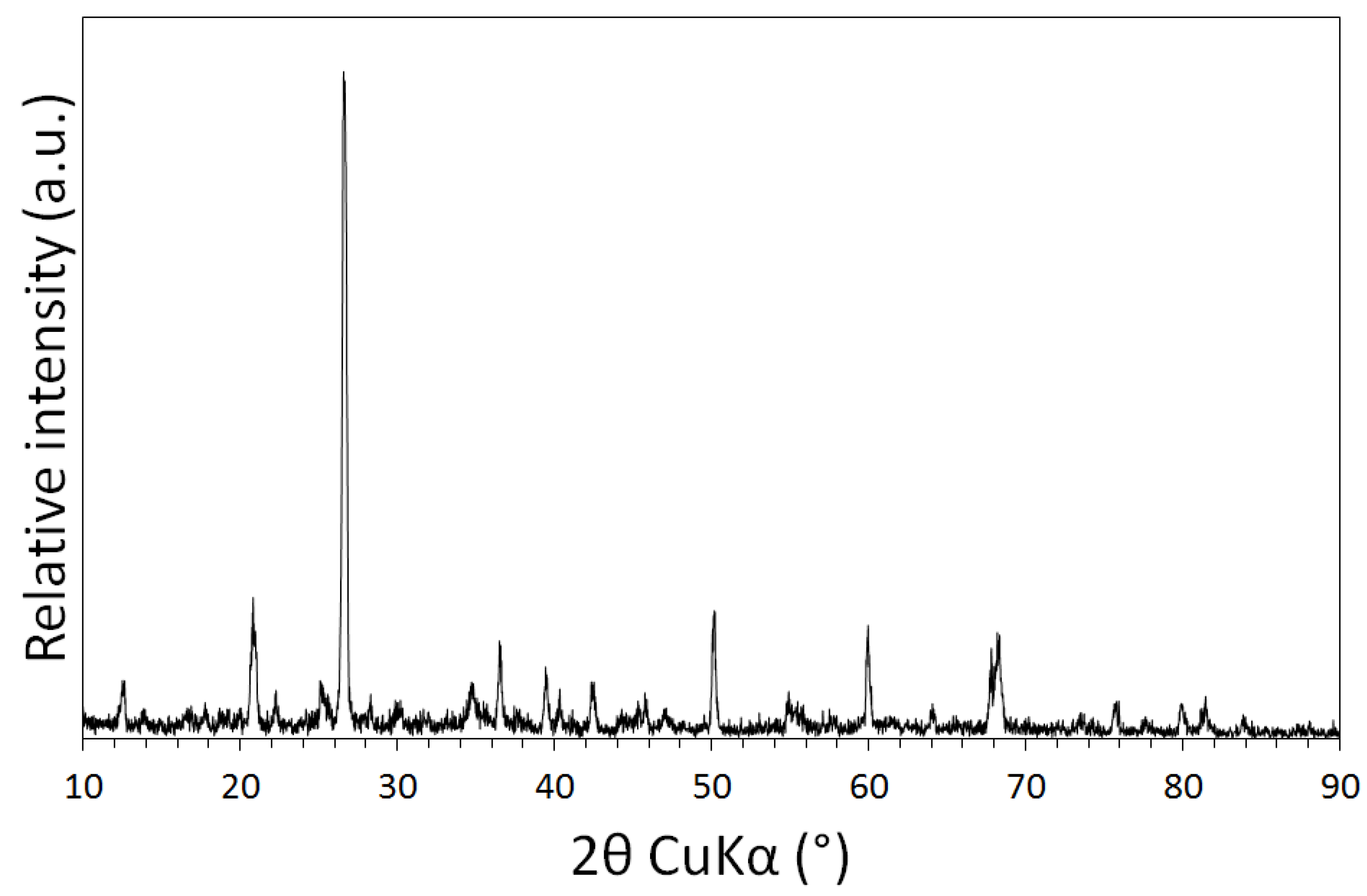
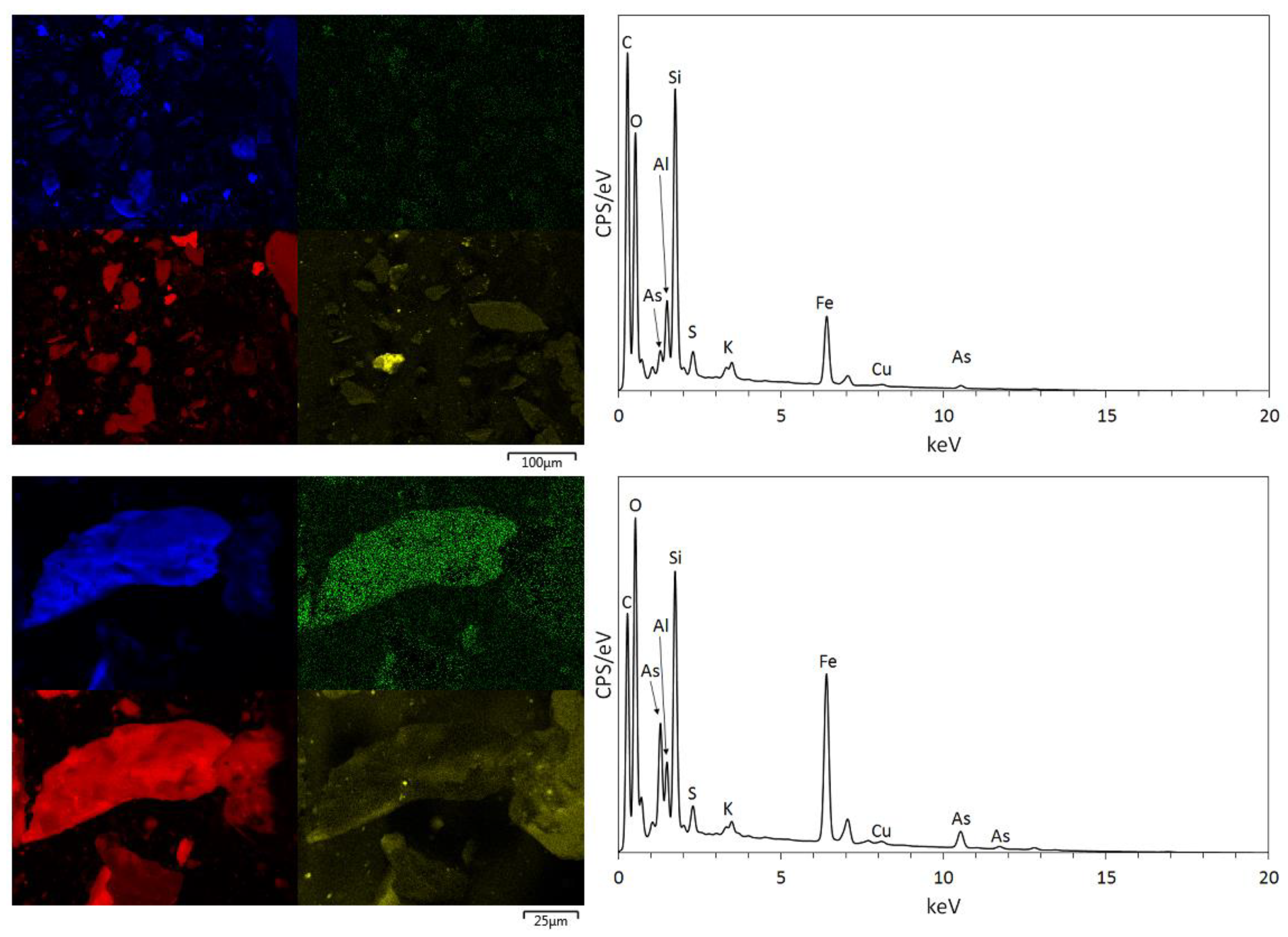
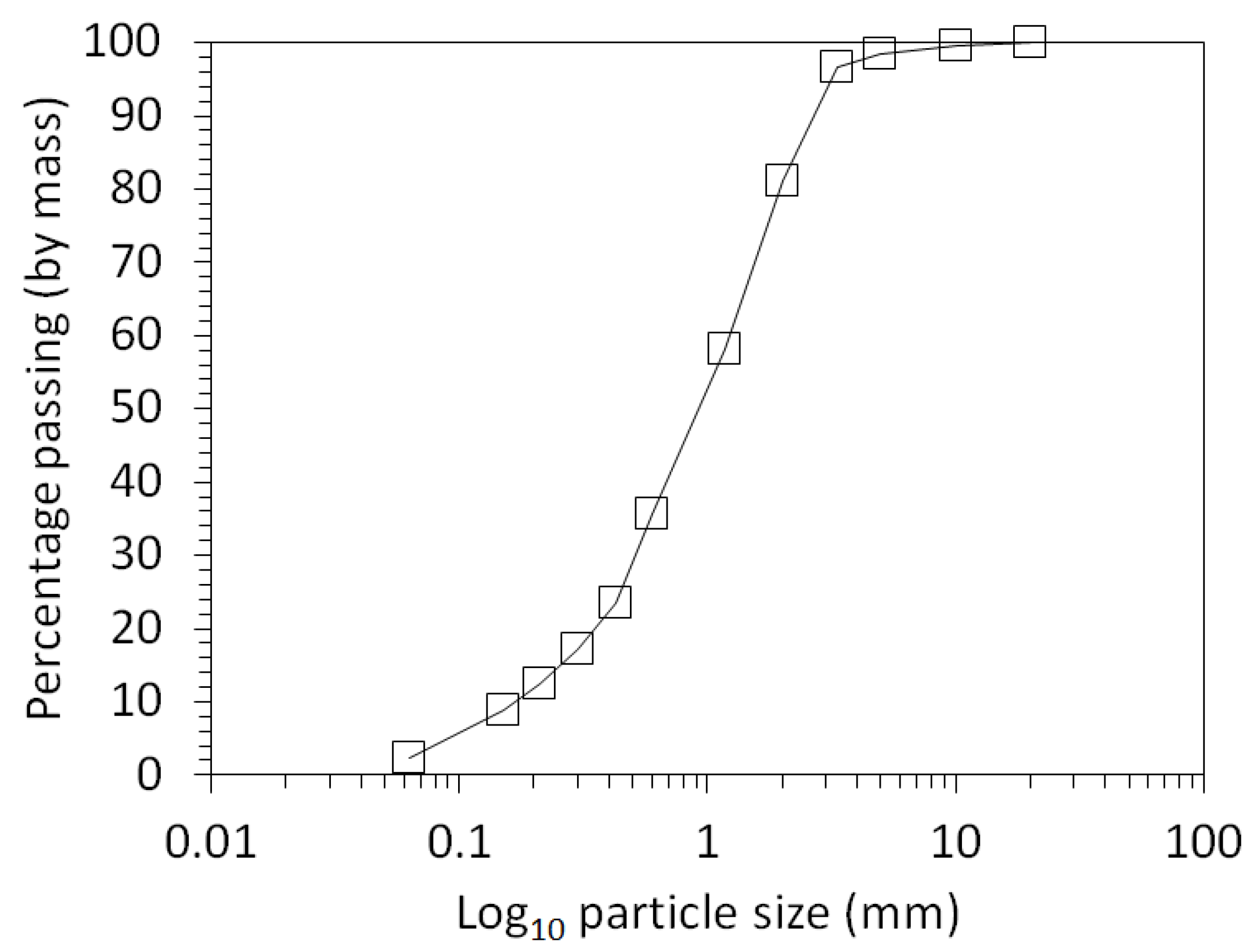
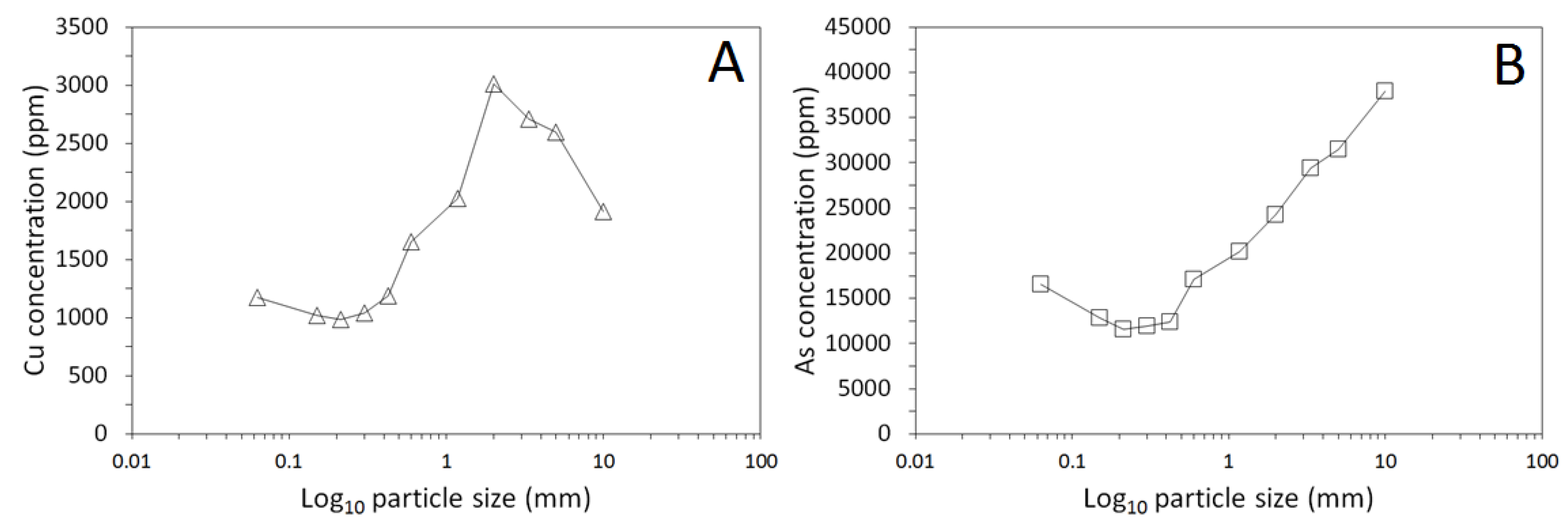
3.1. Physical and Chemical Characterisation of the Mine Tailings
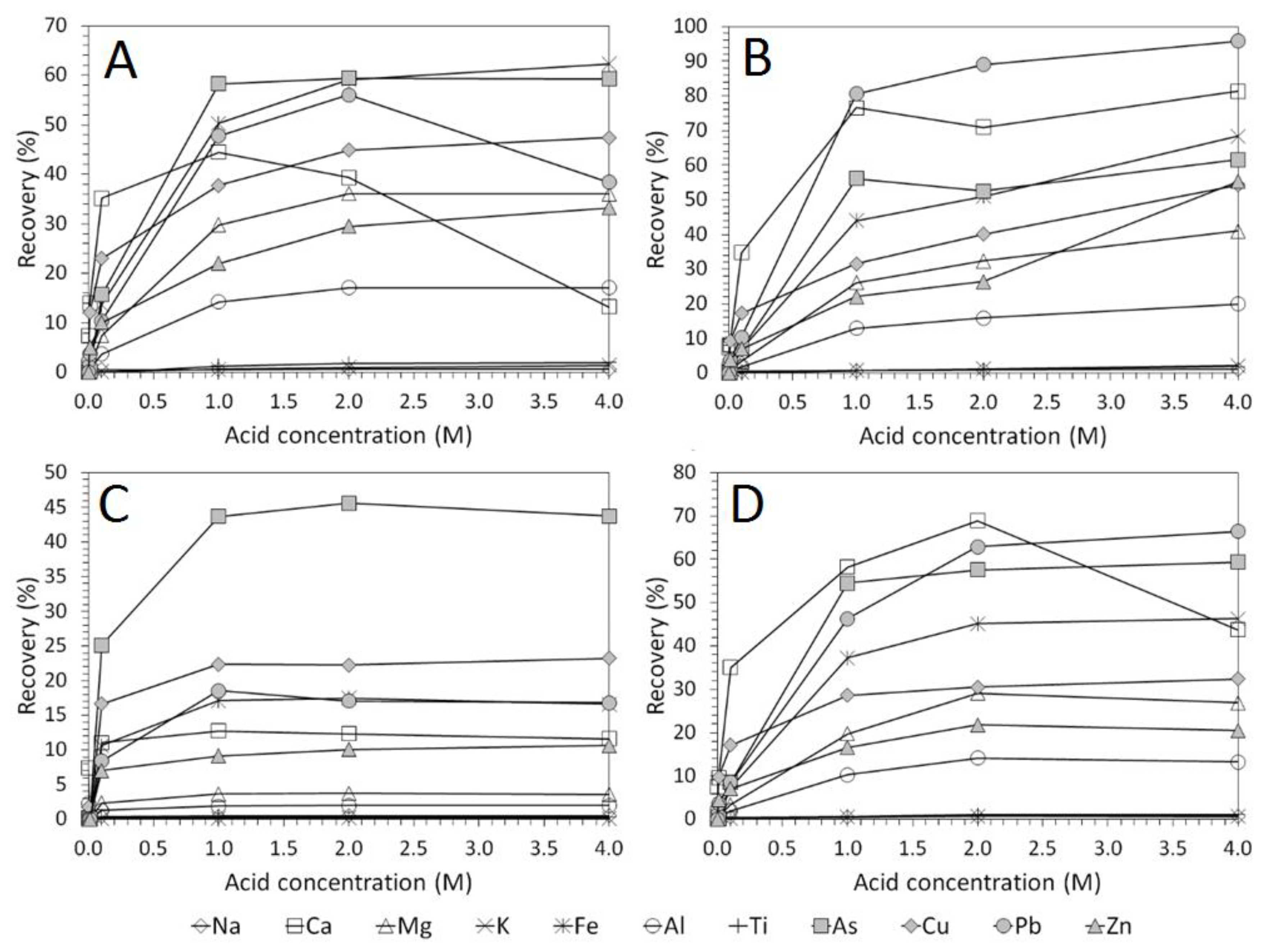
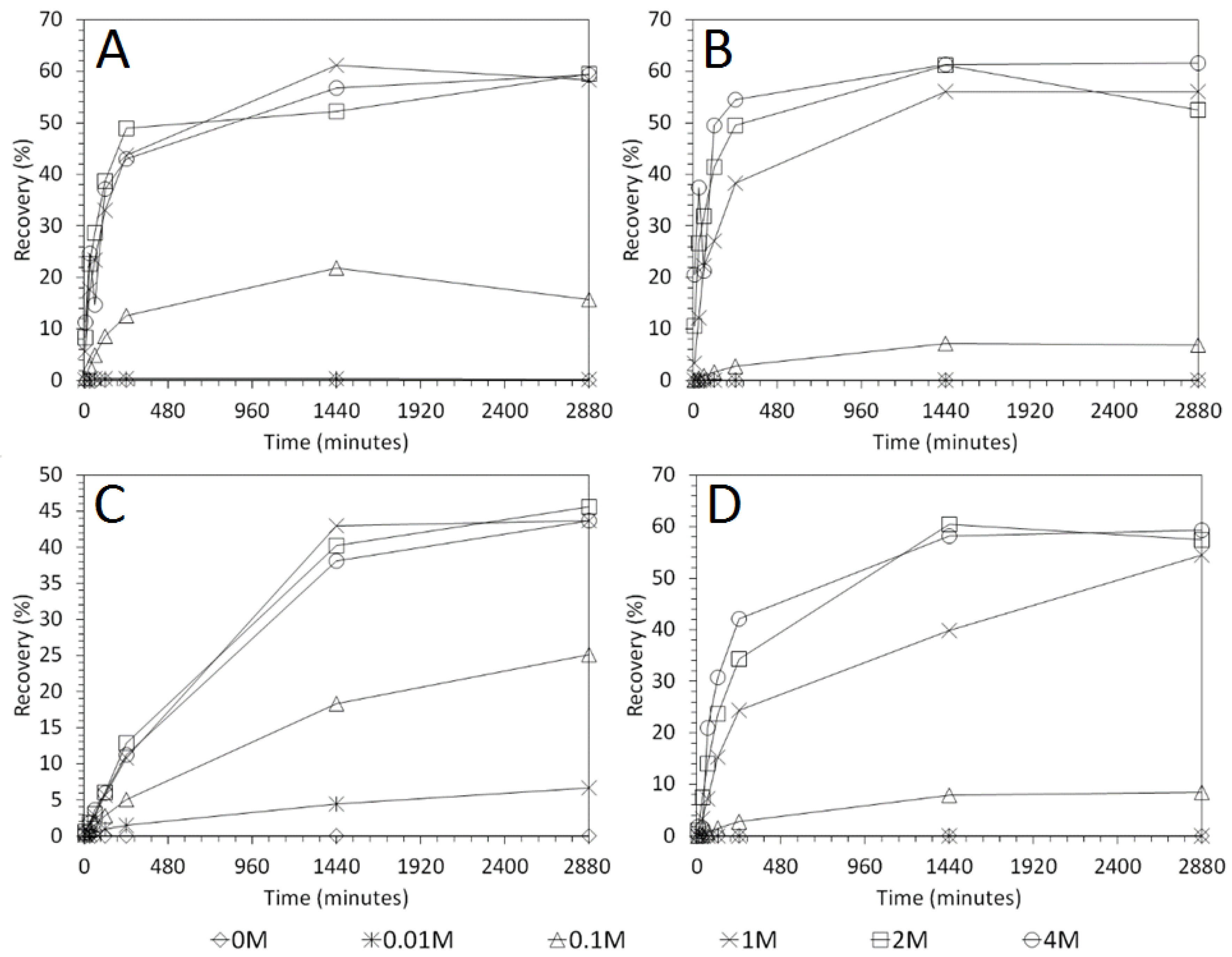
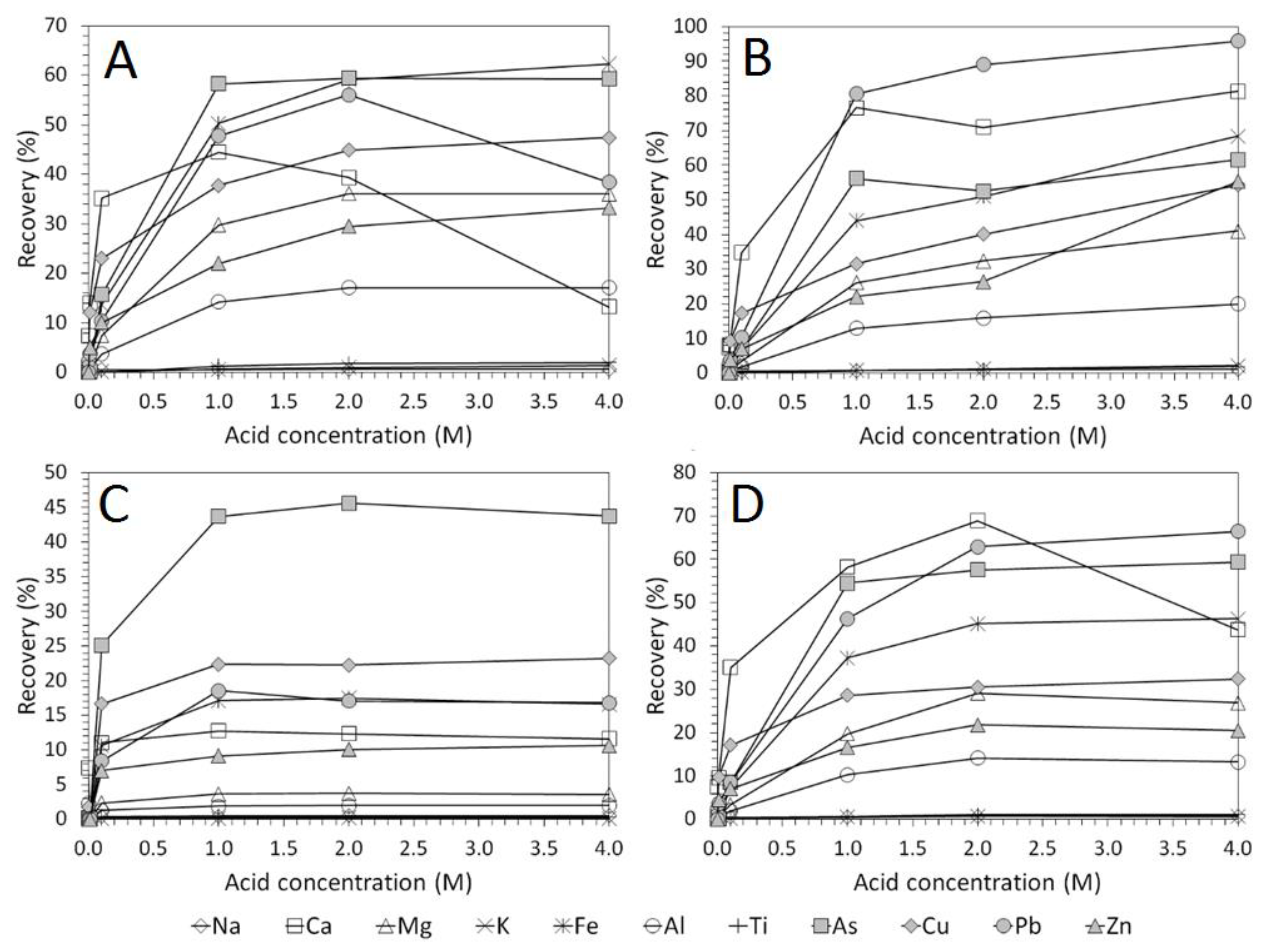
3.2. Influence of Acid Concentration on Cu and As Dissolution
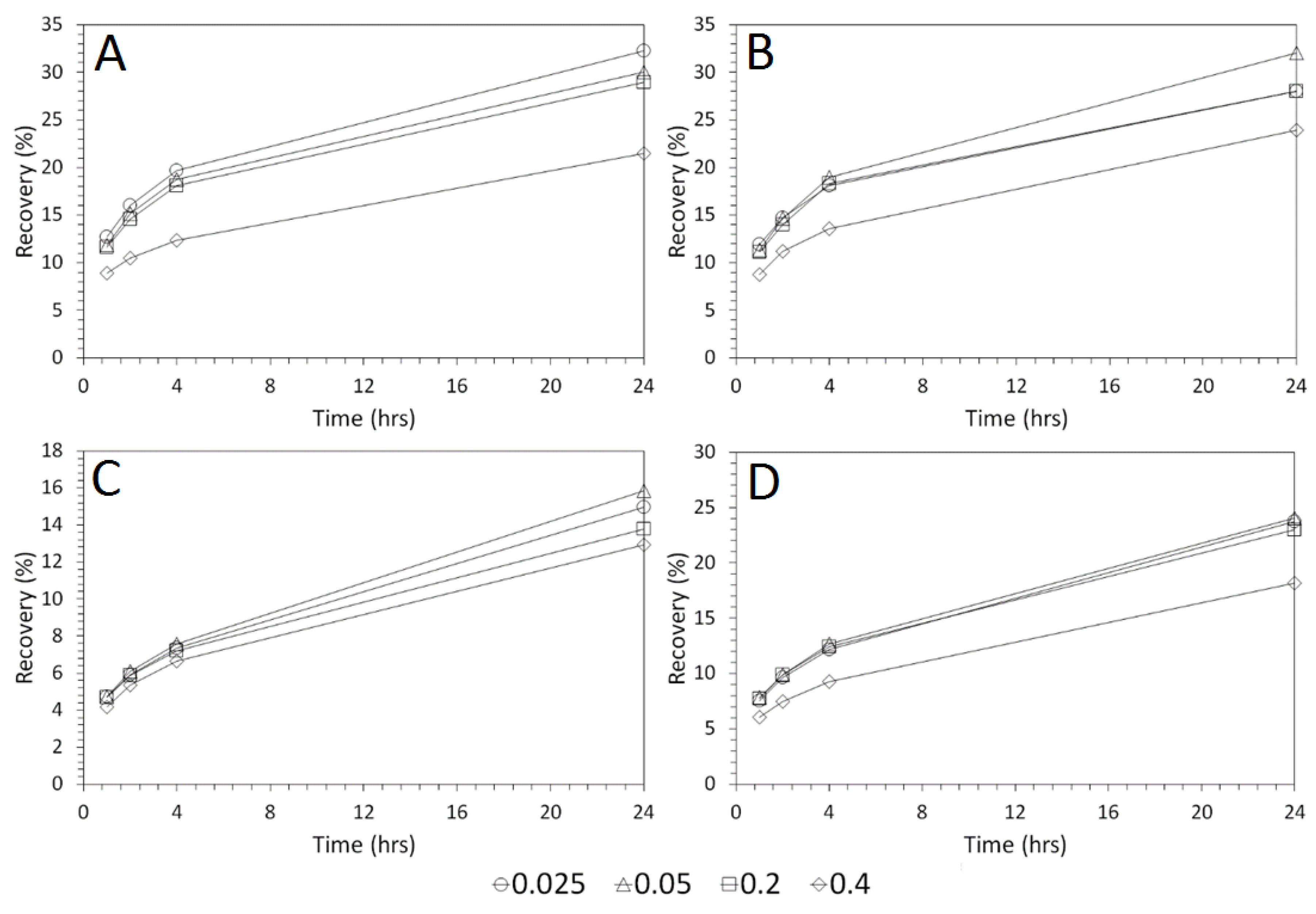
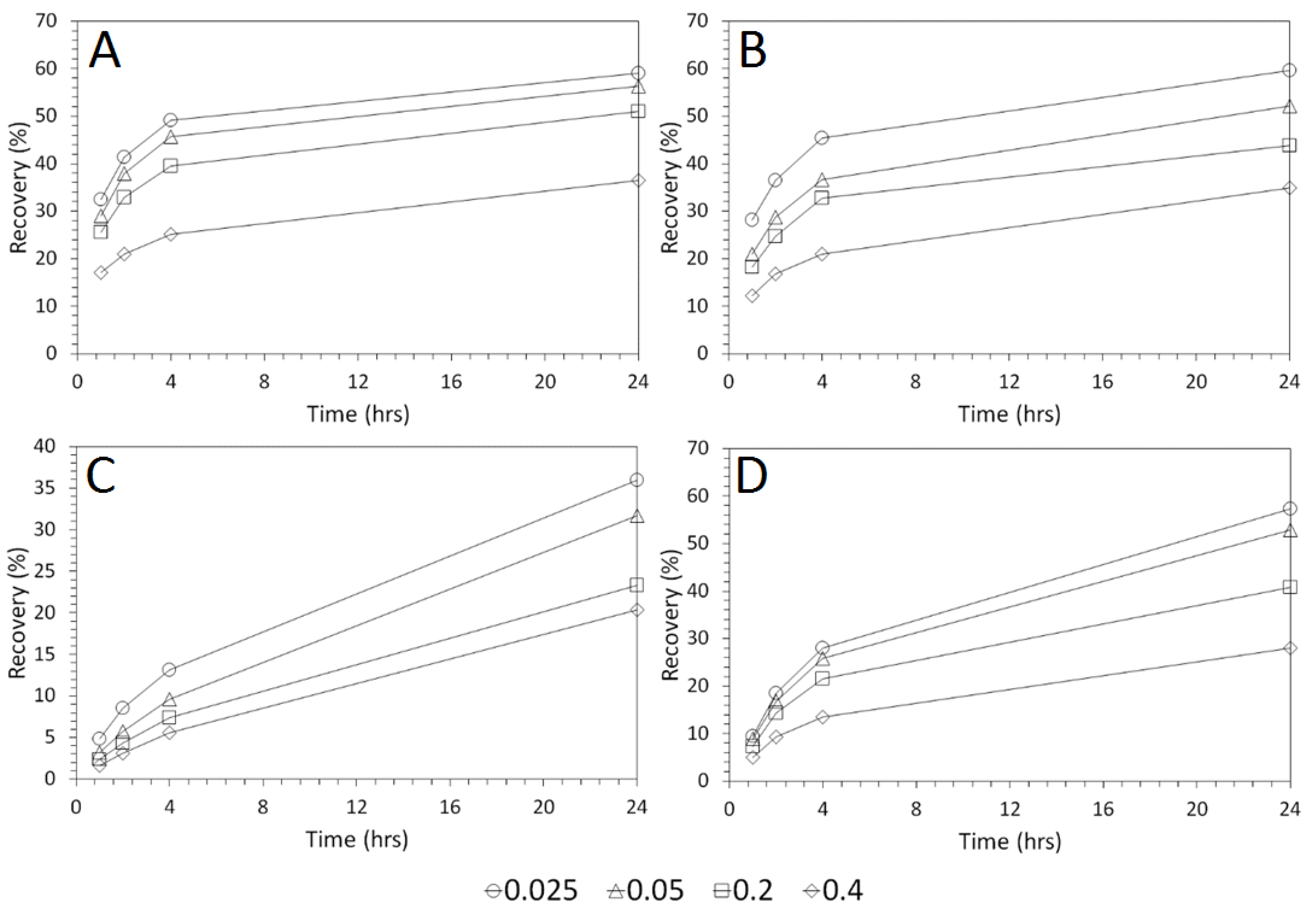
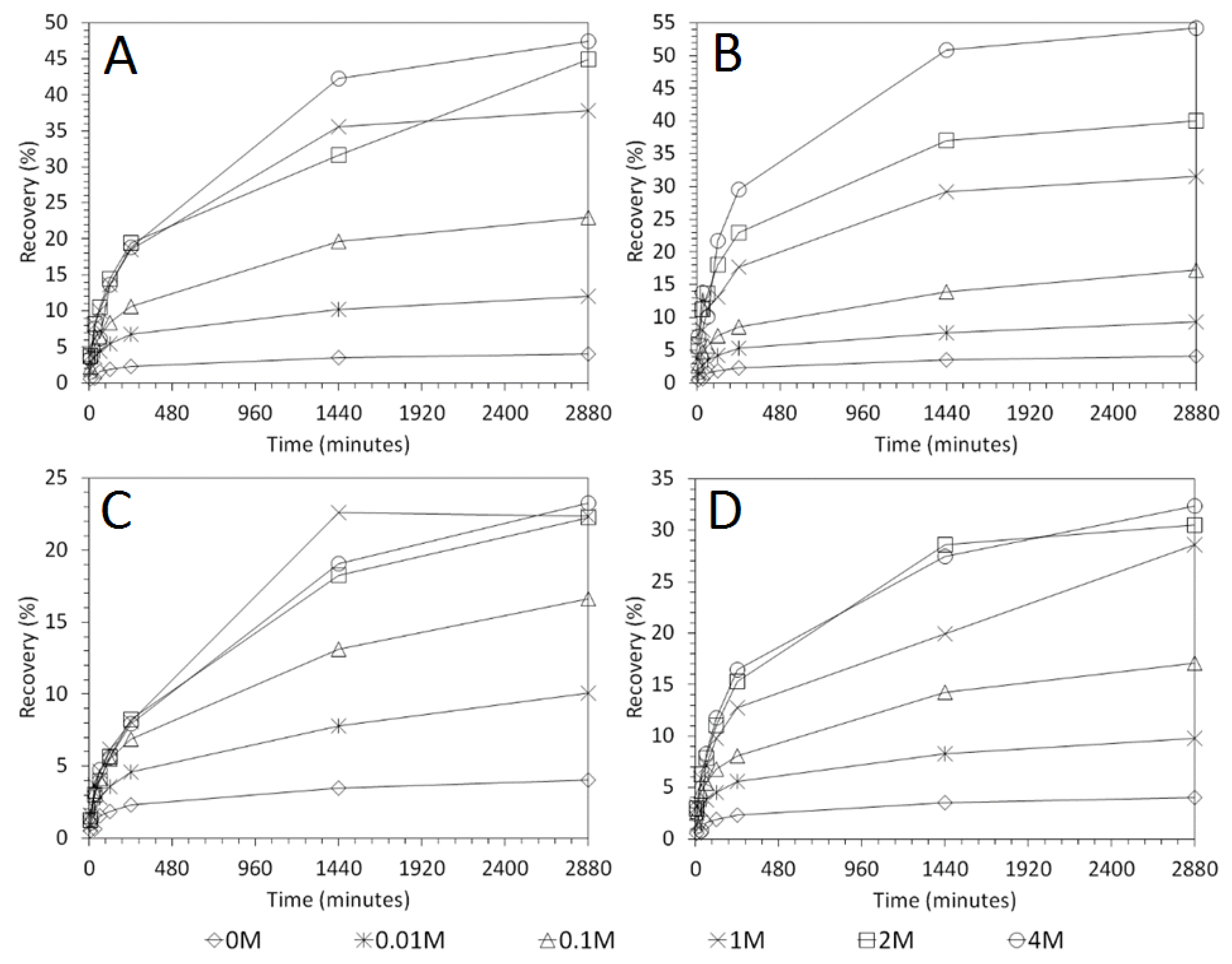
3.3. Influence of Solid–Liquid Ratio for Cu and As Dissolution
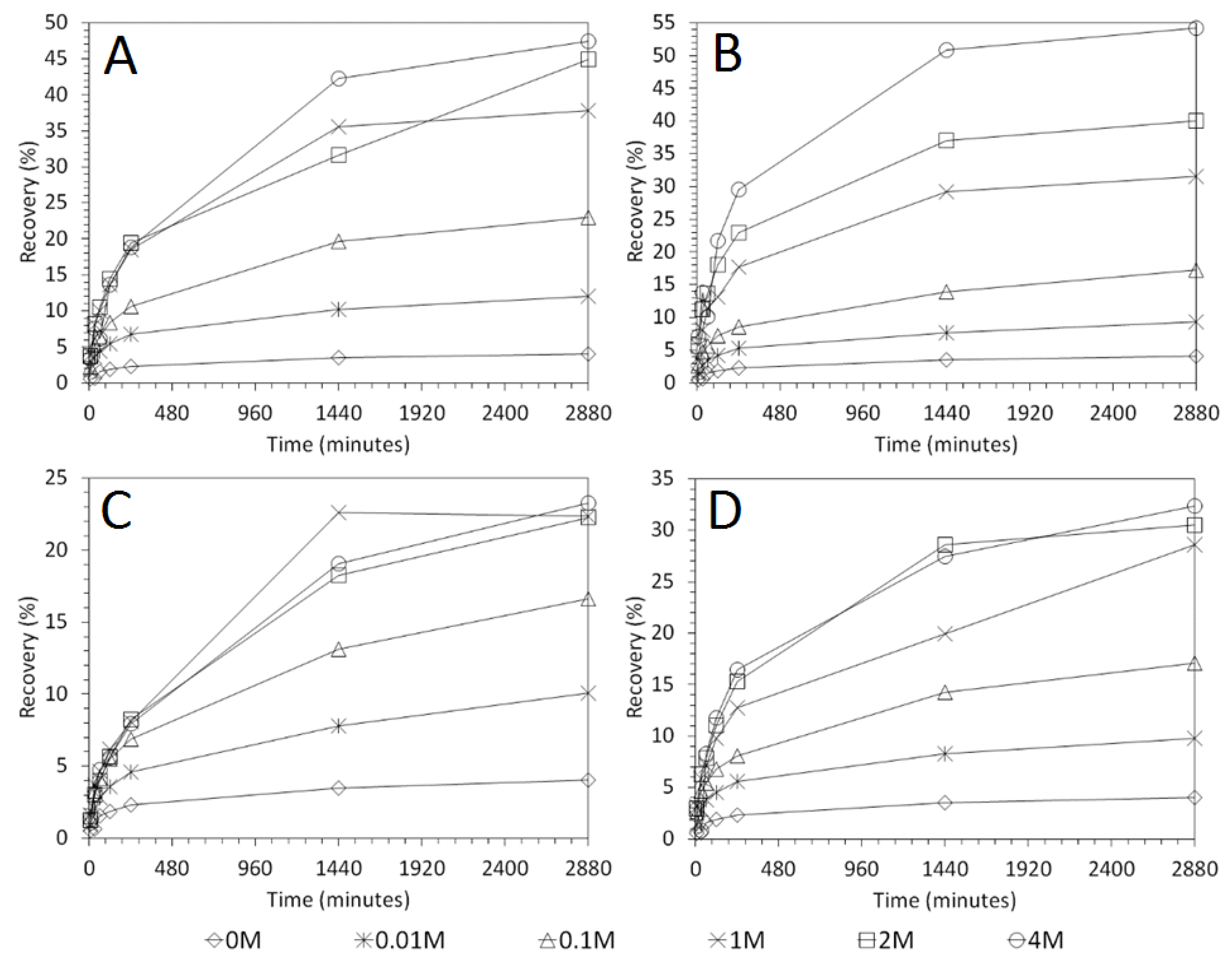
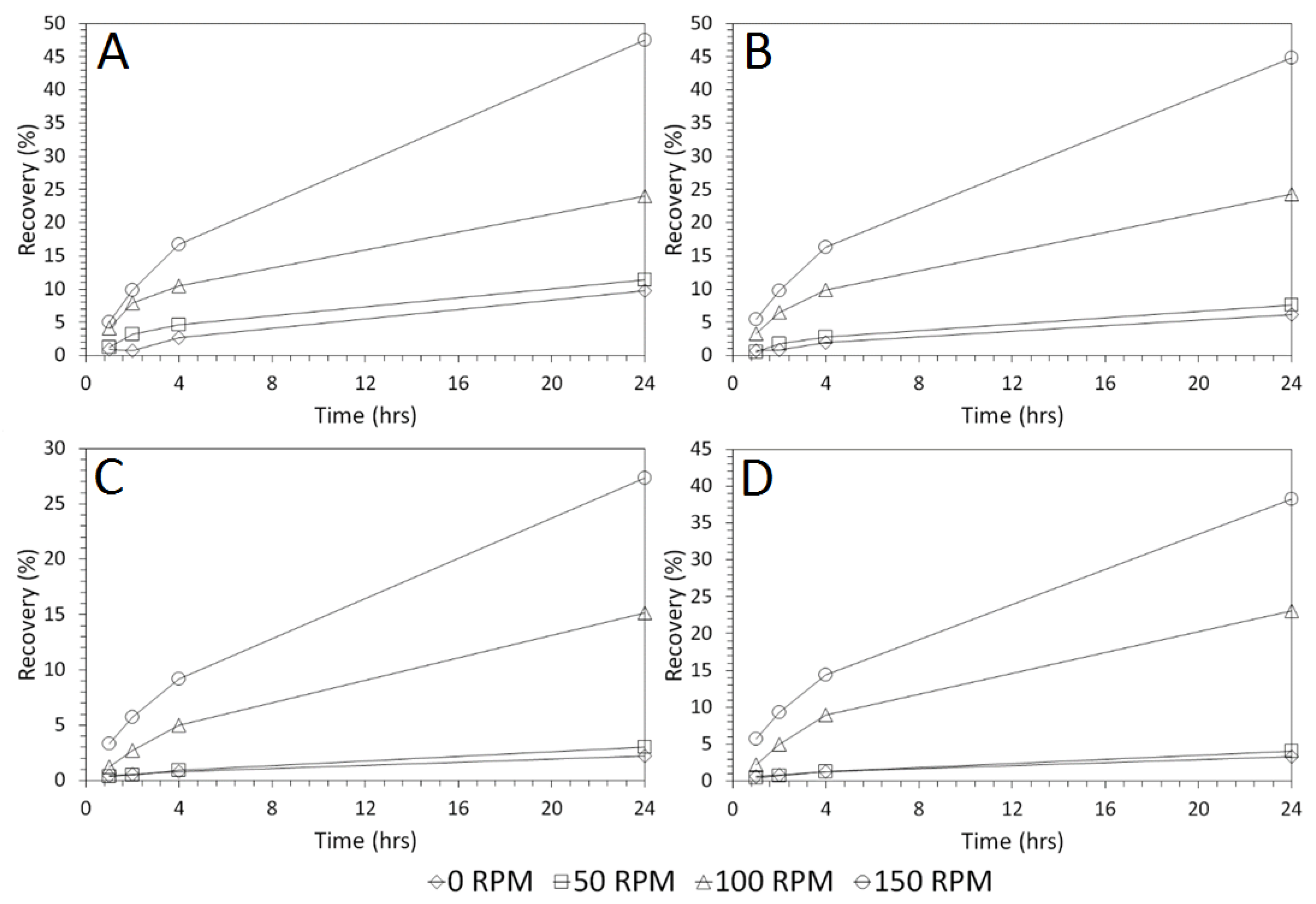
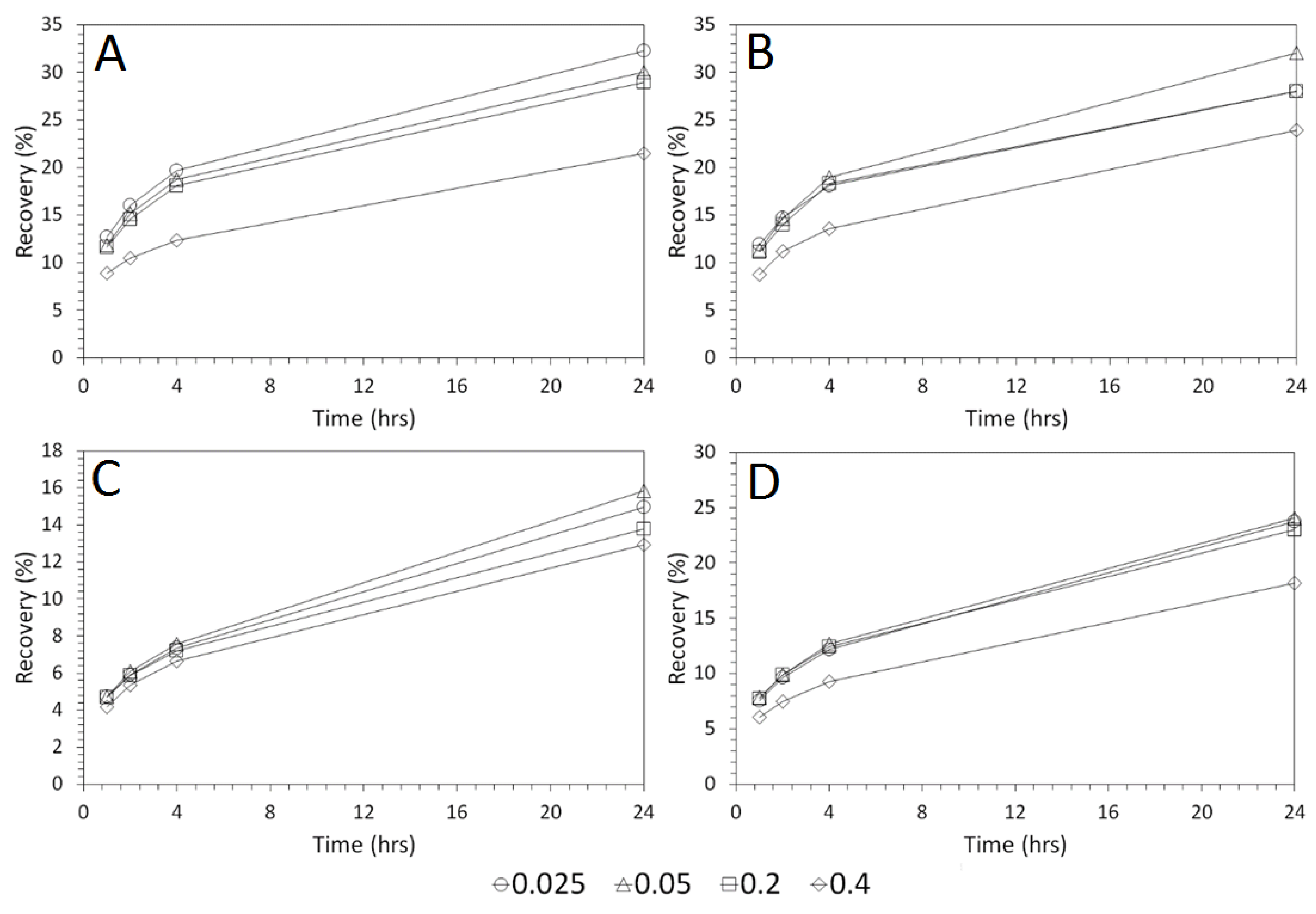
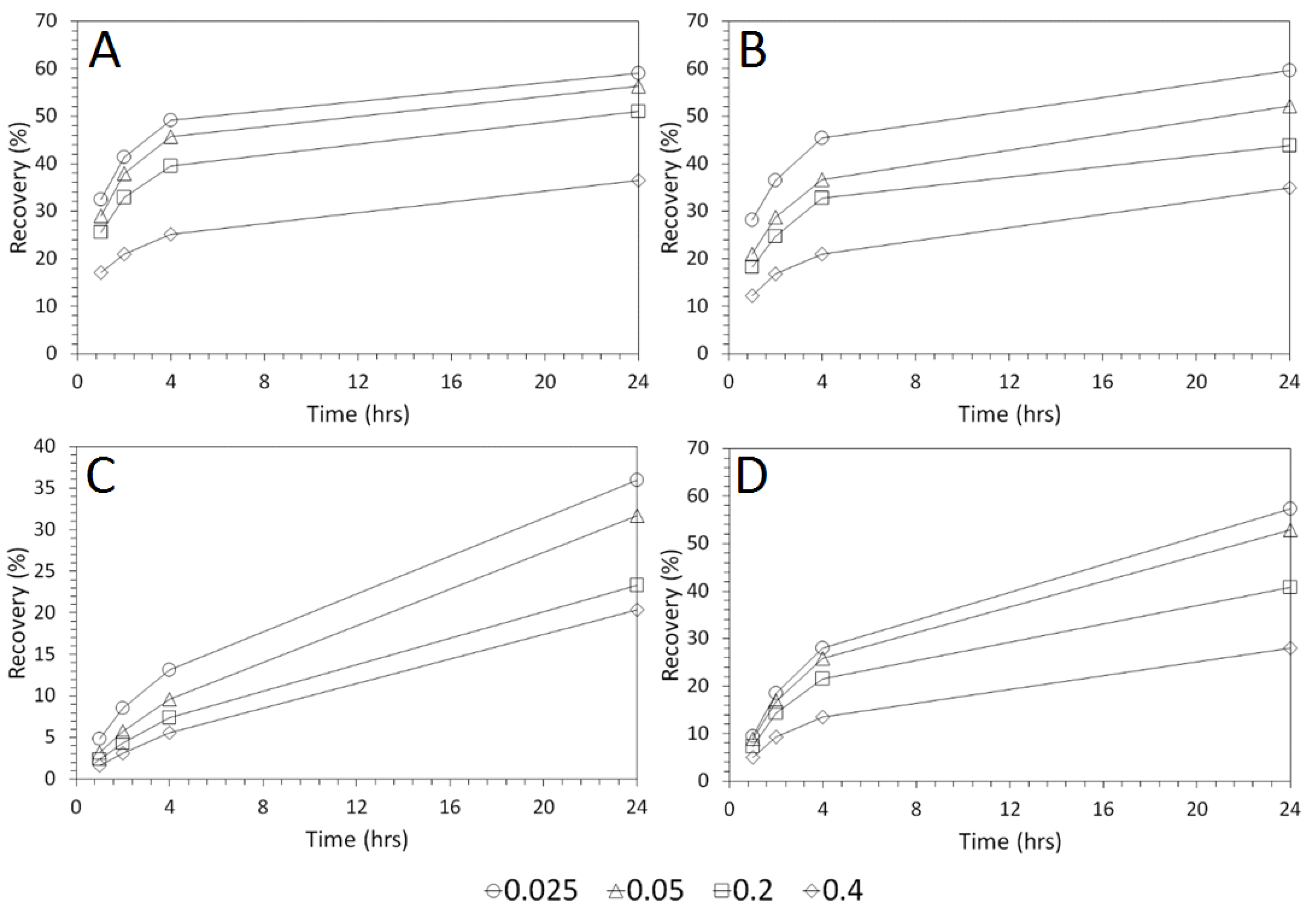
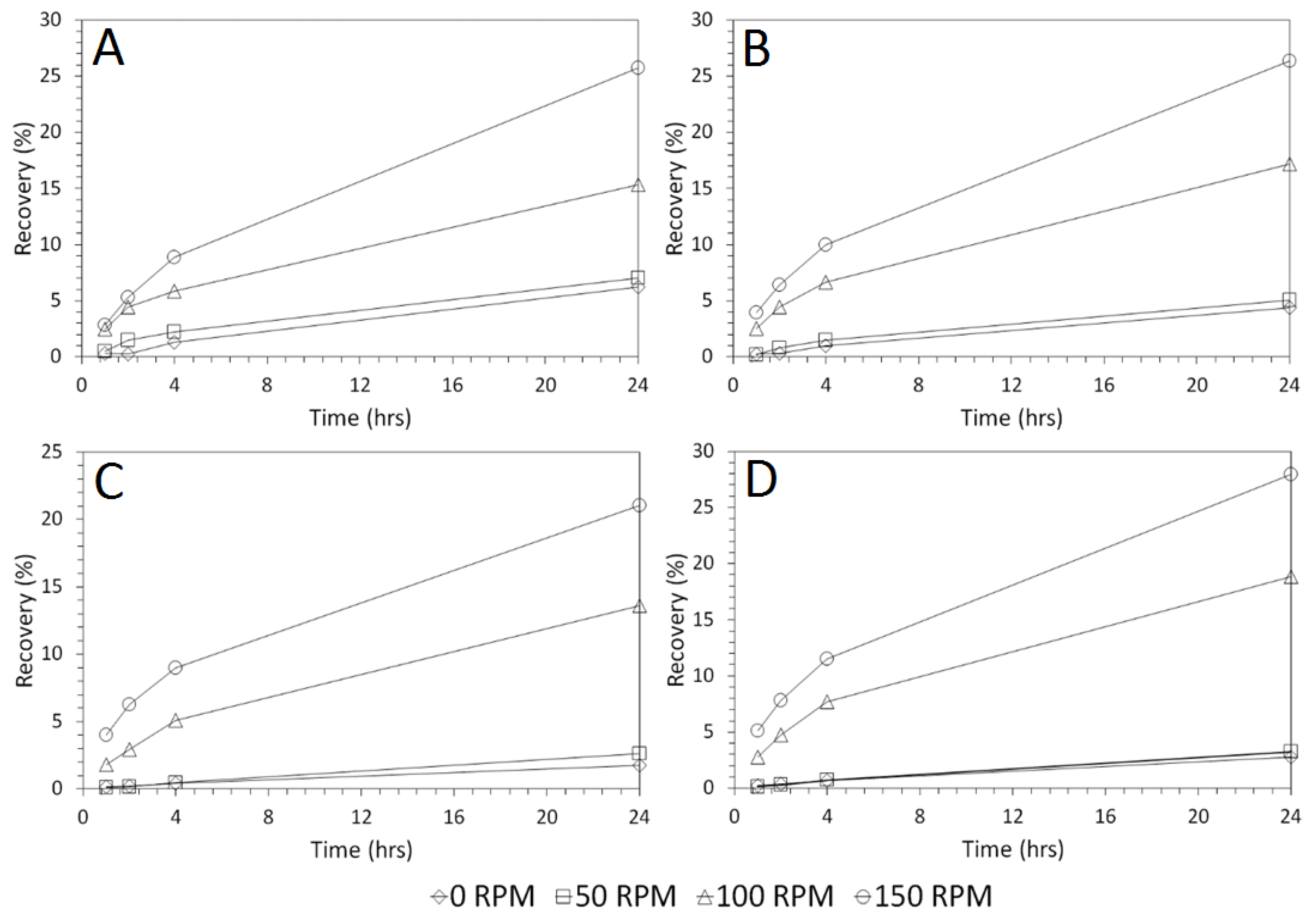
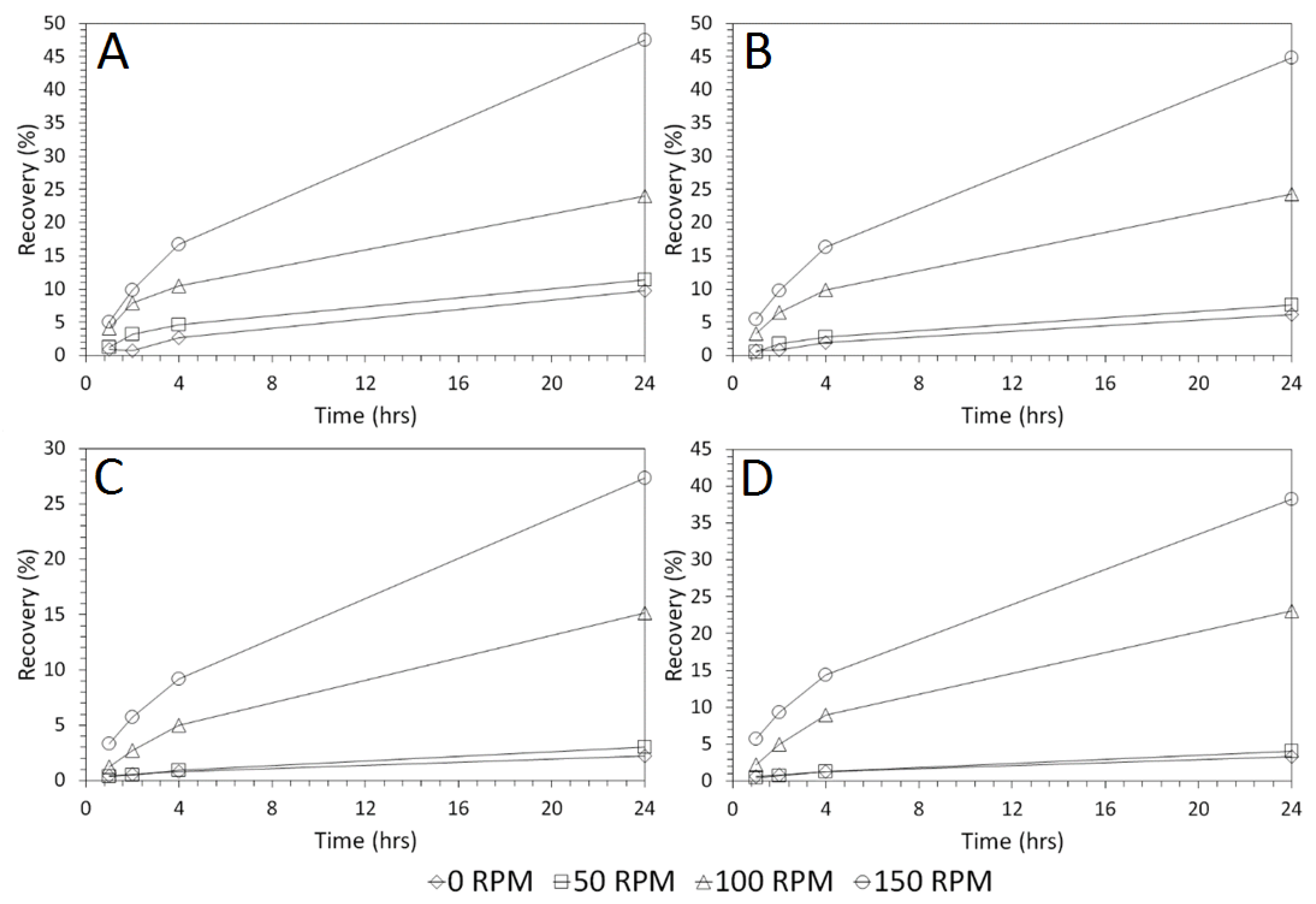
3.4. Influence of Mixing Speed on Cu and As Dissolution
4. Discussion
5. Conclusions
- (a)
- Cu and As dissolution rates were determined to typically increase with an increase in the acid concentration, mixing speed, and liquid to solid ratio.
- (b)
- HCl, H2SO4 and CH3SO3H generally exhibited relatively similar leaching abilities for As despite their different pKa values, with removal percentages after 48 h of 58%, 56%, and 55% recorded for 1 M H2SO4, HCl and CH3SO3H respectively, compared to 44% exhibited by C6H8O7.
- (c)
- H2SO4 was generally shown to be the most effective acid type for Cu removal with 38% removal for 1 M solutions after 48 h, compared to 32%, 29%, and 22% recorded for HCl, CH3SO3H, and C6H8O7 respectively.
- (d)
- Overall the optimum leaching conditions was found to be 1 M acid concentration, 200 RPM mixing speed and a mixing time of 24 h, with only minor improvements in leaching efficacy recorded for concentrations greater than 1 M or time periods greater than 24 h.
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Tordoff, G.M.; Baker, A.J.M.; Willis, A.J. Current approaches to the revegetation and reclamation of metalliferous mine wastes. Chemosphere 2000, 41, 219–228. [Google Scholar] [CrossRef]
- Hudson-Edwards, K.; Jamieson, H.; Lottermoser, B. Mine Wastes: Past, Present and Future. Elements 2011, 7, 375–380. [Google Scholar] [CrossRef]
- Plumlee, G.; Morman, S. Mine Wastes and Human Health. Elements 2011, 7, 399–404. [Google Scholar] [CrossRef]
- Environment Agency (EA). Abandoned Mines and the Water Environment; Science Project SC030136-41; Environment Agency: Bristol, UK, 2008; ISBN 978-1-84432-894-9.
- Crane, R.A.; Sapsford, D.J. Selective formation of copper nanoparticles from acid mine drainage using nanoscale zerovalent iron particles. J. Hazard. Mater. 2018, 347, 252–265. [Google Scholar] [CrossRef] [PubMed]
- Mayes, W.M.; Johnston, D.; Potter, H.A.; Jarvis, A.P. A national strategy for identification, prioritisation and management of pollution from abandoned non-coal mine sites in England and Wales. I. Methodology development and initial results. Sci. Total Environ. 2009, 407, 5435–5447. [Google Scholar] [CrossRef] [PubMed]
- Crane, R.A.; Sapsford, D.J. Towards “Precision Mining” of wastewater: Selective recovery of Cu from acid mine drainage onto diatomite supported nanoscale zerovalent iron particles. Chemosphere 2018, 202, 339–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crane, R.A.; Sinnett, D.E.; Cleall, P.J.; Sapsford, D.J. Physicochemical composition of wastes and co-located environmental designations at legacy mine sites in the south west of England and Wales: Implications for their resource potential. Res. Conserv. Recycl. 2017, 123, 117–134. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.C.; Srivastava, R.R. Leaching of gold from the spent/end-of-life mobile phone-PCBs using “greener reagents”. In The Recovery of Gold from Secondary Sources; Imperial College Press: London, UK, 2016; pp. 7–56. [Google Scholar]
- Jadhao, P.; Chauhan, G.; Pant, K.K.; Nigam, K.D.P. Greener approach for the extraction of copper metal from electronic waste. Waste Manag. 2016, 57, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ge, J.; Chen, R.; Wu, F.; Chen, S.; Zhang, X. Environmental friendly leaching reagent for cobalt and lithium recovery from spent lithium-ion batteries. Waste Manag. 2010, 30, 2615–2621. [Google Scholar] [CrossRef] [PubMed]
- Wasay, S.A.; Barrington, S.; Tokunaga, S. Organic acids for the in situ remediation of soils polluted by heavy metals: Soil flushing in columns. Water Air Soil Pollut. 2001, 127, 301–314. [Google Scholar] [CrossRef]
- Francis, A.J.; Dodge, C.J.; Gillow, J.B. Biodegradation of metal citrate complexes and implications for toxic-metal mobility. Nature 1992, 356, 140–142. [Google Scholar] [CrossRef]
- Renella, G.; Landi, L.; Nannipieri, P. Degradation of low molecular weight organic acids complexed with heavy metals in soil. Geoderma 2004, 122, 311–315. [Google Scholar] [CrossRef]
- Huang, F.Y.; Brady, P.V.; Lindgren, E.R.; Guerra, P. Biodegradation of Uranium−Citrate Complexes: Implications for Extraction of Uranium from Soils. Environ. Sci. Technol. 1998, 32, 379–382. [Google Scholar] [CrossRef]
- Qian, J.; Zhu, X.; Tao, Y.; Zhou, Y.; He, X.; Li, D. Promotion of Ni2+ Removal by masking toxicity to sulfate-reducing bacteria: Addition of citrate. Int. J. Mol. Sci. 2015, 16, 7932–7943. [Google Scholar] [CrossRef] [PubMed]
- Gámez, V.M.; Sierra-Alvarez, R.; Waltz, R.J.; Field, J.A. Anaerobic degradation of citrate under sulfate reducing and methanogenic conditions. Biodegradation 2009, 20, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, B.P. Feasibility of Permeation Grouting for Creating Subsurface Barriers; SAND94-0786.UC-721; Sandia National Laboratories: Albuquerque, NM, USA, 1994. [Google Scholar]
- Ghorbani, Y.; Franzidis, J.P.; Petersen, J. Heap leaching technology—Current state, innovations, and future directions: A. review. Miner. Process. Extr. Metall. Rev. 2016, 37, 73–119. [Google Scholar] [CrossRef] [Green Version]
- Michael, D.G.; Min, W.; Thomas, B.; Patrick, J. Environmental benefits of methanesulfonic acid: Comparative properties and advantages. Green Chem. 1999, 1, 127–140. [Google Scholar]
- Borra, C.R.; Pontikes, Y.; Binnemans, K.; Van Gerven, T. Leaching of rare earths from bauxite residue (red mud). Miner. Eng. 2015, 76, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Burckhard, S.R.; Schwab, A.P.; Banks, M.K. The effects of organic acids on the leaching of heavy metals from mine tailings. J. Hazard. Mater. 1995, 41, 135–145. [Google Scholar] [CrossRef]
- Yuan, S.; Xi, Z.; Jiang, Y.; Wan, J.; Wu, C.; Zheng, Z.; Lu, X. Desorption of copper and cadmium from soils enhanced by organic acids. Chemosphere 2007, 68, 1289–1297. [Google Scholar] [CrossRef] [PubMed]
- Tzeferis, P.G.; Agatzini-Leonardou, S. Leaching of nickel and iron from Greek non-sulphide nickeliferrous ores by organic acid. Hydrometallurgy 1994, 36, 345–360. [Google Scholar] [CrossRef]
- Astuti, W.; Hirajima, T.; Sasaki, K.; Okibe, N. Comparison of effectiveness of citric acid and other acids in leaching of low-grade Indonesian saprolitic ores. Miner. Eng. 2016, 85, 1–16. [Google Scholar] [CrossRef]
- Wanta, K.C.; Perdana, I.; Petrus, H.T.B.M. Evaluation of shrinking core model in leaching process of Pomalaa nickel laterite using citric acid as leachant at atmospheric conditions. In IOP Conference Series: Materials Science and Engineering; IOP Publishing: Bristol, UK, 2016; Volume 162, p. 012018. [Google Scholar]
- Environment Agency (EA). Dissolved Metal Contamination from Mine Wastes–Risk Assessment and Quantification in the Tamar Catchment. Project: SC060095; 2012. Available online: https://www.gov.uk/government/uploads/system/uploads/attachment_data/file/290507/LIT_7320_4babcfpdf (accessed on 8 June 2016).
- Dines, H.G. The Metalliferous Mining Region of South-West England; HMSO Publications: London, UK, 1956; Volume 1, p. 410. [Google Scholar]
- World Heritage Cornwall. Available online: http://www.worldheritagecornwall.com/mines/devon-great-consols.htm (accessed on 21 July 2016).
- ASTM D6009-12. Standard Guide for Sampling Waste Piles; ASTM International: West Conshohocken, PA, USA, 2012; Available online: www.astm.org (accessed on 3 September 2018).
- ISO 11277: Soil Quality—Determination of Particle Size Distribution in Mineral Soil Material—Method by Sieving and Sedimentation; International Organization for Standardization: Geneva, Switzerland, 2009; ISBN 978 0 580 67636 9.
- BS 812: Testing Aggregates of Density, Part 2. Methods of Determination; British Standards Institution: London, UK, 1995; ISBN 0 580 24257 9.
- ASTM D4972-13. Standard Test Method for pH of Soils; ASTM International: West Conshohocken, PA, USA, 2013; Available online: www.astm.org (accessed on 3 September 2018).
- EPA. EPA Method 3052-1. Microwave Assisted Acid Digestion of Siliceous and Organically Based Matrices. 1996. Available online: https://www.epa.gov/sites/production/files/2015-12/documents/3052.pdf (accessed on 8 June 2016).
- Klink, B.; Palumbo, B.; Cave, M.; Wragg, J. Arsenic Dispersal and Bioaccessibility in Mine Contaminated Soils: A Case Study from an Abandoned Arsenic Mine in Devon, UK; British Geological Survey Research Report RR/04/003; British Geological Survey, NERC: Keyworth, Nottingham, UK, 2005; 52p. [Google Scholar]
- Environment Agency. Mitigation of Pollution from Abandoned Metal Mines, Part 2: Review of Resource Recovery Options from the Passive Remediation of Metal-Rich Mine Waters; SC090024/R2; Environment Agency: Bristol, UK, 2012. Available online: https://www.gov.uk/government/uploads/system/uploadsattachment_data/file/291554/scho1111buvo-e-e.pdf (accessed on 8 June 2016).
- Vesborg, P.C.; Jaramillo, T.F. Addressing the terawatt challenge: Scalability in the supply of chemical elements for renewable energy. RSC Adv. 2012, 2, 7933–7947. [Google Scholar] [CrossRef] [Green Version]
- Environment Agency. Guidance on the Use of Soil Screening Values in Ecological Risk Assessment; Science Report SC070009/SR2b; Environment Agency: Bristol, UK, 2008.
- Defra. Development of Category 4 Screening Levels for Assessment of Land Affected by Contamination—Policy Companion Document; Defra: London, UK, 2014.
- Environment Agency. Soil Guideline Values for Arsenic in Soil; Science Report SC050021/Arsenic SGV., s.l.; Environment Agency: Bristol, UK, 2009.
- Environment Agency. Soil Guideline Values for Cadmium in Soil; Science Report SC050021/Cadmium SGV., s.l.; Environment Agency: Bristol, UK, 2009.
- Environment Agency. Soil Guideline Values for Nickel in Soil; Science Report SC050021/Nickel SGV., s.l.; Environment Agency: Bristol, UK, 2009.
- Vracar, R.Z.; Natasa, V.; Kamberovic, Z. Leaching of copper (I) sulphide by sulfuric acid solution with addition of sodium nitrate. Hydrometallurgy 2003, 70, 143–151. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Volume (m3) | Bulk Density (g/cm3) | Mass (Tonnes) [6] | Paste pH | TOC (wt %) | TIC (wt %) |
|---|---|---|---|---|---|
| 198.923 [6] | 1.30 | 258,600 | 3.33 | 0.16 | 0.00 |
| Li | Na | Mg | Al | K | Ca | Ti | Cr | Mn |
| 0.0135 | 0.4312 | 0.5295 | 4.6035 | 0.8871 | 1.1426 | 0.2207 | 0.0315 ‡† | 0.0610 |
| Fe | Ni1 | Cu | Zn | As | Ag | Cd | Sn | Pb |
| 9.9893 | 0.0019 | 0.1833 † | 0.0101 † | 1.9176 ‡ | <DL | 0.0012 † | 0.0290 | 0.0067 |
| Element | Value Per Ton (£) * | Estimated Value in Tailings Pile † |
|---|---|---|
| Cu | 8.50 | 1,511,000 |
| Zn | 0.20 | 36,000 |
| Sn | 4.26 | 757,000 |
| Pb | 0.11 | 19,000 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crane, R.A.; Sapsford, D.J. Towards Greener Lixiviants in Value Recovery from Mine Wastes: Efficacy of Organic Acids for the Dissolution of Copper and Arsenic from Legacy Mine Tailings. Minerals 2018, 8, 383. https://doi.org/10.3390/min8090383
Crane RA, Sapsford DJ. Towards Greener Lixiviants in Value Recovery from Mine Wastes: Efficacy of Organic Acids for the Dissolution of Copper and Arsenic from Legacy Mine Tailings. Minerals. 2018; 8(9):383. https://doi.org/10.3390/min8090383
Chicago/Turabian StyleCrane, Richard A., and Devin J. Sapsford. 2018. "Towards Greener Lixiviants in Value Recovery from Mine Wastes: Efficacy of Organic Acids for the Dissolution of Copper and Arsenic from Legacy Mine Tailings" Minerals 8, no. 9: 383. https://doi.org/10.3390/min8090383