How Can Additives Control the Early Stages of Mineralisation?


Department of Chemistry, Physical Chemistry, University of Konstanz, 78457 Konstanz, Germany
Minerals 2018, 8(5), 179; https://doi.org/10.3390/min8050179
Submission received: 10 April 2018
/
Revised: 10 April 2018
/
Accepted: 23 April 2018
/
Published: 26 April 2018
(This article belongs to the Special Issue Mineral Surface Reactions at the Nanoscale)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:The interactions between additives and mineral precursors and intermediates are at the heart of additive-controlled crystallisation, which is of high importance for various fields. In this commentary, we reflect on potential modes of additive control according to classical nucleation theory on one hand, and from the viewpoint of the so-called pre-nucleation cluster pathway on the other. This includes a brief review of the corresponding literature. While the roles of additives are discussed generally, i.e., without specific chemical or structural details, corresponding properties are outlined where possible. Altogether, our discussion illustrates that “non-classical” nucleation pathways promise an improved understanding of additive-controlled scenarios, which could be utilised in targeted applications in various fields, ranging from scale inhibition to materials chemistry.
1. Introduction
Additive-controlled mineralization, in the broadest sense, is central to various fields [1,2,3], ranging from biomineralisation [4,5,6,7,8] to scale inhibition [9,10,11]. It also touches upon materials chemistry, where the realisation of target-oriented synthetic routes to hybrid materials with advanced properties is every researcher’s dream [12]. In additive-controlled processes, especially the early stages of particle formation are important, as they can determine particle size, morphology, or polymorphism. In other words, the basic question is, how can additives control nucleation?
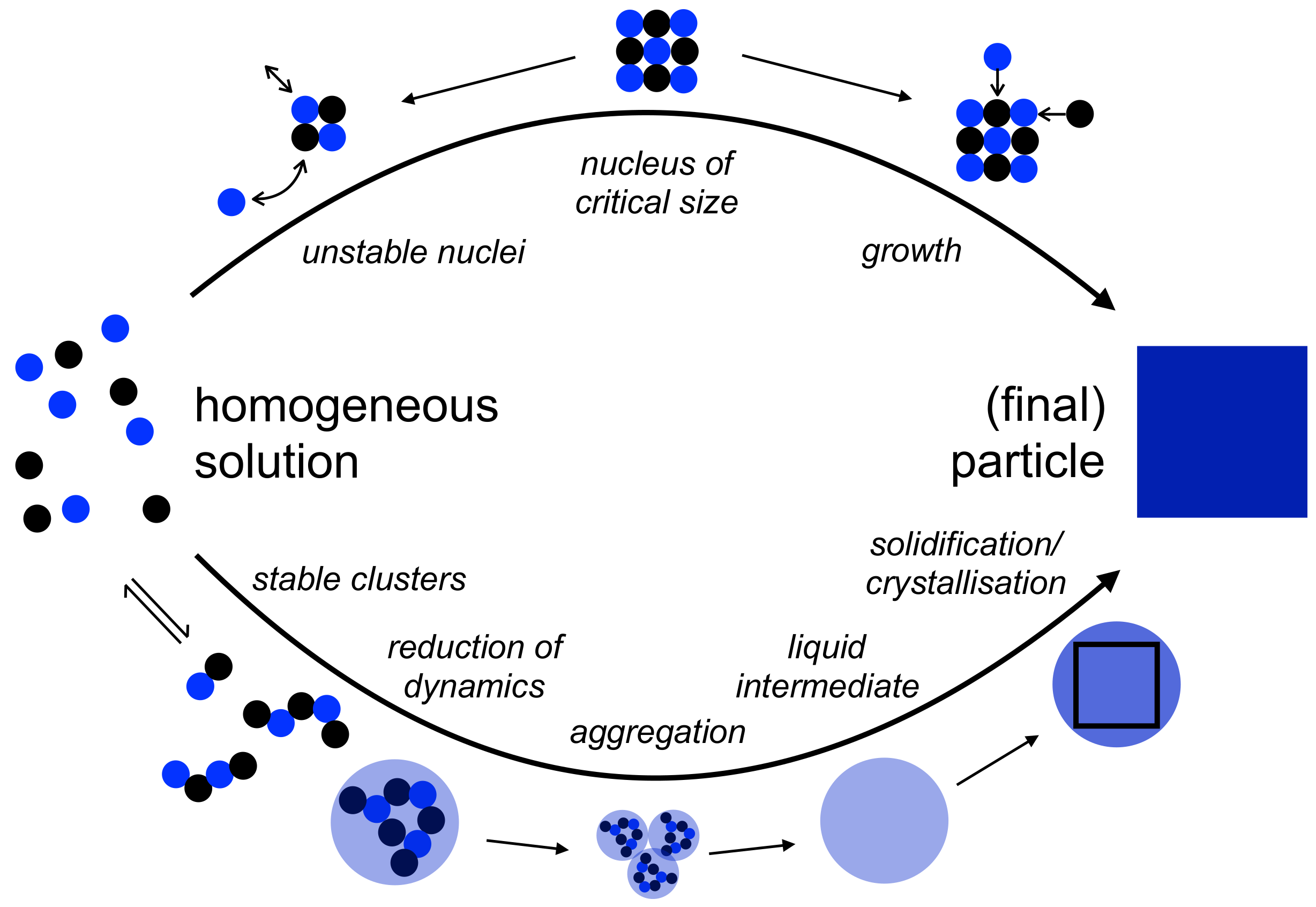
It is obvious that possible controls over the early stages of mineralisation processes are based on interactions of the additives with mineral precursors, intermediates and nascent particles. These may or may not include interfacial interactions. In order to be able to contemplate potential modes of additive control (or, interference) during nucleation processes, it is thus important to consider the structural and thermodynamic speciation of (pre-) nuclei, particle precursors and intermediates, which can interact with the additives and thereby change the nucleation pathway when compared to additive-free scenarios. There are at least two fundamentally distinct ways to look at this. First (Figure 1, top), according to classical nucleation theory (CNT), the monomeric chemical constituents of the nascent particles (i.e., in case of minerals, mostly ions) undergo stochastic collisions in supersaturated solutions, which lead to the formation of thermodynamically unstable pre-critical nuclei. Upon reaching a critical size, the probability of which depends on the level of supersaturation, the transition to post-critical nuclei that can grow without limit, given a sufficient supply of ions, is facilitated. The central notion of CNT is indeed that this nucleus of critical size, which serves as the relevant transition state formed within a metastable state of equilibrium (similar to the activated complex invoked in chemical kinetics [13]), governs the nucleation rate. Importantly, within CNT, pre-critical, critical, and post-critical nuclei are considered to be characterised by a solid-liquid interfacial surface, which is assumed to correspond to that of the macroscopic interface (capillary assumption). In the case where different forms or polymorphs exist, their accessibility depends on the level of supersaturation. If more than one polymorph is accessible, the fastest nucleation rate will determine which form will occur initially. Second (Figure 1, bottom), according to the so-called pre-nucleation cluster (PNC) pathway [14], ions form thermodynamically stable PNCs, which are characterised by a Flory-like [15], broad, decaying cluster size distribution and are highly dynamic. PNCs thus do not have a interfacial surface and must be considered as solutes [16]. Upon reaching a certain ion activity product (IAP), the clusters can change their structural form, leading to a significant decrease of the dynamics and rendering them phase-separated nano-droplets. Driven by the reduction of interfacial surface area, the as-formed nano-droplets undergo aggregation and/or coalescence, yielding larger liquid intermediate phases. These dehydrate and solidify toward amorphous intermediates, which eventually transform into crystals. Here, the major difference when compared to CNT is that the fundamental precursors are thermodynamically stable and form independent of the level of supersaturation. Note again, these PNCs are solutes and thus formally do not have a phase interface. The relevant transition state is then not primarily based on the size of the species forming, but instead on a significant decrease of cluster dynamics, which becomes possible upon exceeding a certain specific IAP, namely the liquid-liquid binodal limit. In other words, liquid–liquid interfacial surfaces between the nascent mineral and the solution are formed initially, which then become solid-liquid interfaces upon dehydration and solidification according to the PNC pathway.
Here we contemplate the possibilities of potential additive controls over nucleation from the viewpoint of CNT on one hand, and according to the PNC pathway on the other. This is done for the generic case of “an additive”, i.e., without specific chemical or structural properties, but where common properties become apparent for designing ion-additive or additive-mineral interactions for realising corresponding control patterns, these are specified as far as possible. We stress that our considerations are theoretical, that is, beyond speculation on principle, and hope to provide researchers with alternative explanations of additive effects for original experimental observations. In general, additives that can influence the early stages of mineralisation range from simple ions and small molecules to macromolecules. Altogether, the discussion below will show that the modes of additive-control over nucleation are rather limited within CNT, but highly diverse from the point of view of the PNC pathway, allowing to explain experimental observations. Last, but not least, we provide such examples of experimental observations that can be rationalised based on the PNC pathway in a straightforward manner but seem incompatible with CNT. In our opinion, this underpins the value of “non-classical” nucleation theories for an improved understanding of additive-controlled nucleation leading to the potential for a broad impact on various scientific fields including biomineralisation, scale inhibition, and materials chemistry.
2. Possible Modes of Additive Control within the Notions of Classical Nucleation Theory
According to CNT, the nucleation rate J can be written as [20];
with a pre-exponential factor, A; a general activation energy, EA; the universal gas constant, R; absolute temperature, T; and the standard free energy associated with the formation of the critical nucleus from free ions, . The pre-exponential factor takes the geometrical shape of the critical nucleus into account, which is often assumed to be spherical as other shapes introduce a significant penalty for nucleation [21]. This pre-factor A also depends on the molecular volume of the nascent nuclei’s monomeric chemical constituents, i.e., for the case of mineralisation, the ions. However, A is independent of supersaturation. The general activation energy, EA, arises from desolvation of the ions or rearrangements within the forming nuclei that may be required for the generation of the critical nucleus, but these contributions are typically neglected, as they are regarded to be minor (and are also difficult to quantify). gives rise to a thermodynamic barrier to nucleation, which is the central quantity derived within CNT;
where β is a geometrical factor (that has its lowest value for a sphere, for which βs = 16π/3), v is the molecular volume of the single ions, σ is the interfacial tension between the macroscopic mineral and the solution, and φ is the so-called affinity. In case of minerals, the affinity can be generally formulated as;
with the solubility product of the nascent phase Ksp and a correspondingly consistent IAP of the relevant ions. Please note that as opposed to the sign convention of ∆G, phase separation is impossible and possible for φ < 0 and φ > 0, respectively. The quotient of IAP and Ksp is the supersaturation ratio S;
Highlighting that the standard free energy of critical nuclei (Equation (2)) depends on supersaturation (Equations (3) and (4)), thereby governing the nucleation rate (Equation (1)). The dependence of the nucleation barrier on supersaturation is the hallmark of CNT; as opposed to generic chemical kinetics that can be understood within the theory of the activated complex [13].
This brief recapitulation of CNT enables us to identify possible effects of additives interacting with (pre-) critical nuclei, thereby influencing nucleation. In doing so, we will also neglect any potential effects of the additives on the kinetic barrier (i.e., EA; Equation (1)) as well as the pre-exponential factor, A, and focus on the thermodynamic barrier (i.e., ; Equations (1) and (2)). In any case, it has to be realised that the concentration of (pre-) critical nuclei is minuscule [21], owing to their excess free energy, so additive concentrations have to be in general relatively high in order to enable interactions with these rare and short-lived transition states to nucleation.
2.1. Additive Incorporation into the Nascent Phase
Additives could be incorporated into forming nuclei, affecting their bulk thermodynamic and structural properties. Since bulk and surface effects can be discussed separately within CNT, we here also assume that additive incorporation into the bulk would not affect the nuclei’s interfacial properties. Whether or not this assumption is justified is certainly debatable; however, as soon as interfacial properties of nuclei are affected by additives, they very likely dominate the effects on the nucleation barrier because the surface free energy (surface tension) is cubed in the numerator when calculating (Equation (2)). In this case, the effects of additive incorporation into the bulk of nuclei are most likely negligible and the combined effects of additive adsorption and incorporation can be discussed based on the interfacial effects alone, which are addressed in the next section.
Additive incorporation into nuclei upon stochastic solute clustering would only occur to any significant extent for a negative standard free energy of interaction between the monomeric ions and the additive. Due to lowering a given IAP* characterising a certain supersaturated state to IAP’ upon ion binding by the additive, this would thus also reduce the supersaturation ratio (Equation (4)) to S’, and with it, the affinity (Equation (3)) to φ’ when compared to the additive-free scenario. On the other hand, additive incorporation into nuclei would, if anything, increase the molecular volume v of the ions due to the additive adsorption to v’, and may introduce deviations from a spherical shape, increasing β to β’. Spontaneous additive incorporation into nuclei thus implies S’ < S*, v’ > v, and β’ > β and increases the thermodynamic barrier to nucleation of the additive-containing phase, ΔG0c’, with respect to that of the additive free phase, ΔG0c, according to;
However, additives that can be incorporated into nuclei would be (weak) nucleation inhibitors mostly based on the lowered supersaturation ratio S’; Equation (5) shows that the formation of additive-containing nuclei is indeed improbable, as the barrier for the formation of additive-free critical nuclei remains lower also at the lowered supersaturation ratio S’. In turn, this consideration implies that from the viewpoint of CNT, the inclusion of additives into crystal lattices [22] rather happens during the subsequent growth stages. Here, the incorporation of additives affects the thermodynamic stability of the nascent phase, and thus Ksp. Since additive incorporation impairs the interactions between the ions in the crystal lattice, this would almost certainly always be a thermodynamic destabilisation, increasing the solubility threshold from Ksp to Ksp’. However, the additive-free phase would still be accessible during homogeneous nucleation, in principle, and the driving force for nucleation of the pure macroscopic phase would remain unchanged (Equation (3)). Altogether, our considerations show that the pure mineral phase would be initially nucleated, homogeneously, and the additive be incorporated during subsequent growth, then inhibiting this process owing to a lowered effective supersaturation due to the increased solubility, Ksp’ [23].
2.2. Interfacial Adsorption of Additives on Nascent Nuclei
As already outlined in the previous section, any potential effects of additives on the interfacial properties of the nascent nuclei would dominate the nucleation behaviour because the interfacial free energy is cubed for calculating the magnitude of (Equation (2)), probably outweighing the contribution of any other effects. The interfacial tension upon additive adsorption, σ’, can be estimated according to the Young equation, which is valid for planar interfaces;
where σ is the interfacial tension between the nucleus and the solution (which is assumed to be equal to that between the macroscopic phases as already mentioned above), σas and σac are the interfacial tensions between the additive and the solution and the additive and the nucleus, respectively, and k is a factor that depends on the aspect ratio of the nucleus [17]. While Equation (6) provides merely an estimate of effects of interfacial adsorption of additives based on oversimplifying assumptions (flat interfaces), it should be noted that corresponding interfacial properties are difficult to determine experimentally, or only with significant errors [24]. Note that these uncertainties would then be cubed (Equation (2)) and enter the exponent for calculating corresponding nucleation rates (Equation (1)), causing even larger quantitative uncertainties in measurable nucleation parameters. Thus, we prefer to focus on a qualitative discussion of corresponding effects here. In any case, for σas < σac, the interfacial tension in the presence of the additive would be increased, σ’ > σ, however, there is then no spontaneous adsorption of the additive and nucleation proceeds homogeneously. For σas = σac, there is the limiting case where the interfacial tension between the nucleus and the solution with and without the additive is equal, σ’ = σ, and the additive would have no effect due to insignificant adsorption. For σas > σac, the additive would spontaneously adsorb to the nucleus and promote nucleation, due to the lowered interfacial tension with the adsorbed additive, σ’ < σ. This highlights that again, significant additive effects can only be expected for spontaneous additive adsorption on the nuclei’s interfacial surface, which always lowers the interfacial free energy, promoting nucleation by analogy to heterogeneous nucleation in presence of surfaces with favourable interfacial properties.
In the case where different polymorphs or forms of a mineral are accessible, these are characterised by distinct solubilities. For example, consider three polymorphs A, B, C with solubilities Ksp(A) < Ksp(B) < Ksp(C), i.e., A, B, and C are stable, metastable, and unstable polymorphs, respectively. It can be argued that the less stable structures B and C have a lower cohesive energy than A, and hence also a lower surface free energy, suggesting that σ(A) > σ(B) > σ(C) [25,26]. In this case, the nucleation rate of the unstable form would be the highest (Equations (1) and (2)). Thus, for a supersaturation ratio at which all polymorphs are accessible, this consideration is a CNT rationalisation of the well-known Ostwald–Volmer rule. In the case where additives bind specifically to one of the polymorphic forms, the interfacial tension of the stable forms could be lowered below that of the un- and meta-stable polymorphs, leading to their direct nucleation instead of ripening according to Ostwald’s rule of stages. In this way, CNT can explain the observation that the nucleation of metastable polymorphs can be poisoned by the presence of additives or impurities; however, it seems impossible to explain scenarios where stable polymorphs cannot be obtained as opposed to metastable forms in presence of certain additives or impurities because specific additive binding can only promote nucleation, and an efficient (post-nucleation) inhibition of ripening towards the stable form would be required. Within a CNT perspective, such observations would then rather imply the complete inhibition of growth by the additive or impurity, not of nucleation.
2.3. Additive Mesophases as Environments for Nucleation
Additives may form mesophases, such as micelles, in the case where they have amphiphilic properties, depending on the additive concentration, as well as that of the ions [27]. When the formation of additive mesophases is induced by the binding of ions that constitute mineral phases, the overall ion concentration within the formed mesophase could be locally increased [28]. Formally, this may lead to a metastable intermediate, which is consistent with two-step nucleation [29,30], but here, rather a “heterogeneous two-step nucleation” scenario.
Whether or not nucleation in this ion-enriched environment proceeds via critical nuclei and according to the notions of CNT or not, however, is debatable. It has to be pointed out that the binding of ions in an additive matrix does not increase the concentration of free ions in the mesophase, and stable bound states are not available for stochastic collisions of free ions toward the formation of critical nuclei according to CNT [31]. This is because in the case where mineral ions can induce the formation of additive mesophases, then the underlying ion-additive interactions are spontaneous and associated with a negative standard free energy. Even if the ion-additive interactions within the mesophase are successively replaced by the counter-ion of the relevant nascent mineral [28], the ion complexes formed are necessarily in a state of even lower free energy and are thus in a free energy trap for the formation of critical nuclei [31]. In this sense, the situation is rather analogous to additive incorporation into the mineral phase, where the main effect is the lowering of supersaturation and a weak inhibition of nucleation, as discussed in Section 2.1. Also, in such mesophases, confinement effects can become important [32,33,34], and the increased viscosity may play an additional role toward inhibition effects. In our opinion, the observed facilitation of nucleation in such phases [28] can be better explained within the notions of “non-classical” nucleation theories as described below (Section 3.2).
3. Additive Control Patterns in “Non-Classical” Nucleation
As opposed to (pre-) critical nuclei, stable PNCs are more abundant species and interactions with additives appear in general more likely. Also, in the multi-step PNC pathway (Figure 1, bottom), interactions between the mineral precursors and intermediates can control the early stages of particle formation through various points of attack, which can allow a better rationalisation of the multiple roles of additives in crystallisation processes [35] as discussed in the following. Generally, it should be noted that according to the PNC pathway, amorphous intermediates form first but may be highly transient. In this sense, also the PNC pathway rationalises the Ostwald–Volmer rule (cf. Section 2.2).
3.1. Adsorption of Free Ions and Pre-Nucleation Clusters
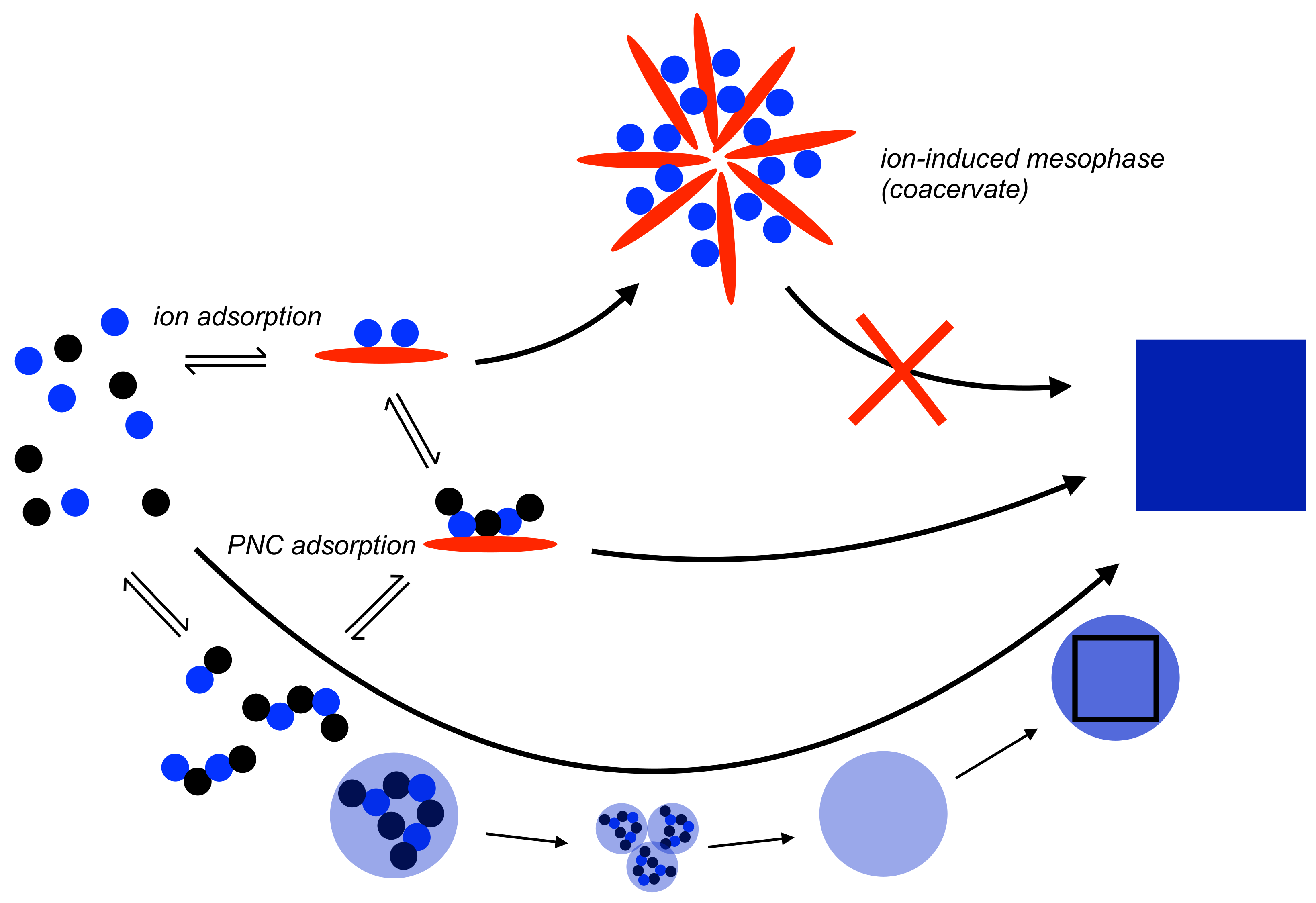
Additives can bind single ions, which occurs to a significant extent only for a corresponding negative standard free energy of interaction (also see Section 2.1). For a very strong interaction and/or high additive concentrations, this can lead to the formation of additive mesophases or coacervates (Figure 2, top) if the additive structure and chemistry allows. These do not provide a pathway to crystals, as the relevant ions are bound in stable states and there is, if any, only moderate supersaturation within the coacervate (only the free ions contribute to the supersaturation ratio, Equation (4)). The chemical functional groups of the additive that allow for interactions with free ions also facilitate PNC binding. Since there is competition between additive binding and counter-ion association, PNC binding becomes more probable for ion-additive interactions that are weaker than cation-anion interactions and/or lower additive concentrations. The pathway toward particles from these states (Figure 2, middle) will be discussed in more detail in the following section.
Additives can influence ion association even without direct binding by modulating the water structure and introducing kosmotropic or chaotropic effects [36,37]. Due to the direct impact on the entropic balance of ion association through the release of hydration waters [38], these effects can increase or weaken ion-ion and/or ion-additive interactions [39], providing additional means to tune the strength of corresponding intermolecular interactions. In combination, the binding of PNCs by additives can thermodynamically stabilise or destabilise the ion associates. In the case where there are polyamorphic forms, as for calcium carbonate [19], the link between pre- and post-nucleation speciation may thus influence the type of polyamorphic solid formed by the presence of additives. In cases where no amorphous polymorphism exists, it is possible that additives induce distinct short-range structural features or motifs in amorphous intermediates (also see Section 3.3).
The binding of PNCs by additives can also have kinetic effects. For instance, it has been proposed that the binding of PNCs can lead to favourable three-dimensional arrangements of ions facilitating the change in the dynamics of the solute clusters underlying liquid-liquid separation [40]. In this case, the presence of the additive would drive phase separation, which may also be associated with the reduction of the IAP corresponding to the locus of the liquid-liquid binodal limit compared to the additive-free case (see blue arrow in Figure 3). The inverse scenario is also possible, where the strong binding of PNCs by additives would lead to configurations that do not allow for the change in speciation toward nano-droplets; however, this case cannot be distinguished from coacervate formation. In the case of weak binding of PNCs by the additive and/or low additive concentrations, inhibition of phase separation via binding of PNCs in configurations that impair nano-droplet formation appear difficult, as phase separation can still occur via non-adsorbed precursors.
3.2. Additives and Liquid Intermediates
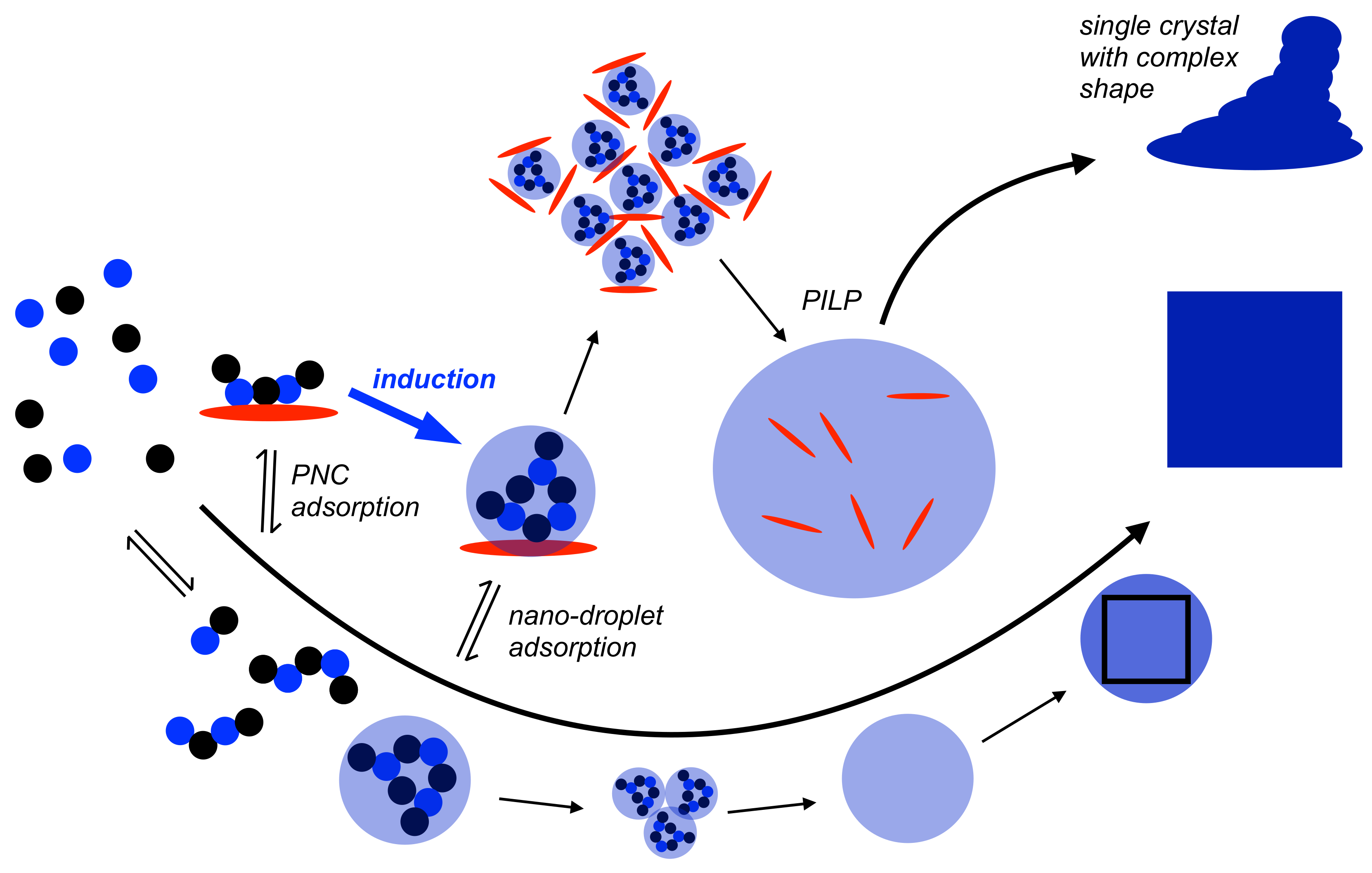
In case of calcium carbonate, the occurrence of liquid intermediates is well-known and their use in bio-inspired materials chemistry has been thoroughly demonstrated [41]. However, the so-called polymer-induced liquid precursors (PILPs) are polymer-stabilised rather than polymer-induced states, and the notions of the PNC pathway can explain their occurrence in a straightforward manner (Figure 3). Since the PNC pathway seems to be more general for particle formation from aqueous solutions than previously thought [14,42], the occurrence of liquid intermediates may be exploited for a number of compounds. For moderate or weak interactions between the additive and PNCs, and/or low additive concentrations [43], the stabilisation of liquid intermediates becomes possible (Figure 3). Consistently, when weaker cation-binding properties of additives are exploited, additive concentrations need to be increased for sufficient PILP stabilisation [44]. However, it appears to be fundamentally required that the additive additionally stabilises the liquid intermediates against dehydration [45], or the nano-droplets against aggregation and/or coalescence by means of colloidal stabilisation [46]. At this point, interfacially active properties of the additives can also become important. These may be influenced by the presence of additional species and control or tune the wettability of the additive-stabilised liquid intermediates on distinct surfaces [47]. This phenomenon can be exploited in target-oriented materials synthesis [48].
3.3. Additives and Nascent Solid Amorphous Intermediates
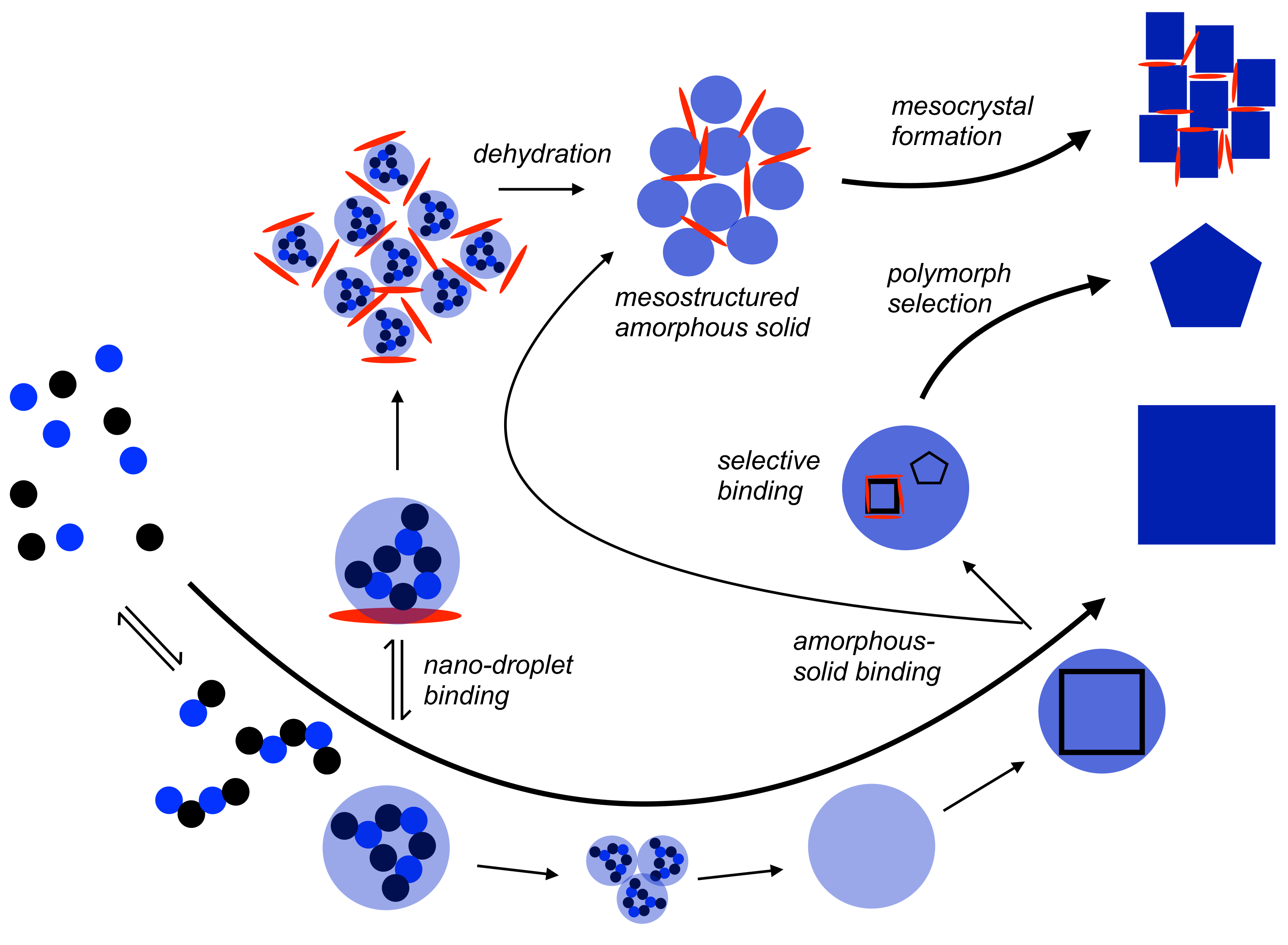
When an additive can bind PNCs and nano-droplets but cannot stabilise the latter against dehydration and/or aggregation/coalescence to any significant extent, the formation of mesostructured amorphous solids is possible (Figure 4). This may be based on either nano-droplet aggregation, or the aggregation of already formed amorphous, solid nanoparticles. While it may be difficult to discriminate between the two possibilities experimentally, it can be argued that a nano-droplet-based pathway would yield amorphous solids structured on shorter length scales (ca. 2–5 nm) than the aggregation of amorphous nanoparticles that emerge later. However, depending on the kinetics of dehydration, aggregation and crystallisation, a complex convoluted pathway involving both liquid and solid amorphous precursors may also be viable. In any case, an insufficient capability of the additive to act against dehydration (or, even a promoted dehydration of the liquid intermediates due to the additive) seems to be required. It is conceivable that tuning the kinetics of interface-coupled re-dissolution/precipitation pathways [50] of this intermediate, or direct crystallisation of the mesostructured amorphous solid via solid-state transformation [51], can then yield mesocrystals [2,52,53] upon crystallisation (Figure 4, top).
Additives could also exhibit specific binding capabilities to certain short-range structural motifs in amorphous intermediates [54]. On the one hand, this may induce the formation of polyamorphic forms that normally do not form under the given conditions, or even induce the formation of distinct short-range structural motifs in amorphous intermediates where polyamorphism does not exist for the pure systems. This could be based on the interfacial binding of additives or structural incorporation. Note, however, that it is difficult to discuss the phenomenon of amorphous polymorphism for phases where additives are structurally incorporated, as the compositions vary and the phenomenon strictly applies only to the pure compounds with unchanged compositions. In any case, the specific binding of distinct short-range structural domains by additives may inhibit their crystallisation, so that other environments crystallise, thereby facilitating polymorph selection (Figure 4, middle) [35,55]. It is also conceivable that specific interactions of certain amorphous short-range structural motifs with additives would trigger the crystallisation of only these environments, enabling a distinct mechanism of polymorph selection from amorphous intermediates.
4. Conclusions
The above discussion shows that classical nucleation theory (CNT) can rationalise only a limited number of additive controls over nucleation, i.e., a rather weak inhibition of nucleation based on the binding of free ions, or the promotion of nucleation due to the lowering of interfacial free energies upon additive binding. The latter may also facilitate polymorph selection for some scenarios. One might argue that distinct additive controls could be rationalised within CNT by strong but unspecific effects on the pre-exponential factor, A, and/or the generic activation energy, EA, (Equation (1)) but this would be a rather hand-waving and actually unqualified argument given that these factors are typically neglected within CNT. In contrast, there are several different mechanisms for additive control during “non-classical” nucleation according to the pre-nucleation cluster (PNC) pathway. These can explain strong inhibition, as well as promotion of nucleation, not only for stable crystalline forms but also for amorphous intermediates. The kinetic stabilisation of liquid and amorphous intermediates can provide pathways to crystals with complex shapes via PILPs, as well as mesocrystals, depending on the capabilities of the additives for, e.g., binding water and thereby tuning the kinetics of the PNC pathway after the emergence of phase separated nanodroplets. Also, the thermodynamic stabilisation and destabilisation of PNCs and amorphous intermediates is possible, which can give rise to the development of distinct short-range structural motifs in the presence of an additive. In turn, specific interactions between additives and distinct short-range structural environments in amorphous intermediates can also provide control over polymorph selection.
Last, but not least, we would like to point out two experimental observations that are difficult to explain based on CNT, but straightforward to understand from the viewpoint of the PNC pathway. Ion potential measurements in combination with THz spectroscopy showed a very strong inhibition of calcium carbonate nucleation by minuscule amounts of polycarboxylates in the low µg/mL regime [45], which was already literature known [35]. While the experiments, for the first time, revealed the locus of the liquid-liquid binodal limit, at which solute PNCs can change their speciation and become phase-separated nanodroplets, the barrier for classical nucleation under corresponding conditions is formidable even in absence of the additive, rendering nucleation via critical nuclei highly improbable [45]. Also, the very strong inhibitory capacity of the low polymer concentration can in our opinion not be explained by CNT. First, owing to the minor polymer concentration, there is no detectable binding of free calcium ions by the polymer and thus, no significant effect on the level of supersaturation that could explain the observed inhibition. Second, as shown in Section 2.2, an increase in interfacial free energy upon additive adsorption—that would be required for inhibition of nucleation from a classical perspective—would render additive adsorption on (pre-) critical nuclei thermodynamically unfavourable (Equation (6)), and nucleation would proceed homogeneously, i.e., without significant additive effects. It is impossible to rationalize the inhibition of nucleation by effects on the interfacial free energy of nuclei upon additive adsorption within CNT. Only spontaneous additive adsorption on (pre-) critical nuclei can have effects on nucleation from the viewpoint of CNT, which always promotes nucleation due to a lowered interfacial tension. The incorporation of the polycarboxylates into liquid intermediates and the inhibition of dehydration due to their superadsorbent properties, on the other hand, explains the highly effective inhibition from the point of view of the PNC pathway as outlined above. In contrast, the same additive molecules can strongly promote the nucleation of iron(III) (oxyhydr)oxides as shown by potentiometric titrations in combination with turbidity measurements at different pH values [40]; this effect, however, is hardly due to the lowering of the interfacial free energy, as it only occurs under conditions where olation PNCs can form in the system [40]. Rather, the three-dimensional configuration of olation PNCs adsorbed on the polymer appears to facilitate the formation of oxo-bridged iron(III) centres in PNCs at much lower concentrations when compared to the additive-free case, which is the fundamental change in reaction mechanism underlying the event of phase separation towards a reduction of cluster dynamics due to the formation of stronger bonds [56]. The orthogonal role of the same additives in distinct systems highlights that additive control patterns are highly dependent on the distinct chemistries, consistent with the PNC pathway, rather than generic physicochemical parameters like supersaturation that are central to CNT. The combination of additives may further bring about synergistic effects [57], which eventually allow the realisation of sophisticated, tuneable control patterns over particle formation. Future research will show if a generalised theory for additive control over particle formation based on the PNC pathway is possible and whether predictable outcomes can be exploited in a target-oriented manner.
Acknowledgments
D.G. is a Research Fellow of the Zukunftskolleg of the University of Konstanz. We thank Julian Gale for stimulating discussions and feedback on the manuscript.
Conflicts of Interest
The author declares no conflict of interest.
References
- Ruiz-Agudo, E.; Putnis, C.V.; Rodriguez-Navarro, C. Reactions between minerals and aqueous solutions. In Mineral Reaction Kinetics: Microstructures, Textures, Chemical and Isotopic Signatures; Mineralogical Society of Great Britain & Ireland: Chantilly, GA, USA, 2017; pp. 419–467. [Google Scholar]
- Cölfen, H.; Antonietti, M. Mesocrystals and Nonclassical Crystallization; John Wiley & Sons, Ltd.: Chichester, UK, 2008. [Google Scholar]
- Meldrum, F.C.; Cölfen, H. Controlling Mineral Morphologies and Structures in Biological and Synthetic Systems. Chem. Rev. 2008, 108, 4332–4432. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.S. ‘Apples’ and ‘oranges’: Comparing the structural aspects of biomineral- and ice-interaction proteins. Curr. Opin. Colloid Interface Sci. 2003, 8, 48–54. [Google Scholar] [CrossRef]
- Evans, J.S. “Liquid-like” biomineralization protein assemblies: A key to the regulation of non-classical nucleation. CrystEngComm 2013, 15, 8388–8394. [Google Scholar] [CrossRef]
- Evans, J. Polymorphs, Proteins, and Nucleation Theory: A Critical Analysis. Minerals 2017, 7, 62. [Google Scholar] [CrossRef]
- Evans, J.S. Principles of Molecular Biology and Biomacromolecular Chemistry. Rev. Min. Geochem. 2003, 54, 31–56. [Google Scholar] [CrossRef]
- Ibsen, C.J.S.; Birkedal, H. Pyrophosphate-Inhibition of Apatite Formation Studied by In Situ X-Ray Diffraction. Minerals 2018, 8, 65. [Google Scholar] [CrossRef]
- Boulahlib-Bendaoud, Y.; Ghizellaoui, S.; Tlili, M. Inhibition of CaCO3 scale formation in ground waters using mineral phosphates. Desal. Water Treat. 2012, 38, 382–388. [Google Scholar] [CrossRef]
- Ruiz-Agudo, E.; Burgos-Cara, A.; Ruiz-Agudo, C.; Ibañez-Velasco, A.; Cölfen, H.; Rodriguez-Navarro, C. A non-classical view on calcium oxalate precipitation and the role of citrate. Nat. Commun. 2017, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Navarro, C.; Benning, L.G. Control of Crystal Nucleation and Growth by Additives. Elements 2013, 9, 203–209. [Google Scholar] [CrossRef]
- Mann, S. Biomimetic Materials Chemistry; VCH: New York, NY, USA, 1996. [Google Scholar]
- Eyring, H. The activated complex in chemical reactions. Chem. Rev. 1935, 17, 65–77. [Google Scholar] [CrossRef]
- Gebauer, D.; Kellermeier, M.; Gale, J.D.; Bergström, L.; Cölfen, H. Pre-nucleation clusters as solute precursors in crystallisation. Chem. Soc. Rev. 2014, 43, 2348–2371. [Google Scholar] [CrossRef] [PubMed]
- Flory, P.J. Molecular size distribution in linear condensation polymers. J. Am. Chem. Soc. 1936, 58, 1877–1885. [Google Scholar] [CrossRef]
- Demichelis, R.; Raiteri, P.; Gale, J.D.; Quigley, D.; Gebauer, D. Stable prenucleation mineral clusters are liquid-like ionic polymers. Nat. Commun. 2011, 2, 590. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Nielsen, M.H.; Freeman, C.L.; Hamm, L.M.; Tao, J.; Lee, J.R.I.; Han, T.Y.J.; Becker, U.; Harding, J.H.; Dove, P.M.; et al. The thermodynamics of calcite nucleation at organic interfaces: Classical vs. non-classical pathways. Faraday Discuss. 2012, 159, 509–523. [Google Scholar] [CrossRef]
- Smeets, P.J.M.; Finney, A.R.; Habraken, W.J.E.M.; Nudelman, F.; Friedrich, H.; Laven, J.; De Yoreo, J.J.; Rodger, P.M.; Sommerdijk, N.A.J.M. A classical view on nonclassical nucleation. Proc. Natl. Acad. Sci. USA 2017, 114, E7882–E7890. [Google Scholar] [CrossRef] [PubMed]
- Cartwright, J.H.E.; Checa, A.G.; Gale, J.D.; Gebauer, D.; Sainz-Díaz, C.I. Calcium carbonate polyamorphism and its role in biomineralization: How many amorphous calcium carbonates are there? Angew. Chem. Int. Ed. 2012, 51, 11960–11970. [Google Scholar] [CrossRef] [PubMed]
- De Yoreo, J.J.; Vekilov, P.G. Principles of crystal nucleation and growth. Rev. Min. Geochem. 2003, 54, 57–93. [Google Scholar] [CrossRef]
- Nielsen, A.E. Kinetics of Precipitation; Pergamon Press: New York, NY, USA, 1964. [Google Scholar]
- Li, H.; Xin, H.L.; Muller, D.A.; Estroff, L.A. Visualizing the 3D Internal Structure of Calcite Single Crystals Grown in Agarose Hydrogels. Science 2009, 326, 1244–1247. [Google Scholar] [CrossRef] [PubMed]
- Davis, K.J.; Dove, P.M.; De Yoreo, J.J. The Role of Mg2+ as an Impurity in Calcite Growth. Science 2000, 290, 1134–1137. [Google Scholar] [CrossRef] [PubMed]
- Dörfler, H.D. Grenzflächen- und Kolloidchemie [Interface and Colloid Chemistry]; VCH: Weinheim, Germany, 1994. [Google Scholar]
- Sun, W.; Jayaraman, S.; Chen, W.; Persson, K.A.; Ceder, G. Nucleation of metastable aragonite CaCO3 in seawater. Proc. Natl. Acad. Sci. USA 2015, 112, 3199–3204. [Google Scholar] [CrossRef] [PubMed]
- Navrotsky, A. Nanoscale Effects on Thermodynamics and Phase Equilibria in Oxide Systems. ChemPhysChem 2011, 12, 2207–2215. [Google Scholar] [CrossRef] [PubMed]
- Israelachvili, J.N. Intermolecular and Surface Forces, 3rd ed.; Academic Press: Burlington, MA, USA, 2011. [Google Scholar]
- Smeets, P.J.M.; Cho, K.R.; Kempen, R.G.E.; Sommerdijk, N.A.J.M.; De Yoreo, J.J. Calcium carbonate nucleation driven by ion binding in a biomimetic matrix revealed by in situ electron microscopy. Nat. Mater. 2015, 14, 394–399. [Google Scholar] [CrossRef] [PubMed]
- Vekilov, P.G. Nucleation. Cryst. Growth Des. 2010, 10, 5007–5019. [Google Scholar] [CrossRef] [PubMed]
- Vekilov, P.G. The two-step mechanism of nucleation of crystals in solution. Nanoscale 2010, 2, 2346–2357. [Google Scholar] [CrossRef] [PubMed]
- De Yoreo, J.J. Crystal nucleation: More than one pathway. Nat. Mater. 2013, 12, 284–285. [Google Scholar] [CrossRef] [PubMed]
- Stephens, C.J.; Ladden, S.F.; Meldrum, F.C.; Christenson, H.K. Amorphous Calcium Carbonate is Stabilized in Confinement. Adv. Funct. Mater. 2010, 20, 2108–2115. [Google Scholar] [CrossRef]
- Kröger, R. Andreas Verch Liquid Cell Transmission Electron Microscopy and the Impact of Confinement on the Precipitation from Supersaturated Solutions. Minerals 2018, 8, 21. [Google Scholar] [CrossRef]
- Mao, L.B.; Xue, L.; Gebauer, D.; Liu, L.; Yu, X.F.; Liu, Y.Y.; Cölfen, H.; Yu, S.H. Anisotropic nanowire growth via a self-confined amorphous template process: A reconsideration on the role of amorphous calcium carbonate. Nano Res. 2016, 9, 1334–1345. [Google Scholar] [CrossRef]
- Gebauer, D.; Cölfen, H.; Verch, A.; Antonietti, M. The multiple roles of additives in CaCO3 crystallization: A quantitative case study. Adv. Mater. 2009, 21, 435–439. [Google Scholar] [CrossRef]
- Rao, A.; Gebauer, D.; Cölfen, H. Modulating Nucleation by Kosmotropes and Chaotropes: Testing the Waters. Crystals 2017, 7, 302. [Google Scholar] [CrossRef]
- Burgos-Cara, A.; Putnis, C.; Rodriguez-Navarro, C.; Ruiz-Agudo, E. Hydration Effects on the Stability of Calcium Carbonate Pre-Nucleation Species. Minerals 2017, 7, 126. [Google Scholar] [CrossRef]
- Kellermeier, M.; Raiteri, P.; Berg, J.K.; Kempter, A.; Gale, J.D.; Gebauer, D. Entropy Drives Calcium Carbonate Ion Association. ChemPhysChem 2016, 17, 3535–3541. [Google Scholar] [CrossRef] [PubMed]
- Sinn, C.G.; Dimova, R.; Antonietti, M. Isothermal Titration Calorimetry of the Polyelectrolyte/Water Interaction and Binding of Ca2+: Effects Determining the Quality of Polymeric Scale Inhibitors. Macromolecules 2004, 37, 3444–3450. [Google Scholar] [CrossRef]
- Scheck, J.; Drechsler, M.; Ma, X.; Stöckl, M.T.; Konsek, J.; Schwaderer, J.B.; Stadler, S.M.; De Yoreo, J.J.; Gebauer, D. Polyaspartic acid facilitates oxolation within iron(III) oxide pre-nucleation clusters and drives the formation of organic-inorganic composites. J. Chem. Phys. 2016, 145, 211917–211919. [Google Scholar] [CrossRef] [PubMed]
- Gower, L.B. Biomimetic model systems for investigating the amorphous precursor pathway and its role in biomineralization. Chem. Rev. 2008, 108, 4551–4627. [Google Scholar] [CrossRef] [PubMed]
- Gebauer, D. Prenucleation Clusters. In Encyclopedia of Nanotechnology; Springer: Dordrecht, The Netherlands, 2015; pp. 1–7. [Google Scholar]
- Jiang, Y.; Gower, L.; Volkmer, D.; Cölfen, H. The existence region and composition of a polymer-induced liquid precursor phase for DL-glutamic acid crystals. Phys. Chem. Chem. Phys. 2012, 14, 914–919. [Google Scholar] [CrossRef] [PubMed]
- Cantaert, B.; Kim, Y.Y.; Ludwig, H.; Nudelman, F.; Sommerdijk, N.A.J.M.; Meldrum, F.C. Think positive: Phase separation enables a positively charged additive to induce dramatic changes in calcium carbonate morphology. Adv. Funct. Mater. 2012, 22, 907–915. [Google Scholar] [CrossRef]
- Sebastiani, F.; Stefan, L.P.W.; Born, B.; Luong, T.Q.; Cölfen, H.; Gebauer, D.; Havenith, M. Water Dynamics from THz Spectroscopy Reveal the Locus of a Liquid-Liquid Binodal Limit in Aqueous CaCO3 Solutions. Angew. Chem. Int. Ed. 2016, 56, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Kellermeier, M.; Gebauer, D.; Melero-García, E.; Drechsler, M.; Talmon, Y.; Kienle, L.; Cölfen, H.; García-Ruiz, J.M.; Kunz, W. Colloidal stabilization of calcium carbonate prenucleation clusters with silica. Adv. Funct. Mater. 2012, 22, 4301–4311. [Google Scholar] [CrossRef]
- Berg, J.K.; Jordan, T.; Binder, Y.; Börner, H.G.; Gebauer, D. Mg2+ tunes the wettability of liquid precursors of CaCO3: Toward controlling mineralization sites in hybrid materials. J. Am. Chem. Soc. 2013, 135, 12512–12515. [Google Scholar] [CrossRef] [PubMed]
- Khouzani, M.F.; Schütz, C.; Durak, G.M.; Fornell, J.; Sort, J.; Salazar-Alvarez, G.; Bergström, L.; Gebauer, D. A CaCO3/nanocellulose-based bioinspired nacre-like material. J. Mater. Chem. A 2017, 5, 16128–16133. [Google Scholar] [CrossRef]
- Bewernitz, M.A.; Gebauer, D.; Long, J.R.; Cölfen, H.; Gower, L.B. A meta-stable liquid precursor phase of calcium carbonate and its interactions with polyaspartate. Faraday Discuss. 2012, 159, 291–312. [Google Scholar] [CrossRef]
- Ruiz-Agudo, E.; Putnis, C.V.; Putnis, A. Coupled dissolution and precipitation at mineral–fluid interfaces. Chem. Geol. 2014, 383, 132–146. [Google Scholar] [CrossRef]
- Ihli, J.; Wong, W.C.; Noel, E.H.; Kim, Y.Y.; Kulak, A.N.; Christenson, H.K.; Duer, M.J.; Meldrum, F.C. Dehydration and crystallization of amorphous calcium carbonate in solution and in air. Nat. Commun. 2014, 5, 3169. [Google Scholar] [CrossRef] [PubMed]
- Sturm, E.V.; Cölfen, H. Mesocrystals: Past, Presence, Future. Crystals 2017, 7, 207. [Google Scholar]
- Sturm, E.V.; Cölfen, H. Mesocrystals: Structural and morphogenetic aspects. Chem. Soc. Rev. 2016, 45, 5821–5833. [Google Scholar] [CrossRef] [PubMed]
- Addadi, L.; Raz, S.; Weiner, S. Taking advantage of disorder: Amorphous calcium carbonate and its roles in biomineralization. Adv. Mater. 2003, 15, 959–970. [Google Scholar] [CrossRef]
- Gebauer, D.; Verch, A.; Börner, H.G.; Cölfen, H. Influence of selected artificial peptides on calcium carbonate precipitation—A quantitative study. Cryst. Growth Des. 2009, 9, 2398–2403. [Google Scholar] [CrossRef]
- Scheck, J.; Wu, B.; Drechsler, M.; Rosenberg, R.; Van Driessche, A.E.S.; Stawski, T.M.; Gebauer, D. The Molecular Mechanism of Iron(III) Oxide Nucleation. J. Phys. Chem. Lett. 2016, 7, 3123–3130. [Google Scholar] [CrossRef] [PubMed]
- Wolf, S.L.P.; Jähme, K.; Gebauer, D. Synergy of Mg2+ and poly(aspartic acid) in additive-controlled calcium carbonate precipitation. CrystEngComm 2015, 17, 6857–6862. [Google Scholar] [CrossRef]
Figure 1.
Schematic illustration of the mechanism of nucleation according to classical nucleation theory (CNT, top) and the pre-nucleation cluster (PNC) pathway (bottom). For different polymorphs or forms, the accessibility depends on the level of supersaturation. The sizes of the different species are system specific and cannot be fully generalized; the critical size (top, middle) for realistic supersaturation levels is typically within tens of ions, i.e., smaller than approximately 3–4 nm in diameter [17]. PNCs are similar in size but thermodynamically stable, and thus significantly more abundant than classical (pre-) critical nuclei. The smallest sizes of phase separated nano-droplets, which directly emerge from the PNC precursors, are thus also in the lower nanometer regime. Upon aggregation and coalescence, dense liquid droplets with sizes up to several hundred nanometers can be formed [18]. Consequently, depending on the kinetics of aggregation and dehydration, which can also be influenced by the presence of additives (see below), the size of solid amorphous intermediates can range from ca. 20 nm to hundreds of micrometer in size [19]. For further explanation, see the text.
Figure 1.
Schematic illustration of the mechanism of nucleation according to classical nucleation theory (CNT, top) and the pre-nucleation cluster (PNC) pathway (bottom). For different polymorphs or forms, the accessibility depends on the level of supersaturation. The sizes of the different species are system specific and cannot be fully generalized; the critical size (top, middle) for realistic supersaturation levels is typically within tens of ions, i.e., smaller than approximately 3–4 nm in diameter [17]. PNCs are similar in size but thermodynamically stable, and thus significantly more abundant than classical (pre-) critical nuclei. The smallest sizes of phase separated nano-droplets, which directly emerge from the PNC precursors, are thus also in the lower nanometer regime. Upon aggregation and coalescence, dense liquid droplets with sizes up to several hundred nanometers can be formed [18]. Consequently, depending on the kinetics of aggregation and dehydration, which can also be influenced by the presence of additives (see below), the size of solid amorphous intermediates can range from ca. 20 nm to hundreds of micrometer in size [19]. For further explanation, see the text.

Figure 2.
Schematic illustration of the mechanism of nucleation according to the PNC pathway (bottom, cf. Figure 1) with potential effects of ion (top) and PNC (middle) adsorption by an additive (red ellipsoid). For strong ion-additive interactions and/or high additive concentrations, the formation of coacervates is expected (if the additive chemistry and structure allows), which inhibit mineral formation entirely. Corresponding size regimes span the whole colloidal domain depending on the type of mesophase formed [27]. For weaker ion-additive interactions and/or low additive concentrations, PNC binding by the additive (middle) becomes probable, and the process can proceed toward particle formation. Possibilities for additive control during the latter process are discussed in more detail in Section 3.2, also see Figure 3. For explanation see the text.
Figure 2.
Schematic illustration of the mechanism of nucleation according to the PNC pathway (bottom, cf. Figure 1) with potential effects of ion (top) and PNC (middle) adsorption by an additive (red ellipsoid). For strong ion-additive interactions and/or high additive concentrations, the formation of coacervates is expected (if the additive chemistry and structure allows), which inhibit mineral formation entirely. Corresponding size regimes span the whole colloidal domain depending on the type of mesophase formed [27]. For weaker ion-additive interactions and/or low additive concentrations, PNC binding by the additive (middle) becomes probable, and the process can proceed toward particle formation. Possibilities for additive control during the latter process are discussed in more detail in Section 3.2, also see Figure 3. For explanation see the text.
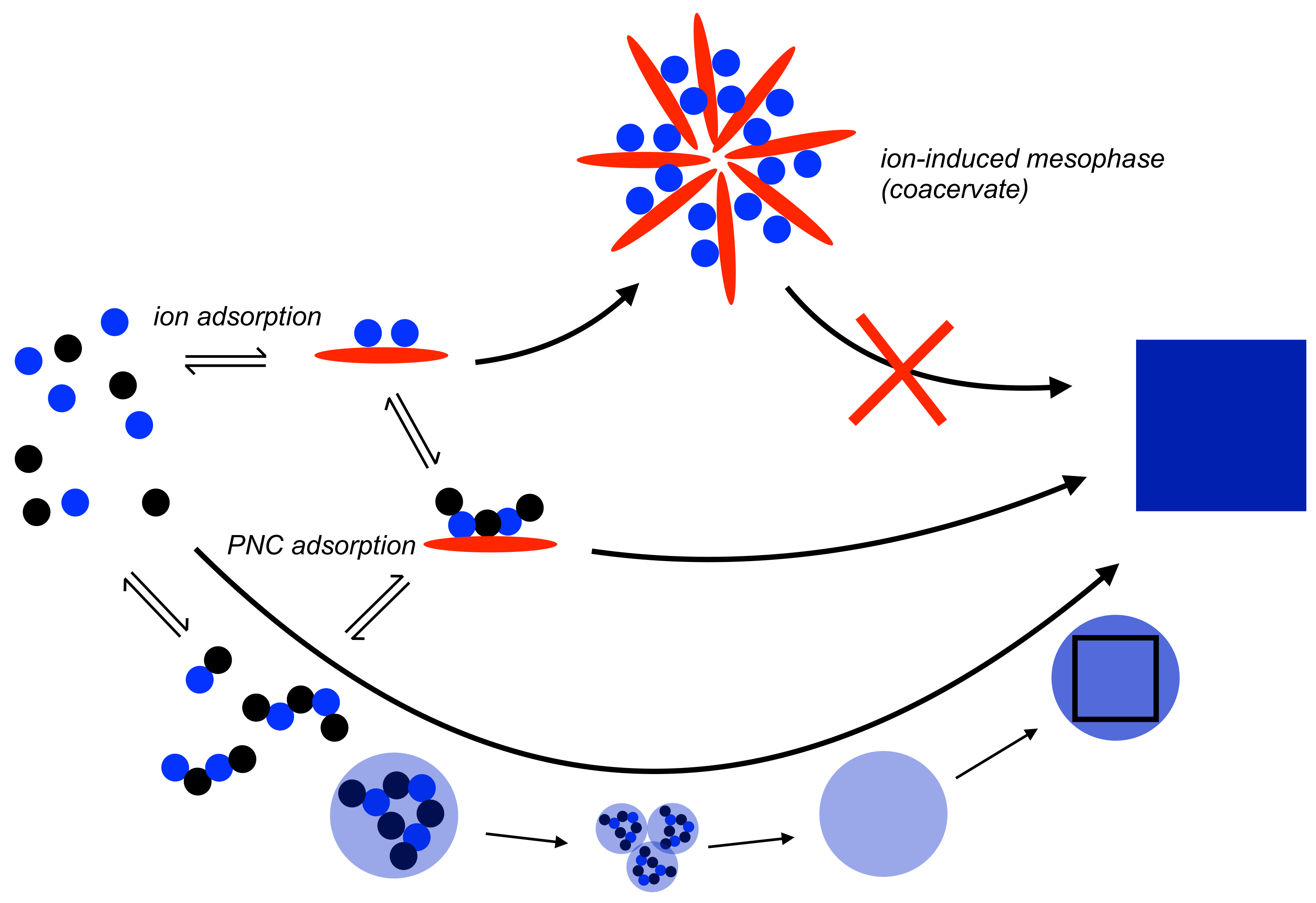
Figure 3.
Schematic illustration of the mechanism of nucleation according to the PNC pathway (bottom, cf. Figure 1) with the effects of PNC and nano-droplet adsorption by an additive (red ellipsoid). For strong ion-additive interactions and low additive concentrations (or medium-to-low-strength interaction but high additive concentrations), the additive becomes incorporated in the liquid intermediates, and may kinetically stabilise these intermediate states that can grow into macroscopic “polymer-induced” liquid precursors (PILPs; note that these species are rather polymer-stabilised than polymer-induced states [45,49]), reaching sizes of hundreds of micrometres that can be observed by means of light microscopy [41]. This pathway is expected for additives that also inhibit dehydration and/or coalescence of nano-droplets. Eventually, single crystals with complex shapes can be obtained in this PILP-mediated process [41]. As discussed in Section 3.1, the binding of PNCs by additives in favourable configurations can also induce liquid-liquid separation as indicated by the bold blue arrow. For further explanation see the text.
Figure 3.
Schematic illustration of the mechanism of nucleation according to the PNC pathway (bottom, cf. Figure 1) with the effects of PNC and nano-droplet adsorption by an additive (red ellipsoid). For strong ion-additive interactions and low additive concentrations (or medium-to-low-strength interaction but high additive concentrations), the additive becomes incorporated in the liquid intermediates, and may kinetically stabilise these intermediate states that can grow into macroscopic “polymer-induced” liquid precursors (PILPs; note that these species are rather polymer-stabilised than polymer-induced states [45,49]), reaching sizes of hundreds of micrometres that can be observed by means of light microscopy [41]. This pathway is expected for additives that also inhibit dehydration and/or coalescence of nano-droplets. Eventually, single crystals with complex shapes can be obtained in this PILP-mediated process [41]. As discussed in Section 3.1, the binding of PNCs by additives in favourable configurations can also induce liquid-liquid separation as indicated by the bold blue arrow. For further explanation see the text.
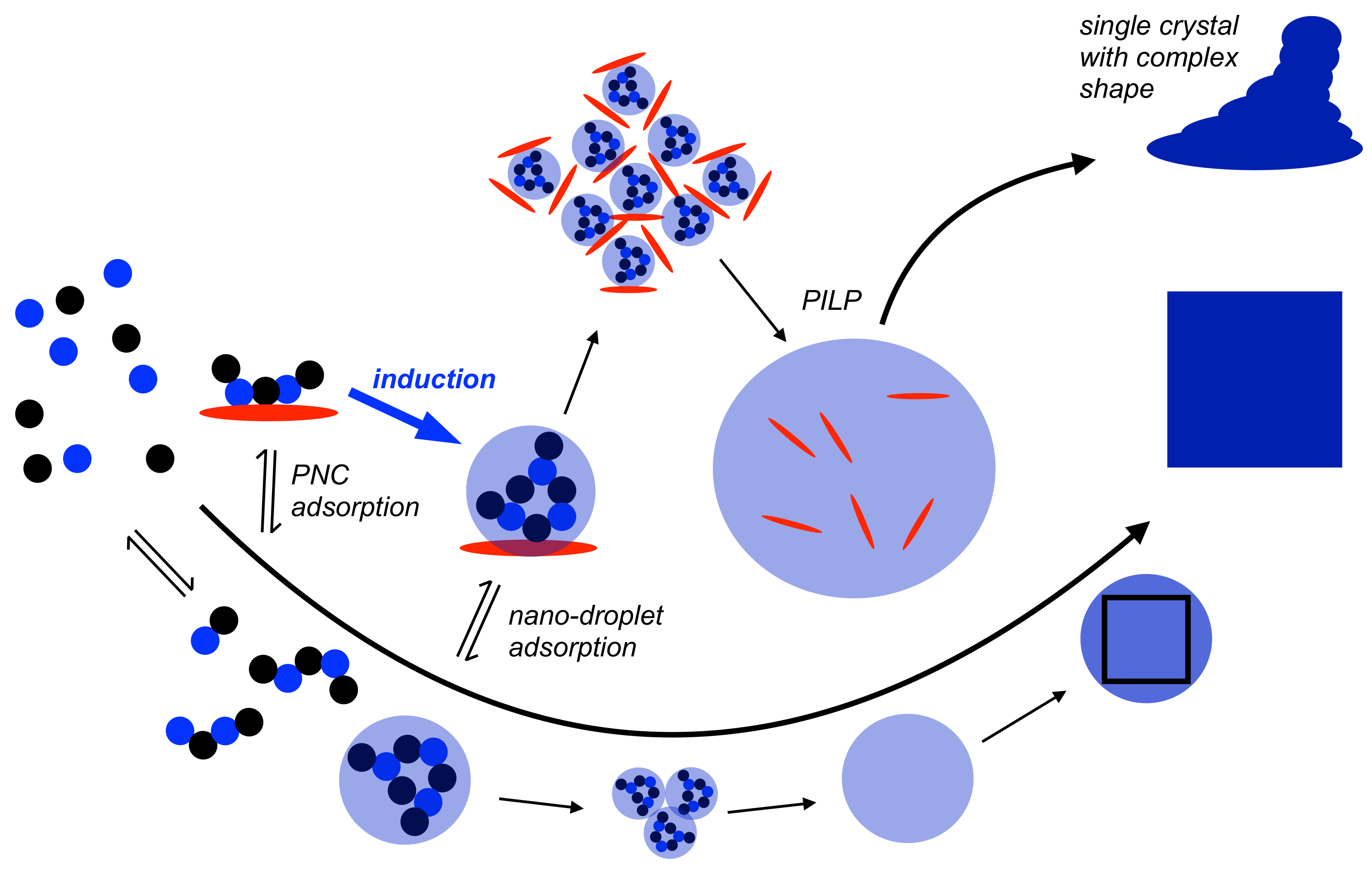
Figure 4.
Schematic illustration of the mechanism of nucleation according to the PNC pathway (bottom, also cf. Figure 1) with potential effects of nano-droplet and amorphous-solid adsorption by additives (red ellipsoid) that do not kinetically stabilise liquid intermediates, at least to any significant extent. For explanation see the text.
Figure 4.
Schematic illustration of the mechanism of nucleation according to the PNC pathway (bottom, also cf. Figure 1) with potential effects of nano-droplet and amorphous-solid adsorption by additives (red ellipsoid) that do not kinetically stabilise liquid intermediates, at least to any significant extent. For explanation see the text.
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Gebauer, D. How Can Additives Control the Early Stages of Mineralisation? Minerals 2018, 8, 179. https://doi.org/10.3390/min8050179
AMA Style
Gebauer D. How Can Additives Control the Early Stages of Mineralisation? Minerals. 2018; 8(5):179. https://doi.org/10.3390/min8050179
Chicago/Turabian StyleGebauer, Denis. 2018. "How Can Additives Control the Early Stages of Mineralisation?" Minerals 8, no. 5: 179. https://doi.org/10.3390/min8050179
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.