Carbonate Apatite Precipitation from Synthetic Municipal Wastewater
by
 , ,
, ,
Jessica Ross
 ,
,
Lu Gao
,
Orysia Meouch
,
Essie Anthony
,
Divya Sutarwala
,
Helina Mamo
and
Sidney Omelon
* Department of Chemical and Biological Engineering, University of Ottawa, Ottawa, ON K1N 6N5, Canada
*
Author to whom correspondence should be addressed.
Minerals 2017, 7(8), 129; https://doi.org/10.3390/min7080129
Submission received: 31 May 2017
/
Revised: 18 July 2017
/
Accepted: 18 July 2017
/
Published: 25 July 2017
(This article belongs to the Special Issue Nucleation of Minerals: Precursors, Intermediates and Their Use in Materials Chemistry)
Abstract
:An important component of phosphorite (phosphate rock) is carbonate apatite, as it is required for phosphorous fertilizer production due to its increased phosphate solubility caused by carbonate substitution in the apatite mineral lattice. High phosphate concentrations in municipal wastewater treatment plants are commonly reduced by precipitating iron phosphate by addition of iron chloride. We investigated the possibility of precipitating carbonate apatite from a potential range of phosphate concentrations that could be available from municipal wastewater treatment plants with anaerobic digestion reactors (5 mM–30 mM). Synthetic phosphate solutions at neutral pH were mixed in batch experiments with a calcium carbonate solution produced by dissolving calcite in contact with carbon dioxide gas, with and without carbonate apatite seed. Batch experiments were used to identify the carbonate apatite supersaturation ranges for homogeneous and heterogeneous nucleation, and the precipitates analyzed with Raman spectroscopy, powder X-ray diffraction, inorganic carbon coulometry, and scanning electron microscopy. Some precipitates contained carbonate weight fractions within the range reported for geological phosphate rock (1.4–6.3 wt %). The precipitates were spherical, poorly crystalline carbonate apatite, suggesting an amorphous precursor transformed to a poorly crystalline carbonate apatite without changing morphology.
1. Introduction
The genesis of phosphorus-rich minerals such as carbonate apatite, which is a component of the valuable P-rich ore known as phosphorite or phosphate rock (PR), was an unanswered geological question for many years. Environmental conditions do not commonly generate inorganic orthophosphate (Pi) concentrations high enough for spontaneous phosphate mineral nucleation events. Theorized mechanisms for PR precipitation included inorganic precipitation and biologically-mediated precipitation [1,2]. One mechanism for Pi concentration in the marine environment is the accumulation and storage of polyphosphates (polyP: (PO3−)n) within sulfide-oxidizing bacteria (genera Thiomargarita [3] and Beggiatoa [4]) during oxic environmental conditions. Periodic flow of anoxic waters into the bacterial mats causes the bacteria to switch their metabolism to a process that breaks down their polyP stores into Pi [5]. The dissolved Pi concentration in these extracellular matrices increases, and was measured to be 300 μM [6]—order of magnitudes above the average ocean Pi concentration of <1 μM [7]. When the Pi concentration increases upon polyP breakdown, the calcium and carbonate ions co-precipitate as apatite within the bacterial mat. The resultant high P content of ancient and modern apatite-rich ores qualifies them as PR. PR is a valuable resource because it is the only available phosphate mineral that can be economically used to produce phosphorus fertilizer, due to its increased solubility caused by its carbonate content [8]. PR is dissolved in sulfuric acid; this wet process produces phosphoric acid for phosphorous fertilizers [9]. The carbonate content of PR ranges from approximately 1.4–6.3 wt % CO32− [10]. PR is a valuable and non-renewable resource [11,12].
A similar biological process of Pi concentration as polyP, followed by Pi release, Pi concentration increase, and precipitation is observed in municipal wastewater treatment plants. Biological phosphorus removal processes have been developed to enhance the uptake of Pi by polyP accumulating organisms [13]. In oxic conditions, the polyP-accumulating bacteria uptake Pi and generate intracellular polyP. Upon anoxic conditions within anaerobic digester reactors, these organisms hydrolyze their intracellular polyP and release Pi into solution. The resulting increased Pi concentration is decreased before treated water is released to the environment to avoid ecological upsets [14]. Soluble di- or trivalent metal chloride salts are added to reduce the Pi concentration by precipitating metal phosphate [15], which is a component of biosolids that are produced by many municipal wastewater treatment processes. Although sometimes applied to soils, the low solubility of iron phosphate in biosolids limits Pi availability to plants [16]. This processing results in a loss of Pi to the food chain.
We wished to investigate if a carbonate apatite could be precipitated from Pi at concentrations available within municipal wastewater treatment plants with anaerobic digestion reactors (ranging from 2.5 to 30 mM Pi), by precipitation with a dissolved calcium carbonate solution. Calcite, a calcium carbonate mineral, in equilibrium with an apatite-precipitation solution does not spontaneously precipitate a carbonate apatite product [17]. Therefore, a more concentrated calcium carbonate solution is required to generate a supersaturated solution with respect to carbonate appetite for our target Pi-concentration ([Pi]) solutions. Calcite solubility was increased by preparing a saturated solution of calcite in potable water, then contacting this slurry with gasses of different CO2 fractions that simulated flue gas CO2 concentrations that could be available at municipal wastewater treatment plants. Some municipal wastewater treatment plants with anaerobic digestion reactors produce a methane-rich biogas, and burn this biogas in an on-site electric power plant [18]. We investigated the possibility of mixing this calcium-bicarbonate-rich solution, which will be referred to as a saturated Ca-CO3 solution, with representative [Pi] to precipitate a carbonate apatite mineral with a carbonate fraction similar to PR, with and without seed.
Seeding a crystallization process is expected to reduce the supersaturation required for precipitation [19]. Both protein-free biological carbonate apatite, and solids produced by homogeneous nucleation experiments were tested. This system is based on synthetic calcium, carbonate, and phosphate sources. The phosphate sources from a wastewater treatment plant contain a myriad of other chemical components which have unknown impacts on carbonate apatite precipitation with this strategy. This work was pursed to determine if the simplest case scenarios could generate carbonate apatite. Precipitation of carbonate apatite from Pi in municipal wastewater treatment streams could generate a potential useful resource for the wet-process phosphorous fertilizer industry.
2. Materials and Methods
2.1. Calcium Carbonate Solution Preparation
ACS grade calcium carbonate (identified as calcite by powder X-ray diffraction and Raman spectroscopy, MP Biomedicals, LLC, Solon, OH, USA) was mixed with potable water (pH 9, 0.2 mM Ca2+, 0.2 mM inorganic carbon, City of Ottawa) at a concentration of 1 g CaCO3/L overnight. N2–CO2 mixed gases with 7% CO2 (representative of natural gas fired power plant flue gas composition [20]), 15% CO2 (representative of the power plant flue gas composition powered by biofuel from a wastewater treatment plant [21]) and 100% CO2 were mixed, then bubbled at a flow rate of ~0.1–0.4 L/min through 1 L of the calcium carbonate slurry in a closed, stirred tank reactor with a gas outlet ducted to a fume hood. The pH and calcium concentrations were monitored as function of time until the values stabilized. The pH was measured with a pH probe (AR10, Orion) and the calcium concentrations measured with colourimetry [22]. Samples were read from a 96 well plate by a spectrophotometer (Epoch Microplate Spectrophotometer, BioTek, Winooski, VT, USA).
2.2. Unseeded Carbonate Apatite Batch Precipitation
Inorganic phosphate solutions used throughout all experiments were prepared by dissolving Na2HPO4 (ACS grade, Sigma-Aldrich, Oakville, ON, Canada) in distilled and deionized water. The inorganic phosphate (Pi) concentrations used ranged from 2.5 mM Pi to 30 mM Pi, based on the Pi concentrations that are available from different municipal wastewater treatment process streams with aerobic and anaerobic sludge treatment. Pi concentrations were measured by colourimetry with the vanadomolybdate assay [23]. The batch precipitation experiments for 2.5, 5, 10, and 20 mM Pi solutions were repeated four times, and the 30 mM Pi solution batch experiment was repeated three times. The experiments are referred to with the initial Pi concentration ([Pi]) before mixing.
The different [Pi] solutions were mixed with an equal volume of a saturated Ca-CO3 solution decanted from calcite in potable water that was mixed with 100% CO2 for a minimum of 20 min contact time. 1:1, 250 mL volumes of the saturated Ca-CO3 and Pi-solutions were mixed in a beaker at room temperature, sealed, and gently shaken. An aliquot was taken of each of the initial solutions prior to mixing to calculate the theoretical initial mixed concentration, and 5 days after mixing. A 5 day contact time was selected to ensure that precursor amorphous phases would recrystallize to apatite, as this transition has been reported to occur within 48 h [24,25]. The initial and final pH values were recorded. The pH was not controlled during these series of experiments. The difference in initial and final [Ca2+] and [Pi] were used to calculate percentage changes, and to calculate the theoretical precipitate molar Ca/P ratio obtained by the reduction in [Ca2+] and [Pi]. Observations regarding whether or not precipitate was immediately visible and if scale formed on the glassware surfaces were noted, as this would indicate whether the saturation state with respect to carbonate apatite was sufficient for heterogeneous nucleation on glass surfaces and other contaminants, or above the critical supersaturation for homogeneous nucleation. After five days, the slurries were vacuum-filtered through a glass filter, rinsed with deionized and distilled water, and vacuum-dried at 40 °C and 5 mm Hg.
The solubility product and the ion activity product are used to calculate the supersaturation of a given salt [26]. Since the exact composition and solubility product of carbonate apatite are unknown, a simplification of the supersaturation index will be used to compare the saturation states. The measured calcium concentration ([Ca2+]) multiplied by the inorganic phosphate concentration ([Ca2+] × [Pi]) will be used as a proxy saturation index, as the dissolved carbonate concentration is challenging to measure accurately. This is an oversimplification of a complex problem. As the carbonate substitution for phosphate or hydroxyl molecules is not known, stoichiometric coefficients will not be used in this saturation index, as used by Fleisch [27].
The goal of this work is to determine if mixing a saturated Ca-CO3 solution produced by calcite in potable water that was in contact with CO2 gas, with different [Pi] solutions generates a supersaturation large enough to spontaneously produce a synthetic PR analogue material through homogeneous nucleation. If possible, then heterogeneous precipitation in the presence of seed would also be possible, because this condition requires a lower supersaturation.
2.3. Seeded Carbonate Apatite Batch Precipitation
Given that the supersaturation required for heterogeneous nucleation is lower than that required for homogeneous nucleation, seeded, batch precipitation tests were undertaken to determine if crystal growth could occur with the lowest and highest supersaturation conditions that generated spontaneous precipitation (5 mM and 30 mM Pi). As the desired final precipitate is a carbonate apatite, it was assumed that biological apatite, which has a higher substituted carbonate fraction in carbonate apatite than PR (5–9 wt % CO32− [28,29]), could be a suitable seed material. The unseeded batch precipitation procedure was followed with the addition of 0.5 or 2 g/L bone mineral in the batch experiment. These seeded experiments were performed in duplicate.
To prepare the bone mineral, bovine bone was crushed, Tris-washed, lyophilized, and defatted in a 2:1 chloroform:methanol solution. The bone powder was deproteinized with bleach, rinsed with distilled and deionized water, and dried in a vacuum oven at less than 100 °C. The final product was ground with a mortar and pestle, and the fraction that passed below a 63 μm sieve was used as seed. A second set of batch precipitation experiments was undertaken, seeded with precipitates produced by an unseeded batch precipitation (referred to as “ppt”). This precipitation was induced by mixing 1 L of 27 mM Pi (similar to a known [Pi] in a local municipal wastewater treatment plant stream) with 1 L of 6 mM Ca2+ solution from CaCO3(s) dissolution in potable water (initial concentration 8 mg/L (0.2 mM) Ca2+, pH 9) in equilibrium with 100% CO2. The precipitate was vacuum filtered, rinsed with distilled and deionized water, and dried in a vacuum oven at 40 °C and 5 mm Hg. This seed was used in the 5 mM initial [Pi] test, as it was expected to provide surface chemistry with a closer match to solid phase favored to grow in these conditions. Precipitate produced from this seeded, batch crystallization experiment was used as seed for a 7 mM initial [Pi] batch crystallization experiment, which is characteristic of the lowest [Pi] in a local municipal wastewater treatment plant stream.
2.4. Precipitate Characterization
As bone mineral is composed of a higher carbonate weight fraction than PR, only unseeded precipitates and precipitates seeded with homogeneously nucleated seed were characterized in order to avoid confounding the new precipitate chemistry with the bone mineral seed chemistry.
2.4.1. Scanning Electron Microscopy
Samples were dried, mounted with carbon tape, and secondary electron images taken (Phenom PRO, Pheonom-World, Eindhoven, The Netherlands) at magnifications of 165×, 500×, and 1000×. The images were scanned at 10 kV and a resolution of 2048 dpi, with an exposure time of 26 s.
2.4.2. Raman Spectroscopy
Raman spectroscopy was undertaken with a WITec Alpha 300 confocal microscope with a 20× lens (WITec GmbH, Ulm, Germany). The laser source used was either 785 nm (XTRA High Power Single Frequency Diode Laser, Toptica Photonics, Victor, NY, USA), 488, or 532 nm (WITec GmbH, Ulm, Germany). Calibration was performed on the 520 nm Raman shift of Si.
Dry powdered samples were placed on a quartz slide, and data was collected with an integration time of 1 s for a minimum of 45 s. The Raman shifts of greatest interest were 960 cm−1 for the Pi identification in apatite, 1070 cm−1 for carbonate in apatite, and 1090 cm−1 for carbonate in calcite. Silicon, synthetic hydroxyapatite (HAP, Sigma-Aldrich, Oakville, ON, Canada), calcite (CaCO3, MP Biomedicals, LLC, Solon, OH, USA), and bone powder were used as standard materials. The resulting spectra were filtered to remove cosmic ray interference. OPUS Spectroscopy software (v 6.5, Bruker Optiks, Ettlingen, Germany) was used to remove the baseline, smooth (9 smoothing points) slightly, and normalize the spectra that were then plotted with OriginPro 9.1 (OriginLab Corporation, Northampton, MA, USA).
2.4.3. Powder X-ray Diffraction
Precipitates were analyzed with a Multiflex D3 (Rigaku Americas Corporation, The Woodlands, TX, USA). The products were prepared on a low-background Si sample-holder, and scanned between 20–45° 2θ at a scan rate of 1°/min. The raw data were compared with ICDD PDF file for hydroxyapatite with PDXL 2 (Version 2.5.1.2, Rigaku Americas Corporation, The Woodlands, TX, USA).
2.4.4. Inorganic Carbon Coulometry
A carbon coulometer (Model #5012) (UIC Inc., Joliet, IL, USA) was used to measure the weight percent carbonate of the precipitates. A measured sample mass was dissolved in a CO2-free chamber with 2 M perchloric acid (HClO4, Sigma-Aldrich, Oakville, ON, Canada), and the CO2 gas evolved from carbonate ions− passed to another chamber to be quantified. Quantification of CO2 (g) is accomplished by a titration reaction that is neutralized by redox reaction. The coulombs required for titration are proportional to the number of CO2 (g) molecules that initiated the titration reaction. The coulometer calculates the mass of carbonate from titrated CO2, allowing for the calculation of the weight percent carbonate.
2.4.5. Solubility
A few milligrams of precipitates were dissolved in 5 mL of distilled and deionized H2O for one week under gentle mechanical shaking so they could reach equilibrium with the solution. The colorimetric methods were used to measure the resulting [Ca2+] and [Pi] that were assumed to be in equilibrium with the precipitates. These values were used to calculate [Ca2+] × [Pi] values, as well as the molar ratio of Ca/P in solution.
3. Results
3.1. Calcium Carbonate Solution Preparation
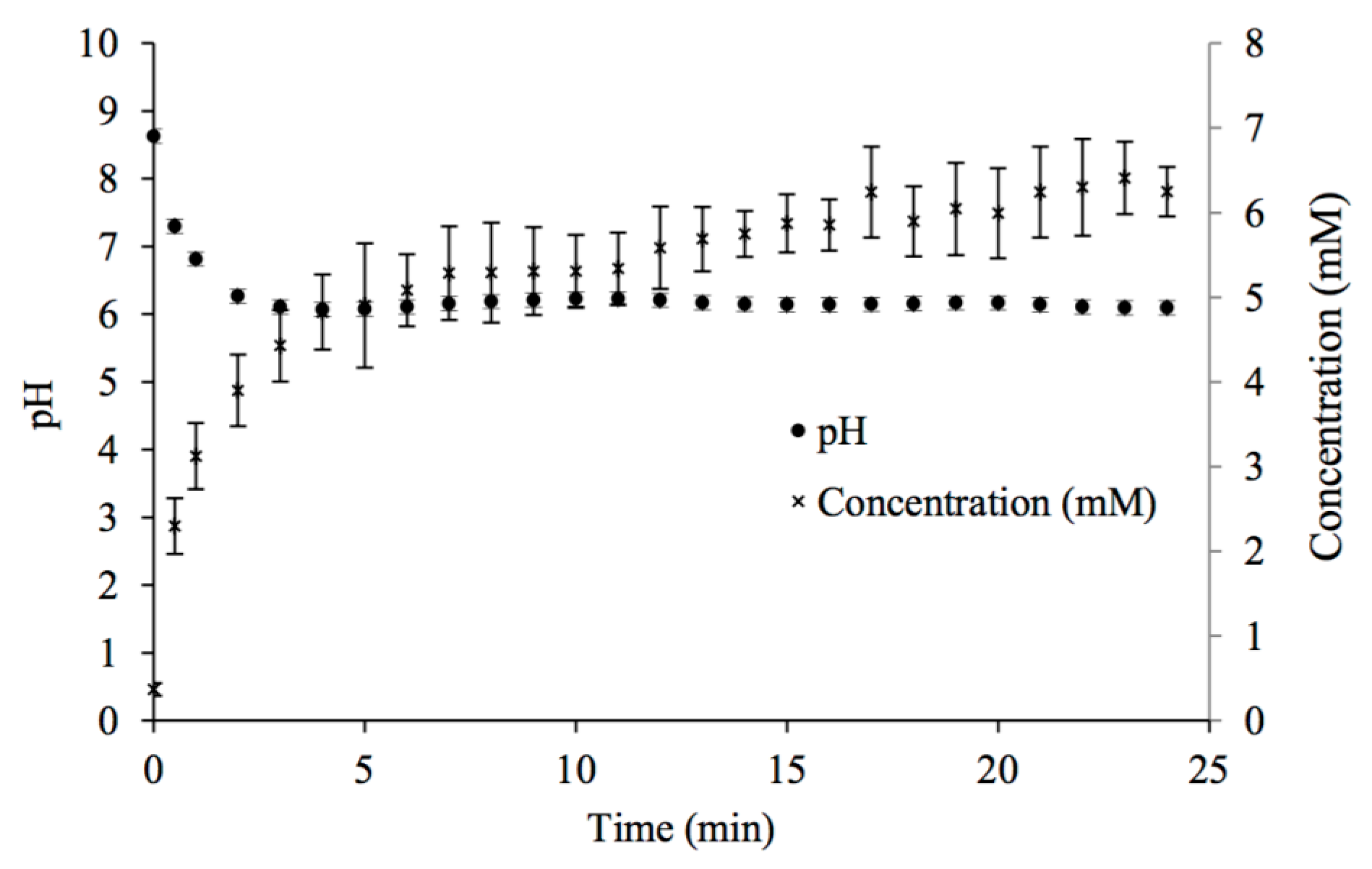
Figure 1 presents an example of initial conditions and the changes in pH and [Ca2+] with time during contact with 100% CO2. Table 1 presents the equilibrium [Ca2+] and pH for the different % CO2 values.
Solutions from the 100% CO2 experiments were used as calcium and carbonate sources in the subsequent carbonate apatite batch precipitation experiments. Ca-CO3 saturated solutions were used as soon as possible after their production, as calcium carbonate precipitates from the decanted solutions as they shift to equilibrium with the atmosphere.
3.2. Unseeded Carbonate Apatite Batch Precipitation
The initial and final concentrations of calcium and Pi in the batch precipitation experiments, as well as the final pH, the theoretical percent calcium and Pi removed from solution, and the theoretical Ca/P in the precipitate are listed in Table 2, Table 3 and Table 4. Before mixing, the average [Ca2+] from CaCO3 solutions used was 6 ± 1 mM, while the target [Pi]s were 2.5, 5, 10, 20, or 30 mM. Note that the sample names reflect the initial [Pi] before mixing with the saturated Ca-CO3 solution.
The concentration changes are shown graphically in Figure 2, where it becomes apparent with the different final [Ca2+] (arrow head) that the limiting reagent is different above and below the case [Pi] = 5 mM. For [Pi] of 5 mM or less, only 56–17% of the initial mixed [Ca2+] precipitated from the solution, whereas 98–94% of the [Ca2+] is reduced when mixed with [Pi] > 10 mM. When the mixed [Pi] is higher than the [Ca2+], the reaction is limited by the [Ca2+]. The pH is likely buffered by the residual [Pi] and carbonate. The different theoretical Ca/P ratios calculated by the loss of these ions from solution also suggest that there is a range in this theoretically calculated chemical composition for the homogeneously nucleated precipitate phases, as the theoretical precipitate Ca/P values are not statistically significantly different (one-way ANOVA, with Bonferroni means comparison).
If the initial [Pi] was 10 mM or higher, the solution became cloudy immediately upon mixing with the Ca-CO3 solution. This indicated that a supersaturation state above the critical supersaturation for homogeneous nucleation was achieved (Scritical, Figure 2). With initial [Pi] = 5 mM, there was no evidence of homogeneous nucleation, but precipitate formed on the beaker walls. This would suggest that the experimental conditions for the metastable zone, below Scritical at an initial [Pi] = 5 mM, and above carbonate apatite saturation were identified. The 2.5 mM initial [Pi] experiment did not produce enough solids to be collected and quantified, but the measurable decrease in both [Ca2+] and [Pi] suggests that heterogeneous precipitation occurred. It was assumed that Scritical in this system lies between an initial [Pi] between 5 and 10 mM before mixing (2.5 and 5 mM after mixing), and [Ca2+] of 3 mM after mixing (dashed vertical line in Figure 2). For this system, Scritical with the simplified index [Ca2+] × [Pi] lies between 7.5 and 15 mM2.
3.3 Seeded Carbonate Apatite Batch Precipitation
The initial and final concentrations of calcium and Pi in the seeded batch precipitation experiments, initial and final pH, percent calcium and Pi removed from solution, and the calculated Ca/P in the precipitate are listed in Table 5, Table 6 and Table 7. The maximum [Ca2+] from Ca-CO3 solutions used was 6 ± 1 mM, while the [Pi] solution concentrations were 5, 7, or 30 mM before mixing with the Ca-CO3 solution.
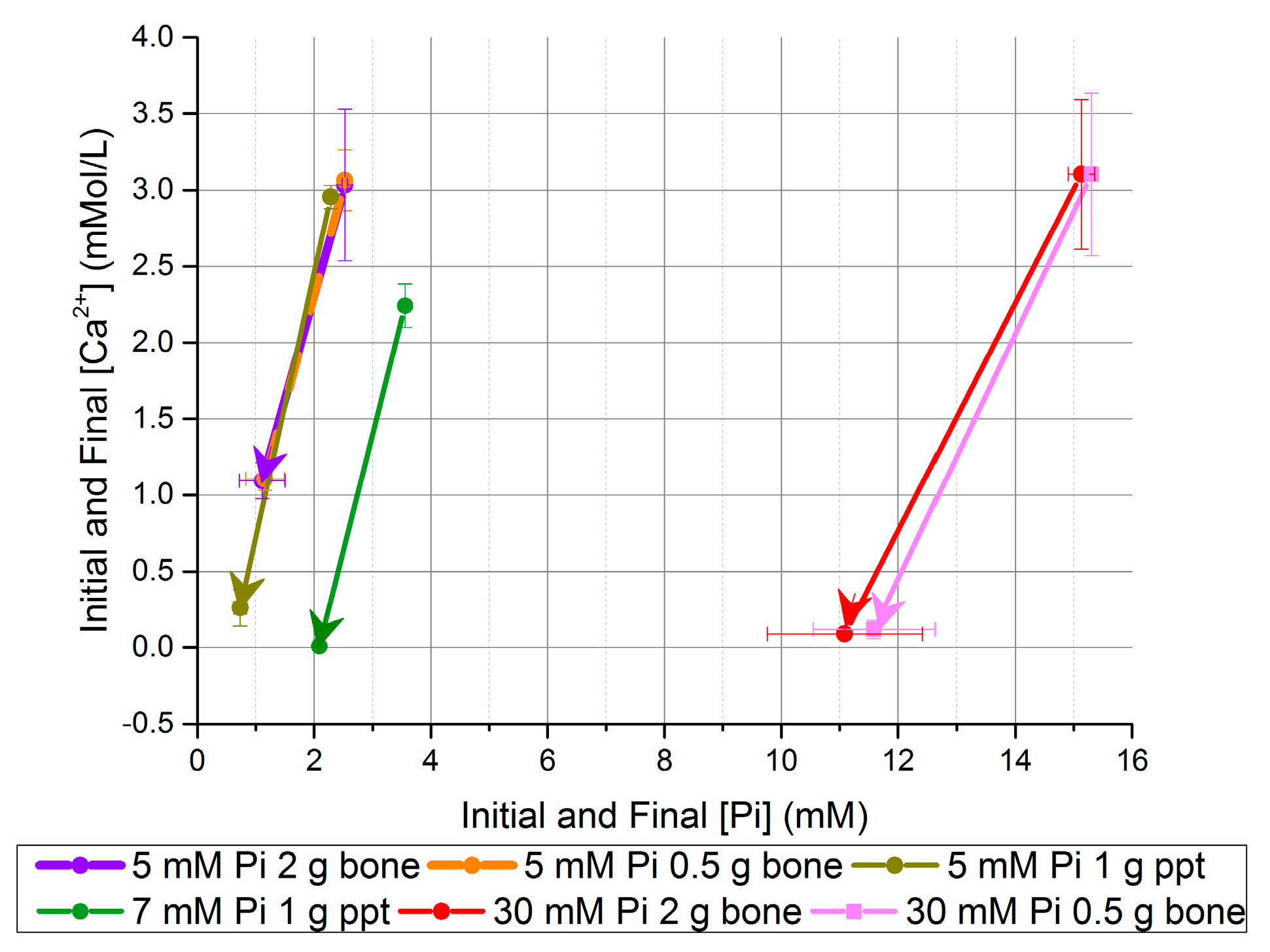
An increase in bone seed concentration did not dramatically increase the percent [Ca2+] or [Pi] reduction, but did reduce the theoretical precipitate Ca/P from 1.44 to 0.95. 5 and 7 mM [Pi] solutions seeded with homogeneously nucleated precipitate (ppt) increased the equilibrium pH and reduced the equilibrium [Ca2+] × [Pi] (Table 8). Figure 3 compares the [Ca2+] and [Pi] changes for the bone mineral- and precipitate-seeded experiments; it highlights the additional [Ca2+] reduction with ppt-seeded experiments, and the small effect of increased bone mineral seed concentration on the equilibrium solution composition.
The simplified saturation index ([Ca2+] × [Pi]) and final pH for the seeded and unseeded experiments are listed in Table 8.
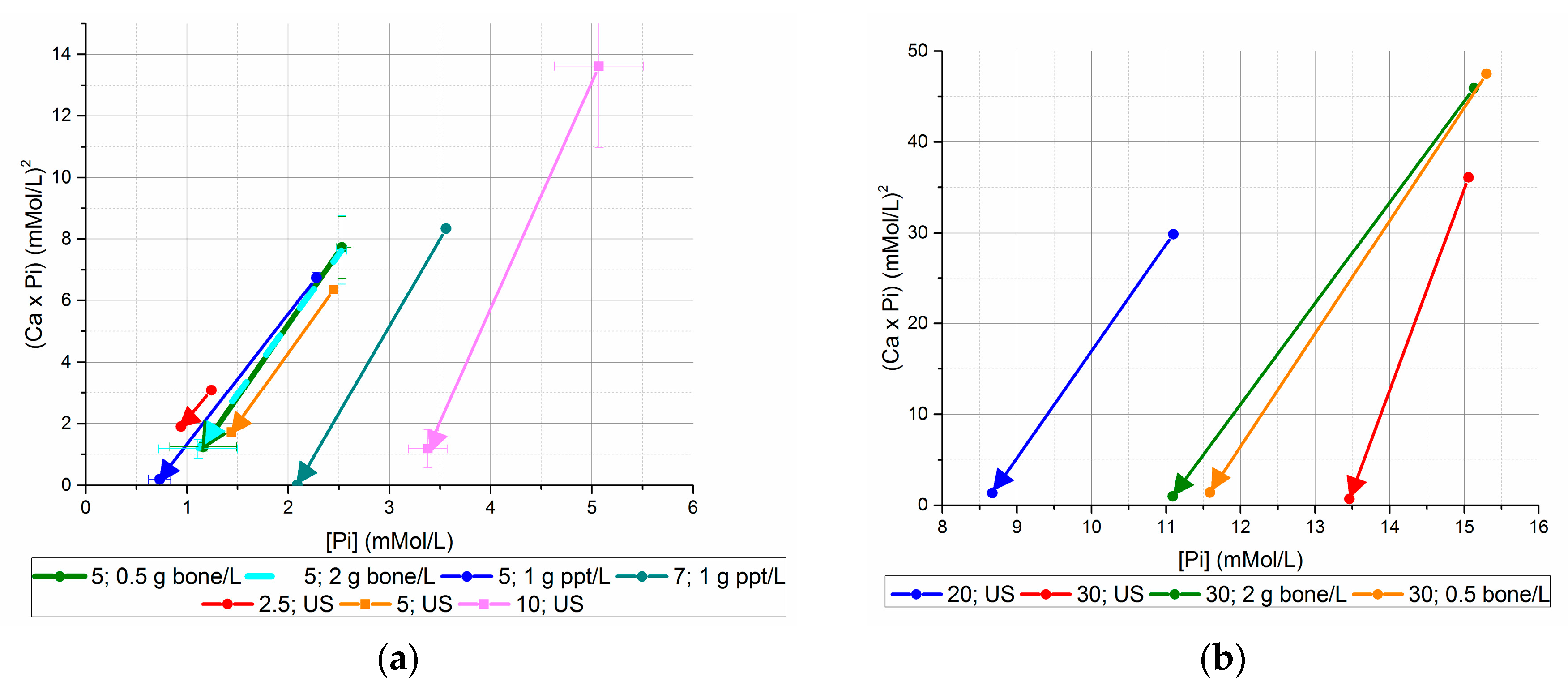
These initial and final [Ca2+] × [Pi] data plotted with respect to the initial and final [Pi] are summarized in Figure 4a,b.
All final [Ca2+] × [Pi] concentrations for bone mineral-seeded and unseeded precipitation experiments were at or higher than 1 mM2 except for the 30 mM initial [Pi], unseeded experiment. Bone mineral seed lowered the final [Ca2+] × [Pi] value for the 5 mM [Pi] experiment, but not the 30 mM [Pi] experiment. The lowest final [Ca2+] × [Pi] values were achieved with the use of precipitate seed from previous unseeded experiments, where the values were 0.2 mM2 or less for initial concentrations of 5 and 7 mM initial [Pi].
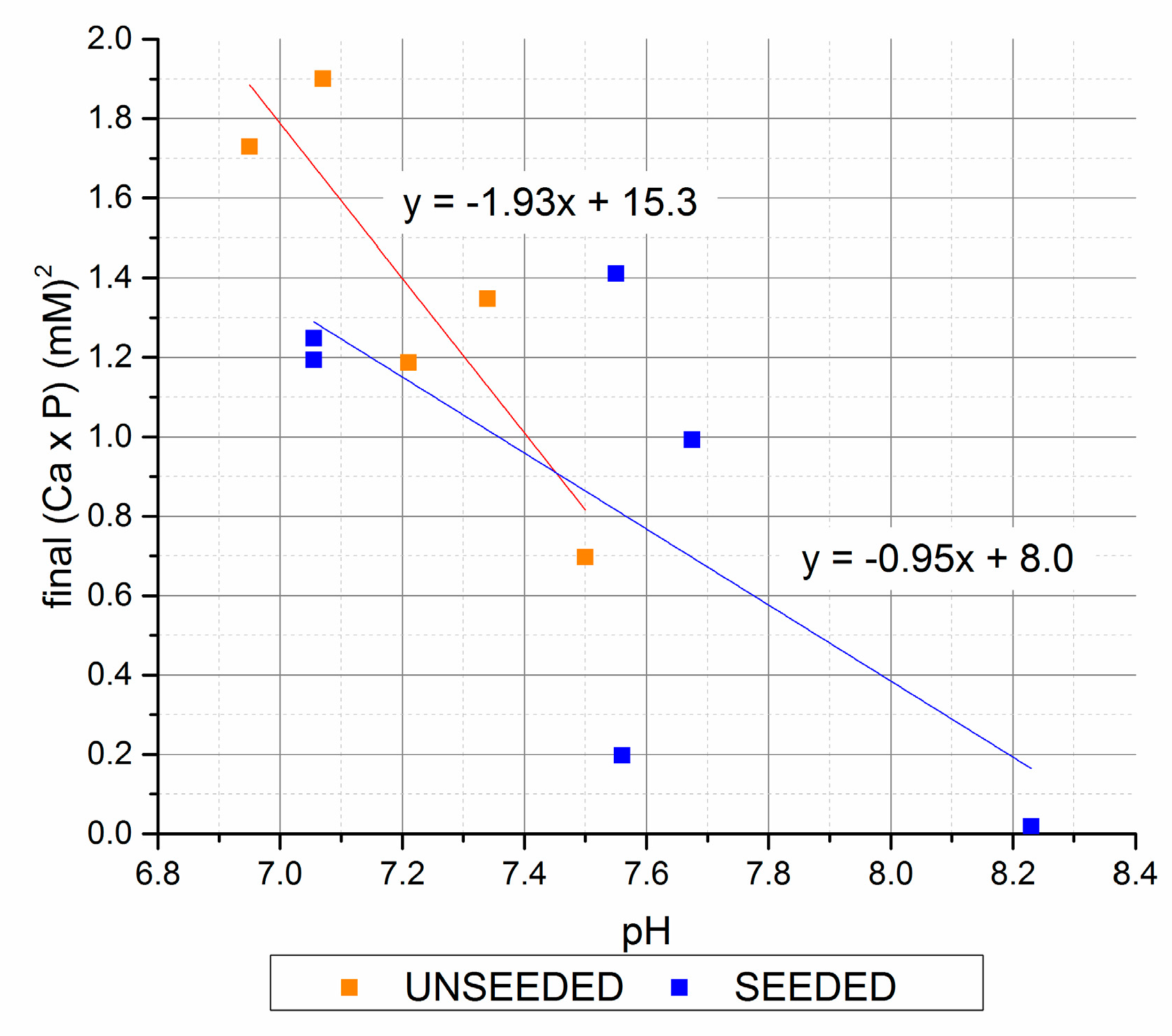
The pH followed an inverse trend of final [Ca2+] × [Pi], with the highest pH values associated with the 7 mM initial [Pi] group with precipitate seed, and the 30 mM initial [Pi] unseeded precipitations. A plot of the final [Ca2+] × [Pi] values vs. pH (Figure 5) shows the correlations for the increase in pH with decrease in [Ca2+] × [Pi]. This reflects the increase in [PO43−] and [CO32−] equilibrium speciation with increasing pH that would be expected to decrease the equilibrium [Pi] above apatite saturation.
The different slopes of the linear regression lines for the seeded and unseeded precipitation cases suggests a possible effect of the seed chemistry on the solution chemistry.
3.4. Precipitate Characterization
3.4.1. Scanning Electron Microscopy
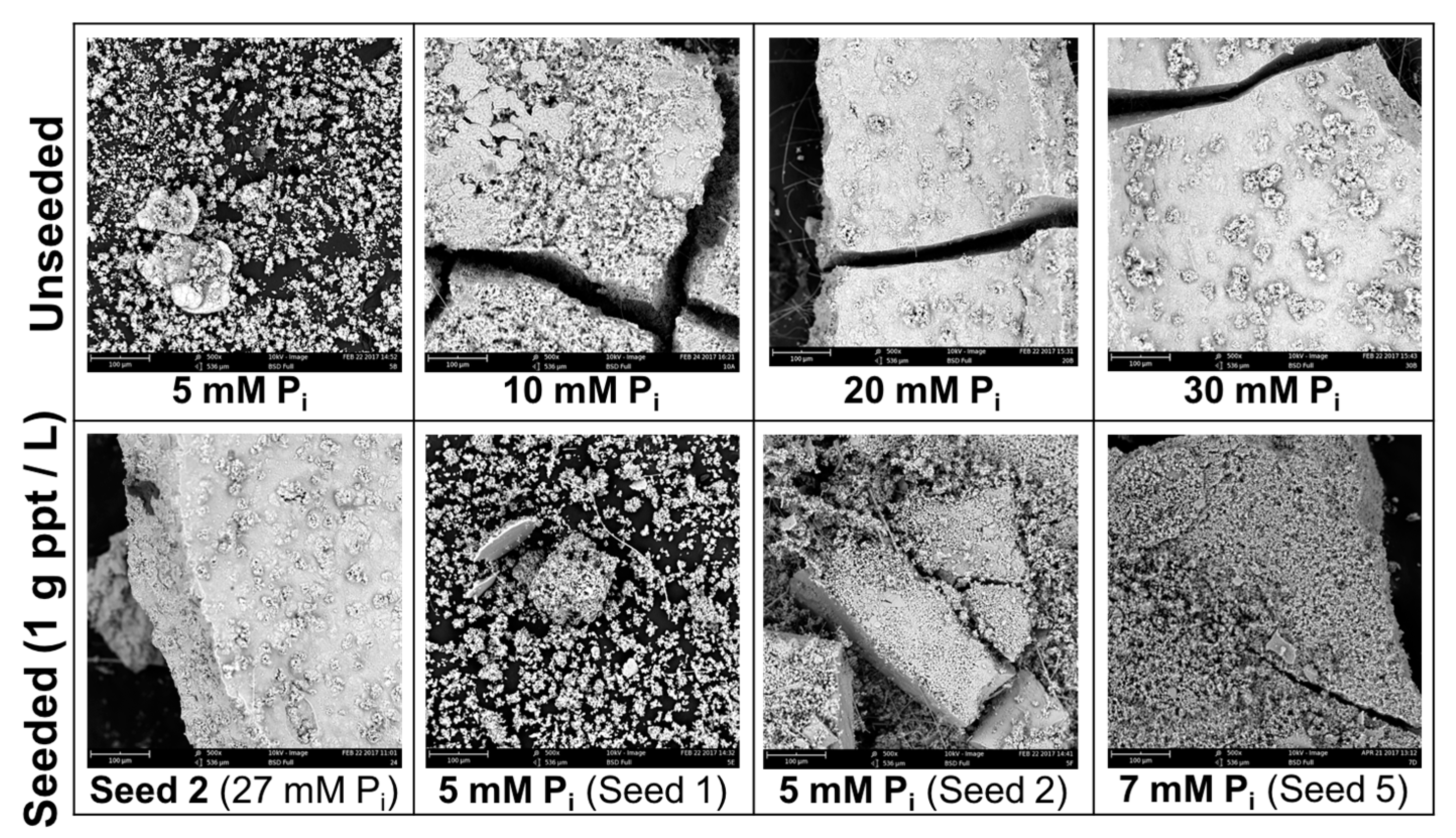
Representative images for 5 and 30 mM initial [Pi] (unseeded) and 7 mM (seeded) taken at 5000× magnification are presented in Figure 6. Products produced at intermediate initial [Pi] were similar to the 30 mM results. Additional SEM images of unseeded and precipitate-seeded experiments are presented in Appendix A, Figure A1.
An example of higher-resolution images of the seeded precipitate from the 7 mM [Pi] experiment are shown in Figure 7. As the crystal habit for synthetic carbonate apatite is plate-like [30], the spherical solids, ranging in diameter from approximately 2 to 7 μm, suggest the preservation of the spherical structure characteristic of an amorphous precursor.
3.4.2. Raman Spectroscopy
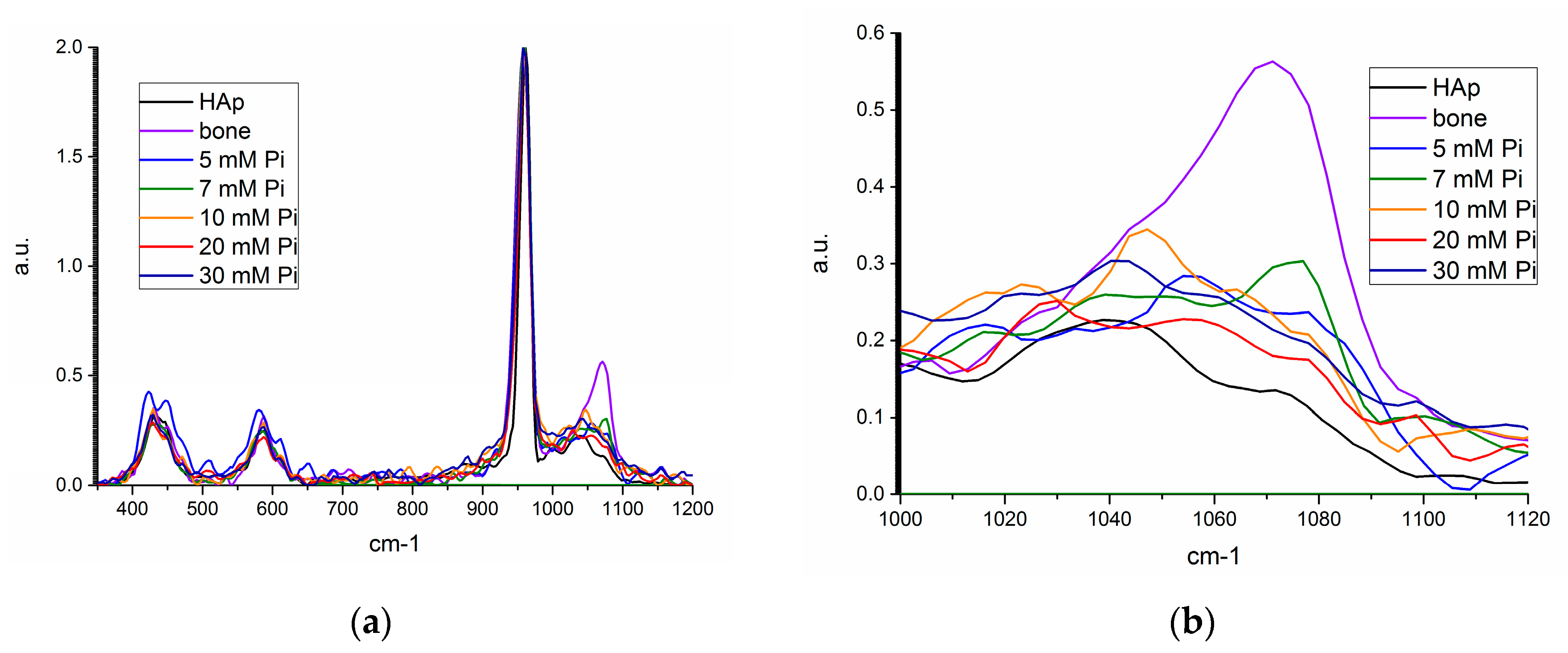
Raman shifts of the unseeded precipitates and the product from the seeded, 7 mM [Pi] precipitation are compared in Figure 8a,b. There was no evidence of an amorphous calcium phosphate (ACP) phase, which has been associated with a ν1 phosphate molecule shift at 951 cm−1, nor a ν1 carbonate shift at 1081 ± 3 cm−1 for ACP precipitating systems that absorbed ambient carbon dioxide [24]. The dominant precipitate Raman shift is for the ν1 phosphate molecule shift at 960 cm−1 of apatite, with shifts similar to the ν3 phosphate molecule attributed at 1041 ± 3 cm−1, and a ν1 carbonate shift at 1073 ± 2 cm−1 [24]. The intensity of the ν1 carbonate shift was largest for bone mineral and the 7 mM precipitate with homogeneously nucleated seed, which is supported by their larger measured carbonate weight fractions.
Raman spectroscopy allowed for the identification of the characteristic P-O shift for phosphate in apatite, and supporting evidence for the measured inorganic carbonate fraction (Section 3.4.4). There was no evidence of calcite, with its ν1 carbonate shift at 1090 cm−1, or another crystalline phosphate mineral phase that is distinguishable from apatite, such as octacalcium phosphate, brushite, or whitlockite.
3.4.3. Powder X-ray Diffraction
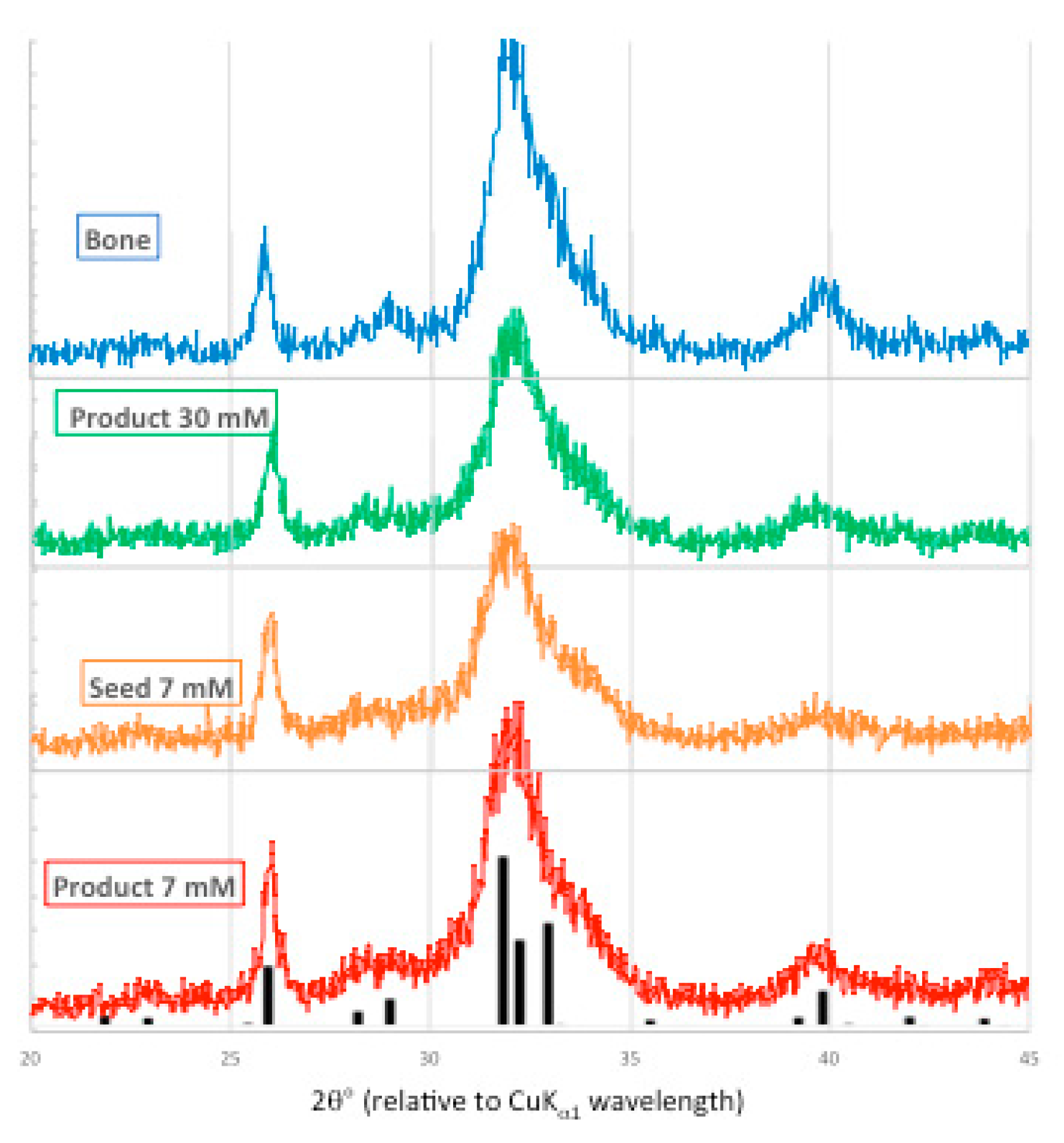
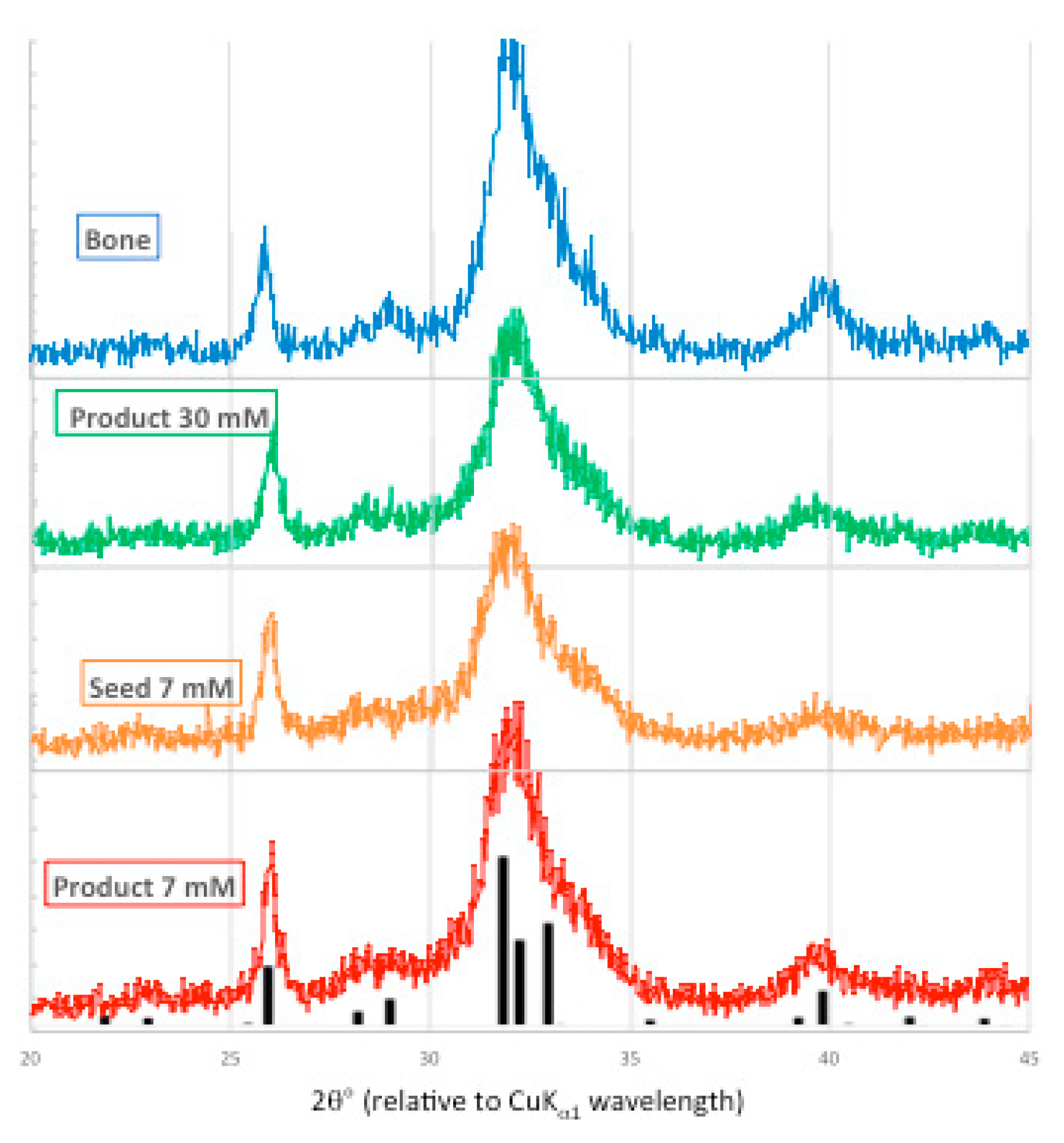
Powder X-ray diffraction patterns generated by bone mineral, the 30 mM Pi unseeded experiment, and the seed and precipitate from the 7 mM [Pi] experiment are presented in Figure 9.
These X-ray diffraction patterns are similar in peak position, and are representative of the low intensity and wide shape of biological apatite. This indicates that the precipitates are composed of small polycrystalline domains and/or are poorly crystalline, and/or highly substituted apatite. For orientation, the ICDD powder diffraction file for hydroxyapatite (01-089-4405) is overlaid on the precipitate product data.
3.4.4. Inorganic Carbon Coulometry
The measured carbonate weight fraction of the unseeded precipitates and the 7 mM [Pi] seeded precipitate are presented in Table 9.
There is a trend of decreasing carbonate weight fraction with increasing initial [Pi] for the unseeded experiments. The carbonate content of the seeded product at 7 mM Pi is higher than the unseeded products. The carbonate weight fraction for the unseeded precipitates are statistically significantly different (one-way ANOVA). The carbonate values for the seeded precipitate and the highest Ca-CO3/Pi ratio (5 mM initial [Pi]) are within the reported carbonate weight fraction of phosphate rock (1.4–6.3 weight % CO32− [10]), and are lower than the percent carbonate measured in bone mineral (5–9 weight % CO32− [28,29]). These data indicate that carbonate apatite with a carbonate content similar to PR precipitates from mixing solutions with a [Pi] at or lower than 5 mM with saturated Ca-CO3 solutions in equilibrium with carbon dioxide gas and calcite, without seed, and a solution with 7 mM [Pi] with seed, at room temperature. The carbonate content is less than that for biological apatite. Future work is required to investigate the effect of seed on this carbonate apatite precipitation process.
3.4.5. Solubility
Bone mineral was determined to be more soluble that the homogeneously nucleated precipitates, with an equilibrium [Ca2+] of 0.34 mM, and [Pi] of 0.13 mM. Therefore, samples that contained bone mineral were not included in the comparison of precipitate solubility (Table 10).
Precipitates grown from previously homogeneously nucleated seed generated the highest average dissolved [Ca2+]. A one-way ANOVA showed that the average dissolved [Ca2+] from the unseeded precipitates did not change significantly with initial [Pi]. Dissolved [Pi] concentrations follow a proportional trend with increasing initial [Pi]. Statistically significant differences between [Pi] means were noted for all groups but not between any two of the 5, 7, and 10 mM Pi groups, and between the 30 and 40 mM Pi groups (Bonferroni test). Ca/P ratios for the unseeded and precipitate-seeded experiments followed an inverse trend with the initial [Pi] from which they were nucleated, and have the same statistically significant differences as the final [Pi]. Within the margins of error, a similar trend is seen with the calculated Ca/P from the [Ca2+] and [Pi] decrease in the precipitation experiments. The average calculated Ca/P values were higher than the measured solubility Ca/P values; this may result from the different solution compositions in which the precipitates were in equilibrium, and the effect of the unmeasured carbonate concentrations.
4. Discussion
The goal of precipitating a carbonate apatite from industrial-scale solutions with uncontrollable Pi concentrations generated by municipal wastewater treatment requires supersaturated conditions for carbonate apatite. This supersaturation condition could be generated by mixing Pi-rich solutions with dissolved calcium carbonate. The calcium concentrations required to precipitate carbonate apatite from a minimum of 5 mM Pi were generated by increasing calcite solubility with dissolved CO2. Calcium concentrations increased and the pH decreased by contacting increasing percentages of CO2 gas with synthetic calcite slurried in potable water, up to 6 ± 1 mM [Ca2+]. This solution, although unstable when removed from contact CO2, homogeneously nucleated and precipitated carbonate apatite from synthetic solutions with 5–30 mM Pi when mixed in a 1:1 volume ratio. Limestone and combustion flue gas could provide these reagents on the scale of municipal wastewater treatment plants; the effects of other components in these materials remain to be tested.
Unseeded precipitation tests with the highest initial [Ca2+] × [Pi] values generally produced the lowest final [Ca2+] × [Pi] values. The seeded tests did not follow this trend; homogeneously nucleated precipitate used as seed resulted in a lower final [Ca2+] × [Pi] value than bone mineral-seeded experiments. It is not known if this is due to the different carbonate content of the seed minerals, and/or could be attributed to the incongruent dissolution of apatite minerals [31,32,33]. Further investigation of the effect of seed and its chemistry is required. Seed dissolution and reprecipitation may also affect the equilibrium [Ca2+] and [Pi]; this possibility requires further precipitation characterization as a function of time. The disparity between the calculated Ca/P precipitate ratios and the measured Ca/P dissolved precipitate ratios for the unseeded and seeded products could be explained by the solution carbonate concentrations; these were not measured, and are the subject of future work. The Ca/P trend is inversely proportional to the initial [Pi], which suggests a lower carbonate fraction in the precipitate that nucleated from higher [Pi]. This was confirmed with decreasing precipitate carbonate content produced from increasing initial [Pi].
pH was not controlled in these precipitation experiments. An inverse correlation between pH and percent Pi removed was observed, which suggests that a carbonate apatite precipitation process for Pi removal could be controlled to meet the effluent [Pi] by increasing the precipitation pH. The different observed relationships between the final pH of unseeded and seeded experiments and percent Pi removed suggest that the seed may have affected the solution chemistry. Dissolution of the known higher carbonate content in bone mineral would also increase pH, [Ca2+], and [Pi]. Future work will include measuring the dissolved carbonate concentrations before and after precipitation.
Precipitates were characterized as carbonate apatite by Raman spectroscopy and X-ray powder diffraction. Inorganic carbon coulometry confirmed the carbonate content of the samples with 5 and 7 mM initial [Pi] to be within reported carbonate bounds of PR. These samples would have had a higher dissolved carbonate to Pi ratio, and this could have contributed to their higher precipitate carbonate content [34].
The spherical shape of the poorly crystalline carbonated apatite represent a possible different precursor pathway than reported by Habraken et al. [35], as they reported that ACP spheres crystallized to hydroxyapatite plates [35]. However, carbonate ions were not present in significant concentrations in their system. In this work, carbonate concentrations ranging from 1.1–1.95 weight percent were measured in unseeded precipitate; the effect of this carbonate fraction on a potential amorphous calcium carbonate phosphate precursor is not known. It is also possible that initial nuclei resulted in a spherical solid formed of small crystal domains due to a fractal growth pattern [36]. The higher weight percent content in the precipitate produced from 7 mM initial [Pi] with seed produced from homogeneous nucleation is interesting, and will be given future attention. It is not known if the seed changed the solution chemistry, and/or if heterogeneous nucleation may favor a different precipitate chemistry.
Citrate-stabilized, spherical ACP particles in phosphate-buffered saline were observed to generate crystalline apatite domains within the ACP spheres [37]. It is possible that the carbonate ions have impacted the restructuring of the initial, spherical amorphous precursor solids as they transform to a poorly crystalline carbonate apatite, without significantly changing morphology. Carbonate apatite precipitated at 60–85 °C temperatures exhibited plate morphology with a c-axis length of 291.8 ± 222.0 at lower carbonate weight percent (3.63 wt %), and a reduced c-axis length of 36.0 ± 28.4 nm for 17.8 weight percent carbonate, as measured by TEM [34]. This result also indicates a carbonate effect on apatite crystal habit, although the carbonate apatite precipitation conditions for this work were different than presented here. Seeding with a homogeneously nucleated precipitate resulted in spherical carbonate apatite solids with a significantly higher carbonate content (2.59%) when precipitated with a 7 mM [Pi] solution. The effect of seeding with homogeneously nucleated solids on the precipitate characteristics and its carbonate content requires further study.
Zou et al. reported a phase transformation from spherical amorphous calcium carbonate (ACC) to spherical vaterite for smaller (~100 nm) and more soluble ACC particles, and a dissolution-reprecipitation pathway for larger ACC spheres that generated calcite rhombohedral crystals [38]. We propose that our homogeneous precipitates initially nucleated as soluble, amorphous calcium carbonate phosphate spheres that underwent a phase transformation to carbonated apatite without creating the plate-like crystal habit of apatite. The surface of this spherical, highly substituted carbonate apatite precipitate may enable epitaxial growth of carbonated apatite with a higher carbonate fraction, but further experiments that include initial precipitate characterization, and tracking the change in precipitate size in seeded experiments are required to confirm this hypothesis.
5. Conclusions
These precipitation conditions were inspired by the availability of waste phosphate-containing solutions from municipal wastewater treatment plants, carbon dioxide gas, calcium carbonate equilibrium, and the apatite lattice that is tolerant of many substitutions. A saturated solution of potable water, calcite, and carbon dioxide generated a sufficiently high calcium concentration to precipitate carbonate apatite from synthetic phosphate solutions at room temperature that are representative of possible phosphate-containing streams from municipal wastewater treatment plants, with or without seed.
When mixed at a 1:1 volume ratio with a CO2-calcium carbonate-saturated solution with a 6 ± 1 mM [Ca2+], the minimum [Pi] to nucleate and grow carbonate apatite was 5 mM [Pi]. Homogeneous nucleation was evident for [Pi] at or greater than 10 mM [Pi]. The unseeded precipitation experiment with highest initial carbonate/phosphate concentration, and the experiment seeded with solids from homogenously nucleated precipitation experiments generated spherical precipitates less than 10 μm in size, with some groups producing carbonate fractions similar to phosphate rock. The precipitate weight fraction of carbonate was higher when precipitation occurred in the presence of seed produced from homogeneous nucleation conditions.
The spherical shape of the precipitates and poorly crystalline structure suggest an amorphous precursor that transformed to a poorly crystalline carbonate apatite without significant morphological change. Future work will involve characterizing the first and subsequent solid phases to nucleate in these conditions between initial mixing and the equilibrium state after 5 days, further investigating the effect of seed on this precipitation reaction, and extending these tests to the complex P-containing solutions from municipal wastewater treatment.
Acknowledgments
The Natural Sciences and Research Council of Canada’s Discovery Grant program is acknowledge for supporting this research. We thank Professor Fabio Variola for use of his Raman spectrometer, Duane Dukart and Christian Kabbe for guidance and support. Bulat Gabidullin is acknowledged for generating the powder X-ray diffraction data. The reviewers are thanked for their helpful feedback, which improved the manuscript.
Author Contributions
J.R., E.A., and D.S. conceived, designed and performed the precipitation experiments, measured the solution chemistry, and undertook the SEM work. O.M. and L.G. designed and performed the calcium carbonate dissolution experiments. S.O. conceived the project, measured carbonate content with H.M. and J.R., and with J.R., wrote the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
Appendix A
SEM summary of products at 500× magnification. Top row are products of unseeded experiments, in order of increasing initial [Pi]. The bottom row shows the seed, and precipitates formed from seeded experiments. Seed 2 was formed by mixing a solution of 27 mM Pi with a CO2-saturated solution of CaCO3, using the same method as the unseeded tests. It was subsequently used as seed for one of the 5 mM Pi tests. The precipitates formed during both 5 mM Pi seeded tests were combined and used as seed (“Seed 5”) for the 7 mM Pi tests.
Figure A1.
SEM images of precipitates from unseeded and seeded experiments, with the corresponding initial [Pi] used to produce the precipitate when mixed with saturated Ca-CO3 solutions.
Figure A1.
SEM images of precipitates from unseeded and seeded experiments, with the corresponding initial [Pi] used to produce the precipitate when mixed with saturated Ca-CO3 solutions.
References
- Kazakov, A. The phosphorite facies and the genesis of phosphorites. USSR Trans. Sci. Inst. Fert. Insetofung 1937, 142, 95–113. [Google Scholar]
- McConnell, D. Precipitation of phosphates in sea water. Econ. Geol. 1965, 60, 1059–1062. [Google Scholar] [CrossRef]
- Schulz, H.N.; Brinkhoff, T.; Ferdelman, T.G.; Marine, M.H.; Teske, A.; Jorgensen, B.B. Dense populations of a giant sulfur bacterium in namibian shelf sediments. Science 1999, 284, 493–495. [Google Scholar] [CrossRef] [PubMed]
- Bruchert, V.; Jorgensen, B.B.; Neumann, K.; Riechmann, D.; Schlosser, M.; Schulz, H. Regulation of bacterial sulfate reduction and hydrogen sulfide fluxes in the central namibian coastal upwelling zone. Geochim. Cosmochim. Acta 2003, 67, 4505–4518. [Google Scholar] [CrossRef]
- Goldhammer, T.; Bruchert, V.; Ferdelman, T.G.; Zabel, M. Microbial sequestration of phosphorus in anoxic upwelling sediments. Nat. Geosci. 2010, 3, 557–561. [Google Scholar] [CrossRef]
- Schulz, H.N.; Schulz, H.D. Large sulfur bacteria and the formation of phosphorite. Science 2005, 307, 416–418. [Google Scholar] [CrossRef] [PubMed]
- Conkright, M.E.; Gregg, W.W.; Levitus, S. Seasonal cycle of phosphate in the open ocean. Deep Sea Res. Part I Oceanogr. Res. Pap. 2000, 47, 159–175. [Google Scholar] [CrossRef]
- Sheldon, R.P. Ancient marine phosphorites. Annu. Rev. Earth Planet. Sci. 1981, 9, 251–284. [Google Scholar] [CrossRef]
- Dawson, C.J.; Hilton, J. Fertiliser availability in a resource-limited world: Production and recycling of nitrogen and phosphorus. Food Policy 2011, 36, S14–S22. [Google Scholar] [CrossRef]
- McArthur, J.M. Francolite geochemistry—Compositional controls during formation, diagenesis, metamorphism and weathering. Geochim. Cosmochim. Acta 1985, 49, 23–35. [Google Scholar] [CrossRef]
- World Fertilizer Trends and Outlook to 2019; Food and Agriculture Organization of the United Nations: Rome, Italy, 2016; Available online: www.fao.org/3/a-i5627e.pdf (accessed on 19 June 2017).
- Cordell, D.; Neset, T.S.S. Phosphorus vulnerability: A qualitative framework for assessing the vulnerability of national and regional food systems to the multi-dimensional stressors of phosphorus scarcity. Glob. Environ. Chang. 2014, 24, 108–122. [Google Scholar] [CrossRef]
- Kuba, T.; Smolders, G.; van Loosdrecht, M.C.M.; Heijnen, J.J. Biological phosphorus removal from wastewater by anaerobic-anoxic sequencing batch reactor. Water Sci. Technol. 1993, 27, 241–252. [Google Scholar]
- Ulrich, A. Taking stock: Phosphorus supply from natural and anthropogenic pools in the 21st century. Sci. Total Environ. 2016, 542, 1005–1007. [Google Scholar] [CrossRef] [PubMed]
- Morse, G.K.; Brett, S.W.; Guy, J.A.; Lester, J.N. Review: Phosphorus removal and recovery technologies. Sci. Total Environ. 1998, 212, 69–81. [Google Scholar] [CrossRef]
- Phosphorus and Nitrogen Removal from Municipal Wastewater: Principles and Practice, 2nd ed.; Sedlak, R.I. (Ed.) Lewis Publishers: Washington, DC, USA, 1991. [Google Scholar]
- Minh, D.P.; Lyczko, N.; Sebei, H.; Nzihou, A.; Sharrock, P. Synthesis of calcium hydroxyapatite from calcium carbonate and different orthophosphate sources: A comparative study. Mater. Sci. Eng. B 2012, 177, 1080–1089. [Google Scholar] [CrossRef]
- McKendry, P. Energy production from biomass (part 2): Conversion technologies. Bioresour. Technol. 2002, 83, 47–54. [Google Scholar] [CrossRef]
- Kashchiev, D.; van Rosmalen, G.M. Review: Nucleation in solutions revisited. Cryst. Res. Technol. 2003, 38, 555–574. [Google Scholar] [CrossRef]
- Songolzadeh, M.; Soleimani, M.; Takht Ravanchi, M.; Songolzadeh, R. Carbon dioxide separation from flue gases: A technological review emphasizing reduction in greenhouse gas emissions. Sci. World J. 2014, 2014, 34. [Google Scholar] [CrossRef] [PubMed]
- Kuo, J.; Dow, J. Biogas Production from Anaerobic Digestion of Food Waste and Relevant Air Quality Implications. J. Air. Waste Manag. Assoc. 2017. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenbach, G. The complexones and their analytical application. Analyst 1955, 80, 713–729. [Google Scholar] [CrossRef]
- Robinson, R.; Roughan, M.E.; Wagstaff, D. Measuring inorganic phosphate without using a reducing agent. Ann. Clin. Biochem. 1971, 8, 168–170. [Google Scholar] [CrossRef]
- Kazanci, M.; Fratzl, P.; Klaushofer, K.; Paschalis, E.P. Complementary information on in vitro conversion of amorphous (precursor) calcium phosphate to hydroxyapatite from raman microscopy and wide-angle X-ray scattering. Calcif. Tissue Int. 2006, 79, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Niu, X.; Chen, S.; Tian, F.; Wang, L.; Feng, Q.; Fan, Y. Hydrolytic conversion of amorphous calcium phosphate into apatite accompanied by sustained calcium and orthophosphate ions release. Mater. Sci. Eng. C Mater. Biol. Appl. 2017, 70, 1120–1124. [Google Scholar] [CrossRef] [PubMed]
- Morse, J.W. Dissolution kinetics of calcium carbonate in sea water, iii. A new method for the study of carbonate reaction kinetics. Am. J. Sci. 1974, 274, 97–107. [Google Scholar] [CrossRef]
- Fleisch, H. Role of nucleation and inhibition in calcification. Clin. Orthop. Relat. Res. 1964, 32, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Penel, G.; Leroy, G.; Rey, C.; Bres, E. Microraman spectral study of the po4 and co3 vibrational modes in synthetic and biological apatites. Calcif. Tissue Int. 1998, 63, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Kuhn, L.T.; Grynpas, M.D.; Rey, C.C.; Wu, Y.; Ackerman, J.L.; Glimcher, M.J. A comparison of the physical and chemical differences between cancellous and cortical bovine bone mineral at two ages. Calcif. Tissue Int. 2008, 83, 146–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moradian-Oldak, J.; Weiner, S.; Addadi, L.; Landis, W.J.; Traub, W. Electron imaging and diffraction study of individual crystals of bone, mineralized tendon and synthetic carbonate apatite. Connect. Tissue Res. 1991, 25, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Jahnke, R.A. The synthesis and solubility of carbonate fluorapatite. Am. J. Sci. 1984, 284, 58–78. [Google Scholar] [CrossRef]
- Perrone, J.; Fourest, B.; Giffaut, E. Surface characterization of synthetic and mineral carbonate fluorapatites. J. Colloid Interface Sci. 2002, 249, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.; Henneman, Z.J.; Nancollas, G.H. Constant composition kinetics study of carbonated apatite dissolution. J. Cryst. Growth 2003, 249, 614. [Google Scholar] [CrossRef]
- Deymier, A.C.; Nair, A.K.; Depalle, B.; Qin, Z.; Arcot, K.; Drouet, C.; Yoder, C.H.; Buehler, M.J.; Thomopoulos, S.; Genin, G.M.; et al. Protein-free formation of bone-like apatite: New insights into the key role of carbonation. Biomaterials 2017, 127, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Habraken, W.J.E.M.; Tao, J.; Brylka, L.J.; Friedrich, H.; Bertinetti, L.; Schenk, A.S.; Verch, A.; Dmitrovic, V.; Bomans, P.H.H.; Frederik, P.M.; et al. Ion-association complexes unite classical and non-classical theories for the biomimetic nucleation of calcium phosphate. Nat. Commun. 2013, 4, 1507. [Google Scholar] [CrossRef] [PubMed]
- Simon, P.; Schwarz, U.; Kniep, R. Hierarchical architecture and real structure in a biomimetic nano-composite of fluorapatite with gelatine: A model system for steps in dentino- and osteogenesis? J. Mater. Chem. 2005, 15, 4992–4996. [Google Scholar] [CrossRef]
- Chatzipanagis, K.; Iafisco, M.; Roncal-Herrero, T.; Bilton, M.; Tampieri, A.; Kroger, R.; Delgado-Lopez, J.M. Crystallization of citrate-stabilized amorphous calcium phosphate to nanocrystalline apatite: A surface-mediated transformation. CrystEngComm 2016, 18, 3170–3173. [Google Scholar] [CrossRef]
- Zou, Z.; Bertinetti, L.; Politi, Y.; Jensen, A.C.S.; Weiner, S.; Addadi, L.; Fratzl, P.; Habraken, W.J.E.M. Opposite particle size effect on amorphous calcium carbonate crystallization in water and during heating in air. Chem. Mater. 2015, 27, 4237–4246. [Google Scholar] [CrossRef]
Figure 1.
Changes in pH and calcium concentration with time in potable water mixed with CaCO3(s) while in contact with 100% CO2 gas at room temperature.
Figure 1.
Changes in pH and calcium concentration with time in potable water mixed with CaCO3(s) while in contact with 100% CO2 gas at room temperature.
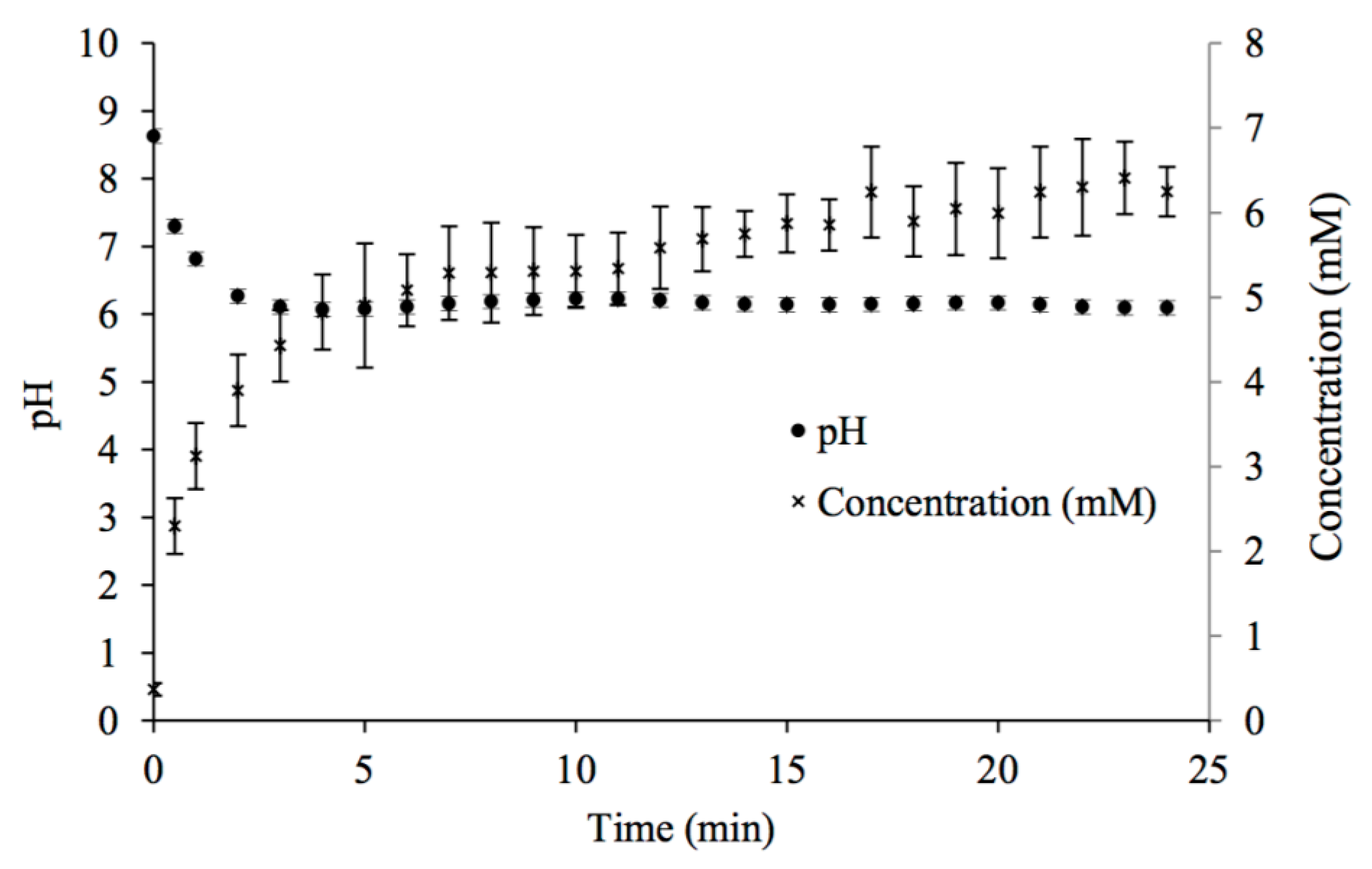
Figure 2.
Change in [Ca2+] and [Pi] after five days in unseeded batch precipitation. The samples are named after the [Pi] before mixing with the Ca-CO3 solution.
Figure 2.
Change in [Ca2+] and [Pi] after five days in unseeded batch precipitation. The samples are named after the [Pi] before mixing with the Ca-CO3 solution.
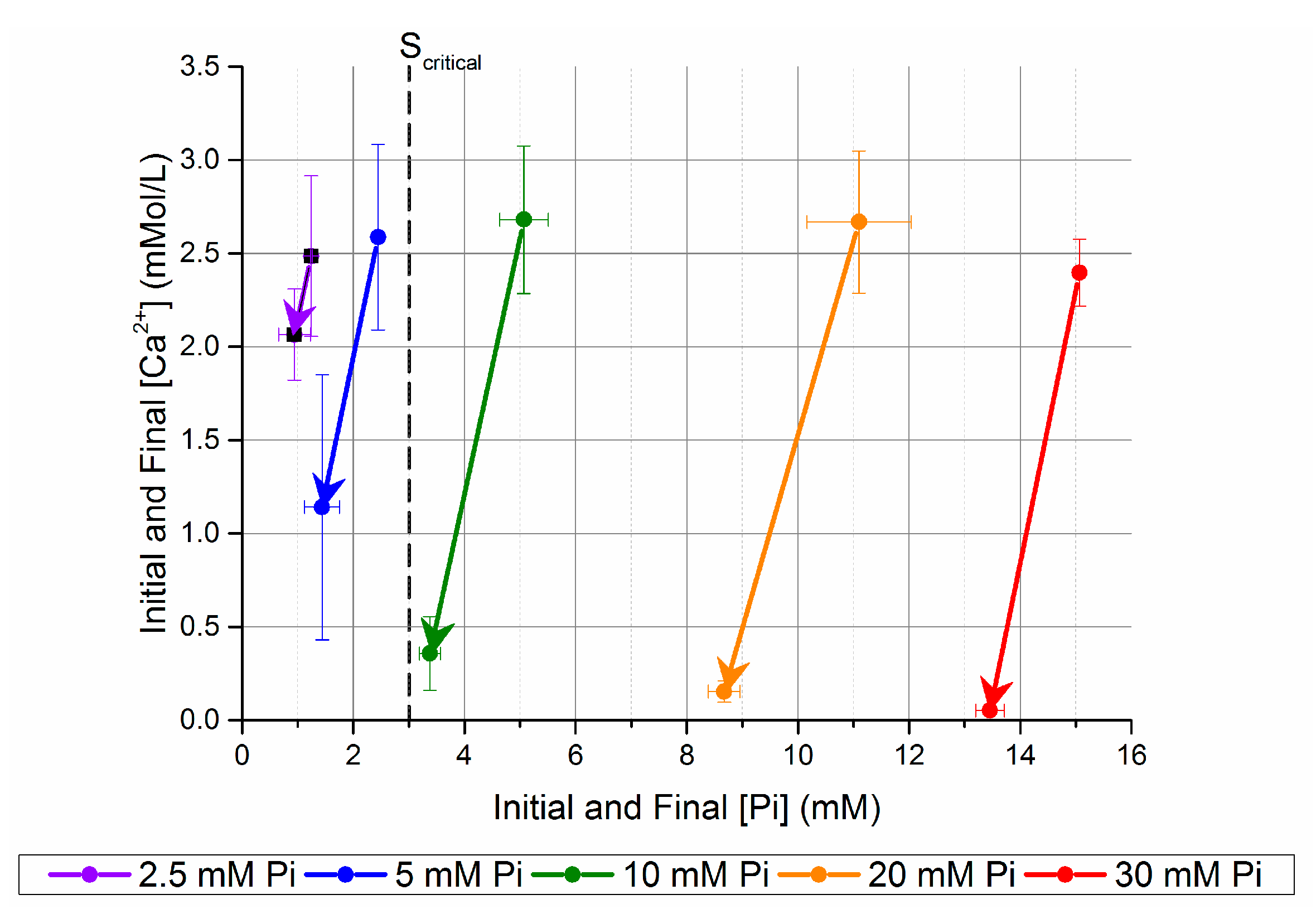
Figure 3.
Change in [Ca2+] and [Pi] after five days in seeded batch precipitation (bone and homogeneously nucleated precipitate (ppt)).
Figure 3.
Change in [Ca2+] and [Pi] after five days in seeded batch precipitation (bone and homogeneously nucleated precipitate (ppt)).
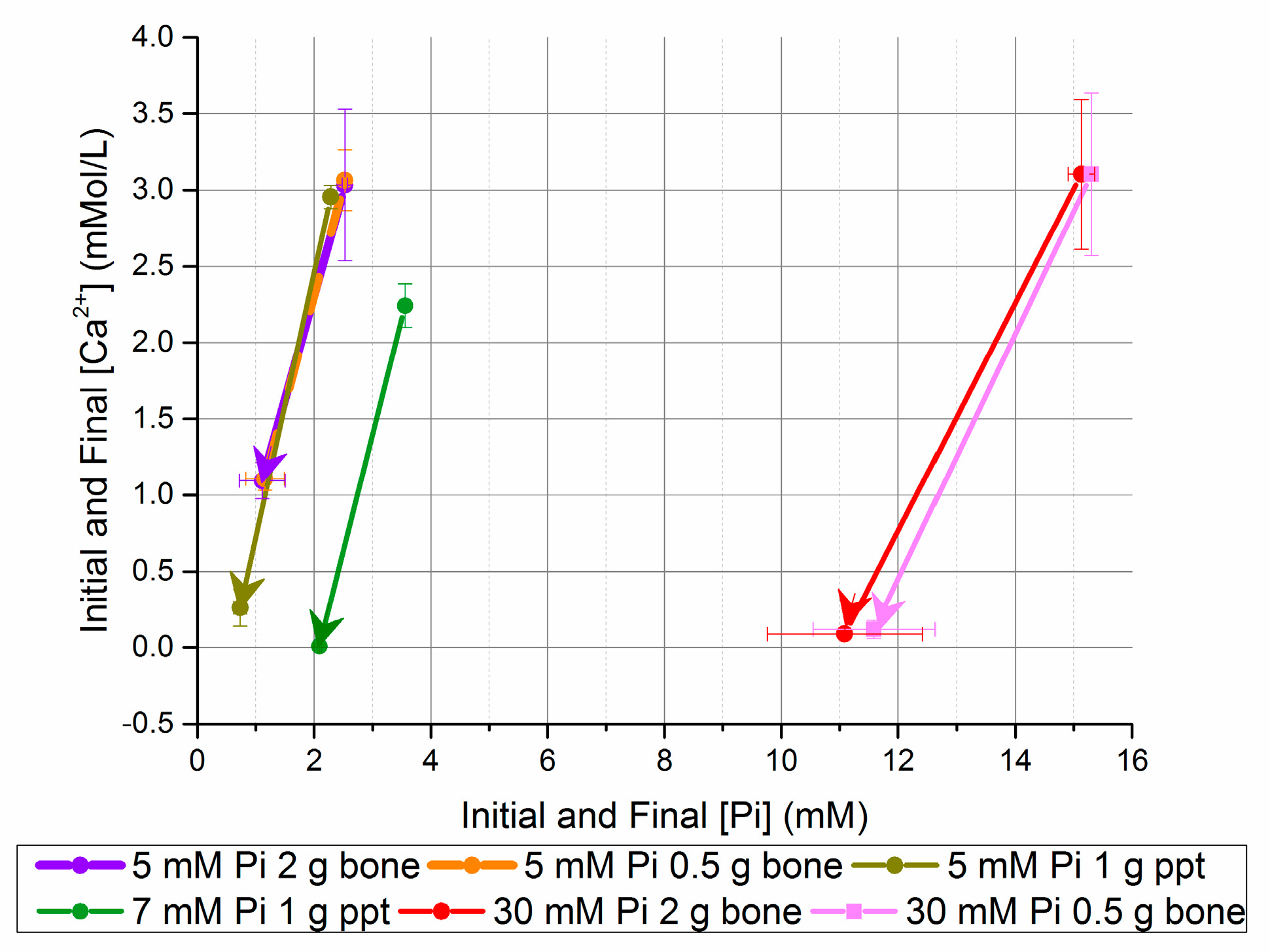
Figure 4.
Initial and final (Ca × P) values, versus initial and final [Pi] ((a) initial [Pi] < 20 mM, (b) initial [Pi] ≥ 20 mM). Using seed from previous unseeded experiments generated lower final [Ca2+] × [Pi] values than bone mineral seed or no seed for equivalent initial [Pi].
Figure 4.
Initial and final (Ca × P) values, versus initial and final [Pi] ((a) initial [Pi] < 20 mM, (b) initial [Pi] ≥ 20 mM). Using seed from previous unseeded experiments generated lower final [Ca2+] × [Pi] values than bone mineral seed or no seed for equivalent initial [Pi].
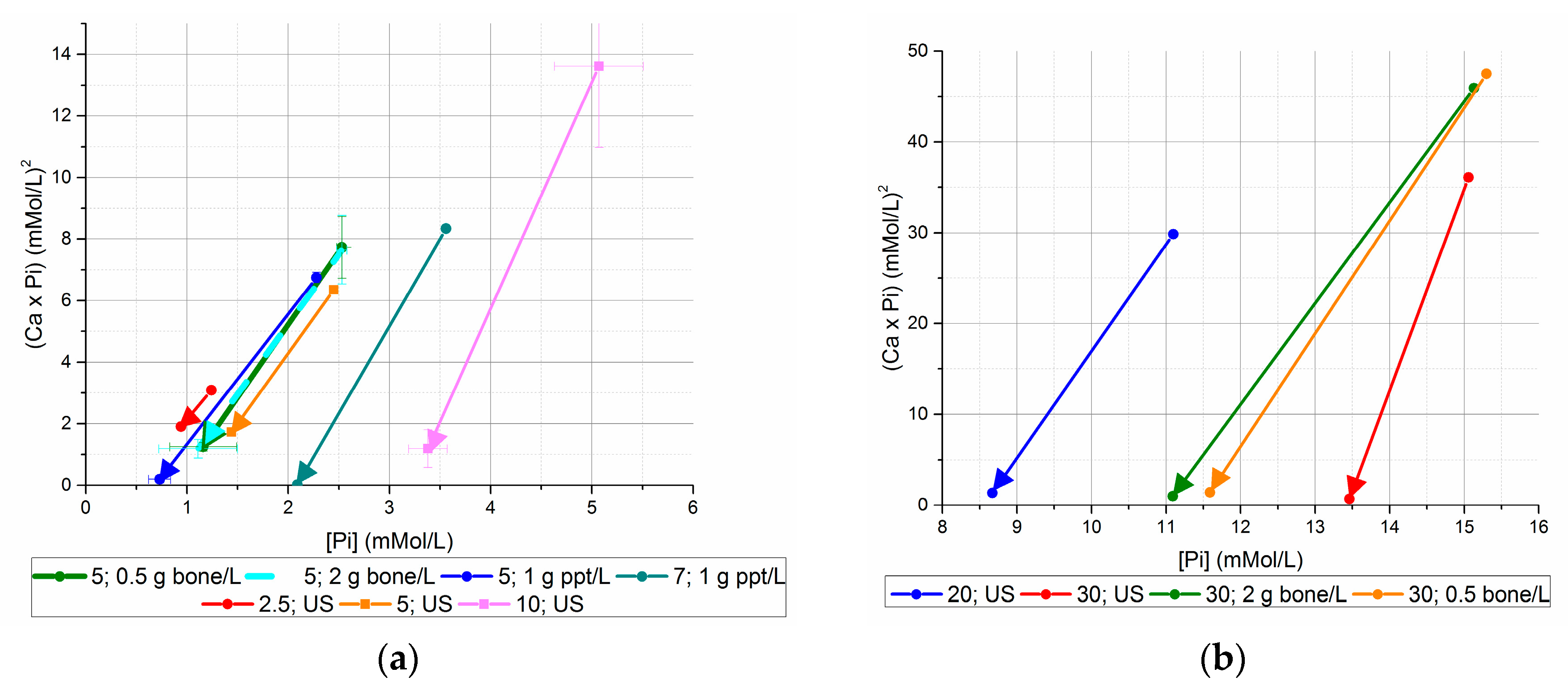
Figure 5.
Final (Ca × P) versus final pH (seeded and unseeded).
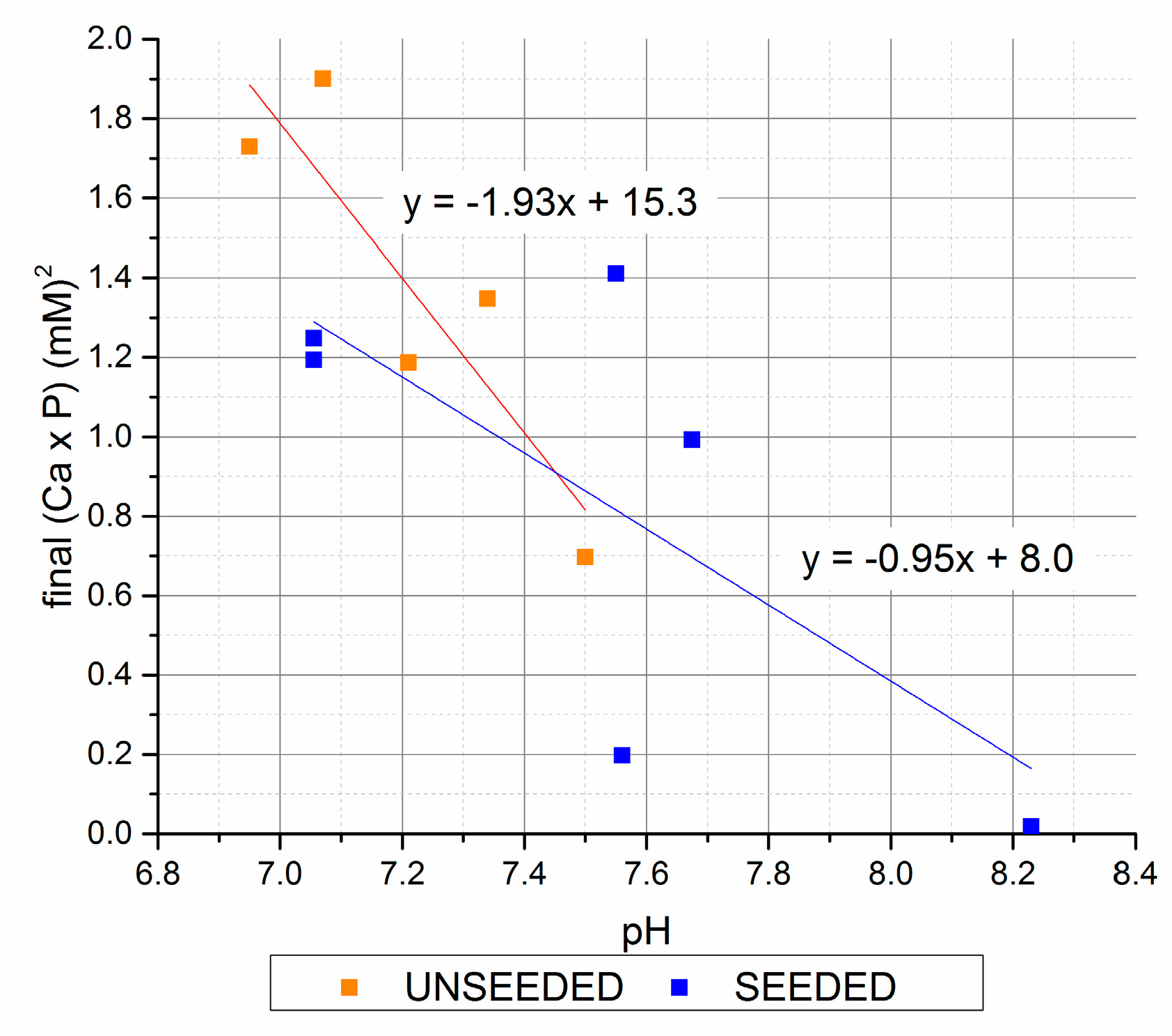
Figure 6.
SEM images of precipitates from unseeded experiments (a) 5 mM; (b) 30 mM [Pi] and precipitate-seeded experiments (c) 7 mM [Pi].
Figure 6.
SEM images of precipitates from unseeded experiments (a) 5 mM; (b) 30 mM [Pi] and precipitate-seeded experiments (c) 7 mM [Pi].

Figure 7.
Spherical precipitates imaged with SEM with increasing magnification (1000×, 3000×, 6000×, and 15,000×).
Figure 7.
Spherical precipitates imaged with SEM with increasing magnification (1000×, 3000×, 6000×, and 15,000×).

Figure 8.
Raman spectra for synthetic hydroxyapatite (HAP, black), bone mineral (violet), unseeded precipitate from 5 (blue), 10 mM (orange), 20 mM (red), and 30 mM (navy), and 7 mM seeded precipitate (green) from (a) 350–1200 cm−1, and (b) 1000–1120 cm−1.
Figure 8.
Raman spectra for synthetic hydroxyapatite (HAP, black), bone mineral (violet), unseeded precipitate from 5 (blue), 10 mM (orange), 20 mM (red), and 30 mM (navy), and 7 mM seeded precipitate (green) from (a) 350–1200 cm−1, and (b) 1000–1120 cm−1.
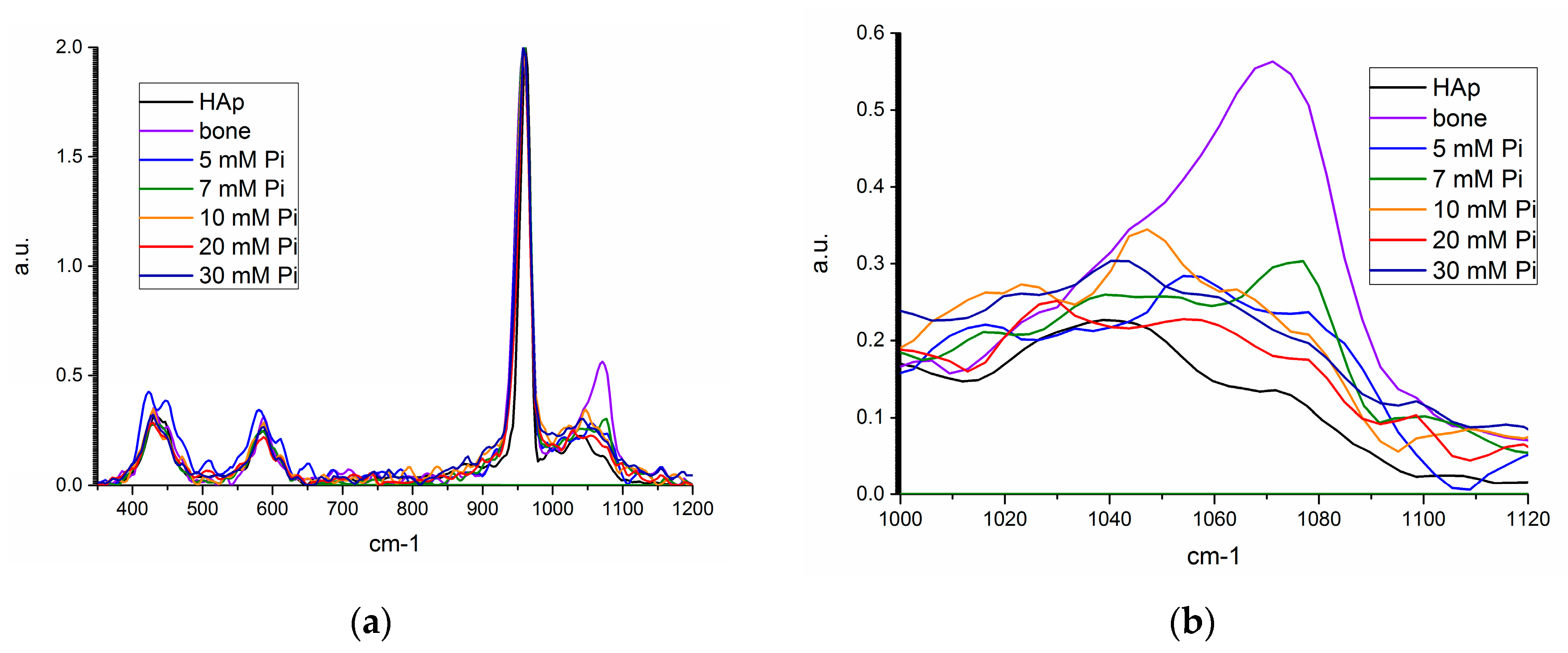
Figure 9.
X-ray powder diffraction patterns from bone mineral (blue), 30 mM [Pi] unseeded experiment (green), 7 mM [Pi] seed (orange) and seeded precipitate (red), overlaid with ICDD 01-089-4405 PDF (hydroxyapatite, black).
Figure 9.
X-ray powder diffraction patterns from bone mineral (blue), 30 mM [Pi] unseeded experiment (green), 7 mM [Pi] seed (orange) and seeded precipitate (red), overlaid with ICDD 01-089-4405 PDF (hydroxyapatite, black).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Effect of percent CO2 on the equilibrium [Ca2+] and pH of saturated Ca-CO3-potable water solutions.
Table 1.
Effect of percent CO2 on the equilibrium [Ca2+] and pH of saturated Ca-CO3-potable water solutions.
| % CO2 | [Ca2+] (mM) | pH |
|---|---|---|
| 0 | 0.37 ± 0.1 | 8.0 |
| 7 ± 2 | 3.6 ± 0.7 | 6.7 ± 0.1 |
| 15 ± 2 | 4.8 ± 0.4 | 6.3 ± 0.1 |
| 100 | 5.7 ± 0.4 | 6.2 ± 0.1 |
Table 2.
Summary of [Ca2+] results for the batch, unseeded precipitation tests.
| [Pi] Solution (mM) | Initial [Ca2+] (mM) | Final [Ca2+] (mM) | Percent [Ca2+] Change |
|---|---|---|---|
| 2.5 | 2.89 ± 0.43 | 2.07 ± 0.24 | −17% |
| 5 | 2.59 ± 0.50 | 1.14 ± 0.71 | −56% |
| 10 | 2.68 ± 0.40 | 0.36 ± 0.20 | −87% |
| 20 | 2.67 ± 0.38 | 0.15 ± 0.06 | −94% |
| 30 | 2.40 ± 0.18 | 0.05 ± 0.01 | −98% |
Table 3.
Summary of [Pi] results for the batch, unseeded precipitation tests.
| [Pi] Solution (mM) | Initial [Pi] (mM) | Final [Pi] (mM) | Percent [Pi] Change |
|---|---|---|---|
| 2.5 | 1.24 ± 0.02 | 0.94 ± 0.28 | −24% |
| 5 | 2.45 ± 0.07 | 1.44 ± 0.32 | −41% |
| 10 | 5.07 ± 0.44 | 3.38 ± 0.19 | −33% |
| 20 | 11.10 ± 0.93 | 8.67 ± 0.29 | −22% |
| 30 | 15.06 ± 0.01 | 13.46 ± 0.26 | −11% |
Table 4.
Summary of pH and theoretical Ca/P results for the batch, unseeded precipitation tests.
| [Pi] Solution (mM) | Final pH | Theoretical Precipitate Ca/P |
|---|---|---|
| 2.5 | 7.07 ± 0.19 | 1.84 ± 0.86 |
| 5 | 6.95 ± 0.21 | 1.47 ± 0.23 |
| 10 | 7.21 ± 0.15 | 1.46 ± 0.43 |
| 20 | 7.34 ± 0.18 | 1.08 ± 0.19 |
| 30 | 7.5 ± 0.08 | 1.48 ± 0.18 |
Table 5.
Summary of [Ca2+] results for the batch, seeded precipitation tests.
| [Pi] Solution (mM) | [Seed] (g/L) & Type | Initial [Ca2+] (mM) | Final [Ca2+] (mM) | Percent [Ca2+] Change |
|---|---|---|---|---|
| 5 1 | 0.50 (bone) | 3.06 | 1.11 | −64% |
| 5 1 | 2.0 (bone) | 3.03 | 1.09 | −64% |
| 5 1 | 1.0 (ppt) | 2.96 | 0.26 | −91% |
| 7 2 | 1.0 (ppt) | 2.24 ± 0.14 | 0.01 ± 0.01 | −100% |
| 30 1 | 0.50 (bone) | 3.10 | 0.12 | −96% |
| 30 1 | 2.0 (bone) | 3.37 | 0.09 | −97% |
1 n = 2, 2 n = 4.
Table 6.
Summary of [Pi] results for the batch, seeded precipitation tests.
| [Pi] Solution (mM) | [Seed] (g/L) & Type | Initial [Pi] (mM) | Final [Pi] (mM) | Percent [Pi] Change |
|---|---|---|---|---|
| 5 1 | 0.50 (bone) | 2.53 | 1.16 | −54% |
| 5 1 | 2.0 (bone) | 2.53 | 1.11 | −56% |
| 5 1 | 1.0 (ppt) | 2.29 | 0.73 | −68% |
| 7 2 | 1.0 (ppt) | 3.56 | 2.9 ± 0.03 | −41% |
| 30 1 | 0.50 (bone) | 15.30 | 11.59 | −24% |
| 30 1 | 2.0 (bone) | 15.13 | 11.09 | −27% |
1 n = 2, 2 n = 4.
Table 7.
Summary of pH and theoretical Ca/P results for the batch, seeded precipitation tests.
| [Pi] Solution (mM) | [Seed] (g/L) & Type | Final pH | Theoretical Precipitate Ca/P |
|---|---|---|---|
| 5 1 | 0.5 (bone) | 7.06 | 1.44 |
| 5 1 | 2 (bone) | 7.06 | 0.95 |
| 5 1 | 1 (ppt) | 7.56 | 1.74 |
| 7 2 | 1 (ppt) | 8.23 ± 0.05 | 1.52 |
| 30 1 | 0.5 (bone) | 7.55 | 0.81 |
| 30 1 | 2 (bone) | 7.67 | 0.86 |
1 n = 2, 2 n = 4.
Table 8.
Measured [Ca2+] × [Pi] values before and after batch precipitation experiments.
| [Pi] Solution (mM) | [Seed] (g/L) & Type | [Ca2+] × [Pi] Initial (mM)2 | [Ca2+] × [Pi] Final (mM)2 | Final pH |
|---|---|---|---|---|
| 2.5 1 | 0 | 3.09 ± 0.51 | 1.90 ± 0.48 | 7.07 ± 0.19 |
| 5 1 | 0 | 6.36 ± 1.38 | 1.73 ± 1.40 | 6.95 ± 0.21 |
| 5 2 | 0.50 (bone) | 7.73 | 1.25 | 7.06 |
| 5 2 | 2.0 (bone) | 7.65 | 1.19 | 7.06 |
| 5 2 | 1.0 (ppt) | 6.75 | 0.20 | 7.56 |
| 7 2 | 1.0 (ppt) | 7.98 | 0.02 | 8.23 ± 0.05 |
| 10 1 | 0 | 13.6 ± 2.64 | 1.19 ± 0.61 | 7.21 ± 0.15 |
| 20 1 | 0 | 29.9 ± 6.71 | 1.35 ± 0.54 | 7.34 ± 0.48 |
| 30 1 | 0 | 36.1 ± 2.69 | 0.70 ± 0.11 | 7.50 ± 0.08 |
| 30 2 | 0.50 (bone) | 47.5 | 1.41 | 7.55 |
| 30 2 | 2.0 (bone) | 45.9 | 0.99 | 7.67 |
1 n = 4, 2 n = 2.
Table 9.
Inorganic carbon coulometry measurement of weight percent carbonate (CO32−)
| [Pi] Solution (mM) | [Seed] g/L | Weight % CO32- |
|---|---|---|
| 5 1 | 0 | 1.95 ± 0.08 |
| 7 2 | 1.0 (ppt) | 2.59 ± 0.09 |
| 10 2 | 0 | 1.21 ± 0.43 |
| 20 2 | 0 | 1.13 ± 0.14 |
| 30 1 | 0 | 1.10 ± 0.04 |
1 n = 3, 2 n = 4
Table 10.
Measured dissolved [Ca2+], [Pi], and Ca/P in distilled and deionized water compared with Ca/P values predicted from the change in precipitating solution composition.
Table 10.
Measured dissolved [Ca2+], [Pi], and Ca/P in distilled and deionized water compared with Ca/P values predicted from the change in precipitating solution composition.
| Initial [Pi] (mM) | [Seed] (g/L) | [Ca2+] (mM) | [Pi] (mM) | Ca/P | Ca/P (Table 4 and Table 7) |
|---|---|---|---|---|---|
| 5 1 | 0 | 0.16 ± 0.02 | 0.16 ± 0.02 | 1.03 ± 0.15 | 1.47 ± 0.23 |
| 5 2 | 1.0 | 0.24 | 0.23 | 1.02 | 1.74 |
| 7 1 | 1.0 | 0.21 ± 0.01 | 0.22 ± 0.01 | 0.97 ± 0.05 | 1.52 ± 0.07 |
| 10 1 | 0 | 0.18 ± 0.03 | 0.23 ± 0.04 | 0.79 ± 0.04 | 1.46 ± 0.43 |
| 20 1 | 0 | 0.18 ± 0.02 | 0.51 ± 0.12 | 0.38 ± 0.14 | 1.08 ± 0.19 |
| 30 3 | 0 | 0.17 ± 0.06 | 0.45 ± 0.09 | 0.39 ± 0.17 | 0.48 ± 0.18 |
1 n=4, 2 n = 2, 3 n = 3
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ross, J.; Gao, L.; Meouch, O.; Anthony, E.; Sutarwala, D.; Mamo, H.; Omelon, S. Carbonate Apatite Precipitation from Synthetic Municipal Wastewater. Minerals 2017, 7, 129. https://doi.org/10.3390/min7080129
AMA Style
Ross J, Gao L, Meouch O, Anthony E, Sutarwala D, Mamo H, Omelon S. Carbonate Apatite Precipitation from Synthetic Municipal Wastewater. Minerals. 2017; 7(8):129. https://doi.org/10.3390/min7080129
Chicago/Turabian StyleRoss, Jessica, Lu Gao, Orysia Meouch, Essie Anthony, Divya Sutarwala, Helina Mamo, and Sidney Omelon. 2017. "Carbonate Apatite Precipitation from Synthetic Municipal Wastewater" Minerals 7, no. 8: 129. https://doi.org/10.3390/min7080129
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.