
Dissolution and Sorption Processes on the Surface of Calcite in the Presence of High Co2+ Concentration


 and
and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Macroscopic Experiments
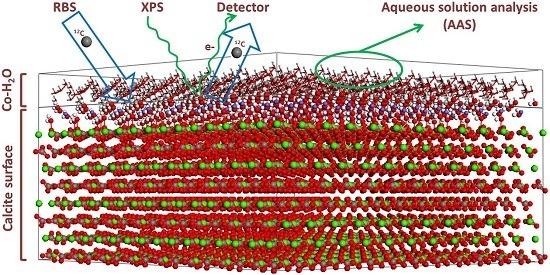
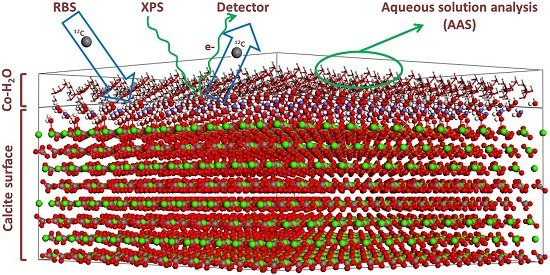
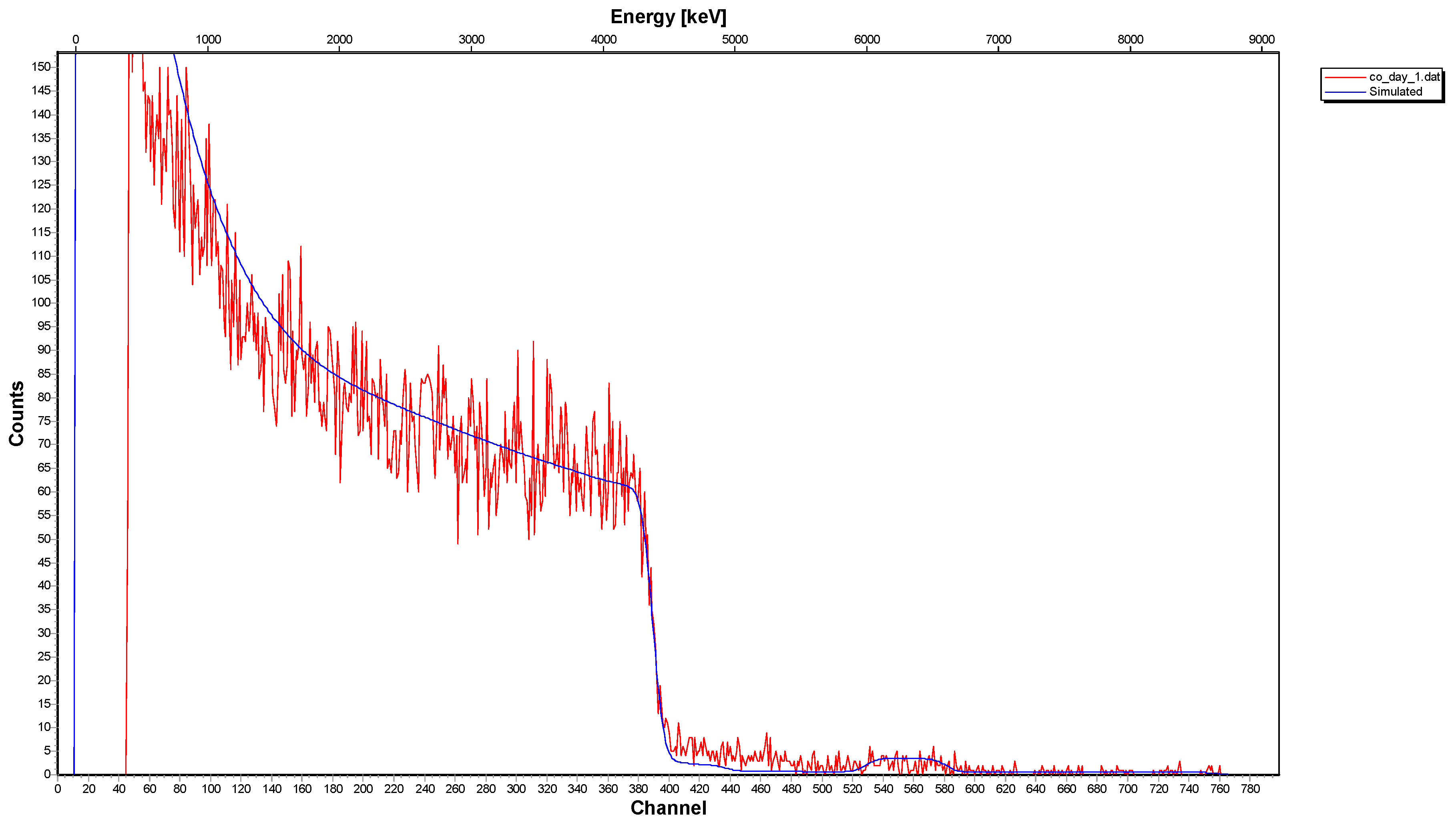
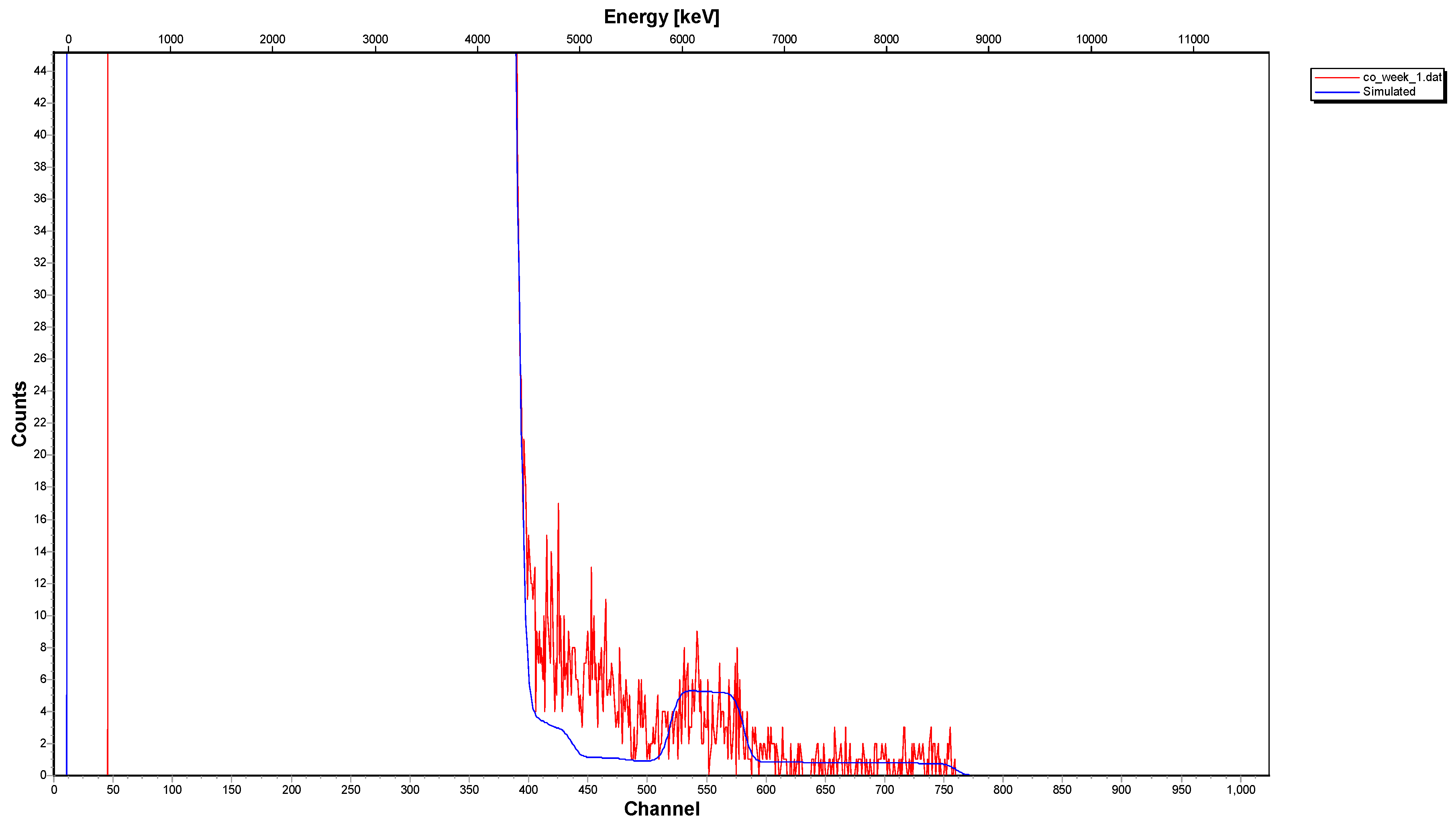
2.2. Surface Spectroscopic Characterization of the Interacted Solids (XPS and 12C-RBS)
3. Results and Discussion
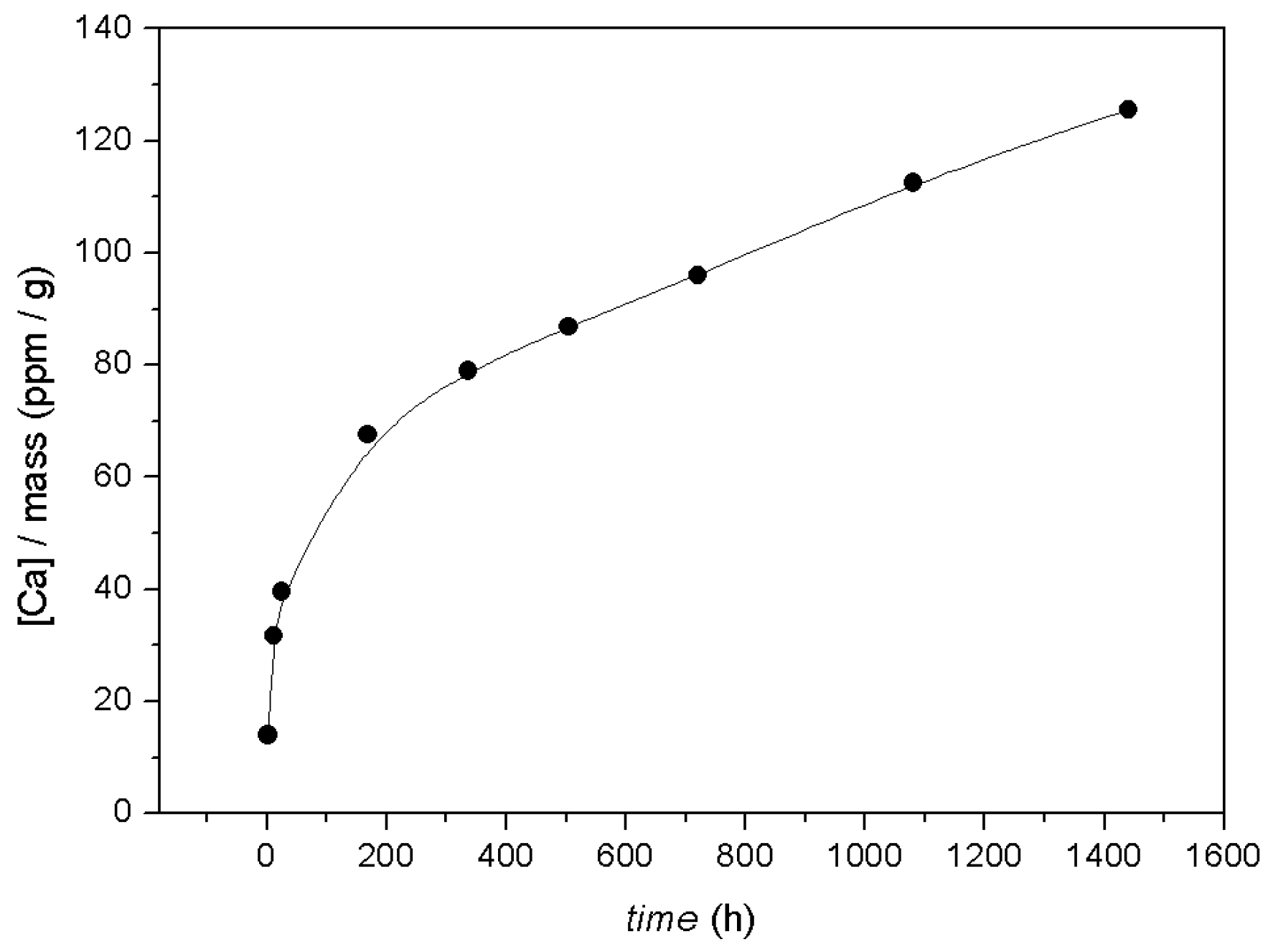
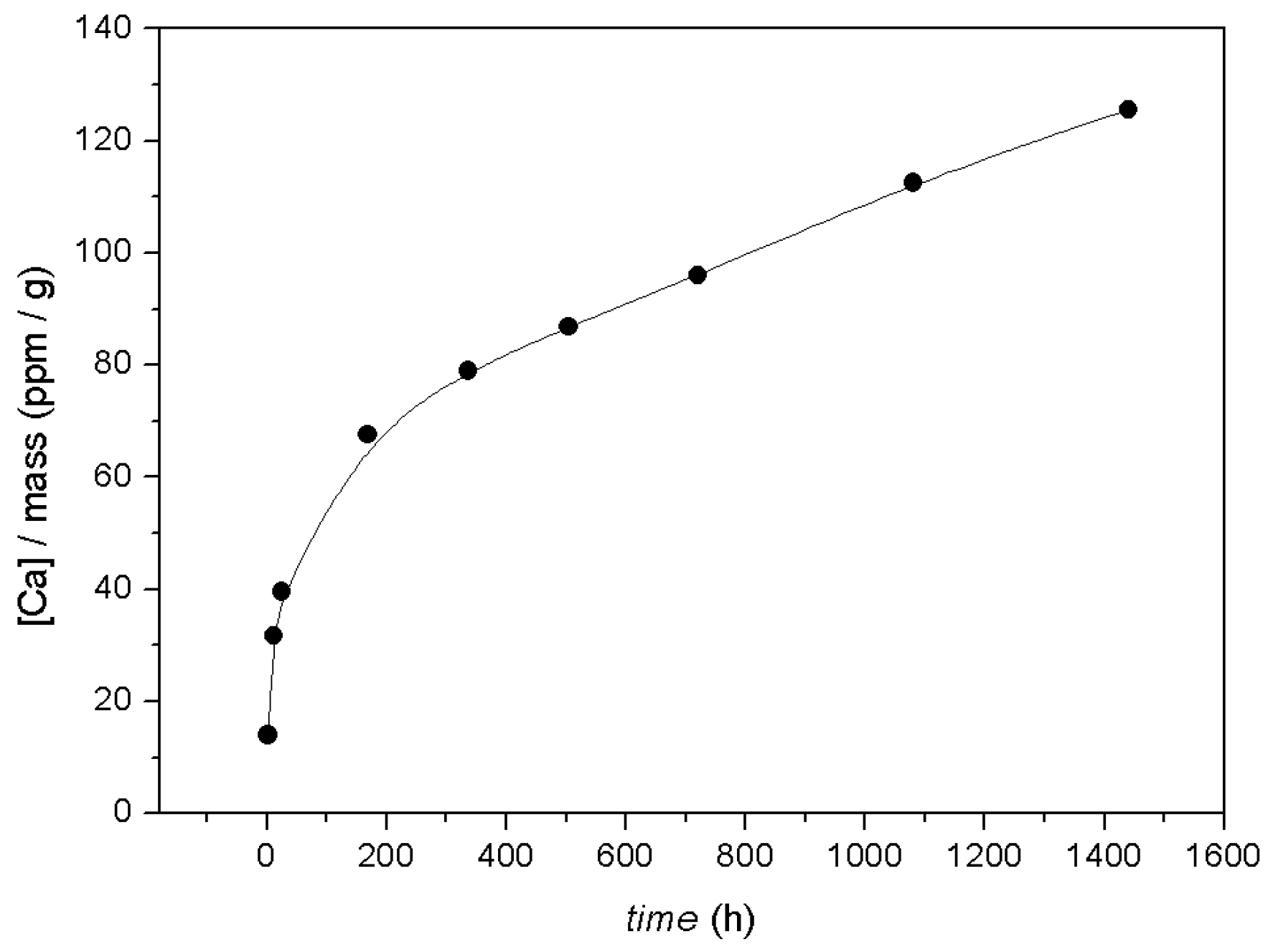
3.1. Macroscopic Data—Dissolution of Calcite in the Presence of Co2+
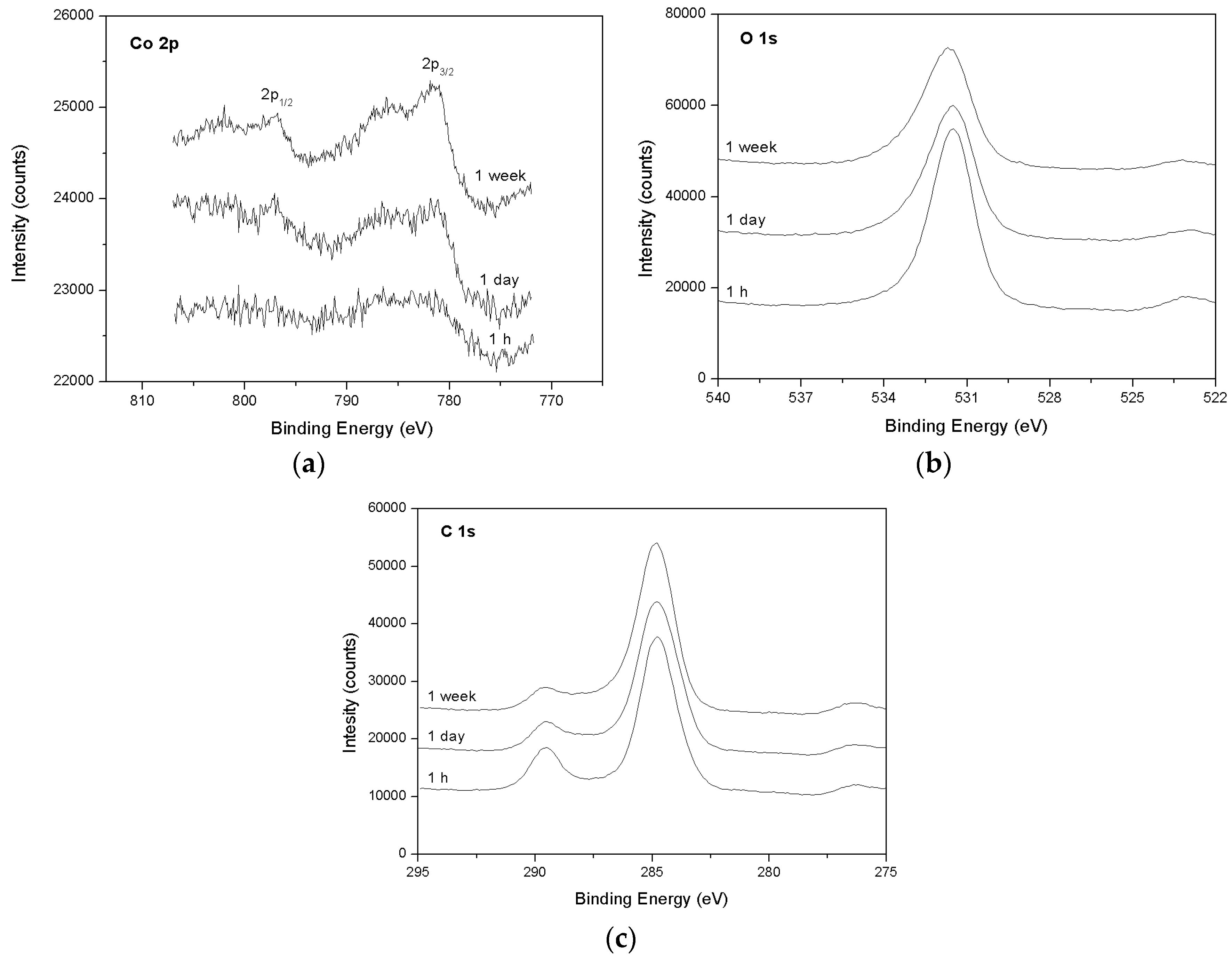
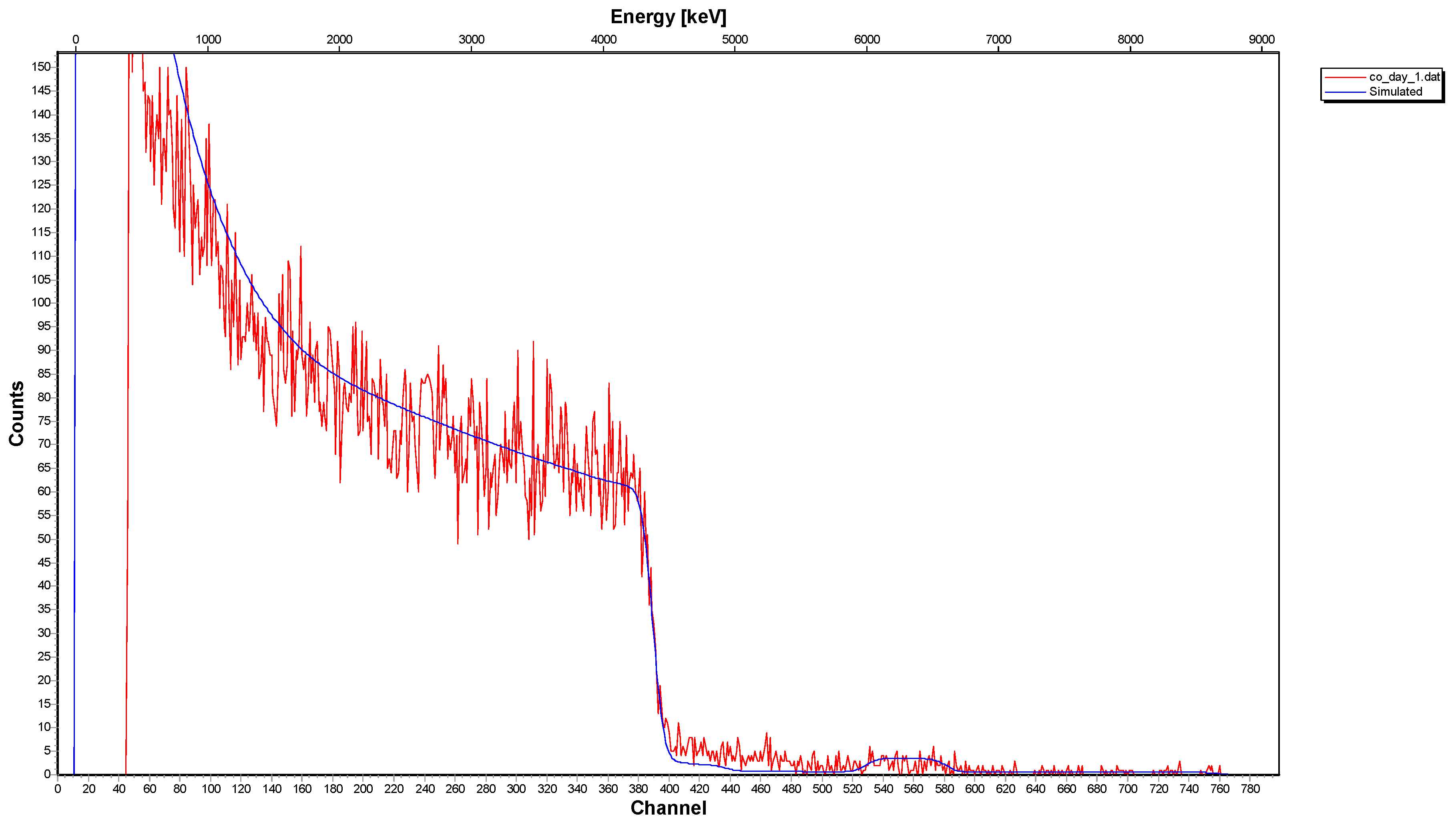
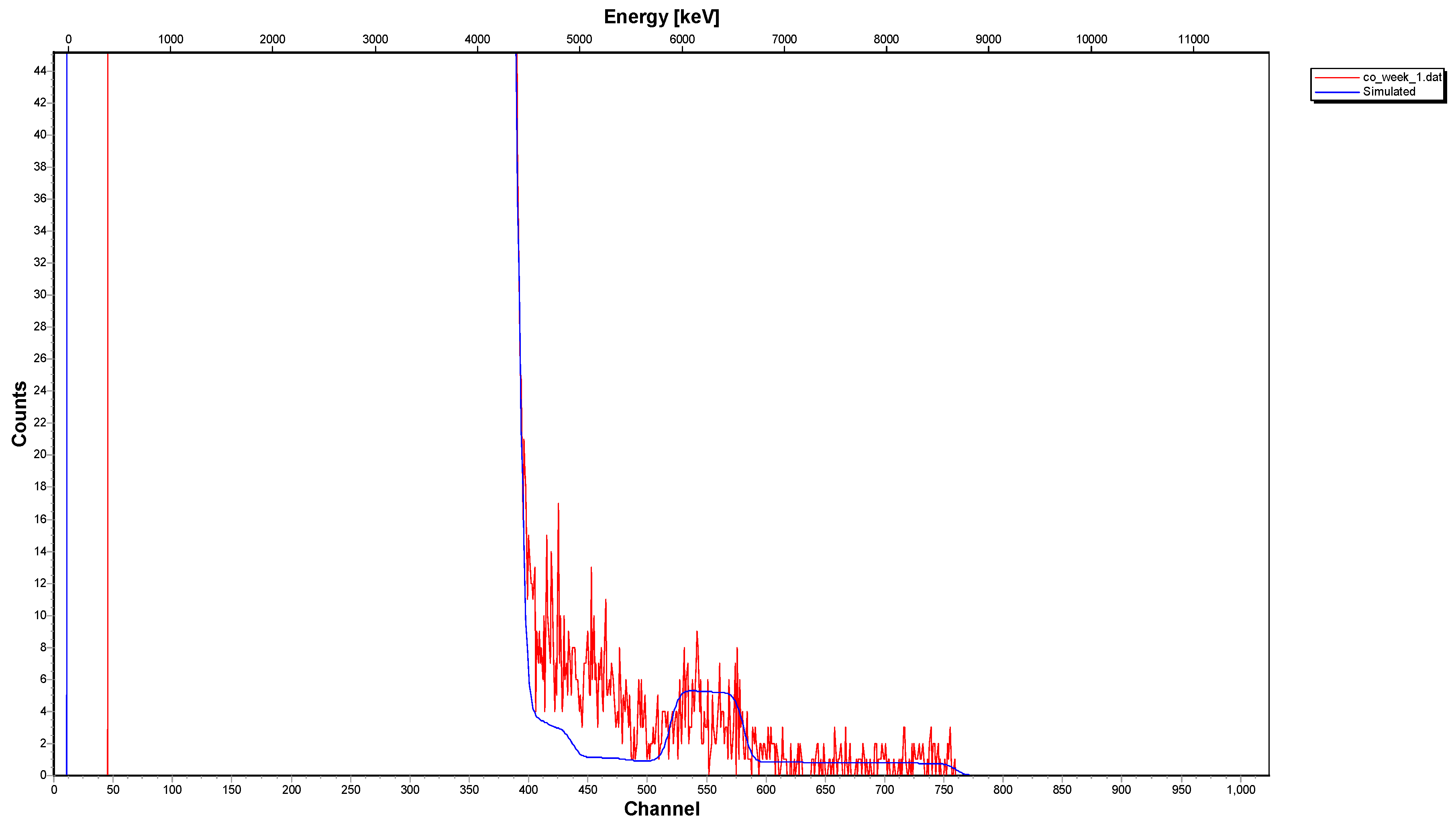
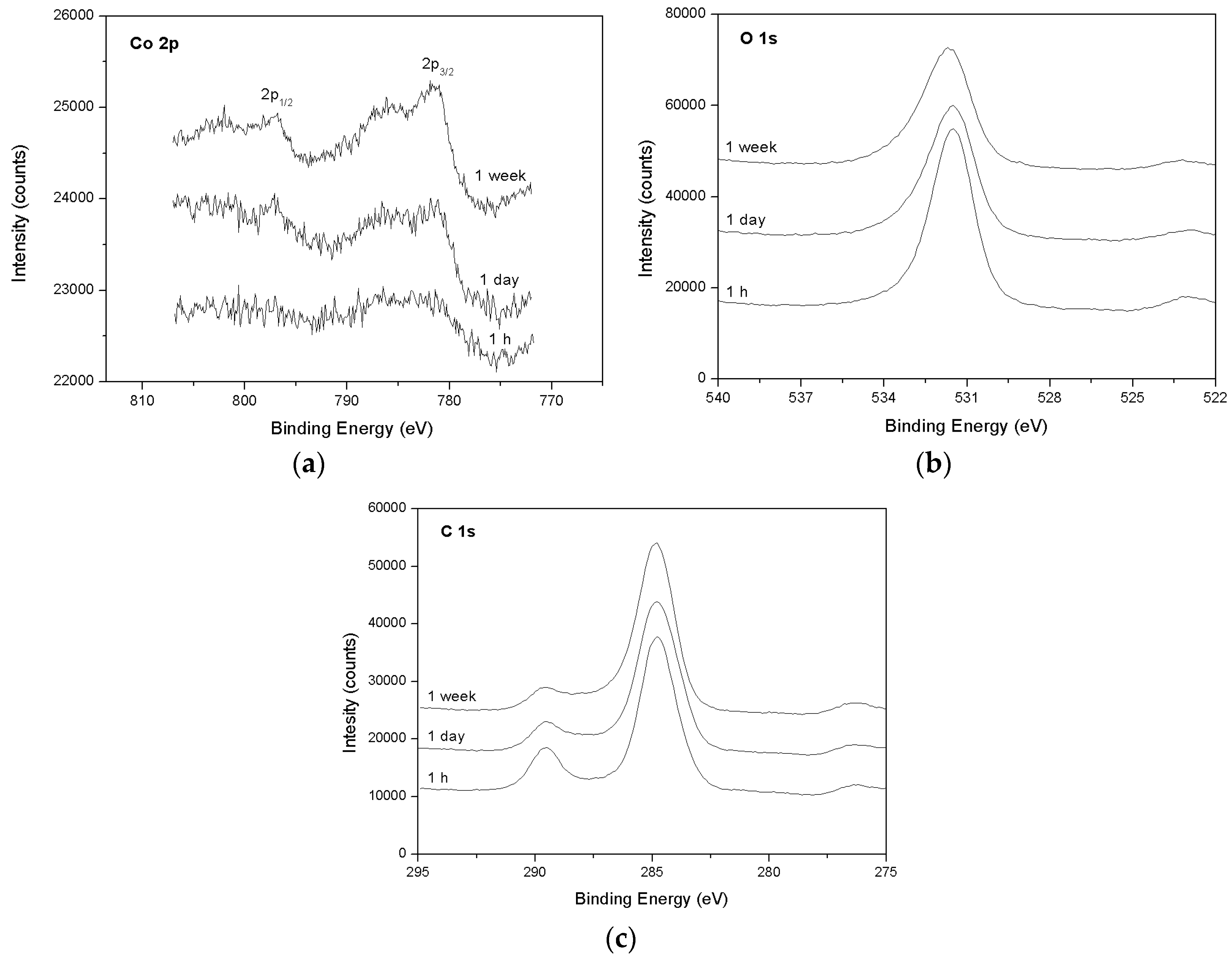
3.2. Spectroscopic Data—Sorption of Co2+ on the Surface of Calcite
4. Conclusions
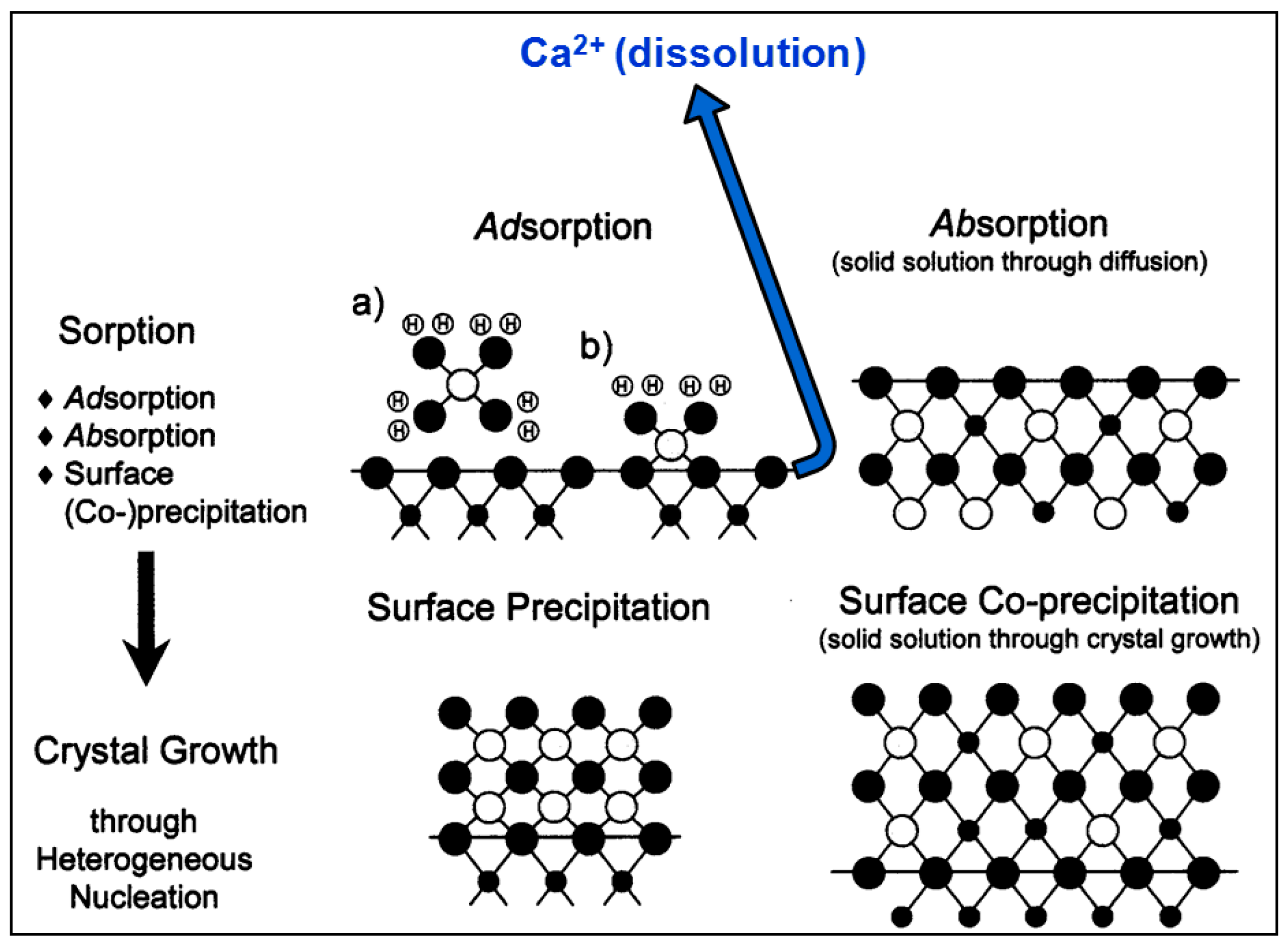
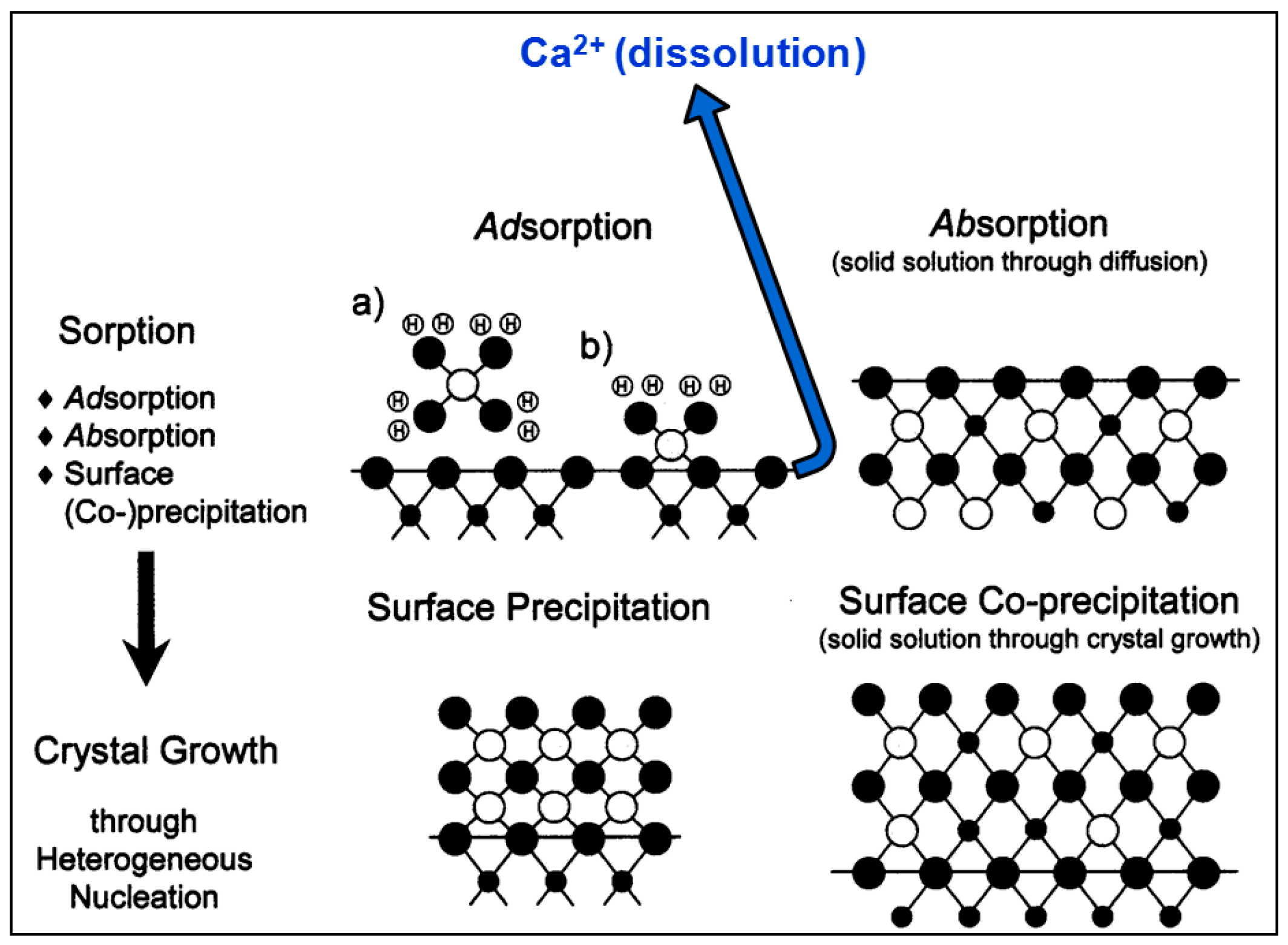
- According to macroscopic results, for both calcite powder and monocrystals interacted in an aqueous solution with a high Co2+ concentration ([Coaq]initial = 1000 ppm, ~17 mM), dissolution occurs on the surface, causing systematic release of Ca2+ into solution. This runs in parallel to surface Co2+ sorption processes.
- The XPS surface study (analyzed at a depth of around 12 nm) confirmed that, indeed, sorption occurs at near-surface layers of calcite, initially by adsorption of Co–OH units and later by surface (co-)precipitation.
- The 12C-RBS measurements on calcite {} indicated that co-precipitation is related to a Co2+-bearing surface layer under the calcite surface, showing a thickness of 270 nm after one day of interaction and reaching 320 nm after one week and/or month.
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AAS | Atomic absorption spectrometry |
| XPS | X-ray photoelectron spectroscopy |
| RBS | Rutherford backscattering spectrometry |
| SEXAFS | Surface-extended X-ray absorption fine structure |
| XSW | X-ray standing wave |
| AFM | Atomic force microscopy |
References
- Smith, I.; Carson, B.L. Trace Metals in the Environment Vol 6. Cobalt an Apraisal of Environmental Exposure; Ann Arbor Science Publishers Inc.: Ann Arbor, MI, USA, 1981; p. 1202. [Google Scholar]
- Agency for Toxic Substances and Disease Registry. Toxicological Profile for Cobalt; U.S. Department of Health and Human Services, Public Health Service: Atlanta, GA, USA, 2004.
- Kim, J.H.; Gibb, H.J.; Howe, P.D.; Sheffer, M. Cobalt and Inorganic Cobalt Compounds; Concise International Chemical Assessment Document; World Health Organization: Geneva, Switzerland, 2006; Volume 69. [Google Scholar]
- Stipp, S.L.; Hochella, M.F.; Parks, G.A.; Leckie, J.O. Cd2+ uptake by calcite, solid-state diffusion, and the formation of solid-solution: Interface processes observed with near-surface sensitive techniques (XPS, LEED, and AES). Geochim. Cosmochim. Acta 1992, 56, 1941–1954. [Google Scholar] [CrossRef]
- Godelitsas, A.; Astilleros, J.M. Dissolution, sorption/(re)precipitation, formation of solid solutions and crystal growth phenomena on mineral surfaces: implications for the removal of toxic metals from the environment. In EMU Notes in Mineralogy—Volume 10: Ion Partitioning in Ambient-Temperature Aqueous Systems; Prieto, M., Stoll, H., Eds.; European Mineralogical Union: Chantilly, VA, USA, 2010. [Google Scholar]
- Godelitsas, A.; Charistos, D.; Dwyer, J.; Tsipis, C.; Filippidis, A.; Hatzidimitriou, A.; Pavlidou, E. Copper (II)-loaded heu-type zeolite crystals: Characterization and evidence of surface complexation with N,N-diethyldithiocarbamate anions. Microporous Mesoporous Mater. 1999, 33, 77–87. [Google Scholar] [CrossRef]
- Morales, J.; Astilleros, J.M.; Jiménez, A.; Göttlicher, J.; Steininger, R.; Fernández-Díaz, L. Uptake of dissolved lead by anhydrite surfaces. Appl. Geochem. 2014, 40, 89–96. [Google Scholar] [CrossRef]
- Prieto, M.; Cubillas, P.; Fernández-González, Á. Uptake of dissolved Cd by biogenic and abiogenic aragonite: A comparison with sorption onto calcite. Geochim. Cosmochim. Acta 2003, 67, 3859–3869. [Google Scholar] [CrossRef]
- Astilleros, J.M.; Fernandez-Diaz, L.; Putnis, A. The role of magnesium in the growth of calcite: An AFM study. Chem. Geol. 2010, 271, 52–58. [Google Scholar] [CrossRef] [Green Version]
- Kornicker, W.A.; Morse, J.W.; Damasceno, R.N. The chemistry of Co2+ interaction with calcite and aragonite surfaces. Chem. Geol. 1985, 53, 229–236. [Google Scholar] [CrossRef]
- Xu, N.; Hochella, M.F.; Brown, G.E., Jr.; Parks, G.A. Co(II) sorption at the calcite-water interface: I. X-ray photoelectron spectroscopic study. Geochim. Cosmochim. Acta 1996, 60, 2801–2815. [Google Scholar] [CrossRef]
- Cheng, L.; Sturchio, N.C.; Bedzyk, M.J. Local structure of Co2+ incorporated at the calcite surface: An X-ray standing wave and sexafs study. Phys. Rev. B 2000, 61, 4877–4883. [Google Scholar] [CrossRef]
- Braybrook, A.L.; Heywood, B.R.; Jackson, R.A.; Pitt, K. Parallel computational and experimental studies of the morphological modification of calcium carbonate by cobalt. J. Cryst. Growth 2002, 243, 336–344. [Google Scholar] [CrossRef]
- Freij, S.J.; Putnis, A.; Astilleros, J. Nanoscale observations of the effect of cobalt on calcite growth and dissolution. J. Cryst. Growth 2004, 267, 288–300. [Google Scholar] [CrossRef] [Green Version]
- Katsikopoulos, D.; Fernández-González, Á.; Prieto, A.C.; Prieto, M. Co-crystallization of Co (II) with calcite: Implications for the mobility of cobalt in aqueous environments. Chem. Geol. 2008, 254, 87–100. [Google Scholar] [CrossRef]
- Lee, Y.J.; Reeder, R.J. The role of citrate and phthalate during Co (II) coprecipitation with calcite. Geochim. Cosmochim. Acta 2006, 70, 2253–2263. [Google Scholar] [CrossRef]
- González-López, J.; Ruiz-Hernández, S.; Fernández-González, Á.; Jiménez, A.; de Leeuw, N.; Grau-Crespo, R. Cobalt incorporation in calcite: Thermochemistry of (Ca,Co)CO3 solid solutions from density functional theory simulations. Geochim. Cosmochim. Acta 2014, 142, 205–216. [Google Scholar] [CrossRef]
- Xu, M.; Ilton, E.; Engelhard, M.; Qafokua, O.; Felmy, A.R.; Rosso, K.M.; Kerisit, S. Heterogeneous growth of cadmium and cobalt carbonate phases at the () calcite surface. Chem. Geol. 2015, 397, 24–36. [Google Scholar] [CrossRef]
- International Organization for Standardization. Surface Chemical Analysis—X-ray Photoelectron Spectrometers—Calibration of Energy Scales; ISO 15472:2001; International Organization for Standardization: Geneva, Switzerland, 2001. [Google Scholar]
- International Organization for Standardization. Surface Chemical Analysis—X-ray Photoelectron Spectrometers—Repeatability and Constancy of Intensity Scale; ISO 24237:2005; International Organization for Standardization: Geneva, Switzerland, 2005. [Google Scholar]
- Cubillas, P.; Kohler, S.; Prieto, M.; Chairat, C.; Oelkers, E.H. Experimental determination of the dissolution rates of calcite, aragonite, and bivalves. Chem. Geol. 2005, 216, 59–77. [Google Scholar] [CrossRef]
- Morse, J.W. The kinetics of calcium carbonate dissolution and precipitation. Rev. Mineral. Geochem. 1983, 11, 227–264. [Google Scholar]
- Parkhurst, D.L.; Appelo, C. User’s Guide to PHREEQC (Version 2): A Computer Program for Speciation, Batch-Reaction, One-Dimensional Transport, and Inverse Geochemical Calculations; U.S. Geological Survey: Reston, VA, USA, 1999.
- Petitto, S.C.; Marsh, E.M.; Carson, G.A.; Langell, M.A. Cobalt oxide surface chemistry: The interaction of CoO(100), Co3O4(110) and Co3O4(111) with oxygen and water. J. Mol. Catal. A Chem. 2008, 281, 49–58. [Google Scholar] [CrossRef]
- Carroll, S.A.; Bruno, J.; Petit, J.-C.; Dran, J.-C. Interactions of U (VI), Nd, and Th (IV) at the calcite-solution interface. Radiochim. Acta 1992, 58, 245–252. [Google Scholar] [CrossRef]
- Godelitsas, A.; Astilleros, J.M.; Hallam, K.; Harissopoulos, S.; Putnis, A. Interaction of calcium carbonates with lead in aqueous solutions. Environ. Sci. Technol. 2003, 37, 3351–3360. [Google Scholar] [CrossRef] [PubMed]
- Godelitsas, A.; Kokkoris, M.; Misaelides, P. Investigation of the interaction of Greek dolomitic marble with metal aqueous solutions using Rutherford backscattering and X-ray photoelectron spectroscopy. J. Radioanal. Nucl. Chem. 2007, 272, 339–344. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Monocrystal Experiments | |||
|---|---|---|---|
| Interaction Time | Surface Area (cm2) | Weigh (g) | Geometric Specific Surface Area (cm2/g) |
| 1 min | 75.90 | 0.085 | 897.15 |
| 5 min | 45.47 | 0.040 | 1142.44 |
| 15min | 81.33 | 0.105 | 774.58 |
| 30 min | 67.62 | 0.068 | 1000.22 |
| 1 h | 73.93 | 0.106 | 697.44 |
| 12 h | 78.92 | 0.115 | 668.06 |
| 1 day | 85.53 | 0.099 | 861.34 |
| 2 days | 111.13 | 0.181 | 613.64 |
| 1 week | 56.23 | 0.064 | 878.66 |
| 1 month | 66.56 | 0.050 | 1344.63 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-López, J.; Fernández-González, Á.; Jiménez, A.; Godelitsas, A.; Ladas, S.; Provatas, G.; Lagogiannis, A.; Pasias, I.N.; Thomaidis, N.S.; Prieto, M. Dissolution and Sorption Processes on the Surface of Calcite in the Presence of High Co2+ Concentration. Minerals 2017, 7, 23. https://doi.org/10.3390/min7020023
González-López J, Fernández-González Á, Jiménez A, Godelitsas A, Ladas S, Provatas G, Lagogiannis A, Pasias IN, Thomaidis NS, Prieto M. Dissolution and Sorption Processes on the Surface of Calcite in the Presence of High Co2+ Concentration. Minerals. 2017; 7(2):23. https://doi.org/10.3390/min7020023
Chicago/Turabian StyleGonzález-López, Jorge, Ángeles Fernández-González, Amalia Jiménez, Athanasios Godelitsas, Spyridon Ladas, Georgios Provatas, Anastasios Lagogiannis, Ioannis N. Pasias, Nikolaos S. Thomaidis, and Manuel Prieto. 2017. "Dissolution and Sorption Processes on the Surface of Calcite in the Presence of High Co2+ Concentration" Minerals 7, no. 2: 23. https://doi.org/10.3390/min7020023