
Mapping of Sulfur Isotopes and Trace Elements in Sulfides by LA-(MC)-ICP-MS: Potential Analytical Problems, Improvements and Implications

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Background
3. Samples and Analytical Methodology
3.1. Geological Background
3.2. Sample Selection and Petrology
3.3. Trace Element Analysis by LA-Q-ICP-MS
3.4. Sulfur Isotope Analysis by LA-MC-ICP-MS
3.5. Data Collection and Processing
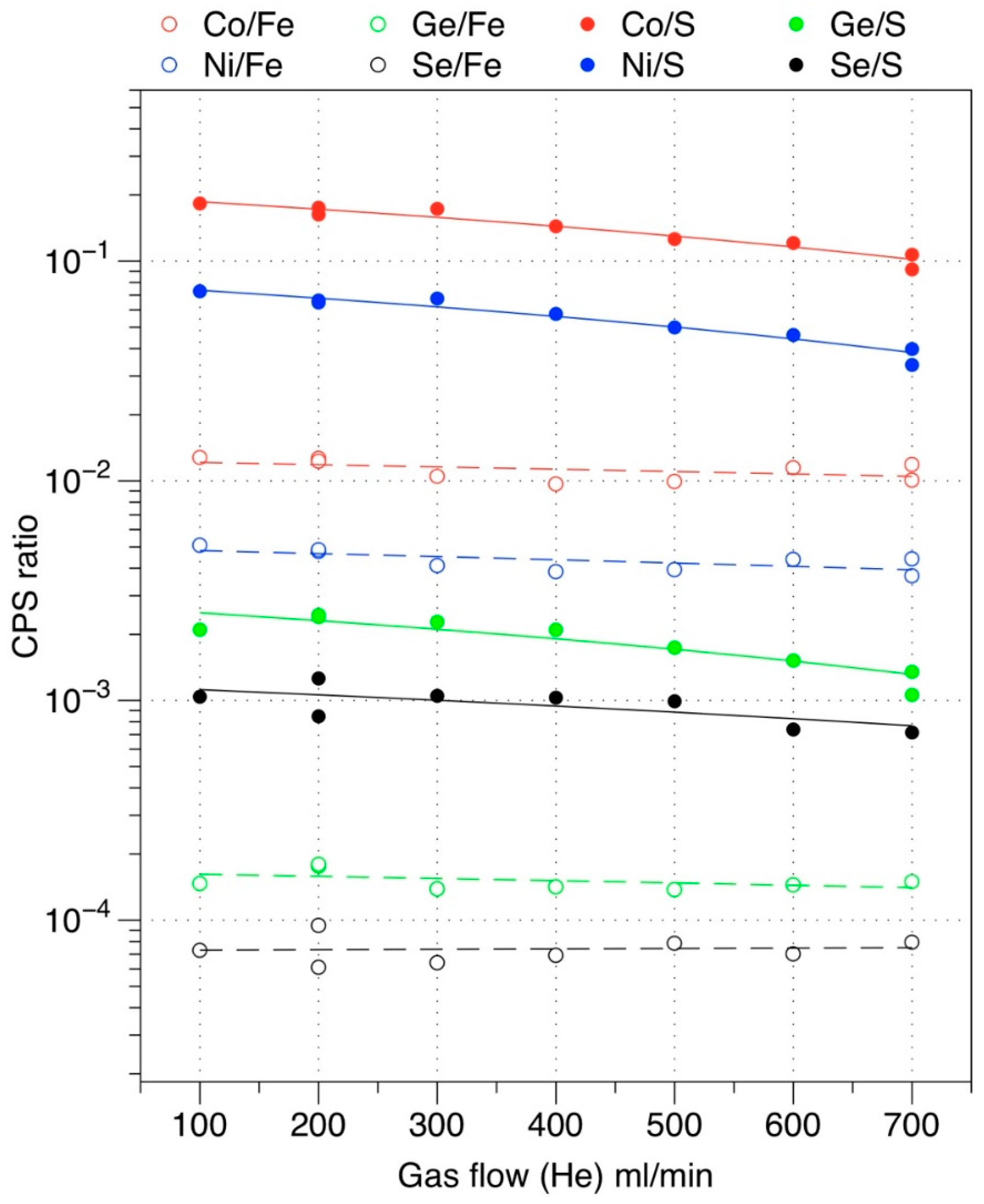
4. Precise Determination of Trace Elements using Fe or S normalization in Pyrite
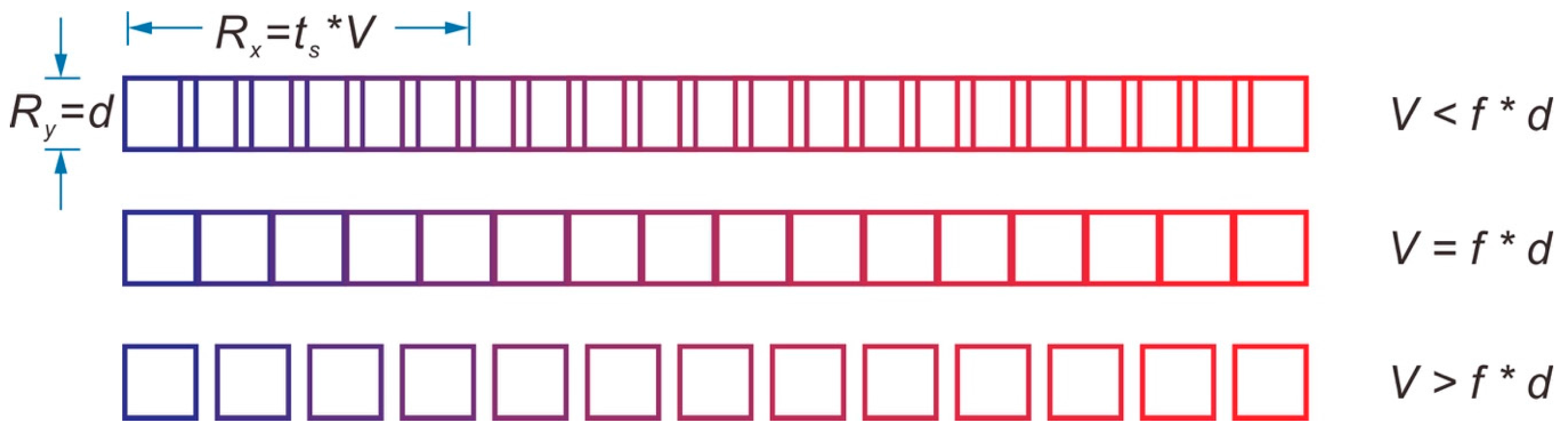
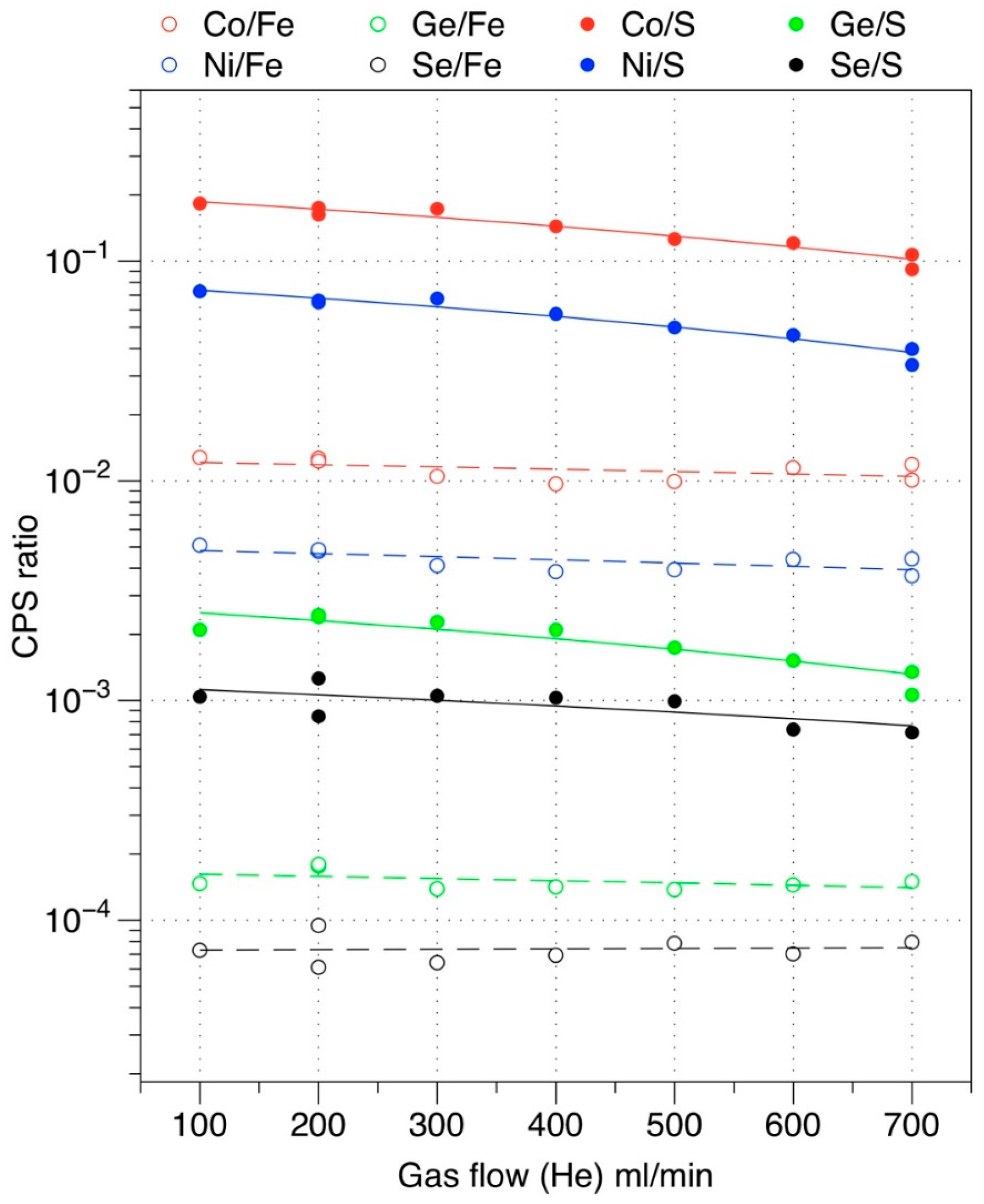
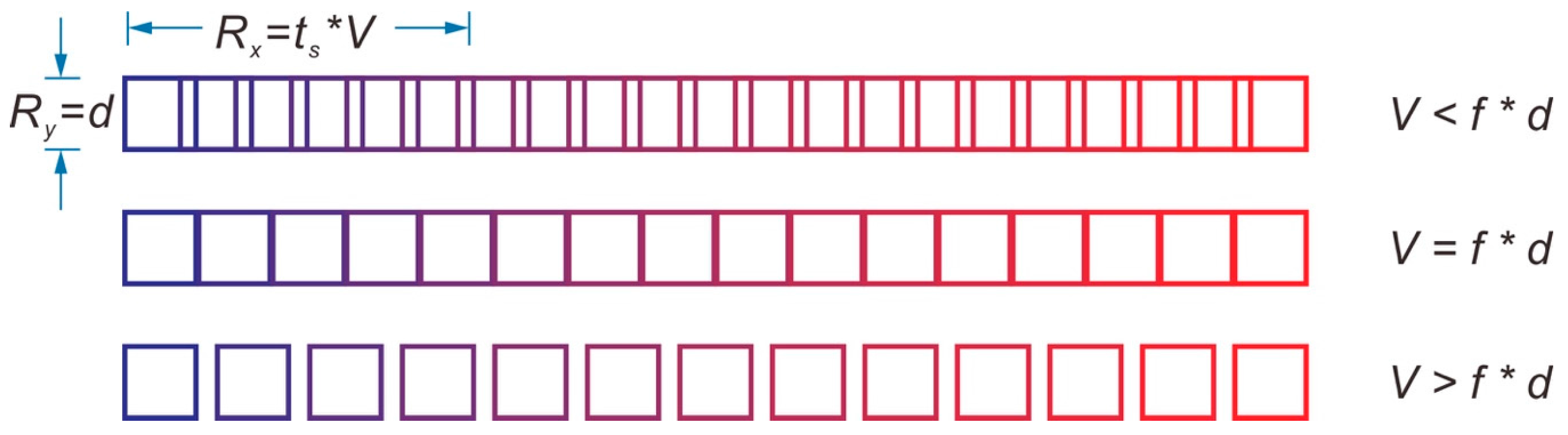
5. Spatial Resolution for Pulsed Laser during ICP-MS Measurement
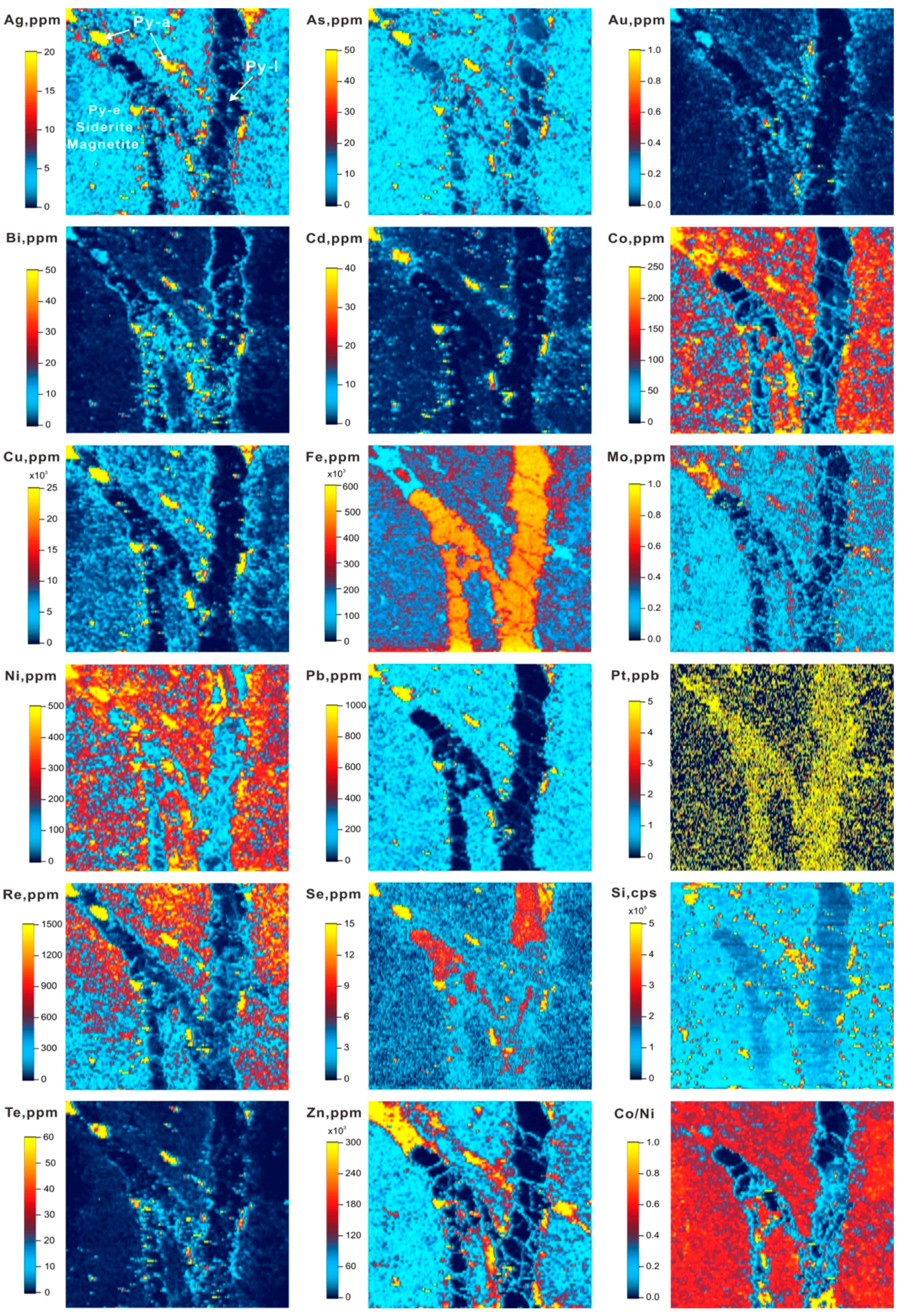
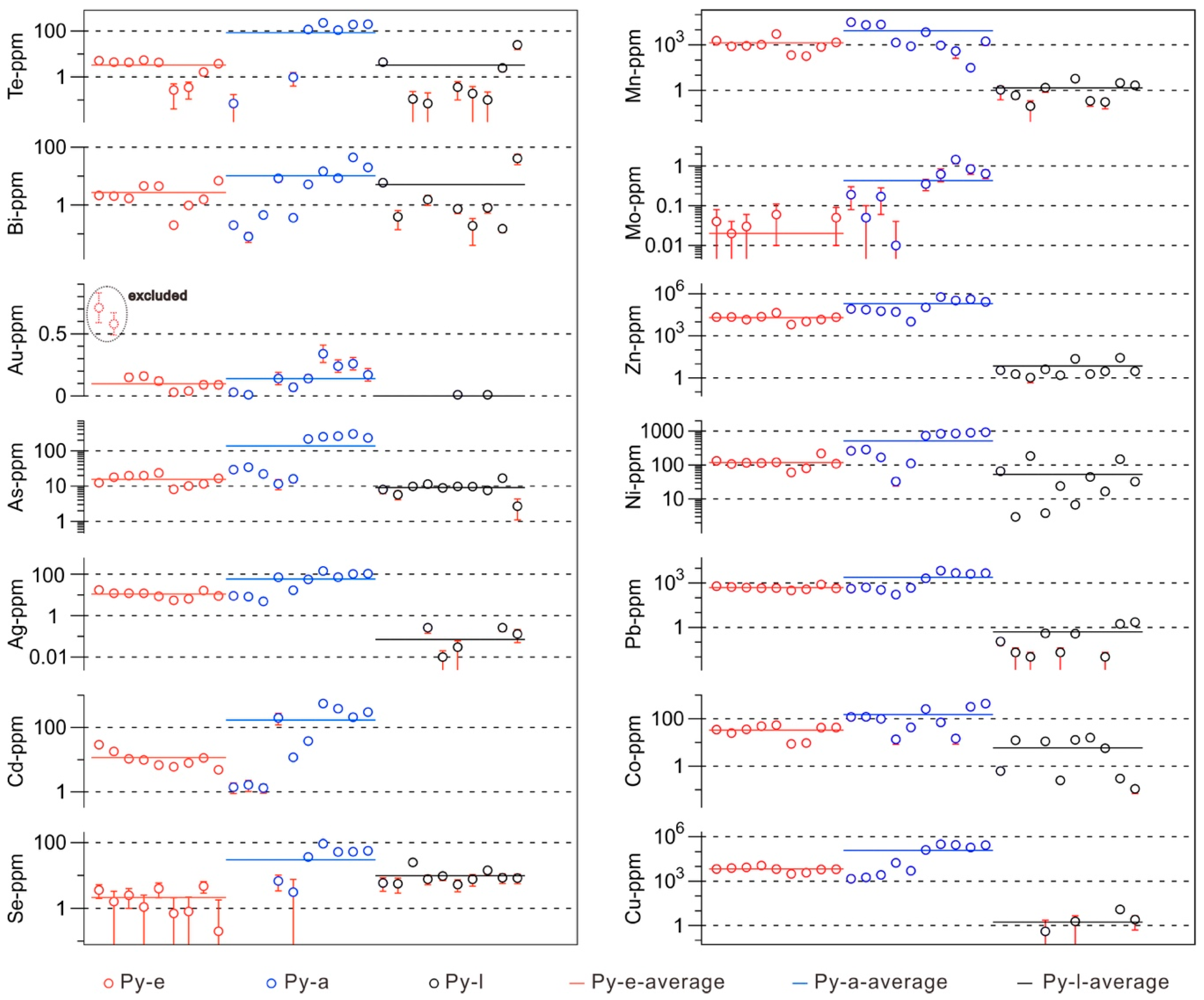
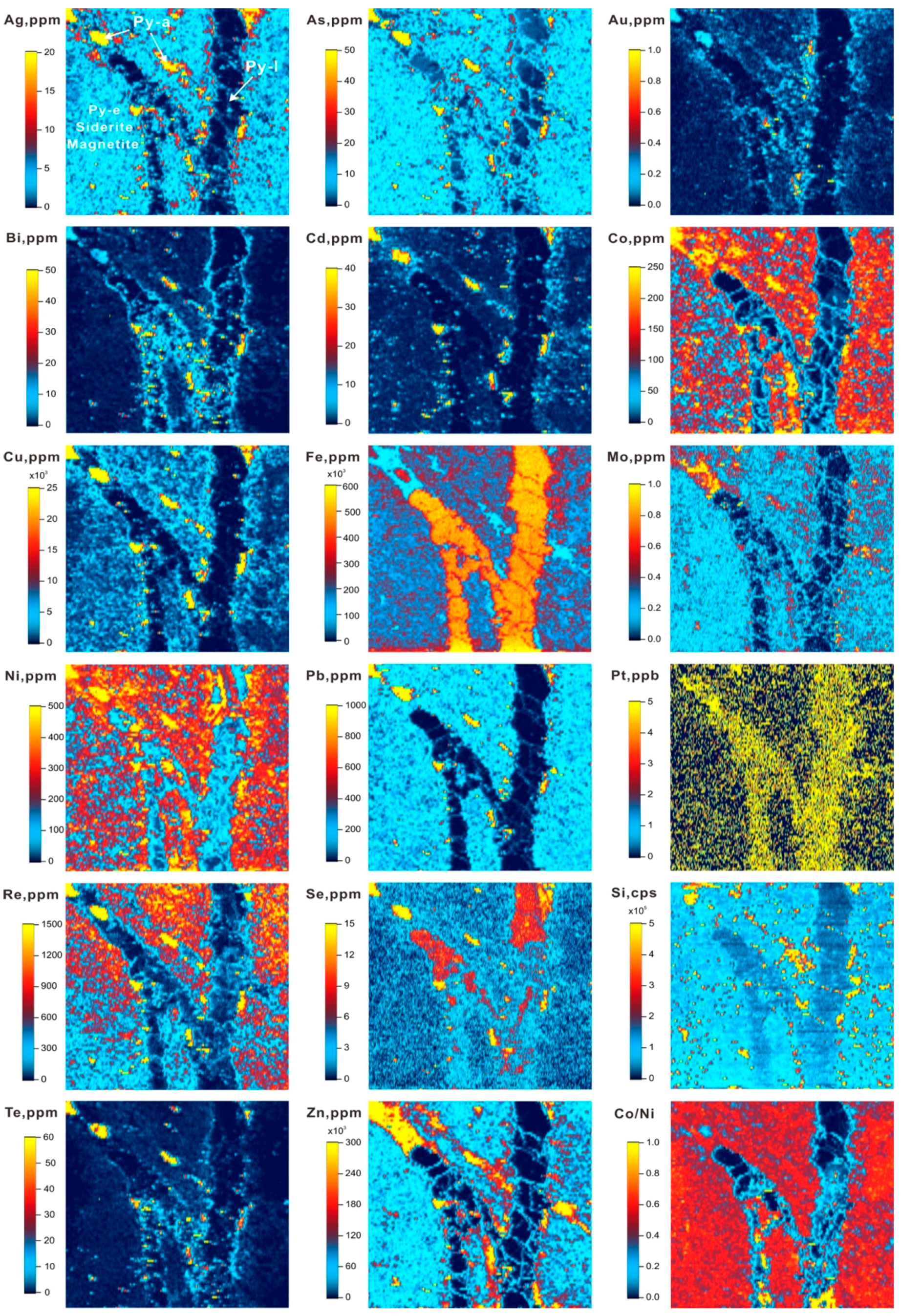
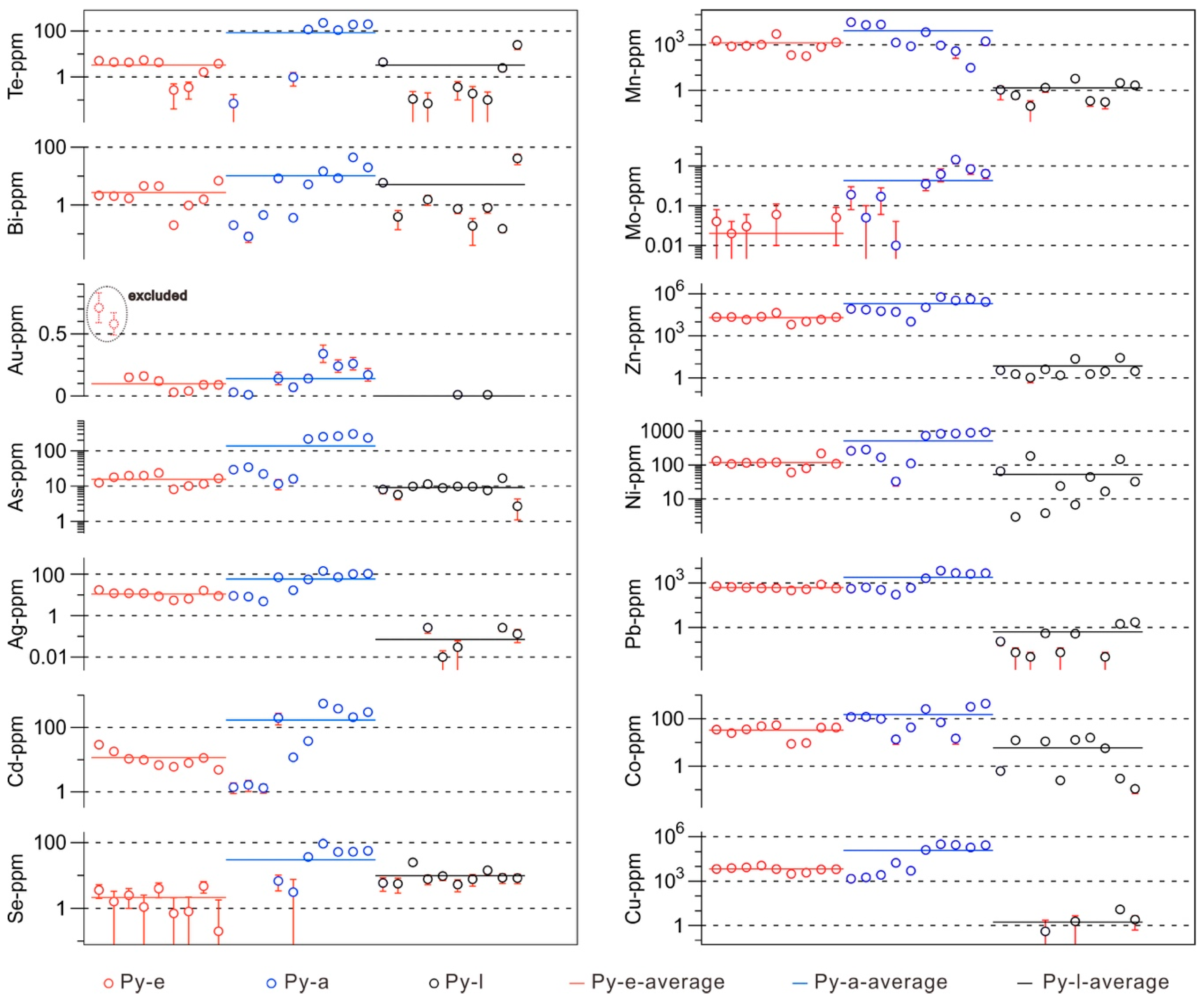
6. Mapping of Trace Elements in Pyrite
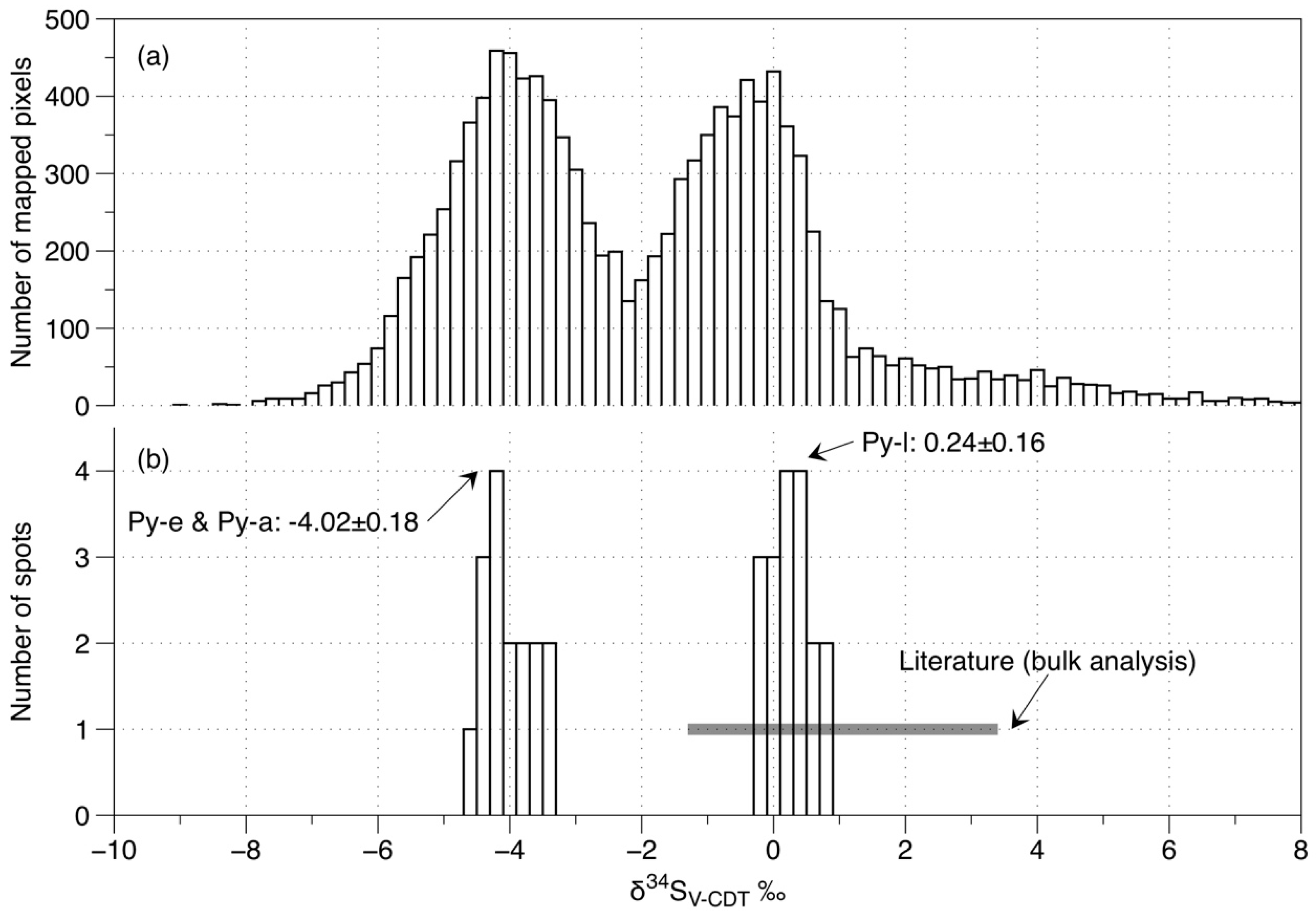
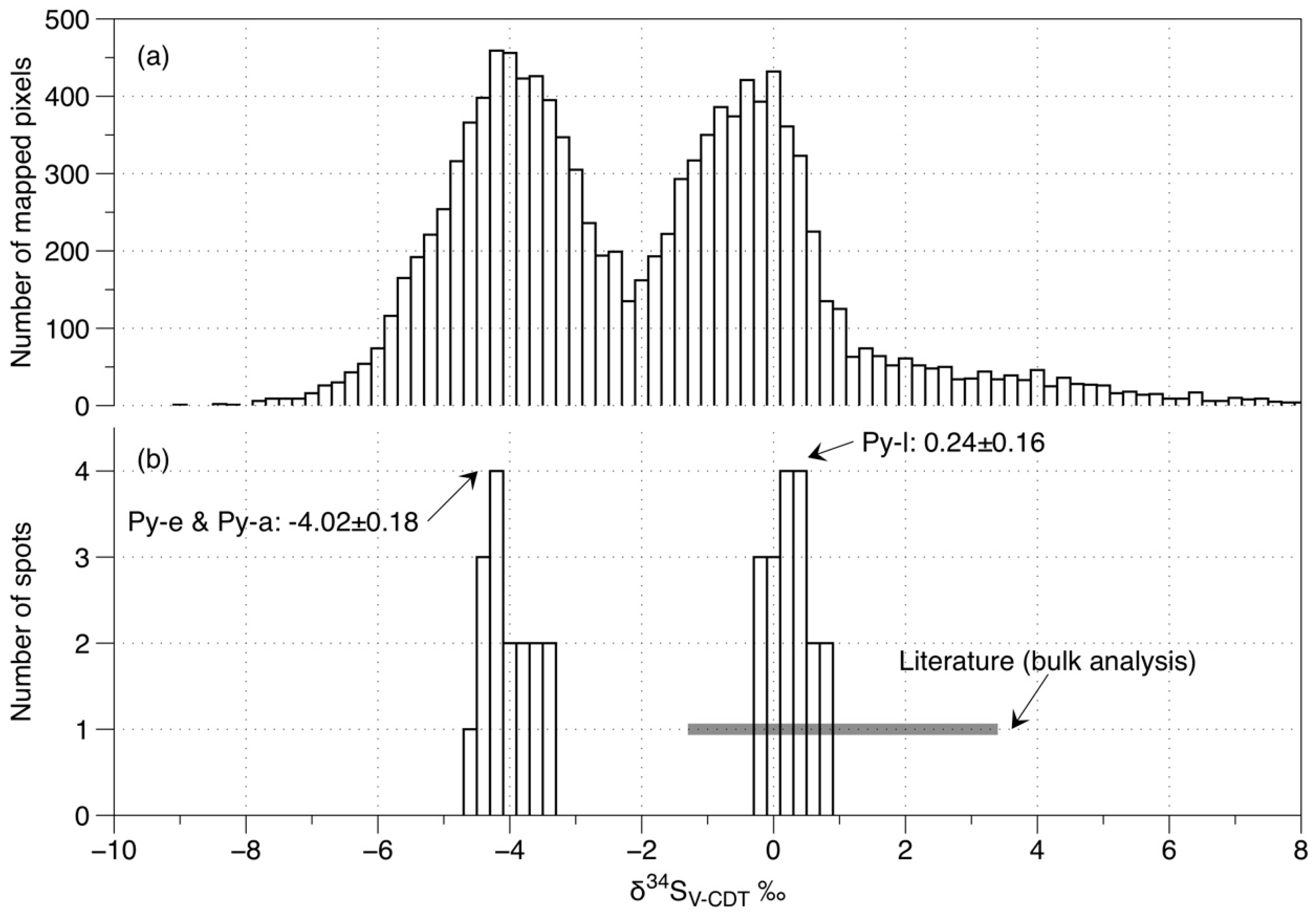
7. Mapping of Sulfur Isotopes in Pyrite
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| LA-(Q/MC)-ICP-MS | Laser Ablation (Quadrupole/Multiple Collector) Inductively Coupled Plasma Mass Spectrometer |
| CPS | Counts per Second |
| DRS | Data Reduction Scheme |
| LOD | Limit of Detection |
| EPMA | Electron Probe Microanalyser |
| SIMS | Secondary Ion Mass Spectrometry |
References
- Ohmoto, H. Systematics of sulfur and carbon isotopes in hydrothermal ore deposits. Econ. Geol. 1972, 67, 551–578. [Google Scholar] [CrossRef]
- Paytan, A.; Mearon, S.; Cobb, K.; Kastner, M. Origin of marine barite deposits: Sr and S isotope characterization. Geology 2002, 30, 747–750. [Google Scholar] [CrossRef]
- Ingham, E.S.; Cook, N.J.; Cliff, J.; Ciobanu, C.L.; Huddleston, A. A combined chemical, isotopic and microstructural study of pyrite from roll-front uranium deposits, Lake Eyre Basin, South Australia. Geochim. Cosmochim. Acta 2014, 125, 440–465. [Google Scholar] [CrossRef]
- Craddock, P.R.; Rouxel, O.J.; Ball, L.A.; Bach, W. Sulfur isotope measurement of sulfate and sulfide by high-resolution MC-ICP-MS. Chem. Geol. 2008, 253, 102–113. [Google Scholar] [CrossRef]
- Fu, J.; Hu, Z.; Zhang, W.; Yang, L.; Liu, Y.; Li, M.; Zong, K.; Gao, S.; Hu, S. In situ sulfur isotopes (δ34S and δ33S) analyses in sulfides and elemental sulfur using high sensitivity cones combined with the addition of nitrogen by laser ablation MC-ICP-MS. Anal. Chim. Acta 2016, 911, 14–26. [Google Scholar] [CrossRef] [PubMed]
- Bendall, C.; Lahaye, Y.; Fiebig, J.; Weyer, S.; Brey, G.P. In situ sulfur isotope analysis by laser ablation MC-ICPMS. Appl. Geochem. 2006, 21, 782–787. [Google Scholar] [CrossRef]
- Paton, C.; Hellstrom, J.; Paul, B.; Woodhead, J.; Hergt, J. Iolite: Freeware for the visualisation and processing of mass spectrometric data. J. Anal. Atomic Spectrom. 2011, 26, 2508–2518. [Google Scholar] [CrossRef]
- Liu, Y.; Gao, S.; Hu, Z.; Gao, C.; Zong, K.; Wang, D. Continental and oceanic crust recycling-induced melt-peridotite interactions in the Trans-North China Orogen: U-Pb Dating, Hf isotopes and trace elements in zircons from mantle xenoliths. J. Petrol. 2010, 51, 537–571. [Google Scholar] [CrossRef]
- Ubide, T.; McKenna, C.A.; Chew, D.M.; Kamber, B.S. High-resolution LA-ICP-MS trace element mapping of igneous minerals: In search of magma histories. Chem. Geol. 2015, 409, 157–168. [Google Scholar] [CrossRef]
- Ulrich, T.; Kamber, B.S.; Jugo, P.J.; Tinkham, D.K. Imaging element-distribution patterns in minerals by laser ablation-inductively coupled plasma-mass spectrometry (LA-ICP-MS). Can. Mineral. 2009, 47, 1001–1012. [Google Scholar] [CrossRef]
- Chew, D.M.; Babechuk, M.G.; Cogné, N.; Mark, C.; O’Sullivan, G.J.; Henrichs, I.A.; Doepke, D.; McKenna, C.A. (LA,Q)-ICPMS trace-element analyses of Durango and McClure Mountain apatite and implications for making natural LA-ICPMS mineral standards. Chem. Geol. 2016, 435, 35–48. [Google Scholar] [CrossRef]
- Cook, N.; Etschmann, B.; Ciobanu, C.; Geraki, K.; Howard, D.; Williams, T.; Rae, N.; Pring, A.; Chen, G.; Johannessen, B.; et al. Distribution and substitution mechanism of Ge in a Ge-(Fe)-Bearing sphalerite. Minerals 2015, 5, 117–132. [Google Scholar] [CrossRef]
- George, L.L.; Cook, N.J.; Ciobanu, C.L. Partitioning of trace elements in co-crystallized sphalerite–galena–chalcopyrite hydrothermal ores. Ore Geol. Rev. 2016, 77, 97–116. [Google Scholar] [CrossRef]
- Agangi, A.; Hofmann, A.; Wohlgemuth-Ueberwasser, C.C. Pyrite zoning as a record of mineralization in the Ventersdorp Contact Reef, Witwatersrand Basin, South Africa. Econ. Geol. 2013, 108, 1243–1272. [Google Scholar] [CrossRef]
- Gagnevin, D.; Menuge, J.F.; Kronz, A.; Barrie, C.; Boyce, A.J. Minor elements in layered sphalerite as a record of fluid origin, mixing, and crystallization in the Navan Zn-Pb ore deposit, Ireland. Econ. Geol. 2014, 109, 1513–1528. [Google Scholar] [CrossRef]
- Large, R.R.; Danyushevsky, L.; Hollit, C.; Maslennikov, V.; Meffre, S.; Gilbert, S.; Bull, S.; Scott, R.; Emsbo, P.; Thomas, H.; et al. Gold and trace element zonation in pyrite using a laser imaging technique: Implications for the timing of gold in orogenic and carlin-style sediment-hosted deposits. Econ. Geol. 2009, 104, 635–668. [Google Scholar] [CrossRef]
- Woodhead, J.D.; Hellstrom, J.; Hergt, J.M.; Greig, A.; Maas, R. Isotopic and elemental imaging of geological materials by laser ablation inductively coupled plasma-mass spectrometry. Geostand. Geoanal. Res. 2007, 31, 331–343. [Google Scholar] [CrossRef]
- Pribil, M.J.; Ridley, W.I.; Emsbo, P. Sulfate and sulfide sulfur isotopes (δ34S and δ33S) measured by solution and laser ablation MC-ICP-MS: An enhanced approach using external correction. Chem. Geol. 2015, 412, 99–106. [Google Scholar] [CrossRef]
- Liu, C.; Bian, X.-P.; Yang, T.; Lin, A.-J.; Jiang, S.-Y. Matrix effects of calcium on high-precision sulfur isotope measurement by multiple-collector inductively coupled plasma mass spectrometry. Talanta 2016, 151, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.; Woodhead, J.D.; Paton, C.; Hergt, J.M.; Hellstrom, J.; Norris, C.A. Towards a method for quantitative LA-ICP-MS imaging of multi-phase assemblages: Mineral identification and analysis correction procedures. Geostand. Geoanal. Res. 2014, 38, 253–263. [Google Scholar] [CrossRef]
- Perkins, W.T.; Pearce, N.J.G.; Westgate, J.A. The development of laser ablation ICP-MS and calibration strategies: Examples from the analysis of trace elements in volcanic glass shards and sulfide minerals. Geostand. Geoanal. Res. 1997, 21, 175–190. [Google Scholar] [CrossRef]
- Gilbert, S.E.; Danyushevsky, L.V.; Goemann, K.; Death, D. Fractionation of sulphur relative to iron during laser ablation-ICP-MS analyses of sulphide minerals: Implications for quantification. J. Anal. Atomic Spectrom. 2014, 29, 1024–1033. [Google Scholar] [CrossRef]
- Zhu, Z.-Y.; Ciobanu, C.L.; Jiang, S.-Y.; Yang, T.; Cook, N.J. Sulfur isotope fractionation in pyrite during laser ablation: Implications for laser ablation multiple collector inductively coupled plasma mass Spectrometry mapping. Chem. Geol. 2016, in press. [Google Scholar]
- Sanborn, M.; Telmer, K. The spatial resolution of LA-ICP-MS line scans across heterogeneous materials such as fish otoliths and zoned minerals. J. Anal. Atomic Spectrom. 2003, 18, 1231–1237. [Google Scholar] [CrossRef]
- Triglav, J.; van Elteren, J.T.; Šelih, V.S. Basic modeling approach to optimize elemental imaging by laser ablation ICPMS. Anal. Chem. 2010, 82, 8153–8160. [Google Scholar] [CrossRef] [PubMed]
- Van Elteren, J.T.; Izmer, A.; Šelih, V.S.; Vanhaecke, F. Novel image metrics for retrieval of the lateral resolution in line scan-based 2D LA-ICPMS imaging via an experimental-modeling approach. Anal. Chem. 2016, 88, 7413–7420. [Google Scholar] [CrossRef] [PubMed]
- Kindness, A. Two-dimensional mapping of copper and zinc in liver sections by laser ablation-inductively coupled plasma mass spectrometry. Clin. Chem. 2003, 49, 1916–1923. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.S.; Zoriy, M.V.; Pickhardt, C. Imaging of copper, zinc, and other elements in thin section of human brain samples (hippocampus) by laser ablation inductively coupled plasma mass spectrometry. Anal. Chem. 2005, 77, 3208–3216. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.S.; Matusch, A.; Depboylu, C.; Dobrowolska, J.; Zoriy, M.V. Quantitative imaging of selenium, copper, and zinc in thin sections of biological tissues (Slugs—Genus Arion) measured by laser ablation inductively coupled plasma mass spectrometry. Anal. Chem. 2007, 79, 6074–6080. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.F.; Liu, X.P.; Wu, Y.C. The Copper Iron Belt of the Middle and Lower Reaches of Yangtze River; Geological Publishing House: Beijing, China, 1991. (In Chinese) [Google Scholar]
- Huang, E.; Zhang, N.; Luo, Z. The genesis of the Chengmenshan and Wushan copper deposits. Miner. Depos. 1990, 9, 291–308. (In Chinese) [Google Scholar]
- Gu, L.; Xu, K. On the carboniferous submarine massive sulphide deposits in the lower reaches of the Changjiang (Yangzi) River. Acta Geol. Sin. 1986, 176–189. (In Chinese) [Google Scholar]
- Jiang, S.Y.; Ding, Q.F.; Yang, S.Y.; Zhu, Z.Y.; Sun, M.Z.; Sun, Y. Discovery and Significance of carbonate mud mounds from cu-polymetallic deposits in the middle and Lower Yangtze metallogenic belt: Examples from the Wushan and Dongguashan deposit. Acta Geol. Sin. 2011, 85, 744–756. (In Chinese) [Google Scholar]
- Mao, J.; Xie, G.; Duan, C.; Pirajno, F.; Ishiyama, D.; Chen, Y. A tectono-genetic model for porphyry–skarn–stratabound Cu-Au-Mo-Fe and magnetite–apatite deposits along the Middle–Lower Yangtze River Valley, Eastern China. Ore Geol. Rev. 2011, 43, 294–314. [Google Scholar] [CrossRef]
- Mao, J.W.; Shao, Y.J.; Xie, G.Q.; Zhang, J.D.; Chen, Y.C. Mineral deposit model for porphyry-skarn polymetallic copper deposits in Tongling ore dense district of Middle-Lower Yangtze Valley metallogenic belt. Miner. Depos. 2009, 28, 109–119. (In Chinese) [Google Scholar]
- Wilson, S.A.; Ridley, W.I.; Koenig, A.E. Development of sulfide calibration standards for the laser ablation inductively-coupled plasma mass spectrometry technique. J. Anal. At. Spectrom. 2002, 17, 406–409. [Google Scholar] [CrossRef]
- Parent, M.; Vanhoe, H.; Moens, L.; Dams, R. Investigation of HfO+ interference in the determination of platinum in a catalytic converter (cordierite) by inductively coupled plasma mass spectrometry. Talanta 1997, 44, 221–230. [Google Scholar] [CrossRef]
- Cook, N.J.; Ciobanu, C.L.; George, L.; Zhu, Z.-Y.; Wade, B.; Ehrig, K. Trace element analysis of minerals in magmatic-hydrothermal ores by laser ablation inductively-coupled plasma mass spectrometry: Approaches and opportunities. Minerals 2016, 6, 111. [Google Scholar]
- Longerich, H.P.; Jackson, S.E.; Günther, D. Inter-laboratory note. Laser ablation inductively coupled plasma mass spectrometric transient signal data acquisition and analyte concentration calculation. J. Anal. Atomic Spectrom. 1996, 11, 899–904. [Google Scholar] [CrossRef]
- Zhao, R.; Xie, Y.H.; Yao, Y.Y. Sulfur isotope study of the copper ore deposit of Chengmenshan and Wushan. Sci. Geol. Sin. 1985, 3, 251–258. (In Chinese) [Google Scholar]


© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, Z.-Y.; Cook, N.J.; Yang, T.; Ciobanu, C.L.; Zhao, K.-D.; Jiang, S.-Y. Mapping of Sulfur Isotopes and Trace Elements in Sulfides by LA-(MC)-ICP-MS: Potential Analytical Problems, Improvements and Implications. Minerals 2016, 6, 110. https://doi.org/10.3390/min6040110
Zhu Z-Y, Cook NJ, Yang T, Ciobanu CL, Zhao K-D, Jiang S-Y. Mapping of Sulfur Isotopes and Trace Elements in Sulfides by LA-(MC)-ICP-MS: Potential Analytical Problems, Improvements and Implications. Minerals. 2016; 6(4):110. https://doi.org/10.3390/min6040110
Chicago/Turabian StyleZhu, Zhi-Yong, Nigel J. Cook, Tao Yang, Cristiana L. Ciobanu, Kui-Dong Zhao, and Shao-Yong Jiang. 2016. "Mapping of Sulfur Isotopes and Trace Elements in Sulfides by LA-(MC)-ICP-MS: Potential Analytical Problems, Improvements and Implications" Minerals 6, no. 4: 110. https://doi.org/10.3390/min6040110