Long-Term Acid-Generating and Metal Leaching Potential of a Sub-Arctic Oil Shale

Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Sample Comminution
2.3. Material Characterisation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Elements | Secondary Target | Measurement Live Time (s) | Tube (kV) | Tube (mA) |
|---|---|---|---|---|
| Na-Mg | Al | 200 | 35 | 17 |
| Al-K | CaF2 | 200 | 40 | 15 |
| Ca-Mn | Fe | 100 | 75 | 8 |
| Fe-Ga | Ge | 100 | 75 | 8 |
| Ge-Y | Zr | 100 | 100 | 6 |
| Zr-U | Al2O3 (Barkla) | 100 | 100 | 6 |
2.4. Sample Acidification and Leaching
3. Results
3.1. Material Characterisation
| Shale | Major Minerals (Quartz SiO2, Muscovite KAl2(AlSi3O10)(F,OH)2) | Minor Minerals (Anorthite CaAl2Si2O8, Halite NaCl, Orthoclase KAlSi3O8) | Acidity-releasing minerals (Pyrite FeS2, Marcasite FeS2) | Neutralizing Minerals (Dolomite CaMg(CO3)2) |
|---|---|---|---|---|
| A | Quartz 46%, Muscovite 52% | Pyrite 1%, Marcasite 1% | ||
| B | Quartz 47%, Muscovite 42% | Anorthite 11% | Pyrite 1% | |
| C | Quartz 45%, Muscovite 49% | Anorthite 11% | Pyrite <1% | |
| D | Quartz 53%, Muscovite 43% | Halite 3% | Pyrite 1% | Dolomite 1% |
| E | Quartz 74%, Muscovite 25% | Pyrite 1% | ||
| F | Quartz 56%, Muscovite 44% | Pyrite 1% | ||
| G | Quartz 54%, Muscovite 45% | Pyrite 1% | ||
| H | Quartz 52%, Muscovite 40% | Orthoclase 4% | Pyrite 1% | Dolomite 3% |
| Shale | Na2O | MgO | Al2O3 | SiO2 | S | K2O | CaO | TiO2 | Fe2O3 |
|---|---|---|---|---|---|---|---|---|---|
| Shale A | <1.0 | <1.0 | 9.8 | 84.6 | <1.0 | 1.15 | 0.68 | 0.30 | 2.46 |
| Shale B | <1.0 | <1.0 | 10.5 | 83.7 | <1.0 | 1.29 | 0.54 | 0.32 | 2.66 |
| Shale C | <1.0 | <1.0 | 9.9 | 85.0 | <1.0 | 1.12 | 0.69 | 0.28 | 2.00 |
| Shale D | <1.0 | <1.0 | 10.2 | 83.6 | <1.0 | 1.12 | 1.53 | 0.28 | 2.19 |
| Shale E | <1.0 | <1.0 | <0.1 | 96.4 | <1.0 | 0.79 | 0.39 | 0.23 | 1.07 |
| Shale F | <1.0 | <1.0 | 9.4 | 86.0 | <1.0 | 1.02 | 0.49 | 0.26 | 1.83 |
| Shale G | <1.0 | <1.0 | 9.5 | 85.6 | <1.0 | 1.04 | 0.65 | 0.27 | 1.93 |
| Shale H | <1.0 | <1.0 | 13.1 | 77.5 | <1.0 | 1.63 | 3.20 | 0.40 | 3.01 |
| Shale | V | Cr | Mn | Ni | Cu | Zn | As | Se | Ba | Pb |
|---|---|---|---|---|---|---|---|---|---|---|
| Shale A | 168 | 96 | 93 | 79 | 123 | 133 | 19 | 12 | 1827 | 38 |
| Shale B | 162 | 103 | 85 | 58 | 126 | 84 | 16 | 12 | 1890 | 43 |
| Shale C | 305 | 96 | 63 | 66 | 110 | 80 | 24 | 13 | 2150 | 31 |
| Shale D | 255 | 89 | 115 | 104 | 125 | 251 | 20 | 12 | 2069 | 30 |
| Shale E | 265 | 66 | 48 | 75 | 96 | 80 | 23 | 13 | 2051 | <5 |
| Shale F | 249 | 82 | 72 | 94 | 113 | 146 | 17 | 13 | 1881 | 28 |
| Shale G | 198 | 82 | 76 | 102 | 106 | 152 | 20 | 15 | 2024 | 21 |
| Shale H | 289 | 116 | 205 | 163 | 144 | 244 | 34 | 16 | 2212 | 32 |
| CSQG (mg/kg) | 130 | 64 | N/A | 50 | 63 | 200 | 12 | 1 | 750 | 70 |
| Sites failing CSQG | A-H | A-H | N/A | A-H | A-H | D, H | A-H | A-H | A-H | Nil |
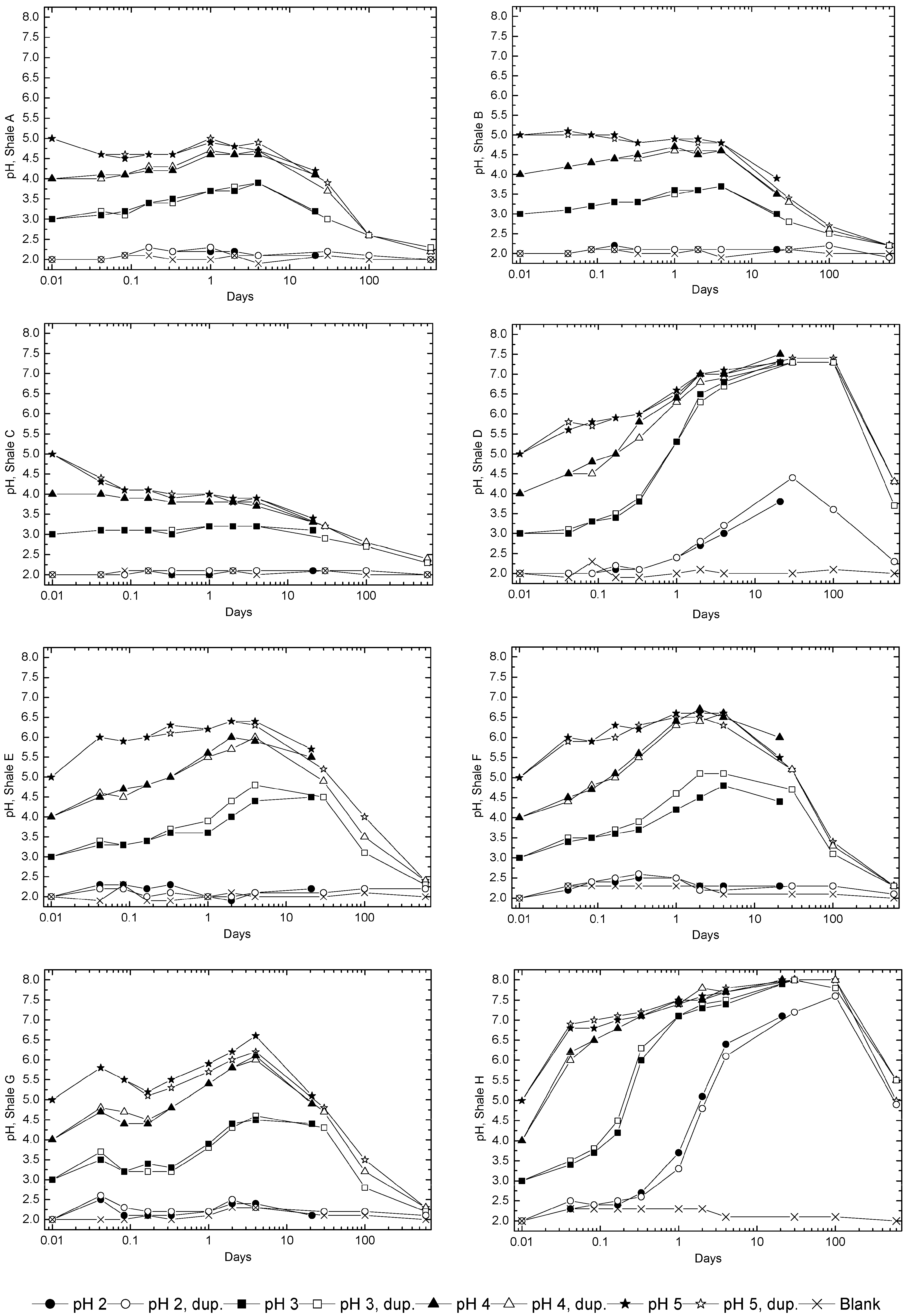
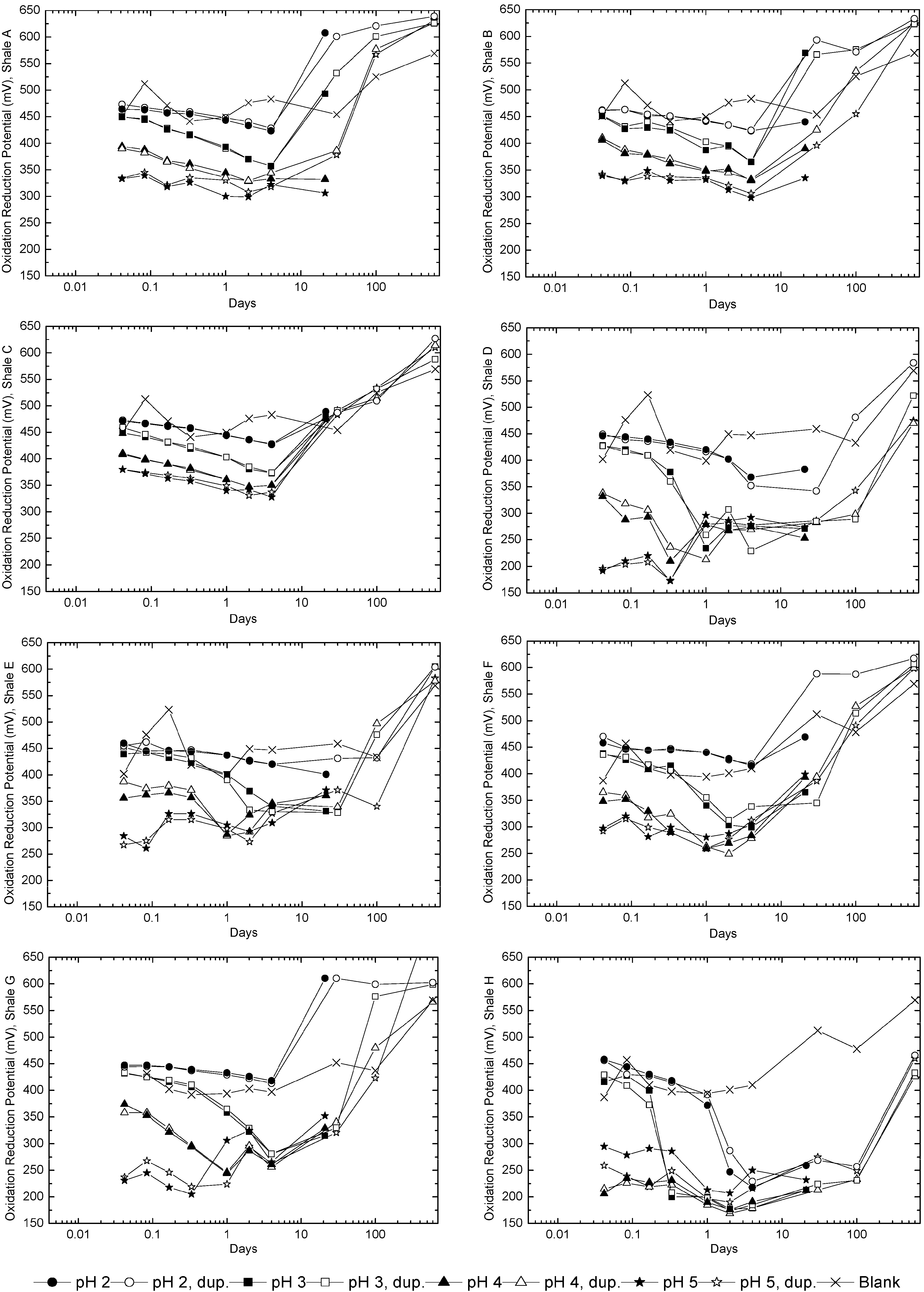
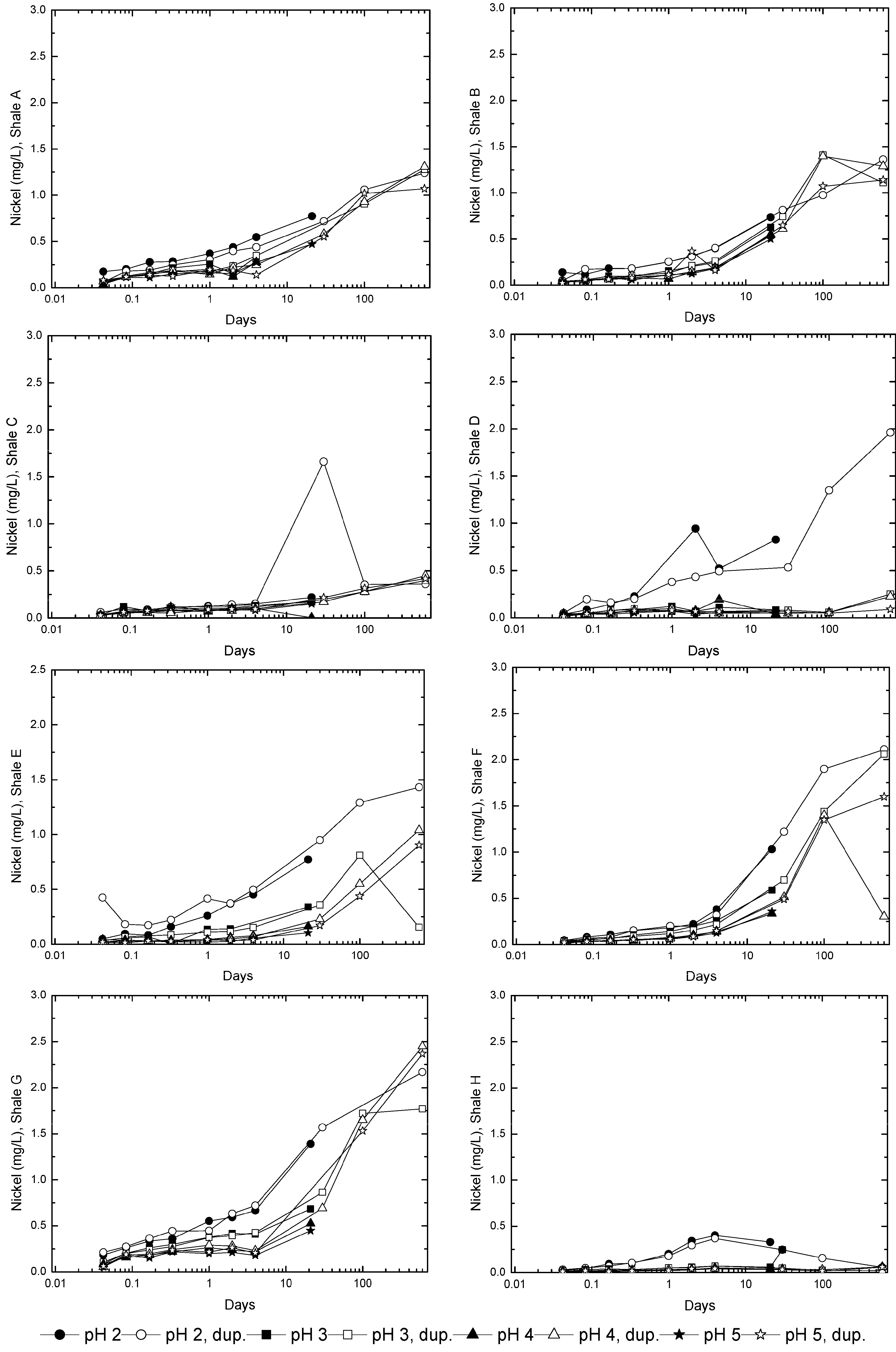
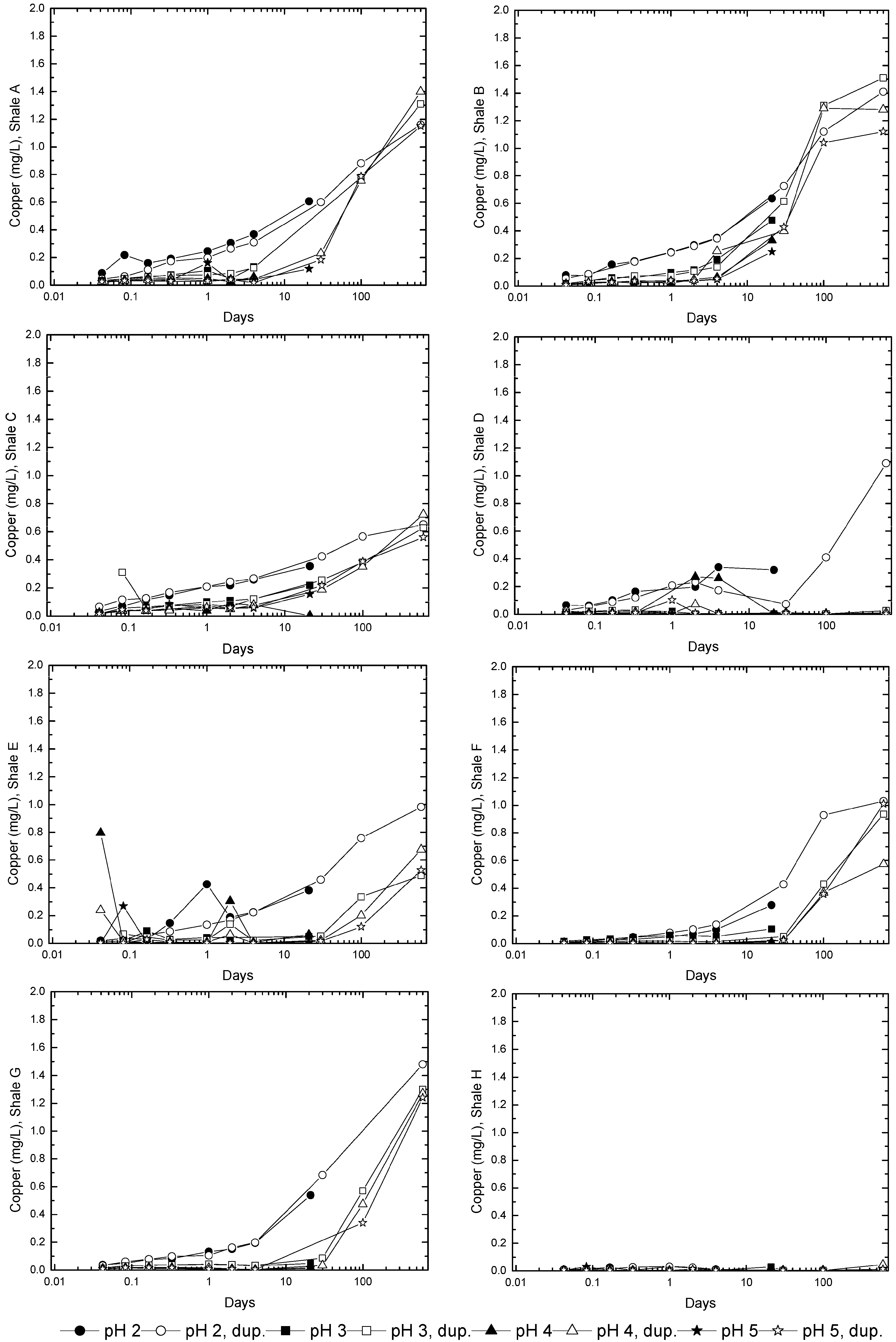
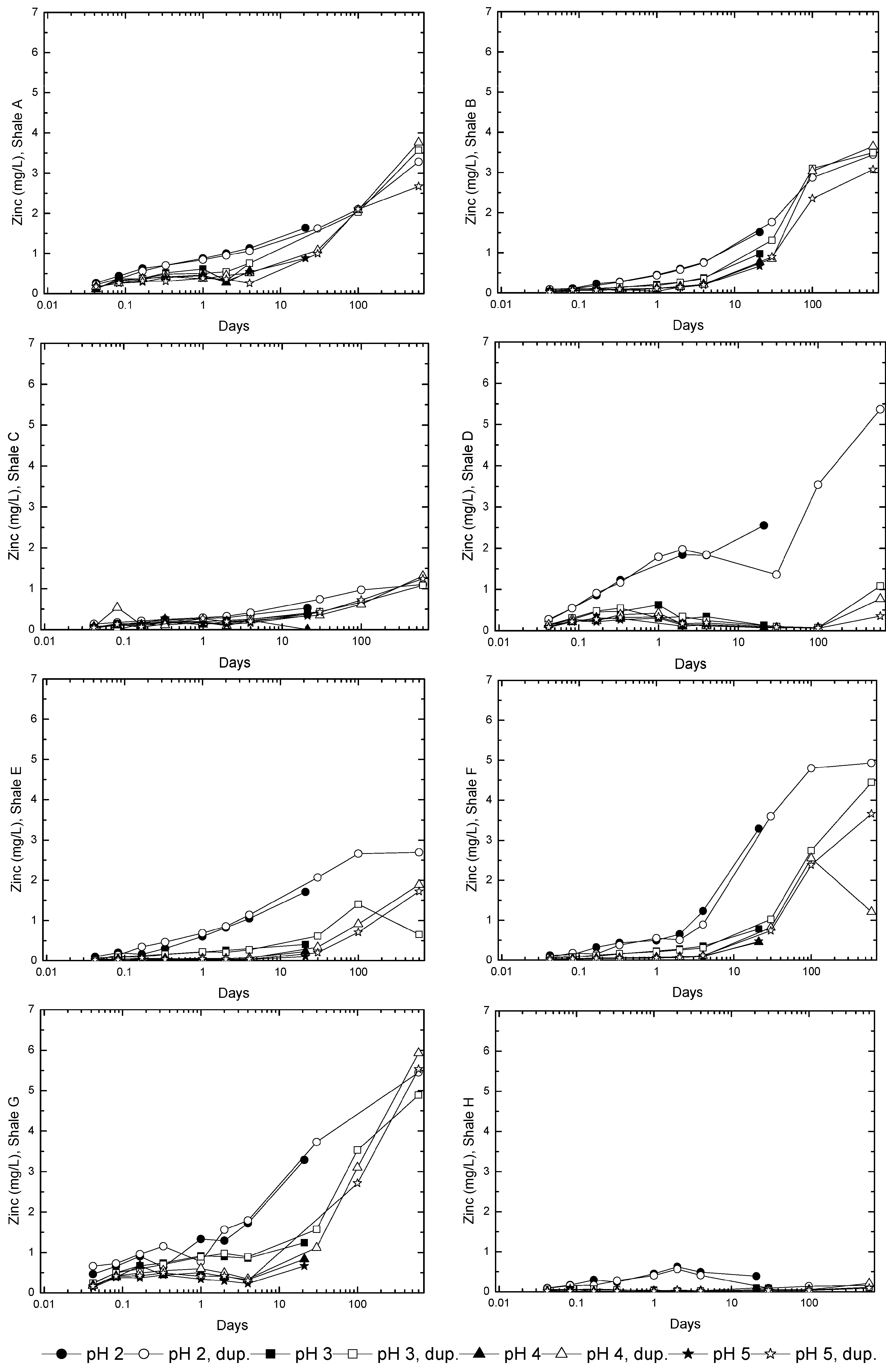
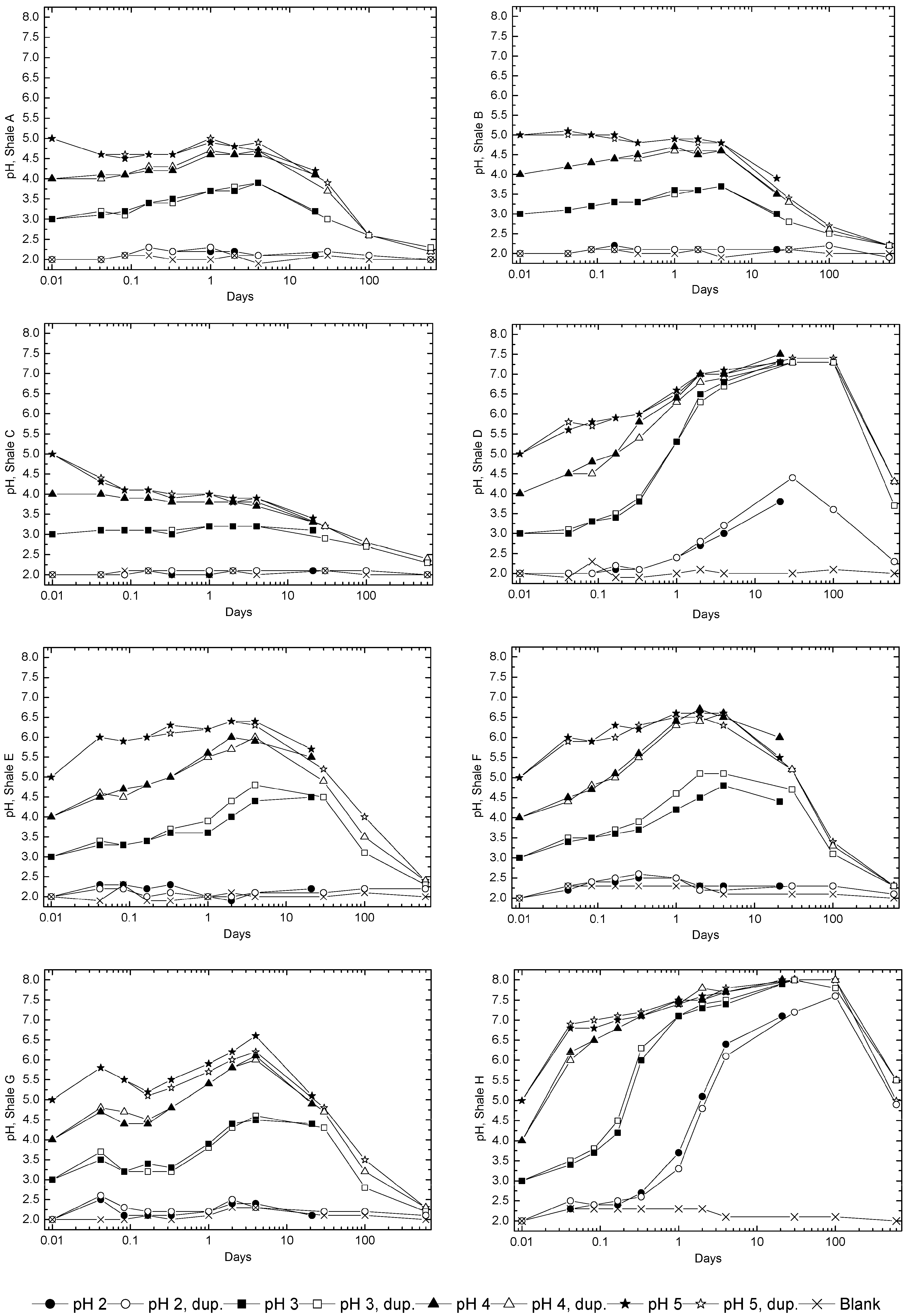
3.2. Sample Acidification and Leaching
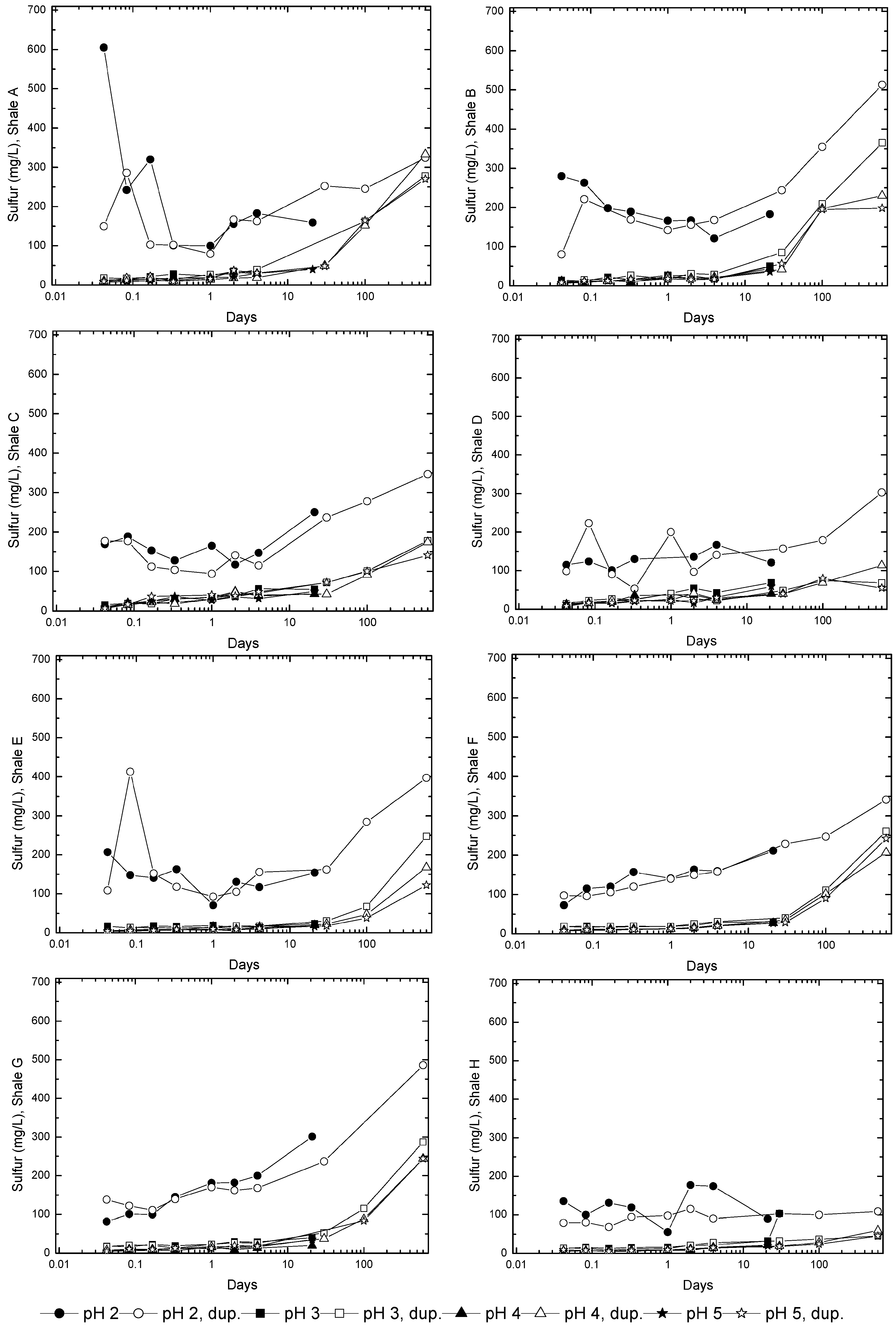
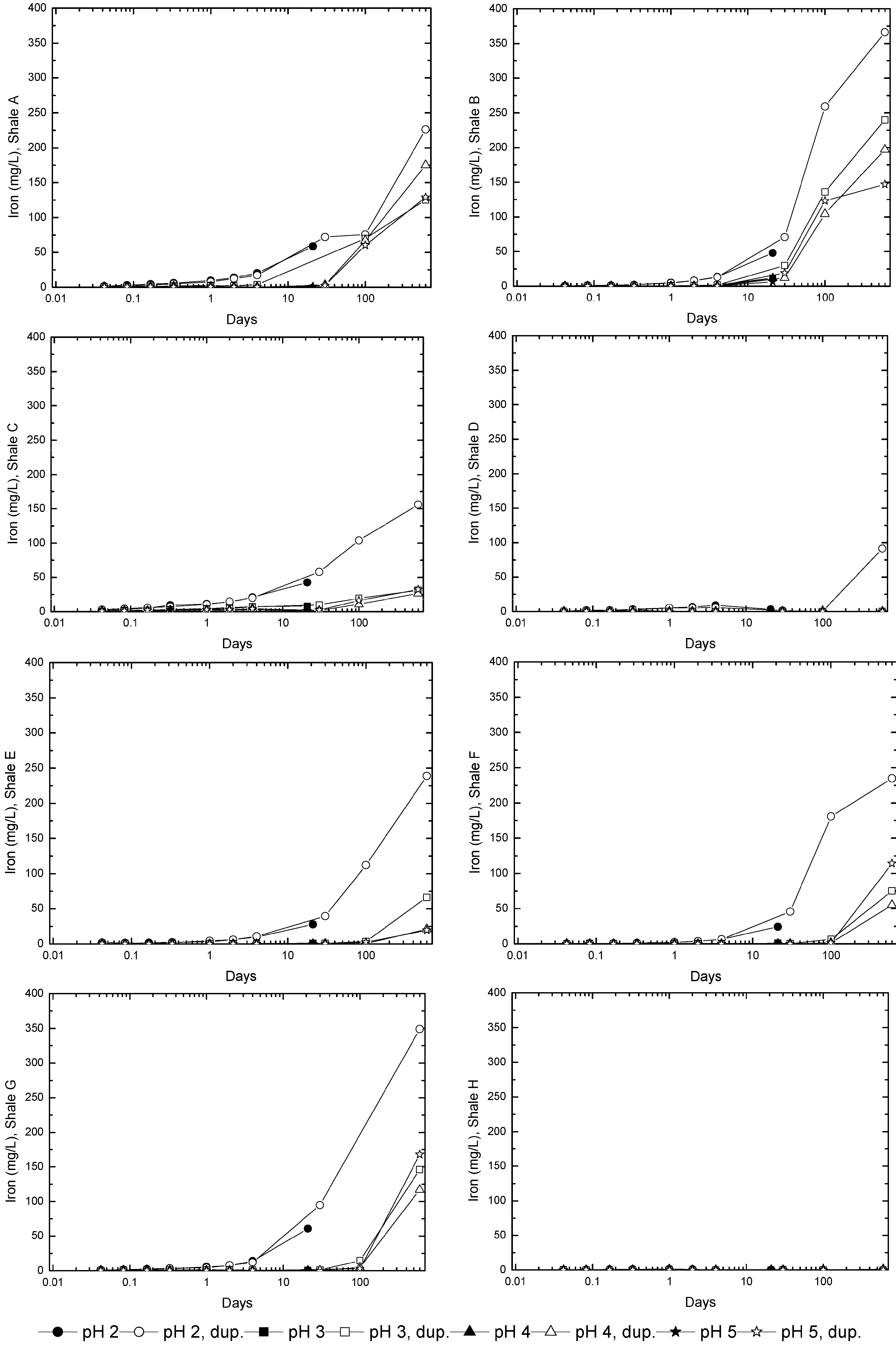
3.3. Evolution of Sulfur and Metal Concentrations
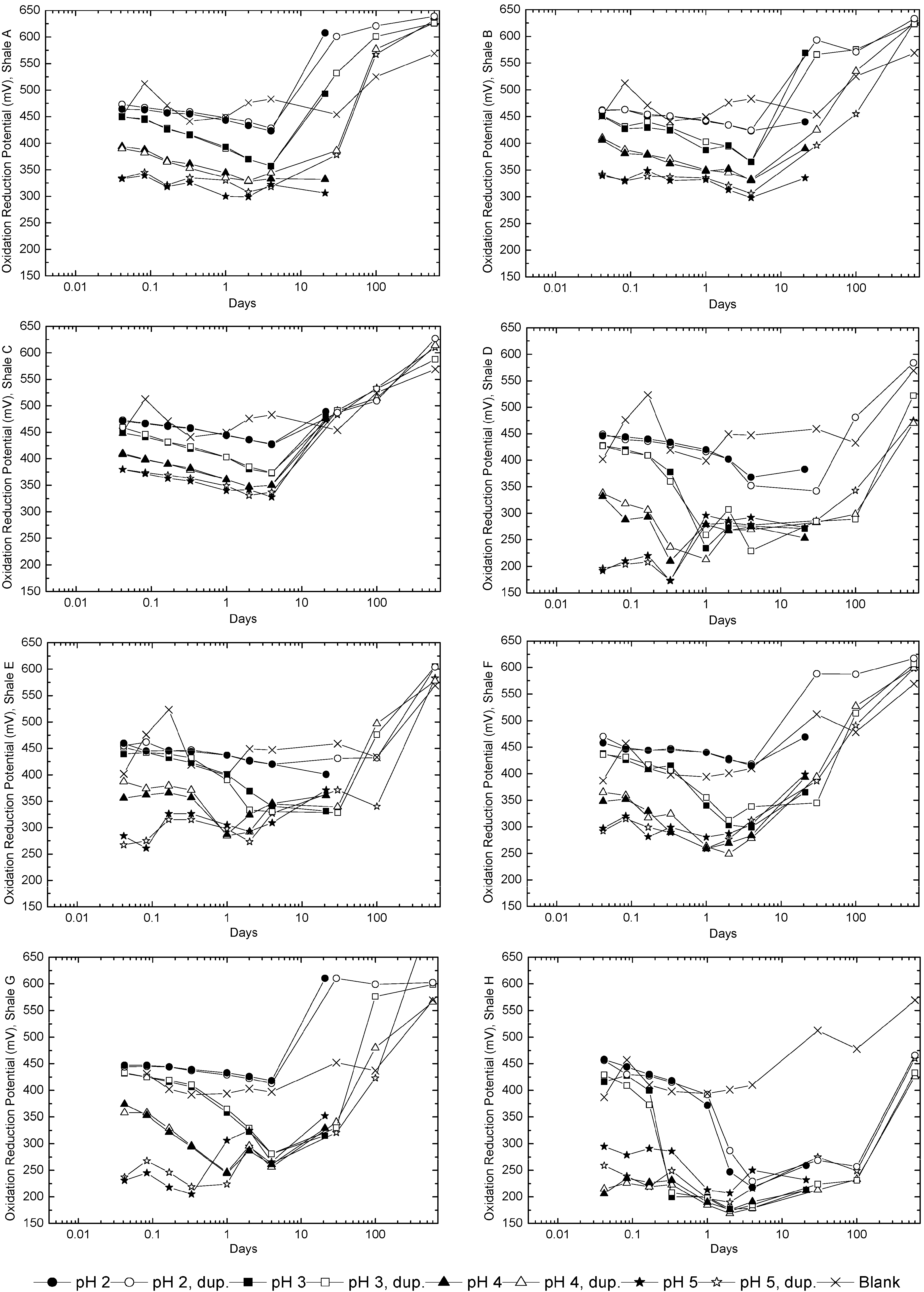
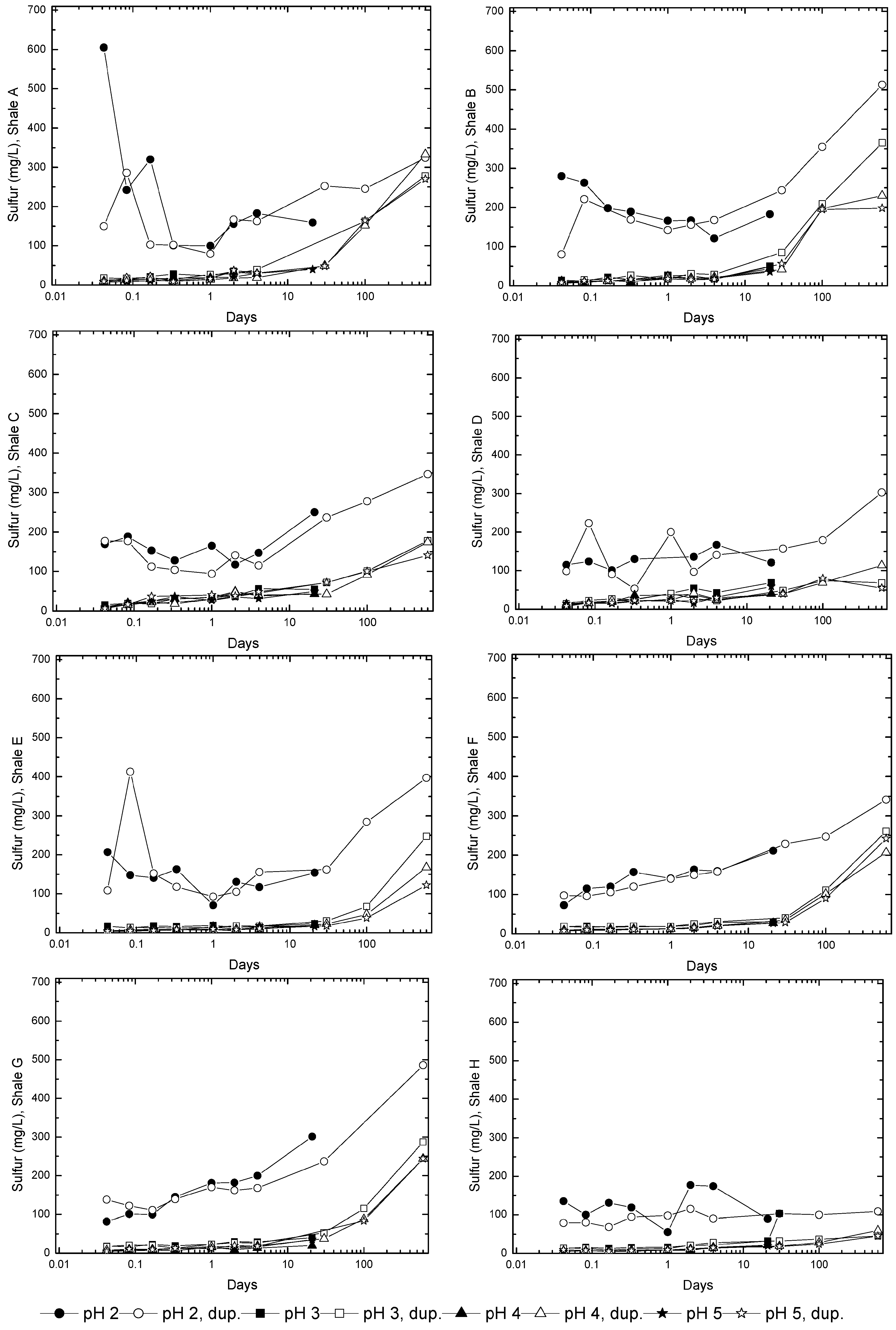

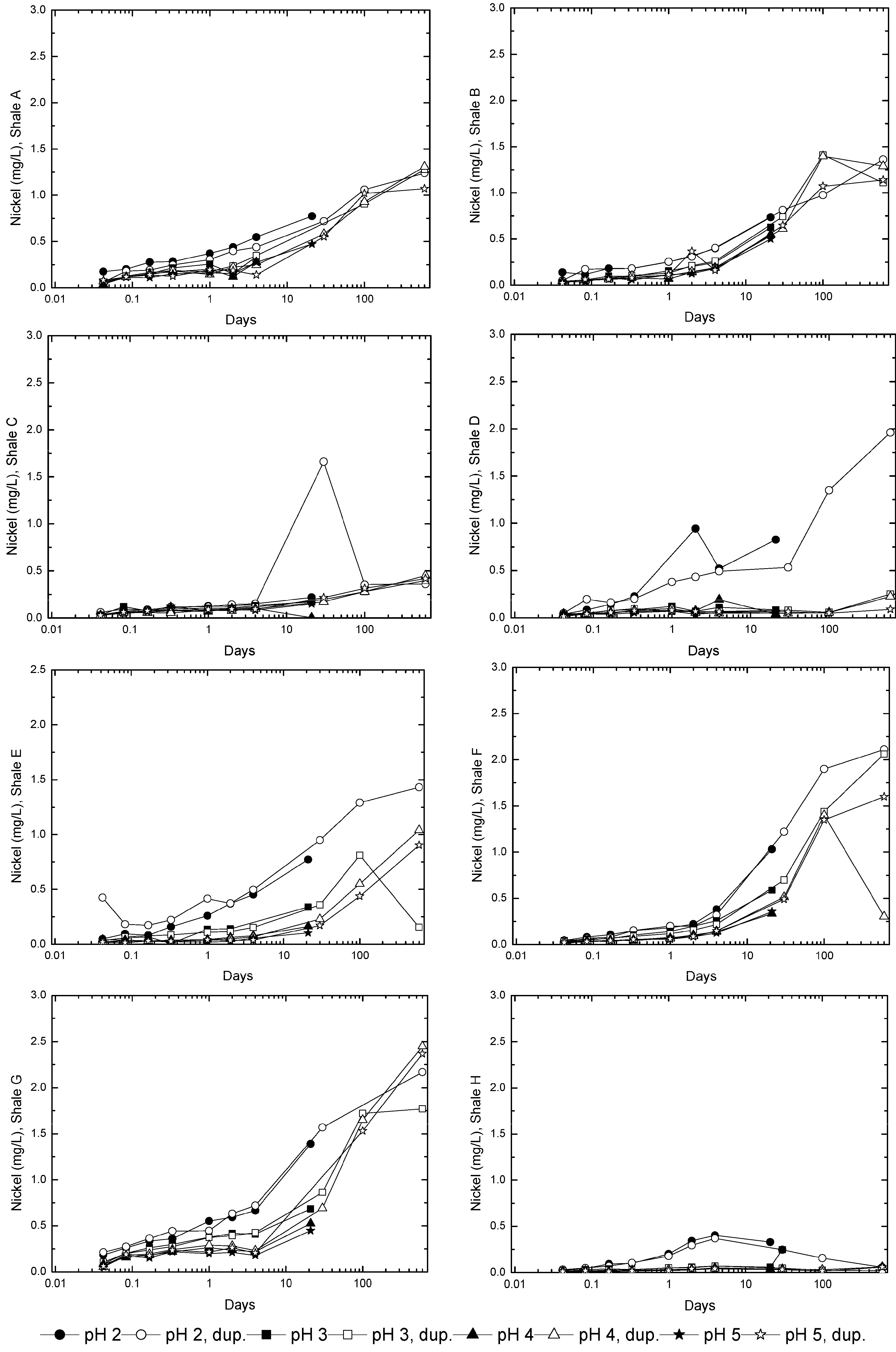
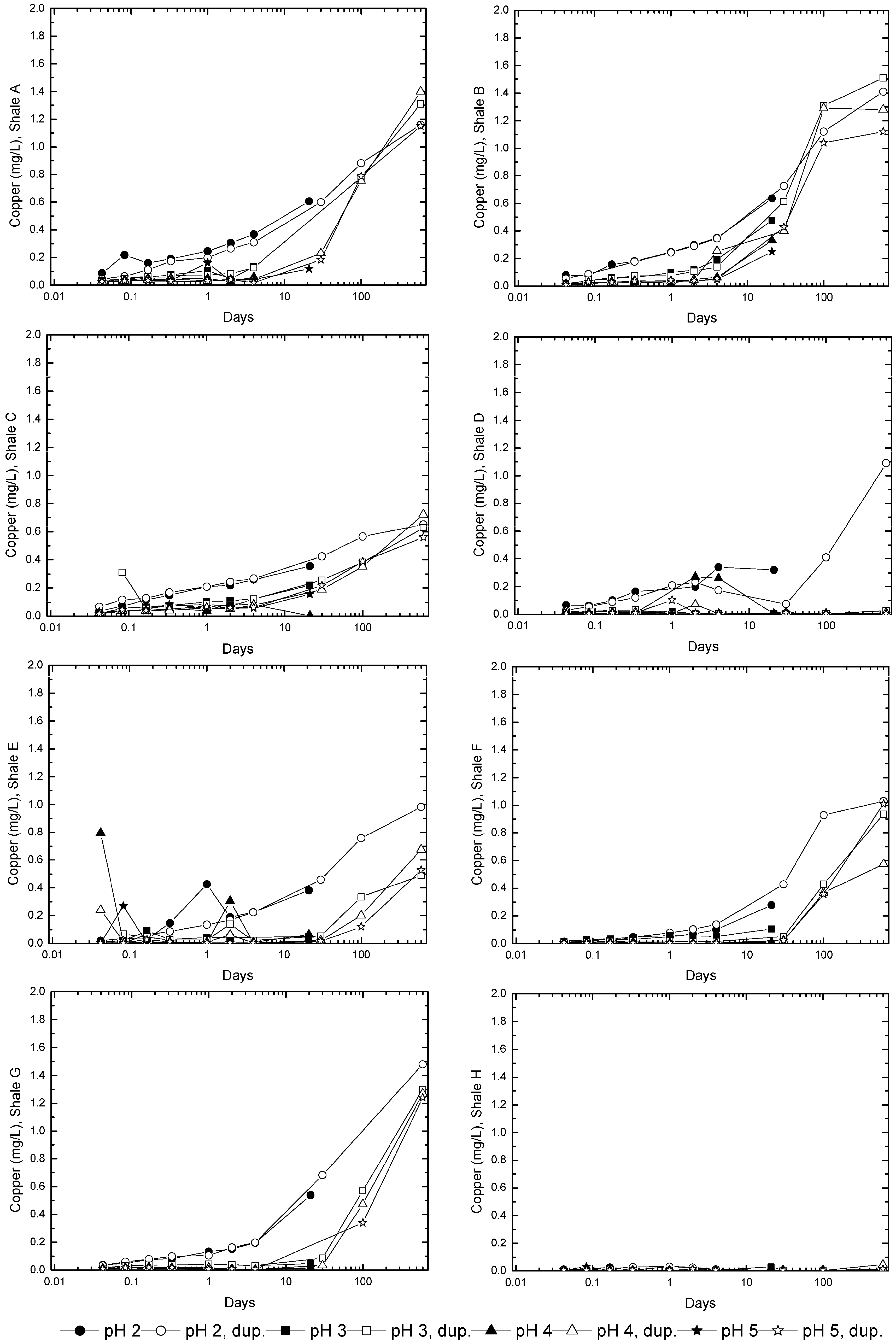
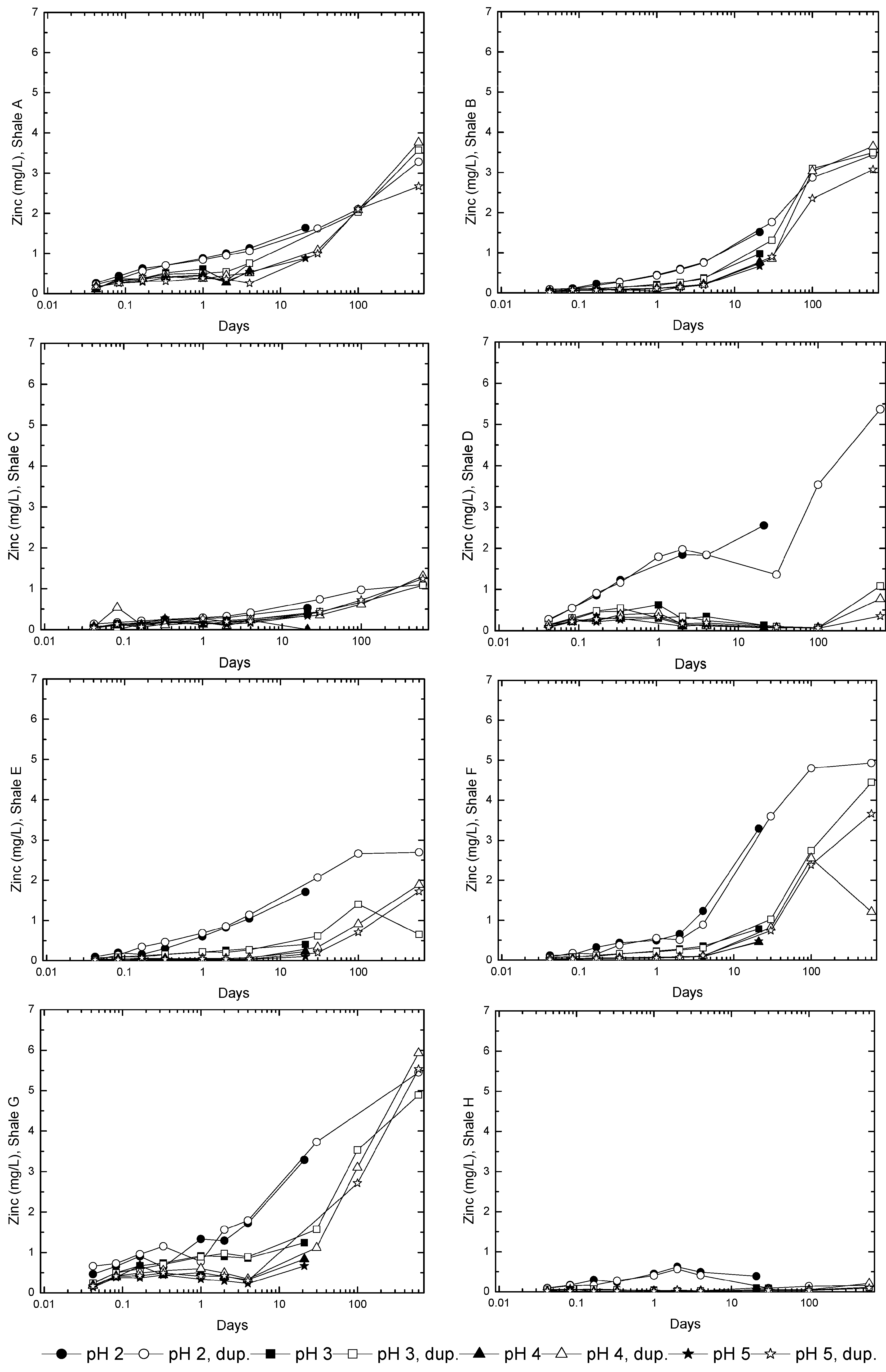
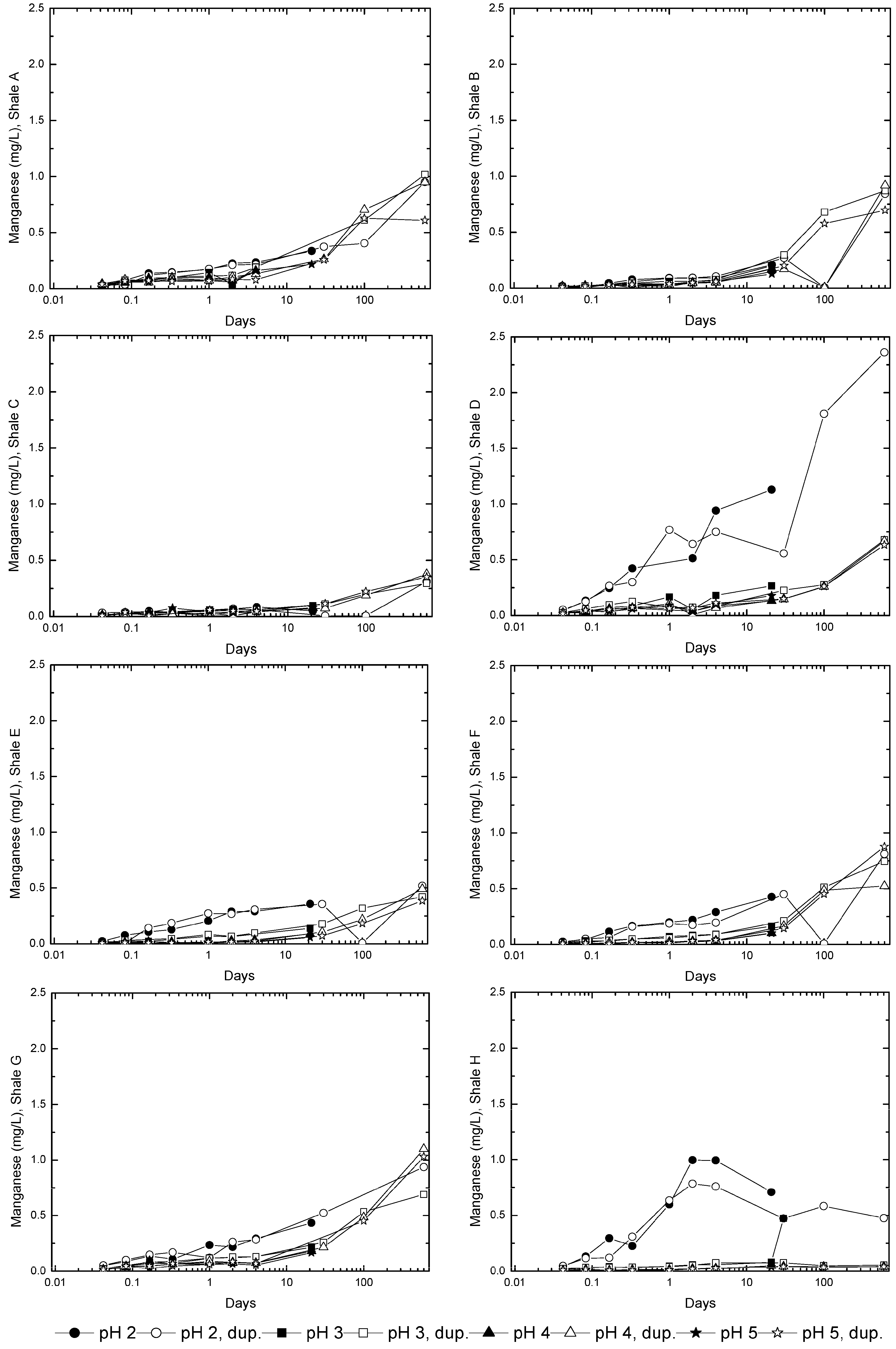
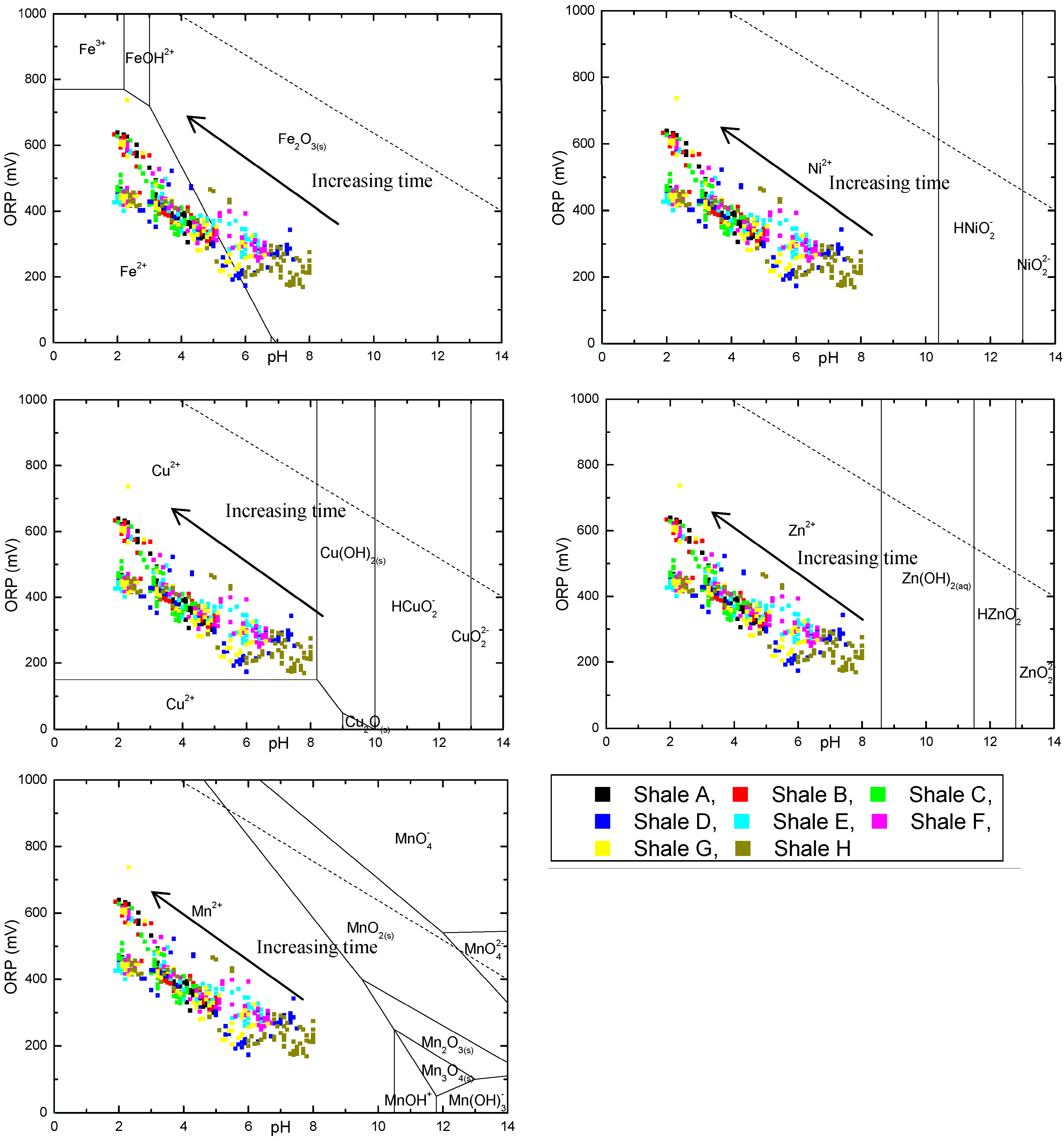
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Neculita, C.M.; Zagury, G.J.; Bussiere, B. Passive treatment of acid mine drainage in bioreactors using sulfate-reducing bacteria: Critical review and research needs. J. Environ. Qual. 2007, 36, 1–16. [Google Scholar] [CrossRef]
- Pareuil, P.; Penilla, S.; Ozkan, N.; Bordas, F.; Bollinger, J.C. Influence of reducing conditions on metallic elements released from various contaminated soil samples. Environ. Sci. Technol. 2008, 42, 7615–7621. [Google Scholar] [CrossRef]
- Akcil, A.; Koldas, S. Acid Mine Drainage (AMD): Causes, treatment and case studies. J. Clean Prod. 2006, 14, 1139–1145. [Google Scholar] [CrossRef]
- Tabak, H.H.; Scharp, R.; Burckle, J.; Kawahara, F.K.; Govind, R. Advances in biotreatment of acid mine drainage and biorecovery of metals: 1. Metal precipitation for recovery and recycle. Biodegradation 2003, 14, 423–436. [Google Scholar] [CrossRef]
- Pandey, P.K.; Sharma, R.; Roy, M.; Pandey, M. Toxic mine drainage from Asia’s biggest copper mine at Malanjkhand, India. Environ. Geochem. Health 2007, 29, 237–248. [Google Scholar] [CrossRef]
- Luis, A.T.; Teixeira, P.; Almeida, S.F.P.; Ector, L.; Matos, J.X.; da Silva, E.A.F. Impact of acid mine drainage (AMD) on water quality, stream sediments and periphytic diatom communities in the surrounding streams of Aljustrel mining area (Portugal). Water Air Soil Pollut. 2009, 200, 147–167. [Google Scholar] [CrossRef]
- Askaer, L.; Schmidt, L.B.; Elberling, B.; Asmund, G.; Jonsdottir, I.S. Environmental impact on an arctic soil-plant system resulting from metals released from coal mine waste in Svalbard (78 degrees N). Water Air Soil Pollut. 2008, 195, 99–114. [Google Scholar] [CrossRef]
- El Khalil, H.; El Hamiani, O.; Bitton, G.; Ouazzani, N.; Boularbah, A. Heavy metal contamination from mining sites in South Morocco: Monitoring metal content and toxicity of soil runoff and groundwater. Environ. Monit. Assess. 2008, 136, 147–160. [Google Scholar]
- Khan, M.S.; Zaidi, A.; Wani, P.A.; Oves, M. Role of plant growth promoting rhizobacteria in the remediation of metal contaminated soils. Environ. Chem. Lett. 2009, 7, 1–19. [Google Scholar] [CrossRef]
- Brake, S.S.; Dannelly, H.K.; Connors, K.A.; Hasiotis, S.T. Influence of water chemistry on the distribution of an acidophilic protozoan in an acid mine drainage system at the abandoned Green Valley coal mine, Indiana, USA. Appl. Geochem. 2001, 16, 1641–1652. [Google Scholar] [CrossRef]
- Lee, J.S.; Chon, H.T. Hydrogeochemical characteristics of acid mine drainage in the vicinity of an abandoned mine, Daduk Creek, Korea. J. Geochem. Explor. 2006, 88, 37–40. [Google Scholar] [CrossRef]
- Migaszewski, Z.M.; Galuszka, A.; Paslawski, P.; Starnawska, E. An influence of pyrite oxidation on generation of unique acidic pit water: A case study, podwisniowka quarry, Holy Cross Mountains (south-central Poland). Pol. J. Environ. Stud. 2007, 16, 407–421. [Google Scholar]
- Tutu, H.; McCarthy, T.S.; Cukrowska, E. The chemical characteristics of acid mine drainage with particular reference to sources, distribution and remediation: The Witwatersrand Basin, South Africa as a case study. Appl. Geochem. 2008, 23, 3666–3684. [Google Scholar] [CrossRef]
- Smith, R.M.; Sobek, A.A.; Arkele, T.; Sencindiver, J.C.; Freeman, J.K. Extensive Overburden Potentials for Soil and Water Quality; EPA-600/2-76-185; U.S. Environmental Protection Agency (EPA): Washington, DC, USA, 1976.
- McDonald, D.M.; Webb, J.A.; Taylor, J. Chemical stability of acid rock drainage treatment sludge and implications for sludge management. Environ. Sci. Technol. 2006, 40, 1984–1990. [Google Scholar] [CrossRef]
- Sapsford, D.J.; Bowell, R.J.; Dey, M.; Williams, K.P. Humidity cell tests for the prediction of acid rock drainage. Miner. Eng. 2009, 22, 25–36. [Google Scholar]
- Ardau, C.; Blowes, D.W.; Ptacek, C.J. Comparison of laboratory testing protocols to field observations of the weathering of sulfide-bearing mine tailings. J. Geochem. Explor. 2009, 100, 182–191. [Google Scholar]
- Carbone, C.; Dinelli, E.; Marescotti, P.; Gasparotto, G.; Lucchetti, G. The role of AMD secondary minerals in controlling environmental pollution: Indications from bulk leaching tests. J. Geochem. Explor. 2013, 132, 188–200. [Google Scholar] [CrossRef]
- Wobrauschek, P. Total reflection X-ray fluorescence analysis—A review. X-Ray Spectrom. 2007, 36, 289–300. [Google Scholar] [CrossRef]
- Zou, H.F.; Xu, S.K.; Fang, Z.L. Determination of chromium in environmental samples by flame AAS with flow injection on-line coprecipitation. At. Spectrosc. 1996, 17, 112–118. [Google Scholar]
- Capota, P.; Baiulescu, G.E.; Constantin, M. The analysis of environmental samples by ICP-AES. Chem. Anal. 1996, 41, 419–427. [Google Scholar]
- Ammann, A.A. Inductively coupled plasma mass spectrometry (ICP-MS): A versatile tool. J. Mass Spectrom. 2007, 42, 419–427. [Google Scholar] [CrossRef]
- Townsend, A.T. The accurate determination of the first row transition metals in water, urine, plant, tissue and rock samples by sector field ICP-MS. J. Anal. At. Spectrom. 2000, 15, 307–314. [Google Scholar] [CrossRef]
- Wang, H.; Bigham, J.A.; Tuovinen, O.H. Oxidation of marcasite and pyrite by iron-oxidizing bacteria and archaea. Hydrometallurgy 2007, 88, 127–131. [Google Scholar]
- Gurung, S.R.; Stewart, R.B.; Gregg, P.E.H.; Bolan, N.S. An assessment of requirements of neutralising materials of partially oxidised pyritic mine waste. Aust. J. Soil Res. 2000, 38, 329–344. [Google Scholar] [CrossRef]
- Canadian Council of Ministers of the Environment (CCME). Canadian Water Quality Guidelines for the Protection of Aquatic Life: Summary Table, Updated 7.1, December 2007; Canadian Council of Ministers of the Environment (CCME): Winnipeg, MB, Canada, 2007. Available online: https://www.halifax.ca/environment/documents/CWQG.PAL.summaryTable7.1.Dec2007.pdf (accessed on 20 January 2014).
- Chandra, A.P.; Gerson, A.R. The mechanisms of pyrite oxidation and leaching: A fundamental perspective. Surf. Sci. Rep. 2010, 65, 293–315. [Google Scholar] [CrossRef]
- Chuan, M.C.; Shu, G.Y.; Liu, J.C. Solubility of heavy metals in a contaminated soil: Effects of redox potential and pH. Water Air Soil Pollut. 1996, 90, 543–556. [Google Scholar] [CrossRef]
- Takeno, N. Atlas of Eh-pH Diagrams: Intercomparison of Thermodynamic Databases; Open File Report No. 419. Geological Survey of Japan: Tsukuba, Japan, 2005. Available online: http://www.gsj.jp/GDB/openfile/files/no0419/openfile419e.pdf (accessed on 20 January 2014).
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mumford, K.A.; Pitt, B.; Townsend, A.T.; Snape, I.; Gore, D.B. Long-Term Acid-Generating and Metal Leaching Potential of a Sub-Arctic Oil Shale. Minerals 2014, 4, 293-312. https://doi.org/10.3390/min4020293
Mumford KA, Pitt B, Townsend AT, Snape I, Gore DB. Long-Term Acid-Generating and Metal Leaching Potential of a Sub-Arctic Oil Shale. Minerals. 2014; 4(2):293-312. https://doi.org/10.3390/min4020293
Chicago/Turabian StyleMumford, Kathryn A., Brendan Pitt, Ashley T. Townsend, Ian Snape, and Damian B. Gore. 2014. "Long-Term Acid-Generating and Metal Leaching Potential of a Sub-Arctic Oil Shale" Minerals 4, no. 2: 293-312. https://doi.org/10.3390/min4020293