
Antibody Aggregation: Insights from Sequence and Structure

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
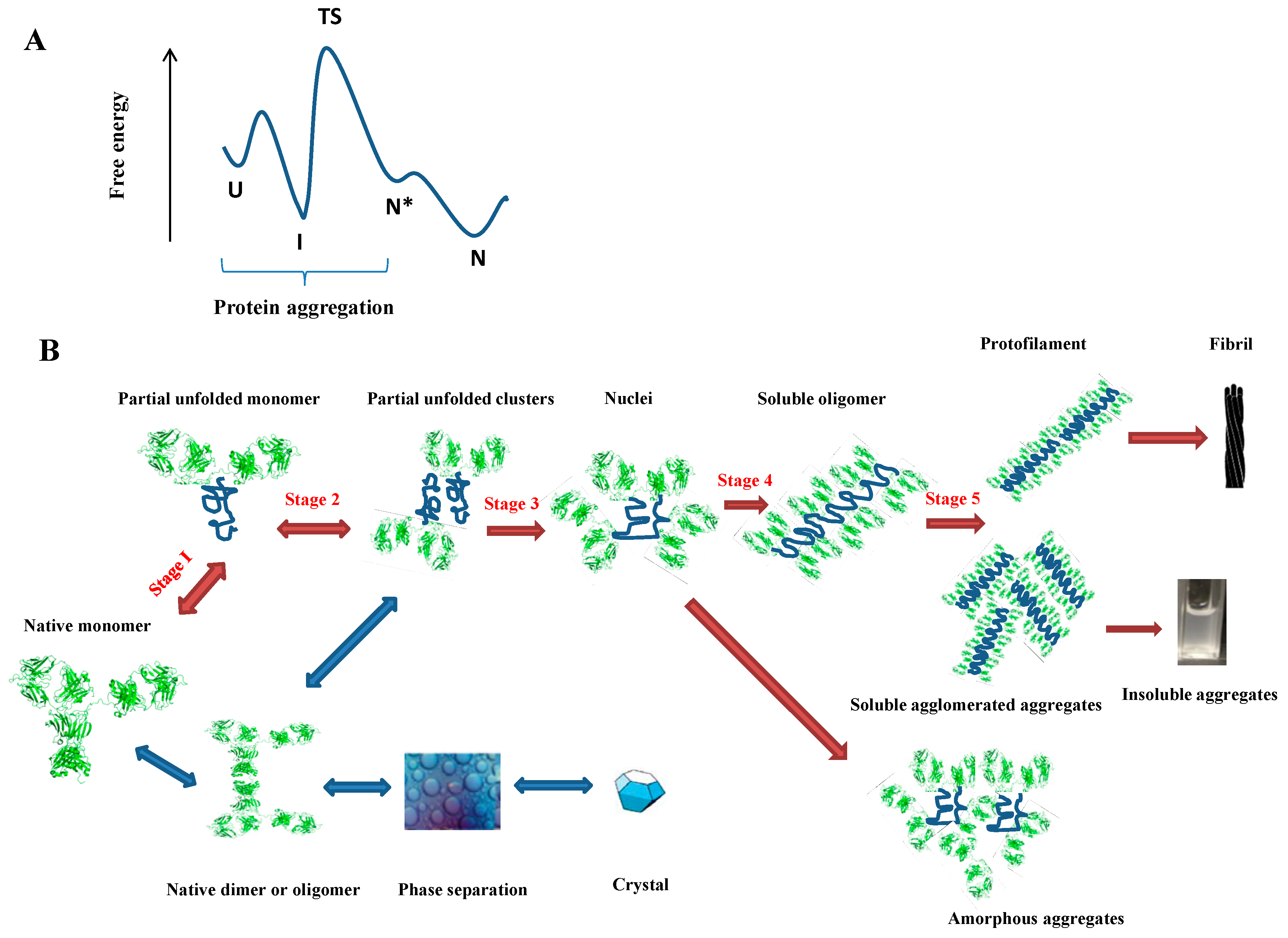
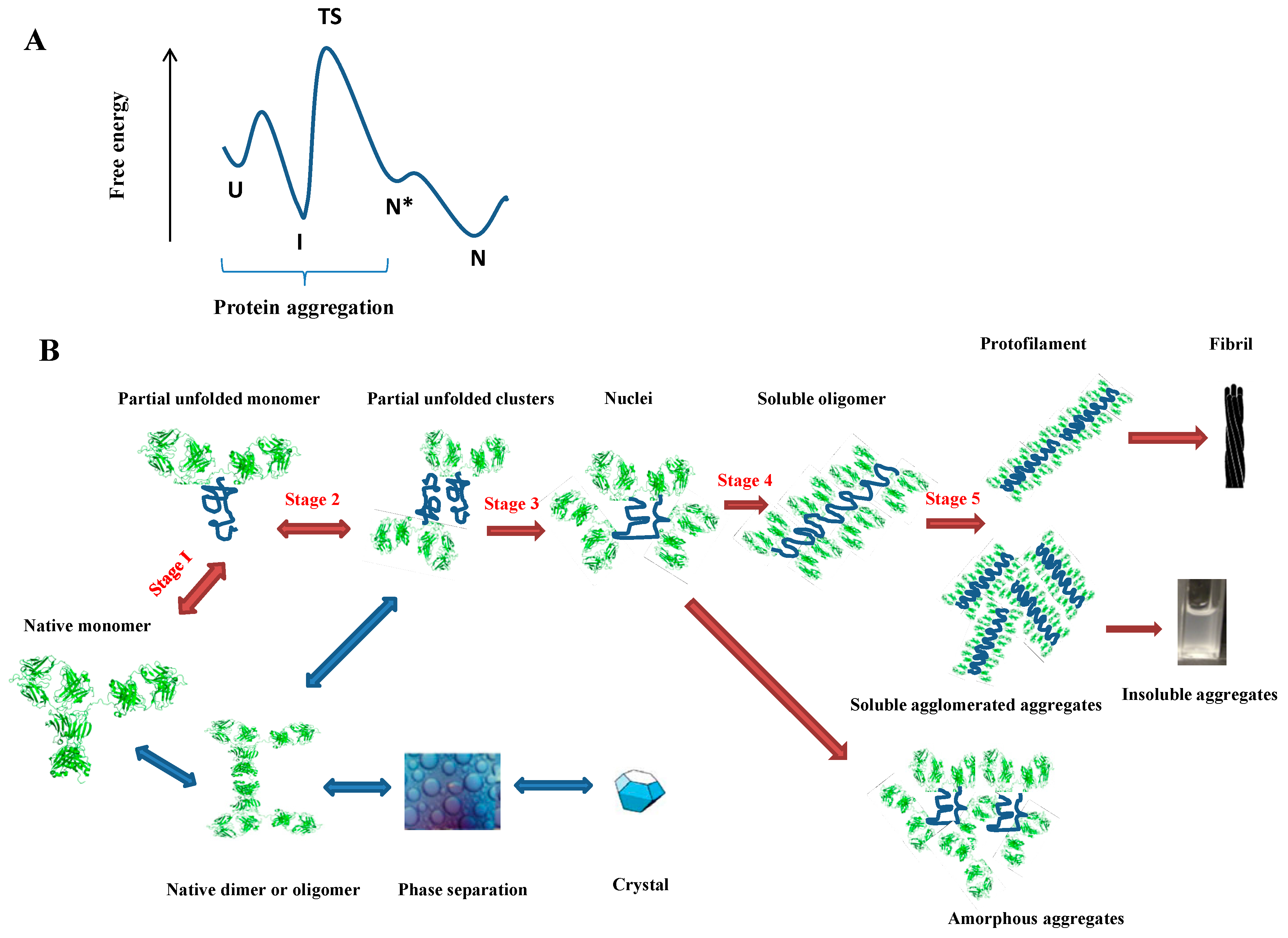
2. Protein Aggregation
2.1. Why Does a Protein Aggregate?
2.2. How Does a Protein Aggregate?
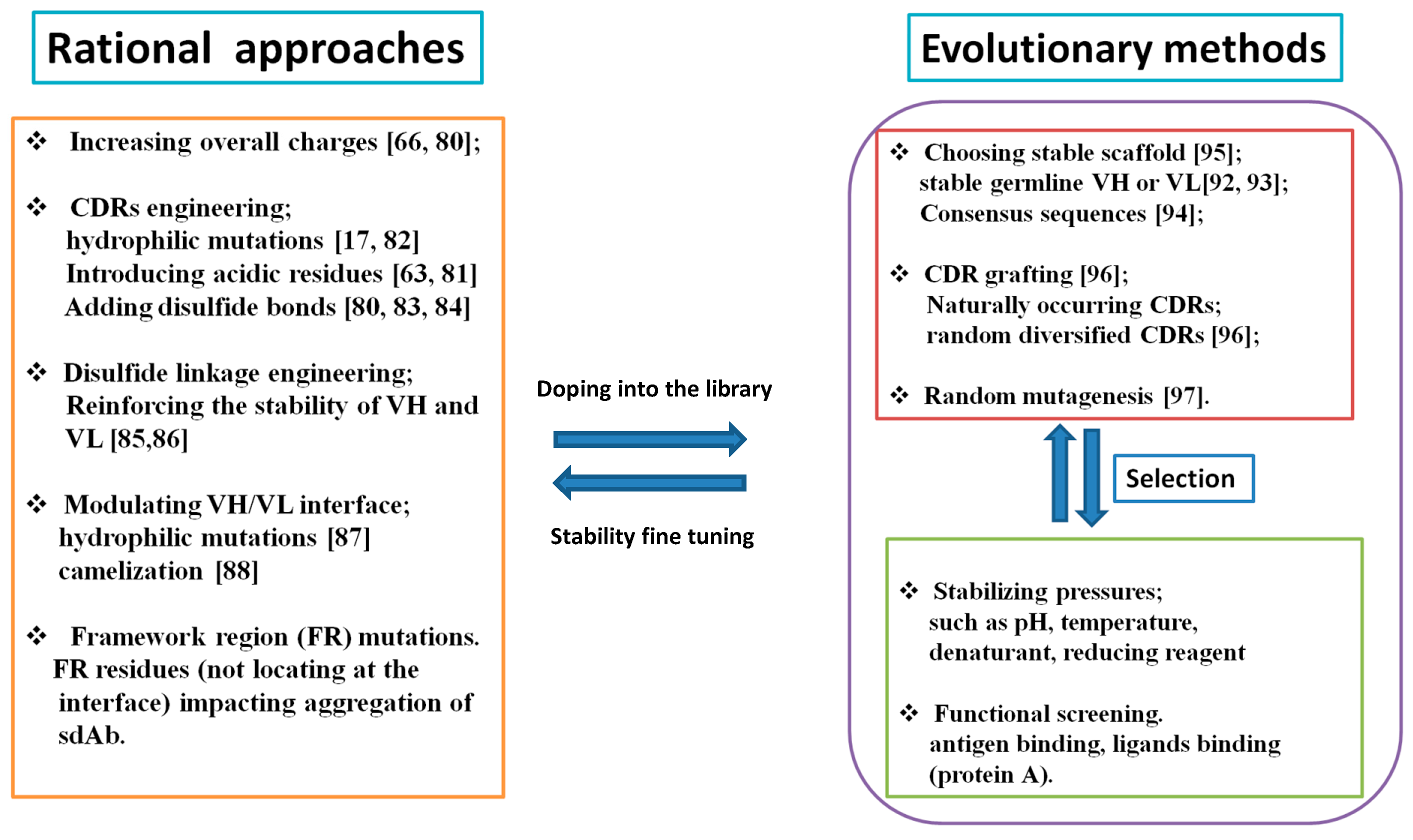
2.3. How to Mitigate Protein Aggregation?
2.4. Computational Methods for Studying or Predicting Protein Aggregation
3. Antibody Aggregation
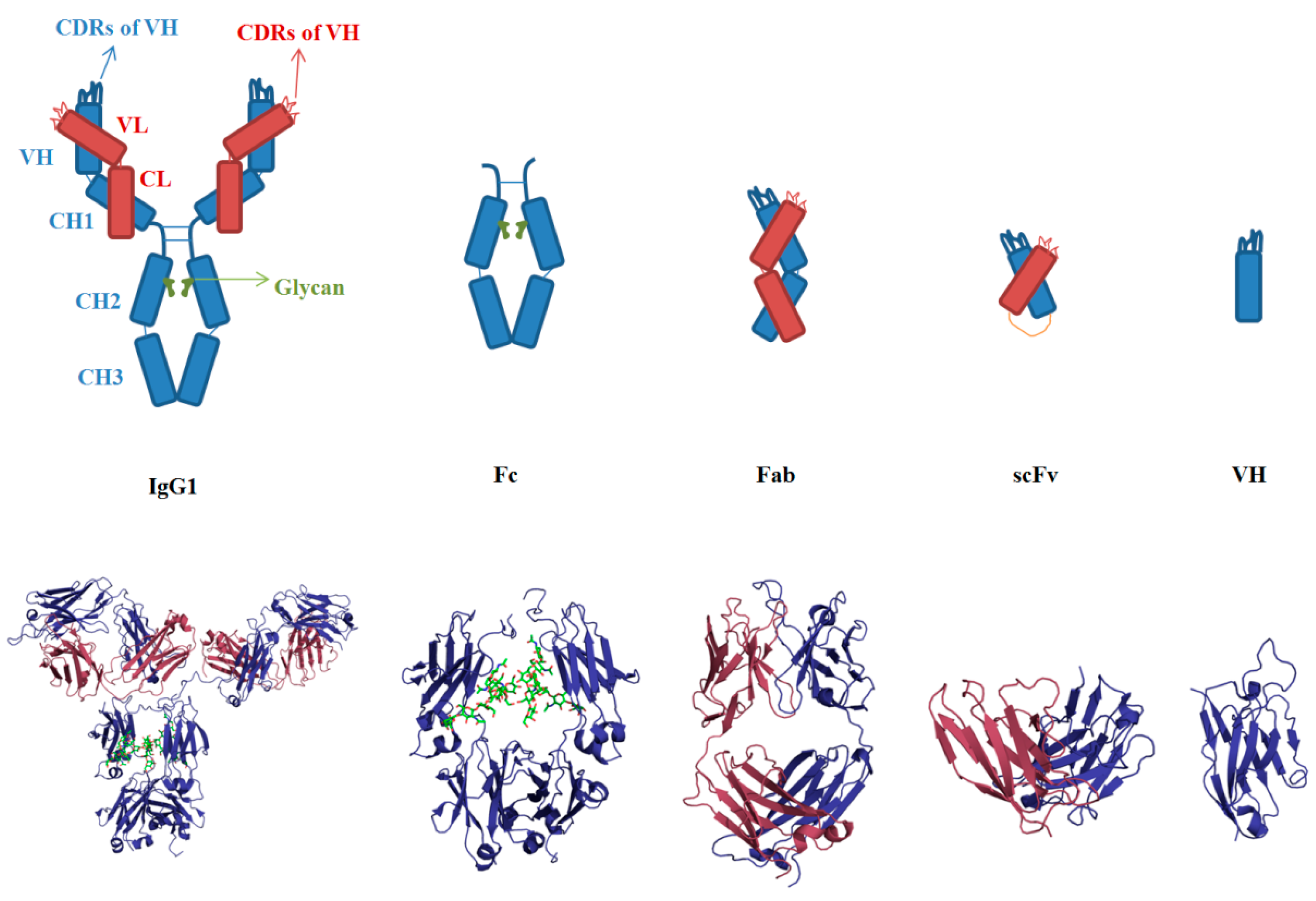
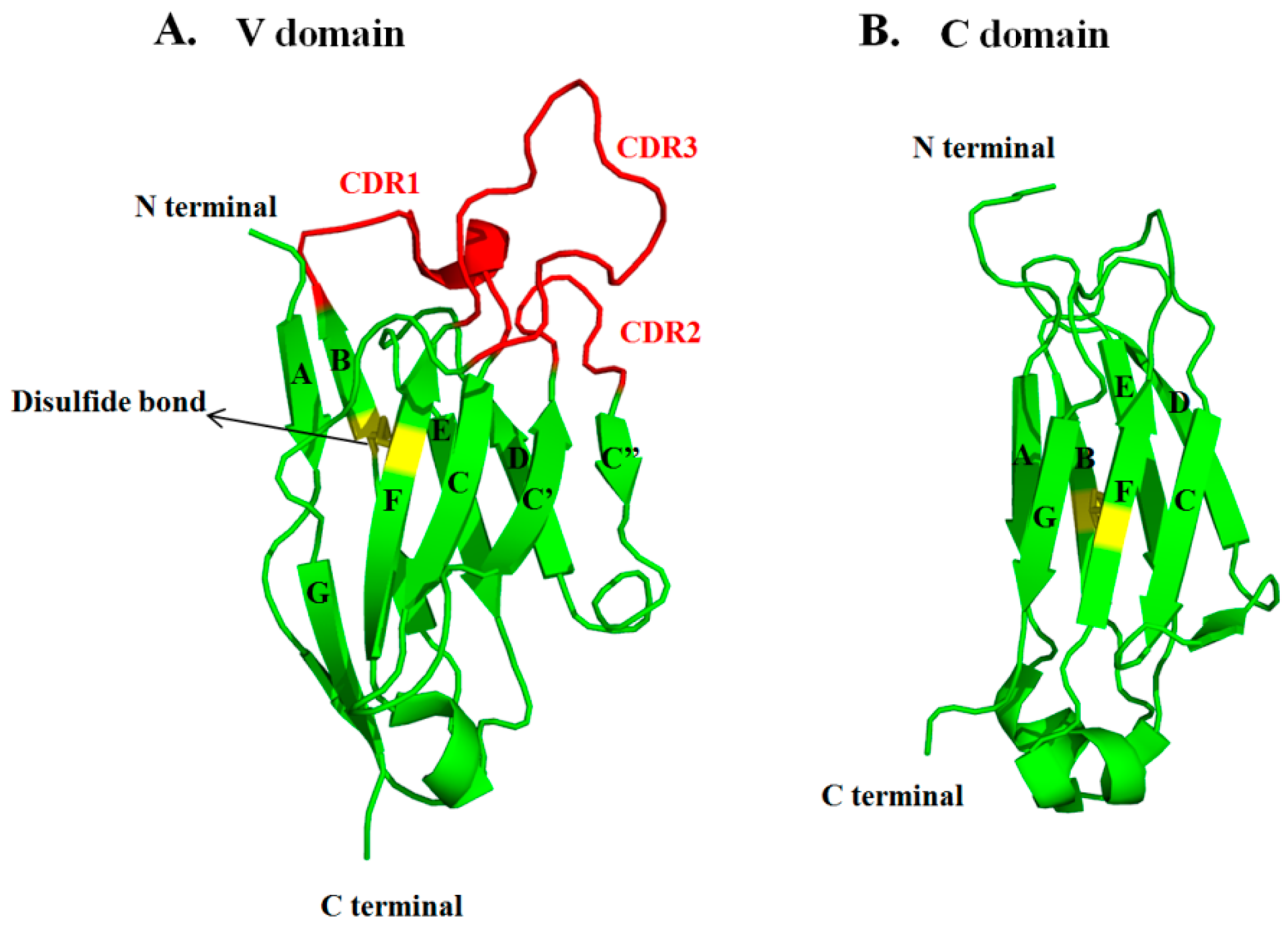
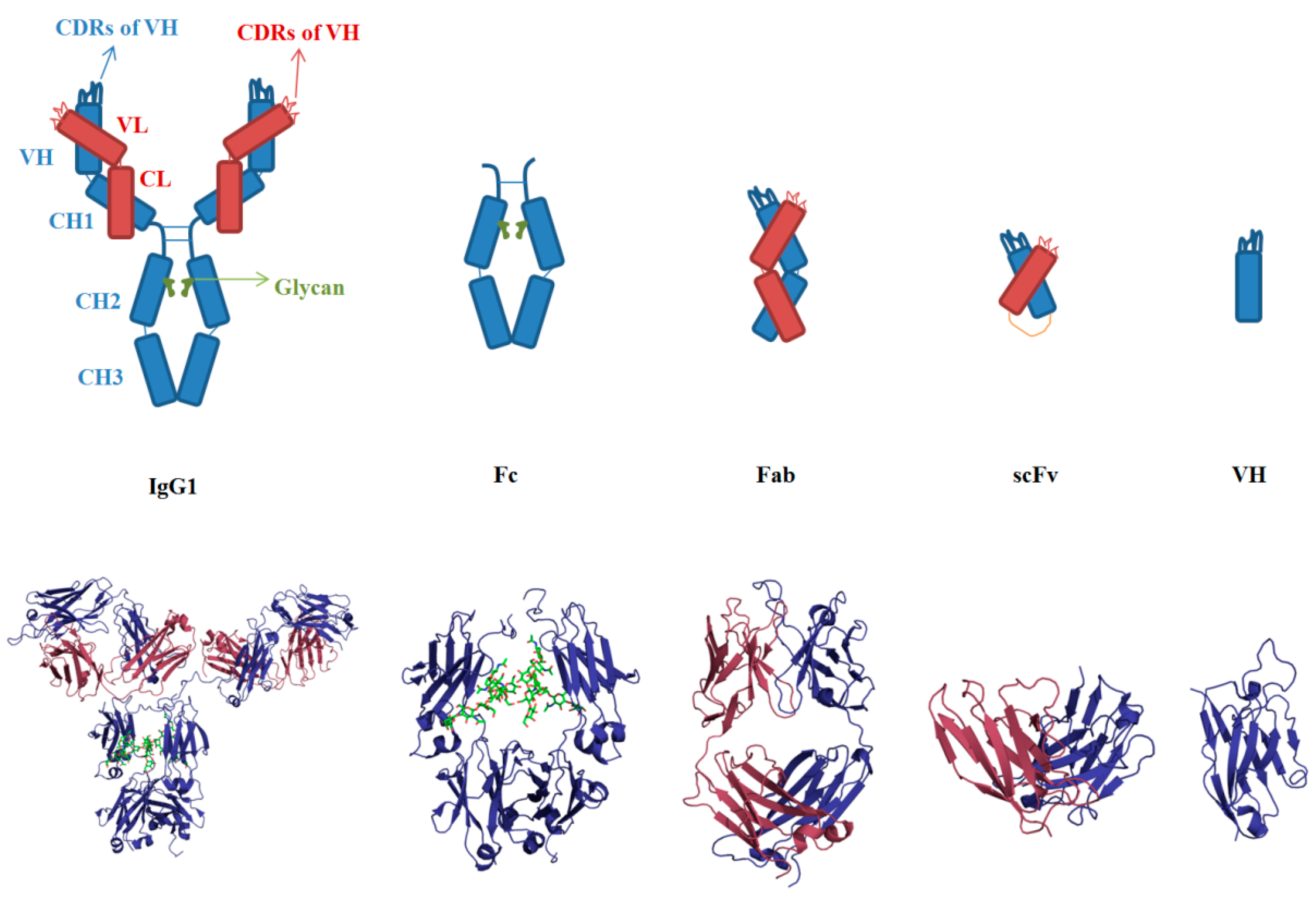
3.1. Sequences and Structures of mAbs
3.2. Aggregation of Full-Length IgG
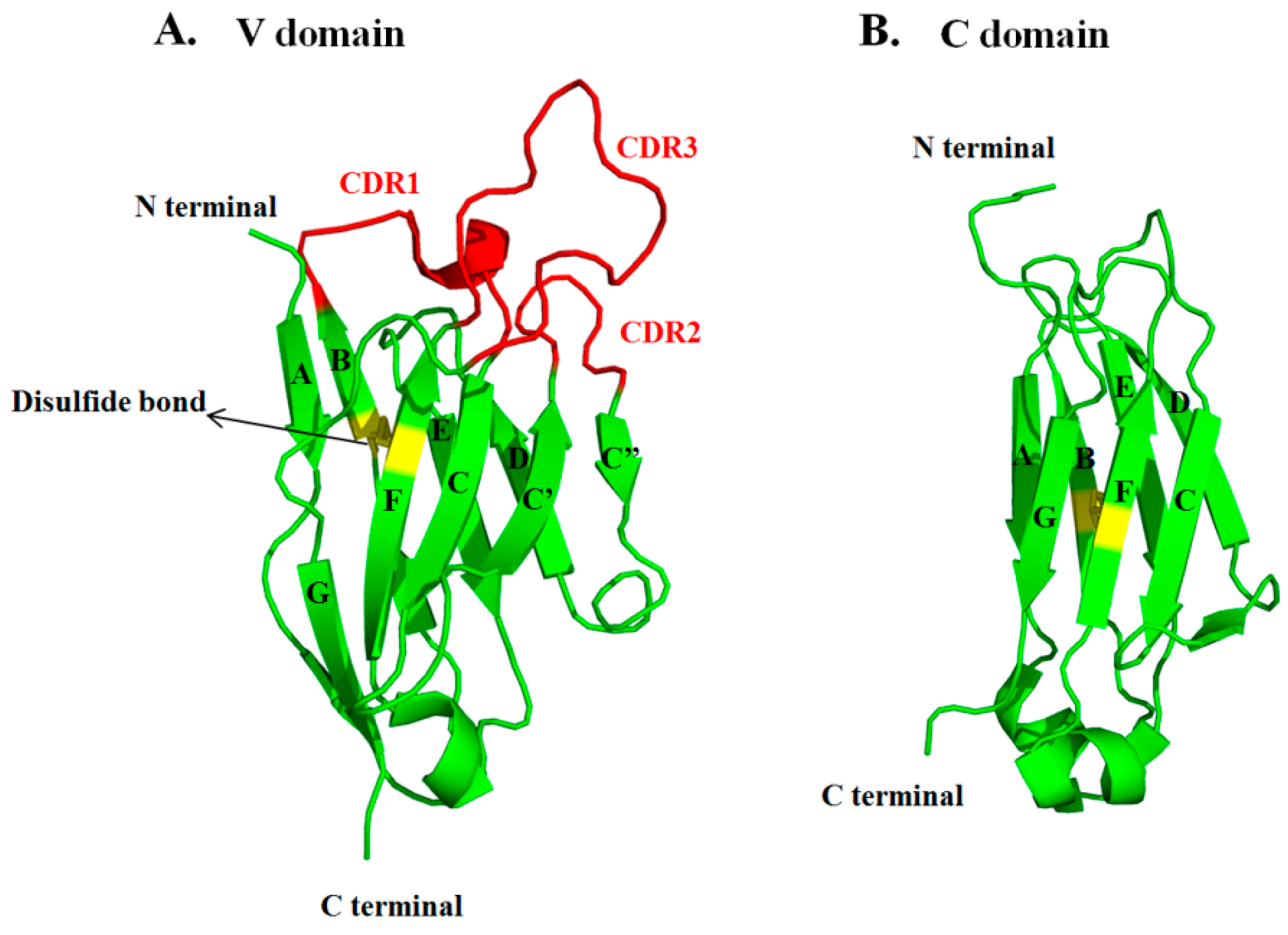
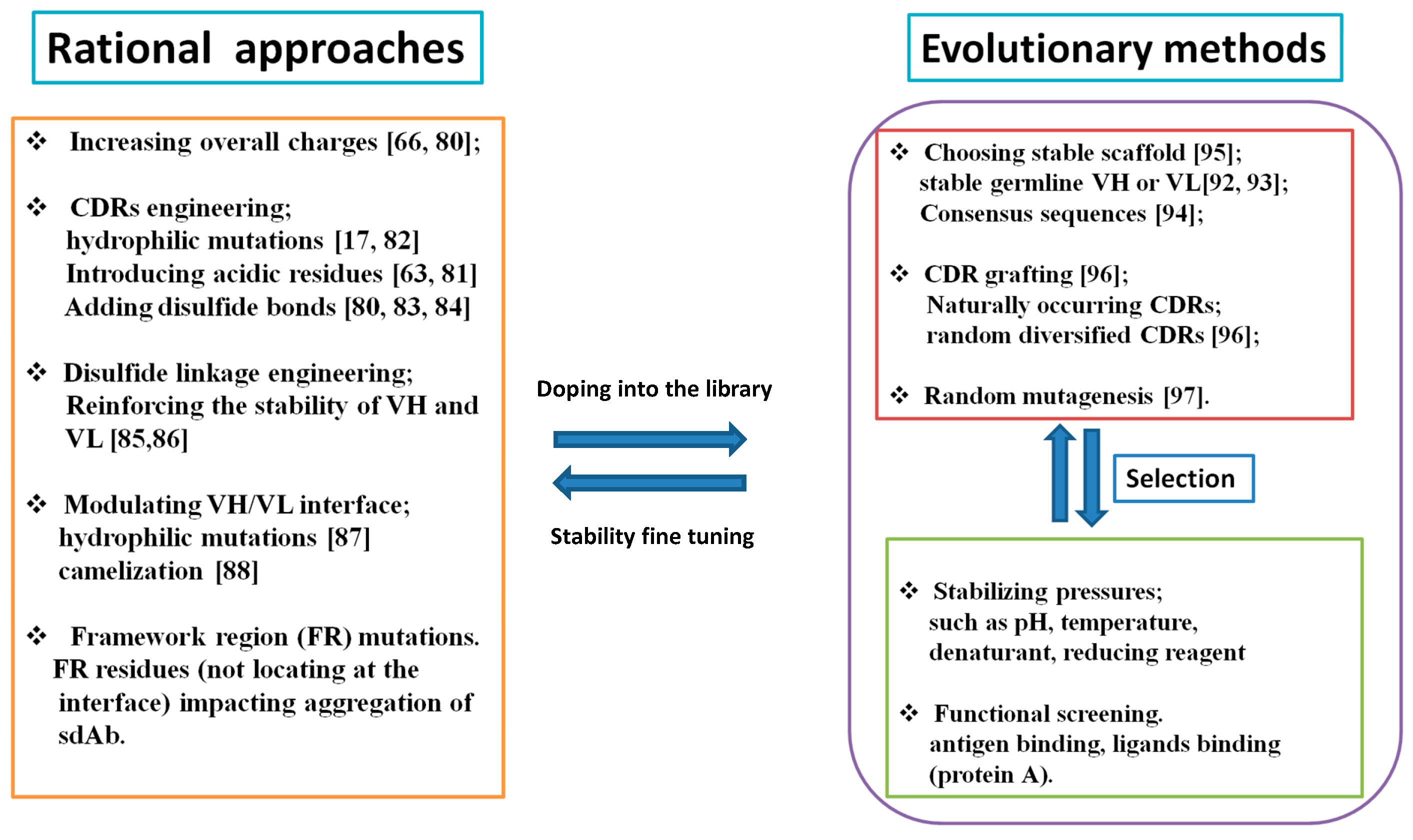
3.3. VH and VL
3.4. scFv
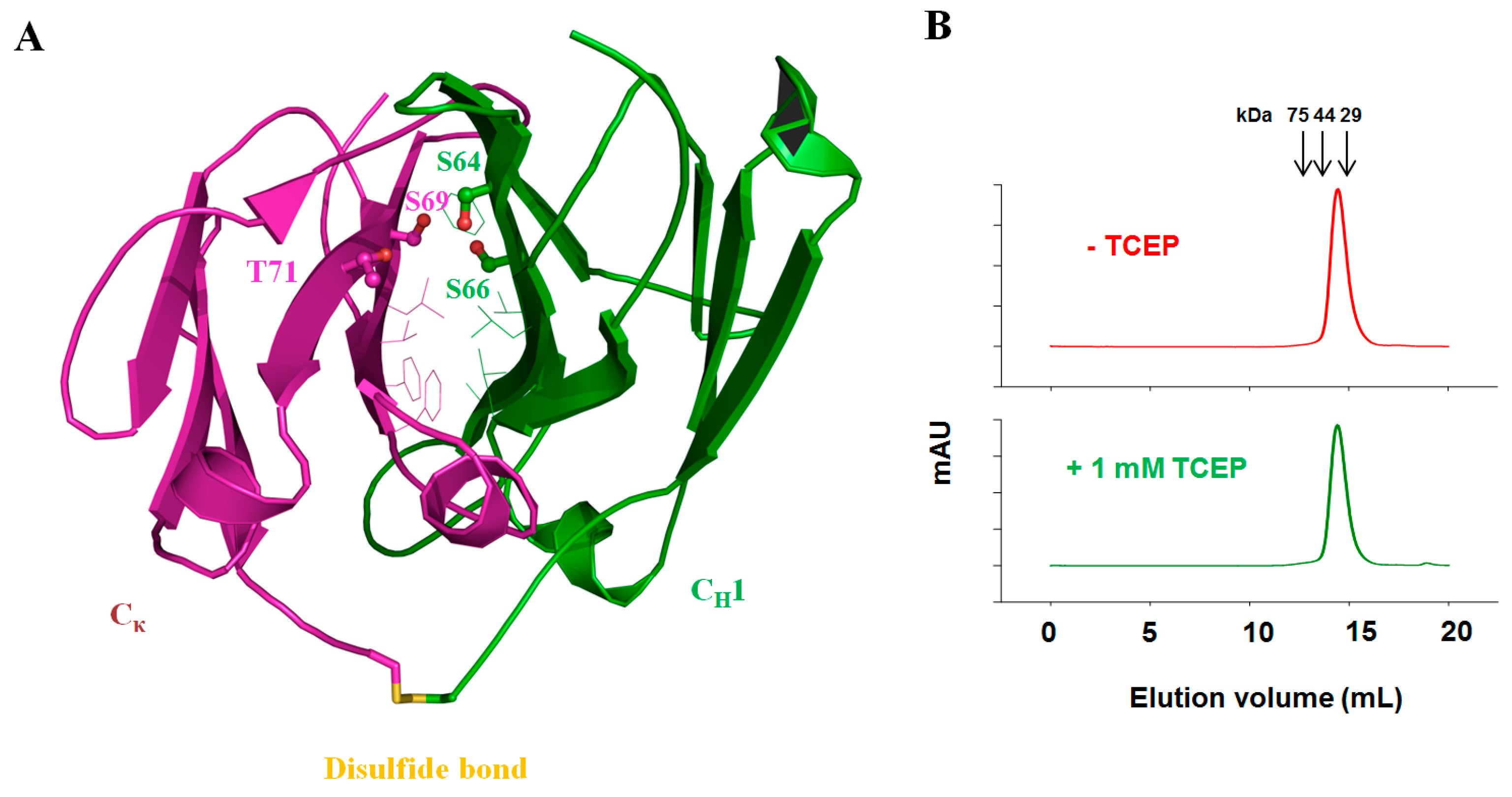
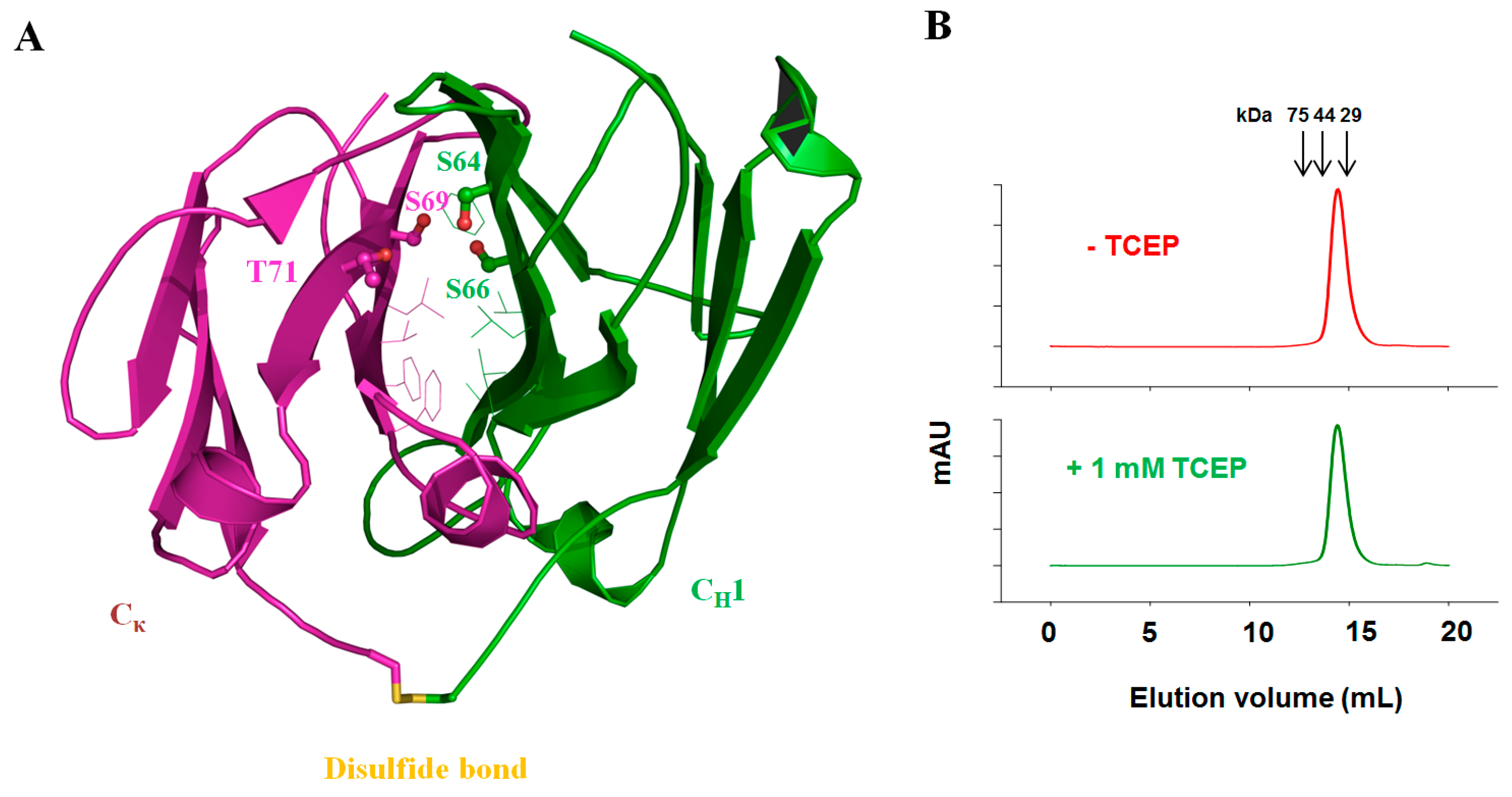
3.5. Fab
3.6. Fc
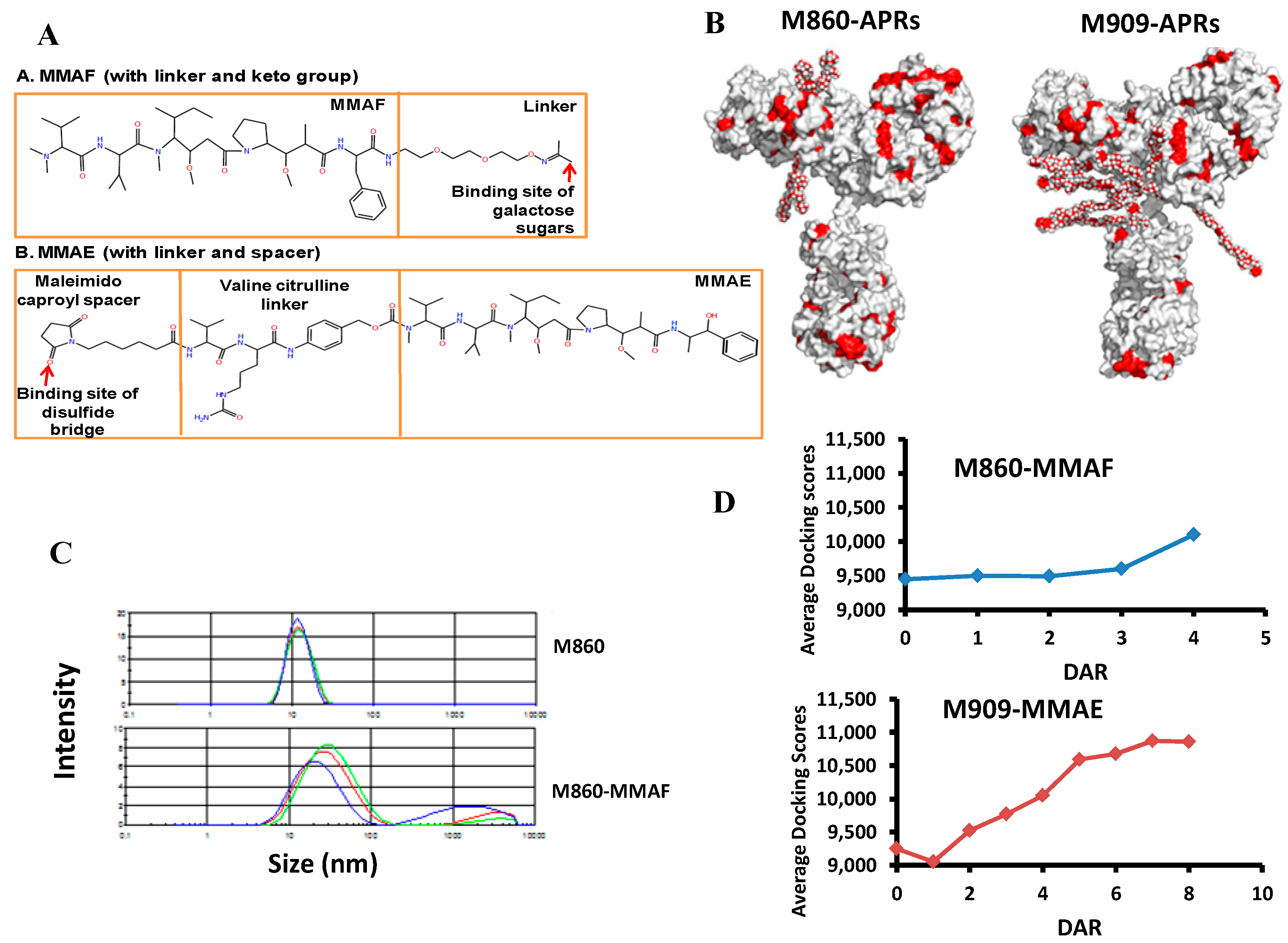
4. Aggregation of Antibody Drug Conjugates
5. Conclusions and Outlooks
Acknowledgments
Conflicts of Interest
References
- Creighton, T.E. Protein Folding; WH Freeman and Company: New York, NY, USA, 1992. [Google Scholar]
- Lindorff-Larsen, K.; Piana, S.; Dror, R.O.; Shaw, D.E. How fast-folding proteins fold. Science 2011, 334, 517–520. [Google Scholar] [CrossRef] [PubMed]
- Dobson, C.M.; Karplus, M. The fundamentals of protein folding: Bringing together theory and experiment. Curr. Opin. Struct. Biol. 1999, 9, 92–101. [Google Scholar] [CrossRef]
- James, L.C.; Tawfik, D.S. Conformational diversity and protein evolution—A 60-year-old hypothesis revisited. Trends Biochem. Sci. 2003, 28, 361–368. [Google Scholar] [CrossRef]
- Chiti, F.; Dobson, C.M. Amyloid formation by globular proteins under native conditions. Nat. Chem. Biol. 2009, 5, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Dobson, C.M. Protein folding and misfolding. Nature 2003, 426, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.J. Therapeutic protein aggregation: Mechanisms, design, and control. Trends Biotechnol. 2014, 32, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Dobson, C.M. The structural basis of protein folding and its links with human disease. Philos. Trans. R. Soc. Lond. Ser. B 2001, 356, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Taddei, N.; Baroni, F.; Capanni, C.; Stefani, M.; Ramponi, G.; Dobson, C.M. Kinetic partitioning of protein folding and aggregation. Nat. Struct. Biol. 2002, 9, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Reichert, J.M. Monoclonal antibodies as innovative therapeutics. Curr. Pharm. Biotechnol. 2008, 9, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Reichert, J.M.; Rosensweig, C.J.; Faden, L.B.; Dewitz, M.C. Monoclonal antibody successes in the clinic. Nat. Biotechnol. 2005, 23, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- Lowe, D.; Dudgeon, K.; Rouet, R.; Schofield, P.; Jermutus, L.; Christ, D. Aggregation, stability, and formulation of human antibody therapeutics. Adv. Protein Chem. Struct. Biol. 2011, 84, 41–61. [Google Scholar] [PubMed]
- Perchiacca, J.M.; Tessier, P.M. Engineering aggregation-resistant antibodies. Annu. Rev. Chem. Biomol. Eng. 2012, 3, 263–286. [Google Scholar] [CrossRef] [PubMed]
- Nishi, H.; Miyajima, M.; Nakagami, H.; Noda, M.; Uchiyama, S.; Fukui, K. Phase separation of an IgG1 antibody solution under a low ionic strength condition. Pharm. Res. 2010, 27, 1348–1360. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Nguyen, M.D.; Andya, J.D.; Shire, S.J. Reversible self-association increases the viscosity of a concentrated monoclonal antibody in aqueous solution. J. Pharm. Sci. 2005, 94, 1928–1940. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.S.; Richardson, D.C. Natural beta-sheet proteins use negative design to avoid edge-to-edge aggregation. Proc. Natl. Acad. Sci. USA 2002, 99, 2754–2759. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.J.; Luo, J.; O’Neil, K.T.; Kang, J.; Lacy, E.R.; Canziani, G.; Baker, A.; Huang, M.; Tang, Q.M.; Raju, T.S.; et al. Structure-based engineering of a monoclonal antibody for improved solubility. Protein Eng. Des. Sel. 2010, 23, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Das, T.K.; Singh, S.K.; Kumar, S. Potential aggregation prone regions in biotherapeutics: A survey of commercial monoclonal antibodies. mAbs 2009, 1, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Nishi, H.; Miyajima, M.; Wakiyama, N.; Kubota, K.; Hasegawa, J.; Uchiyama, S.; Fukui, K. Fc domain mediated self-association of an IgG1 monoclonal antibody under a low ionic strength condition. J. Biosci. Bioeng. 2011, 112, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Nezlin, R. Interactions between immunoglobulin G molecules. Immunol. Lett. 2010, 132, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Shire, S.J. Formulation and manufacturability of biologics. Curr. Opin. Biotechnol. 2009, 20, 708–714. [Google Scholar] [CrossRef] [PubMed]
- Daugherty, A.L.; Mrsny, R.J. Formulation and delivery issues for monoclonal antibody therapeutics. Adv. Drug Deliv. Rev. 2006, 58, 686–706. [Google Scholar] [CrossRef] [PubMed]
- De Groot, A.S.; Scott, D.W. Immunogenicity of protein therapeutics. Trends Immunol. 2007, 28, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K. Impact of product-related factors on immunogenicity of biotherapeutics. J. Pharm. Sci. 2011, 100, 354–387. [Google Scholar] [CrossRef] [PubMed]
- Hotzel, I.; Theil, F.P.; Bernstein, L.J.; Prabhu, S.; Deng, R.; Quintana, L.; Lutman, J.; Sibia, R.; Chan, P.; Bumbaca, D.; et al. A strategy for risk mitigation of antibodies with fast clearance. mAbs 2012, 4, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Tsumoto, K.; Ejima, D.; Kita, Y.; Arakawa, T. Review: Why is arginine effective in suppressing aggregation? Protein Pept. Lett. 2005, 12, 613–619. [Google Scholar] [CrossRef] [PubMed]
- Manning, M.C.; Matsuura, J.E.; Kendrick, B.S.; Meyer, J.D.; Dormish, J.J.; Vrkljan, M.; Ruth, J.R.; Carpenter, J.F.; Sheftert, E. Approaches for increasing the solution stability of proteins. Biotechnol. Bioeng. 1995, 48, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.J. Non-native protein aggregation kinetics. Biotechnol. Bioeng. 2007, 98, 927–938. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Kroe-Barrett, R.; Singh, S.; Robinson, A.S.; Roberts, C.J. Competing aggregation pathways for monoclonal antibodies. FEBS Lett. 2014, 588, 936–941. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Alvarado, M.; Merkel, J.S.; Regan, L. A systematic exploration of the influence of the protein stability on amyloid fibril formation in vitro. Proc. Natl. Acad. Sci. USA 2000, 97, 8979–8984. [Google Scholar] [CrossRef] [PubMed]
- Vermeer, A.W.; Norde, W. The thermal stability of immunoglobulin: Unfolding and aggregation of a multi-domain protein. Biophys. J. 2000, 78, 394–404. [Google Scholar] [CrossRef]
- Fink, A.L. Protein aggregation: Folding aggregates, inclusion bodies and amyloid. Fold. Des. 1998, 3, R9–R23. [Google Scholar] [CrossRef]
- Vendruscolo, M.; Paci, E. Protein folding: Bringing theory and experiment closer together. Curr. Opin. Struct. Biol. 2003, 13, 82–87. [Google Scholar] [CrossRef]
- De Simone, A.; Dhulesia, A.; Soldi, G.; Vendruscolo, M.; Hsu, S.T.; Chiti, F.; Dobson, C.M. Experimental free energy surfaces reveal the mechanisms of maintenance of protein solubility. Proc. Natl. Acad. Sci. USA 2011, 108, 21057–21062. [Google Scholar] [CrossRef] [PubMed]
- Sahin, E.; Grillo, A.O.; Perkins, M.D.; Roberts, C.J. Comparative effects of pH and ionic strength on protein-protein interactions, unfolding, and aggregation for IgG1 antibodies. J. Pharm. Sci. 2010, 99, 4830–4848. [Google Scholar] [CrossRef] [PubMed]
- Wang, W. Protein aggregation and its inhibition in biopharmaceutics. Int. J. Pharm. 2005, 289, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Russel, W.B.; Saville, D.A.; Schowalter, W.R. Colloidal Dispersions; Cambridge University Press: Cambridge, UK, 1992. [Google Scholar]
- Geng, S.B.; Cheung, J.K.; Narasimhan, C.; Shameem, M.; Tessier, P.M. Improving monoclonal antibody selection and engineering using measurements of colloidal protein interactions. J. Pharm. Sci. 2014, 103, 3356–3363. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.J. Kinetics of Irreversible Protein Aggregation: Analysis of Extended Lumry-Eyring Models and Implications for Predicting Protein Shelf Life. J. Phys. Chem. B 2003, 107, 1194–1207. [Google Scholar] [CrossRef]
- Lee, C.C.; Perchiacca, J.M.; Tessier, P.M. Toward aggregation-resistant antibodies by design. Trends Biotechnol. 2013, 31, 612–620. [Google Scholar] [CrossRef] [PubMed]
- Booth, D.R.; Sunde, M.; Bellotti, V.; Robinson, C.V.; Hutchinson, W.L.; Fraser, P.E.; Hawkins, P.N.; Dobson, C.M.; Radford, S.E.; Blake, C.C.; et al. Instability, unfolding and aggregation of human lysozyme variants underlying amyloid fibrillogenesis. Nature 1997, 385, 787–793. [Google Scholar] [CrossRef] [PubMed]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Broome, B.M.; Hecht, M.H. Nature disfavors sequences of alternating polar and non-polar amino acids: Implications for amyloidogenesis. J. Mol. Biol. 2000, 296, 961–968. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, R.; Istrail, S.; King, J. Frequencies of amino acid strings in globular protein sequences indicate suppression of blocks of consecutive hydrophobic residues. Protein Sci.: Publ. Protein Soc. 2001, 10, 1023–1031. [Google Scholar] [CrossRef] [PubMed]
- Cellmer, T.; Bratko, D.; Prausnitz, J.M.; Blanch, H.W. Protein aggregation in silico. Trends Biotechnol. 2007, 25, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Hall, C.K.; Voegler, A.C. Effect of denaturant and protein concentrations upon protein refolding and aggregation: A simple lattice model. Protein Sci. Publ. Protein Soc. 1998, 7, 2642–2652. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Nussinov, R. Molecular dynamics simulations of alanine rich beta-sheet oligomers: Insight into amyloid formation. Protein Sci. Publ. Protein Soc. 2002, 11, 2335–2350. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.G.; Jongejan, J.A.; Laman, J.D.; de Leeuw, S.W. Quantitative prediction of amyloid fibril growth of short peptides from simulations: Calculating association constants to dissect side chain importance. J. Am. Chem. Soc. 2008, 130, 15772–15773. [Google Scholar] [CrossRef] [PubMed]
- De Groot, N.S.; Pallares, I.; Aviles, F.X.; Vendrell, J.; Ventura, S. Prediction of “hot spots” of aggregation in disease-linked polypeptides. BMC Struct. Biol. 2005, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monsellier, E.; Ramazzotti, M.; de Laureto, P.P.; Tartaglia, G.G.; Taddei, N.; Fontana, A.; Vendruscolo, M.; Chiti, F. The distribution of residues in a polypeptide sequence is a determinant of aggregation optimized by evolution. Biophys. J. 2007, 93, 4382–4391. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chen, H.; Lai, L. Identification of amyloid fibril-forming segments based on structure and residue-based statistical potential. Bioinformatics 2007, 23, 2218–2225. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, N.J.; Kumar, S.; Wang, X.; Helk, B.; Singh, S.K.; Trout, B.L. Aggregation in protein-based biotherapeutics: Computational studies and tools to identify aggregation-prone regions. J. Pharm. Sci. 2011, 100, 5081–5095. [Google Scholar] [CrossRef] [PubMed]
- Chennamsetty, N.; Helk, B.; Voynov, V.; Kayser, V.; Trout, B.L. Aggregation-prone motifs in human immunoglobulin G. J. Mol. Biol. 2009, 391, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Chennamsetty, N.; Voynov, V.; Kayser, V.; Helk, B.; Trout, B.L. Design of therapeutic proteins with enhanced stability. Proc. Natl. Acad. Sci. USA 2009, 106, 11937–11942. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, H.W., Jr.; Cavacini, L. Structure and function of immunoglobulins. J. Allergy Clin. Immunol. 2010, 125, S41–S52. [Google Scholar] [CrossRef] [PubMed]
- Alamyar, E.; Giudicelli, V.; Duroux, P.; Lefranc, M.P. Antibody V and C domain sequence, structure, and interaction analysis with special reference to IMGT(R). Methods Mol. Biol. 2014, 1131, 337–381. [Google Scholar] [PubMed]
- Ionescu, R.M.; Vlasak, J.; Price, C.; Kirchmeier, M. Contribution of variable domains to the stability of humanized IgG1 monoclonal antibodies. J. Pharm. Sci. 2008, 97, 1414–1426. [Google Scholar] [CrossRef] [PubMed]
- Latypov, R.F.; Hogan, S.; Lau, H.; Gadgil, H.; Liu, D. Elucidation of acid-induced unfolding and aggregation of human immunoglobulin IgG1 and IgG2 Fc. J. Biol. Chem. 2012, 287, 1381–1396. [Google Scholar] [CrossRef] [PubMed]
- Weiss, W.F.; Young, T.M.; Roberts, C.J. Principles, approaches, and challenges for predicting protein aggregation rates and shelf life. J. Pharm. Sci. 2009, 98, 1246–1277. [Google Scholar] [CrossRef] [PubMed]
- Telikepalli, S.N.; Kumru, O.S.; Kalonia, C.; Esfandiary, R.; Joshi, S.B.; Middaugh, C.R.; Volkin, D.B. Structural characterization of IgG1 mAb aggregates and particles generated under various stress conditions. J. Pharm. Sci. 2014, 103, 796–809. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, M.V.; Laurence, J.S.; Siahaan, T.J. The role of thiols and disulfides on protein stability. Curr. Protein Pept. Sci. 2009, 10, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, A.; Clementel, V.; Woods, R.; Harn, N.; Bowen, M.A.; Mo, W.; Popovic, B.; Bishop, S.M.; Dall’Acqua, W.; Minter, R.; et al. Engineering a therapeutic IgG molecule to address cysteinylation, aggregation and enhance thermal stability and expression. mAbs 2013, 5, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Dudgeon, K.; Rouet, R.; Kokmeijer, I.; Schofield, P.; Stolp, J.; Langley, D.; Stock, D.; Christ, D. General strategy for the generation of human antibody variable domains with increased aggregation resistance. Proc. Natl. Acad. Sci. USA 2012, 109, 10879–10884. [Google Scholar] [CrossRef] [PubMed]
- Kayser, V.; Chennamsetty, N.; Voynov, V.; Forrer, K.; Helk, B.; Trout, B.L. Glycosylation influences on the aggregation propensity of therapeutic monoclonal antibodies. Biotechnol. J. 2011, 6, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Krapp, S.; Mimura, Y.; Jefferis, R.; Huber, R.; Sondermann, P. Structural analysis of human IgG-Fc glycoforms reveals a correlation between glycosylation and structural integrity. J. Mol. Biol. 2003, 325, 979–989. [Google Scholar] [CrossRef]
- Schaefer, J.V.; Plückthun, A. Engineering Aggregation Resistance in IgG by Two Independent Mechanisms: Lessons from Comparison of Pichia pastoris and Mammalian Cell Expression. J. Mol. Biol. 2012, 417, 309–335. [Google Scholar] [CrossRef] [PubMed]
- Pepinsky, R.B.; Silvian, L.; Berkowitz, S.A.; Farrington, G.; Lugovskoy, A.; Walus, L.; Eldredge, J.; Capili, A.; Mi, S.; Graff, C.; et al. Improving the solubility of anti-LINGO-1 monoclonal antibody Li33 by isotype switching and targeted mutagenesis. Protein Sci.: Publ. Protein Soc. 2010, 19, 954–966. [Google Scholar] [CrossRef] [PubMed]
- Nezlin, R.; Ghetie, V. Interactions of immunoglobulins outside the antigen-combining site. Adv. Immunol. 2004, 82, 155–215. [Google Scholar] [PubMed]
- Nezlin, R. Internal movements in immunoglobulin molecules. Adv. Immunol. 1990, 48, 1–40. [Google Scholar] [PubMed]
- Tiller, K.E.; Tessier, P.M. Advances in Antibody Design. Annu. Rev. Biomed. Eng. 2015, 17, 191–216. [Google Scholar] [CrossRef] [PubMed]
- Rouet, R.; Lowe, D.; Christ, D. Stability engineering of the human antibody repertoire. FEBS Lett. 2014, 588, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Singh, S.; Zeng, D.L.; King, K.; Nema, S. Antibody structure, instability, and formulation. J. Pharm. Sci. 2007, 96, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Tischenko, V.M.; Abramov, V.M.; Zav’yalov, V.P. Investigation of the cooperative structure of Fc fragments from myeloma immunoglobulin G. Biochemistry 1998, 37, 5576–5581. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Ichimura, N.; Hamaguchi, K. Effects of ammonium sulfate on the unfolding and refolding of the variable and constant fragments of an immunoglobulin light chain. Biochemistry 1988, 27, 1670–1677. [Google Scholar] [CrossRef] [PubMed]
- Zav’yalov, V.P.; Tishchenko, V.M. Mechanisms of generation of antibody diversity as a cause for natural selection of homoiothermal animals in the process of evolution. Scand. J. Immunol. 1991, 33, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Gong, R.; Ying, T.; Prabakaran, P.; Zhu, Z.; Feng, Y.; Dimitrov, D.S. Discovery of novel candidate therapeutics and diagnostics based on engineered human antibody domains. Curr. Drug Discov. Technol. 2014, 11, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Hussack, G.; Keklikian, A.; Alsughayyir, J.; Hanifi-Moghaddam, P.; Arbabi-Ghahroudi, M.; van Faassen, H.; Hou, S.T.; Sad, S.; MacKenzie, R.; Tanha, J. A VL single-domain antibody library shows a high-propensity to yield non-aggregating binders. Protein Eng. Des. Sel. 2012, 25, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Hamaguchi, K. The role of the intrachain disulfide bond in the conformation and stability of the constant fragment of the immunoglobulin light chain. J. Biochem. 1979, 86, 1433–1441. [Google Scholar] [PubMed]
- Chothia, C.; Janin, J. Principles of protein-protein recognition. Nature 1975, 256, 705–708. [Google Scholar] [CrossRef] [PubMed]
- Arbabi-Ghahroudi, M.; To, R.; Gaudette, N.; Hirama, T.; Ding, W.; MacKenzie, R.; Tanha, J. Aggregation-resistant VHs selected by in vitro evolution tend to have disulfide-bonded loops and acidic isoelectric points. Protein Eng. Des. Sel. 2009, 22, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Perchiacca, J.M.; Ladiwala, A.R.A.; Bhattacharya, M.; Tessier, P.M. Aggregation-resistant domain antibodies engineered with charged mutations near the edges of the complementarity-determining regions. Protein Eng. Des. Sel. 2012, 25, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Perchiacca, J.M.; Bhattacharya, M.; Tessier, P.M. Mutational analysis of domain antibodies reveals aggregation hotspots within and near the complementarity determining regions. Proteins 2011, 79, 2637–2647. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, A.S.; Avila, D.; Hughes, M.; Hughes, A.; McKinney, E.C.; Flajnik, M.F. A new antigen receptor gene family that undergoes rearrangement and extensive somatic diversification in sharks. Nature 1995, 374, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.; Riechmann, L. Single antibody domains as small recognition units: Design and in vitro antigen selection of camelized, human VH domains with improved protein stability. Protein Eng. 1996, 9, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Kandalaft, H.; Ding, W.; Ryan, S.; van Faassen, H.; Hirama, T.; Foote, S.J.; MacKenzie, R.; Tanha, J. Disulfide linkage engineering for improving biophysical properties of human VH domains. Protein Eng. Des. Sel. 2012, 25, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; To, R.; Kandalaft, H.; Ding, W.; van Faassen, H.; Luo, Y.; Schrag, J.D.; St-Amant, N.; Hefford, M.; Hirama, T.; et al. Antibody light chain variable domains and their biophysically improved versions for human immunotherapy. mAbs 2014, 6, 219–235. [Google Scholar] [CrossRef] [PubMed]
- Barthelemy, P.A.; Raab, H.; Appleton, B.A.; Bond, C.J.; Wu, P.; Wiesmann, C.; Sidhu, S.S. Comprehensive Analysis of the Factors Contributing to the Stability and Solubility of Autonomous Human VH Domains. J. Biol. Chem. 2008, 283, 3639–3654. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.; Riechmann, L. ‘Camelising’ human antibody fragments: NMR studies on VH domains. FEBS Lett. 1994, 339, 285–290. [Google Scholar] [CrossRef]
- Langedijk, A.C.; Honegger, A.; Maat, J.; Planta, R.J.; van Schaik, R.C.; Pluckthun, A. The nature of antibody heavy chain residue H6 strongly influences the stability of a VH domain lacking the disulfide bridge. J. Mol. Biol. 1998, 283, 95–110. [Google Scholar] [CrossRef] [PubMed]
- Gorlani, A.; Hulsik, D.L.; Adams, H.; Vriend, G.; Hermans, P.; Verrips, T. Antibody engineering reveals the important role of J segments in the production efficiency of llama single-domain antibodies in Saccharomyces cerevisiae. Protein Eng. Des. Sel. 2012, 25, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Spada, S.; Honegger, A.; Pluckthun, A. Reproducing the natural evolution of protein structural features with the selectively infective phage (SIP) technology. The kink in the first strand of antibody kappa domains. J. Mol. Biol. 1998, 283, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Ewert, S.; Huber, T.; Honegger, A.; Pluckthun, A. Biophysical properties of human antibody variable domains. J. Mol. Biol. 2003, 325, 531–553. [Google Scholar] [CrossRef]
- Nilson, B.H.; Solomon, A.; Bjorck, L.; Akerstrom, B. Protein L from Peptostreptococcus magnus binds to the kappa light chain variable domain. J. Biol. Chem. 1992, 267, 2234–2239. [Google Scholar] [PubMed]
- Steipe, B.; Schiller, B.; Pluckthun, A.; Steinbacher, S. Sequence statistics reliably predict stabilizing mutations in a protein domain. J. Mol. Biol. 1994, 240, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Ignatovich, O.; Jespers, L.; Tomlinson, I.M.; de Wildt, R.M.T. Creation of the Large and Highly Functional Synthetic Repertoire of Human VH and Vκ Domain Antibodies. In Single Domain Antibodies: Methods and Protocols; Saerens, D., Muyldermans, S., Eds.; Humana Press: Totowa, NJ, USA, 2012; pp. 39–63. [Google Scholar]
- Chen, W.; Zhu, Z.; Feng, Y.; Xiao, X.; Dimitrov, D.S. Construction of a Large Phage-Displayed Human Antibody Domain Library with a Scaffold Based On a Newly Identified Highly Soluble, Stable Heavy Chain Variable Domain. J. Mol. Biol. 2008, 382, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Colby, D.W.; Garg, P.; Holden, T.; Chao, G.; Webster, J.M.; Messer, A.; Ingram, V.M.; Wittrup, K.D. Development of a Human Light Chain Variable Domain (VL) Intracellular Antibody Specific for the Amino Terminus of Huntingtin via Yeast Surface Display. J. Mol. Biol. 2004, 342, 901–912. [Google Scholar] [CrossRef] [PubMed]
- Gil, D.; Schrum, A.G. Strategies to stabilize compact folding and minimize aggregation of antibody-based fragments. Adv. Biosci. Biotechnol. 2013, 4, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Worn, A.; Pluckthun, A. Stability engineering of antibody single-chain Fv fragments. J. Mol. Biol. 2001, 305, 989–1010. [Google Scholar] [CrossRef] [PubMed]
- Pluckthun, A. Mono- and bivalent antibody fragments produced in Escherichia coli: Engineering, folding and antigen binding. Immunol. Rev. 1992, 130, 151–188. [Google Scholar] [CrossRef] [PubMed]
- Glockshuber, R.; Malia, M.; Pfitzinger, I.; Pluckthun, A. A comparison of strategies to stabilize immunoglobulin Fv-fragments. Biochemistry 1990, 29, 1362–1367. [Google Scholar] [CrossRef] [PubMed]
- Tan, P.H.; Sandmaier, B.M.; Stayton, P.S. Contributions of a highly conserved VH/VL hydrogen bonding interaction to scFv folding stability and refolding efficiency. Biophys. J. 1998, 75, 1473–1482. [Google Scholar] [CrossRef]
- Zhu, Z.; Presta, L.G.; Zapata, G.; Carter, P. Remodeling domain interfaces to enhance heterodimer formation. Protein Sci. Publ. Protein Soc. 1997, 6, 781–788. [Google Scholar] [CrossRef] [PubMed]
- Hakansson, M.; Linse, S. Protein reconstitution and 3D domain swapping. Curr. Protein Pept. Sci. 2002, 3, 629–642. [Google Scholar] [CrossRef] [PubMed]
- Robinson, C.R.; Sauer, R.T. Optimizing the stability of single-chain proteins by linker length and composition mutagenesis. Proc. Natl. Acad. Sci. USA 1998, 95, 5929–5934. [Google Scholar] [CrossRef] [PubMed]
- Arndt, K.M.; Muller, K.M.; Pluckthun, A. Factors influencing the dimer to monomer transition of an antibody single-chain Fv fragment. Biochemistry 1998, 37, 12918–12926. [Google Scholar] [CrossRef] [PubMed]
- Nelson, A.L.; Reichert, J.M. Development trends for therapeutic antibody fragments. Nat. Biotechnol. 2009, 27, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Kumru, O.S.; Yi, L.; Wang, Y.J.; Zhang, J.; Kim, J.H.; Joshi, S.B.; Middaugh, C.R.; Volkin, D.B. Effect of ionic strength and pH on the physical and chemical stability of a monoclonal antibody antigen-binding fragment. J. Pharm. Sci. 2013, 102, 2520–2537. [Google Scholar] [CrossRef] [PubMed]
- Roux, K.H.; Tankersley, D.L. A view of the human idiotypic repertoire. Electron microscopic and immunologic analyses of spontaneous idiotype-anti-idiotype dimers in pooled human IgG. J. Immunol. 1990, 144, 1387–1395. [Google Scholar] [PubMed]
- Kanai, S.; Liu, J.; Patapoff, T.W.; Shire, S.J. Reversible self-association of a concentrated monoclonal antibody solution mediated by Fab-Fab interaction that impacts solution viscosity. J. Pharm. Sci. 2008, 97, 4219–4227. [Google Scholar] [CrossRef] [PubMed]
- Peters, S.J.; Smales, C.M.; Henry, A.J.; Stephens, P.E.; West, S.; Humphreys, D.P. Engineering an Improved IgG4 Molecule with Reduced Disulfide Bond Heterogeneity and Increased Fab Domain Thermal Stability. J. Biol. Chem. 2012, 287, 24525–24533. [Google Scholar] [CrossRef] [PubMed]
- Teerinen, T.; Valjakka, J.; Rouvinen, J.; Takkinen, K. Structure-based stability engineering of the mouse IgG1 Fab fragment by modifying constant domains. J. Mol. Biol. 2006, 361, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Bardhi, A.; Feng, Y.; Wang, Y.; Qi, Q.; Li, W.; Zhu, Z.; Dyba, M.A.; Ying, T.; Jiang, S.; et al. Improving the CH1-CK heterodimerization and pharmacokinetics of 4Dm2m, a novel potent CD4-antibody fusion protein against HIV-1. mAbs 2016, 8, 761–774. [Google Scholar] [CrossRef] [PubMed]
- Gong, R.; Wang, Y.; Feng, Y.; Zhao, Q.; Dimitrov, D.S. Shortened engineered human antibody CH2 domains: Increased stability and binding to the human neonatal Fc receptor. J. Biol. Chem. 2011, 286, 27288–27293. [Google Scholar] [CrossRef] [PubMed]
- DeLano, W.L.; Ultsch, M.H.; de Vos, A.M.; Wells, J.A. Convergent solutions to binding at a protein-protein interface. Science 2000, 287, 1279–1283. [Google Scholar] [CrossRef] [PubMed]
- Kolenko, P.; Dohnálek, J.; Dušková, J.; Skálová, T.; Collard, R.; Hašek, J. New insights into intra- and intermolecular interactions of immunoglobulins: Crystal structure of mouse IgG2b-Fc at 2·1-Å resolution. Immunology 2009, 126, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Kong, L.; Connelly, S.; Dendle, J.M.; Liu, Y.; Wilson, I.A.; Powers, E.T.; Kelly, J.W. Stabilizing the CH2 Domain of an Antibody by Engineering in an Enhanced Aromatic Sequon. ACS Chem. Biol. 2016, 11, 1852–1861. [Google Scholar] [CrossRef] [PubMed]
- Fast, J.L.; Cordes, A.A.; Carpenter, J.F.; Randolph, T.W. Physical instability of a therapeutic Fc fusion protein: Domain contributions to conformational and colloidal stability. Biochemistry 2009, 48, 11724–11736. [Google Scholar] [CrossRef] [PubMed]
- Gong, R.; Vu, B.K.; Feng, Y.; Prieto, D.A.; Dyba, M.A.; Walsh, J.D.; Prabakaran, P.; Veenstra, T.D.; Tarasov, S.G.; Ishima, R.; et al. Engineered human antibody constant domains with increased stability. J. Biol. Chem. 2009, 284, 14203–14210. [Google Scholar] [CrossRef] [PubMed]
- Ying, T.; Chen, W.; Feng, Y.; Wang, Y.; Gong, R.; Dimitrov, D.S. Engineered soluble monomeric IgG1 CH3 domain: Generation, mechanisms of function, and implications for design of biological therapeutics. J. Biol. Chem. 2013, 288, 25154–25164. [Google Scholar] [CrossRef] [PubMed]
- Wozniak-Knopp, G.; Stadlmann, J.; Ruker, F. Stabilisation of the Fc fragment of human IgG1 by engineered intradomain disulfide bonds. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody therapy of cancer. Nat. Rev. Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Zolot, R.S.; Basu, S.; Million, R.P. Antibody-drug conjugates. Nat. Rev. Drug Discov. 2013, 12, 259–260. [Google Scholar] [CrossRef] [PubMed]
- Beckley, N.S.; Lazzareschi, K.P.; Chih, H.-W.; Sharma, V.K.; Flores, H.L. Investigation into Temperature-Induced Aggregation of an Antibody Drug Conjugate. Bioconjug. Chem. 2013, 24, 1674–1683. [Google Scholar] [CrossRef] [PubMed]
- Wakankar, A.A.; Feeney, M.B.; Rivera, J.; Chen, Y.; Kim, M.; Sharma, V.K.; Wang, Y.J. Physicochemical Stability of the Antibody-Drug Conjugate Trastuzumab-DM1: Changes due to Modification and Conjugation Processes. Bioconjug. Chem. 2010, 21, 1588–1595. [Google Scholar] [CrossRef] [PubMed]
- Acchione, M.; Kwon, H.; Jochheim, C.M.; Atkins, W.M. Impact of linker and conjugation chemistry on antigen binding, Fc receptor binding and thermal stability of model antibody-drug conjugates. mAbs 2012, 4, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Kumar, S.; Chipley, M.; Marcq, O.; Gupta, D.; Jin, Z.; Tomar, D.S.; Swabowski, C.; Smith, J.; Starkey, J.A.; et al. Characterization and Higher-Order Structure Assessment of an Interchain Cysteine-Based ADC: Impact of Drug Loading and Distribution on the Mechanism of Aggregation. Bioconjug. Chem. 2016, 27, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Ramakrishnan, B.; Li, J.; Wang, Y.; Feng, Y.; Prabakaran, P.; Colantonio, S.; Dyba, M.A.; QasbaM, P.K.; Dimitrov, D.S. Site-specific antibody-drug conjugation through an engineered glycotransferase and a chemically reactive sugar. mAbs 2014, 6, 1190–1200. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Shen, J.; Streaker, E.D.; Lockwood, M.; Zhu, Z.; Low, P.S.; Dimitrov, D.S. A folate receptor beta-specific human monoclonal antibody recognizes activated macrophage of rheumatoid patients and mediates antibody-dependent cell-mediated cytotoxicity. Arthritis Res. Ther. 2011, 13. [Google Scholar] [CrossRef] [PubMed]
- Chothia, C. Hydrophobic bonding and accessible surface area in proteins. Nature 1974, 248, 338–339. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Kumar, S.; Prashad, A.; Starkey, J.; Singh, S.K. Assessment of Physical Stability of an Antibody Drug Conjugate by Higher Order Structure Analysis: Impact of Thiol- Maleimide Chemistry. Pharm. Res. 2014, 31, 1710–1723. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.Y.; Wilhelm, S.D.; Audette, C.; Jones, G.; Leece, B.A.; Lazar, A.C.; Goldmacher, V.S.; Singh, R.; Kovtun, Y.; Widdison, W.C.; et al. Synthesis and Evaluation of Hydrophilic Linkers for Antibody-Maytansinoid Conjugates. J. Med. Chemi. 2011, 54, 3606–3623. [Google Scholar] [CrossRef] [PubMed]
- Lyon, R.P.; Bovee, T.D.; Doronina, S.O.; Burke, P.J.; Hunter, J.H.; Neff-LaFord, H.D.; Jonas, M.; Anderson, M.E.; Setter, J.R.; Senter, P.D. Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index. Nat. Biotech. 2015, 33, 733–735. [Google Scholar] [CrossRef] [PubMed]
- Adem, Y.T.; Schwarz, K.A.; Duenas, E.; Patapoff, T.W.; Galush, W.J.; Esue, O. Auristatin Antibody Drug Conjugate Physical Instability and the Role of Drug Payload. Bioconjug. Chem. 2014, 25, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.Y.; Salas-Solano, O.; Valliere-Douglass, J.F. Conformation and Dynamics of Interchain Cysteine-Linked Antibody-Drug Conjugates as Revealed by Hydrogen/Deuterium Exchange Mass Spectrometry. Anal. Chem. 2014, 86, 2657–2664. [Google Scholar] [CrossRef] [PubMed]
- Schneidman-Duhovny, D.; Inbar, Y.; Nussinov, R.; Wolfson, H.J. PatchDock and SymmDock: Servers for rigid and symmetric docking. Nucleic Acids Res. 2005, 33, W363–W367. [Google Scholar] [CrossRef] [PubMed]
- Harding, F.A.; Stickler, M.M.; Razo, J.; DuBridge, R.B. The immunogenicity of humanized and fully human antibodies: Residual immunogenicity resides in the CDR regions. mAbs 2010, 2, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Hwang, W.Y.; Foote, J. Immunogenicity of engineered antibodies. Methods 2005, 36, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Van Walle, I.; Gansemans, Y.; Parren, P.W.; Stas, P.; Lasters, I. Immunogenicity screening in protein drug development. Expert Opin. Biol. Ther. 2007, 7, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, A.M.; Elliott, S. Glycoengineering: The effect of glycosylation on the properties of therapeutic proteins. J. Pharm. Sci. 2005, 94, 1626–1635. [Google Scholar] [CrossRef] [PubMed]
- Veronese, F.M.; Mero, A. The impact of PEGylation on biological therapies. BioDrugs Clin. Immunother. Biopharm. Gene Ther. 2008, 22, 315–329. [Google Scholar] [CrossRef]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, W.; Prabakaran, P.; Chen, W.; Zhu, Z.; Feng, Y.; Dimitrov, D.S. Antibody Aggregation: Insights from Sequence and Structure. Antibodies 2016, 5, 19. https://doi.org/10.3390/antib5030019
Li W, Prabakaran P, Chen W, Zhu Z, Feng Y, Dimitrov DS. Antibody Aggregation: Insights from Sequence and Structure. Antibodies. 2016; 5(3):19. https://doi.org/10.3390/antib5030019
Chicago/Turabian StyleLi, Wei, Ponraj Prabakaran, Weizao Chen, Zhongyu Zhu, Yang Feng, and Dimiter S. Dimitrov. 2016. "Antibody Aggregation: Insights from Sequence and Structure" Antibodies 5, no. 3: 19. https://doi.org/10.3390/antib5030019