1. Introduction
Antibodies are immunoglobulin molecules that recognize antigens naturally circulating or displayed on the exterior of the cell and mediate humoral immunity but do not naturally have an effect on intracellular antigens. However, in principle, antibodies or antibody fragments could target intracellular molecules if they were able to gain access to the inside of cells while retaining their binding ability. The first demonstration that whole antibody could work in cells was made when an anti-RAS whole antibody was micro-injected into cells and reverted the RAS-mediated phenotype [
1]. However, whole antibody (comprising two heavy and two light chains covalently linked by disulphide bonds) cannot remain stable in cells because reduction of the disulphide bonds would occur and a whole antibody is unlikely to form from
de novo synthesis in the reducing environment. Developments in antibody engineering in recent years have offered solutions to this stability problem, and now a range of antibody fragment formats exist that are suitable for intracellular function, pioneered by Cattaneo and Marasco and colleagues [
2,
3].
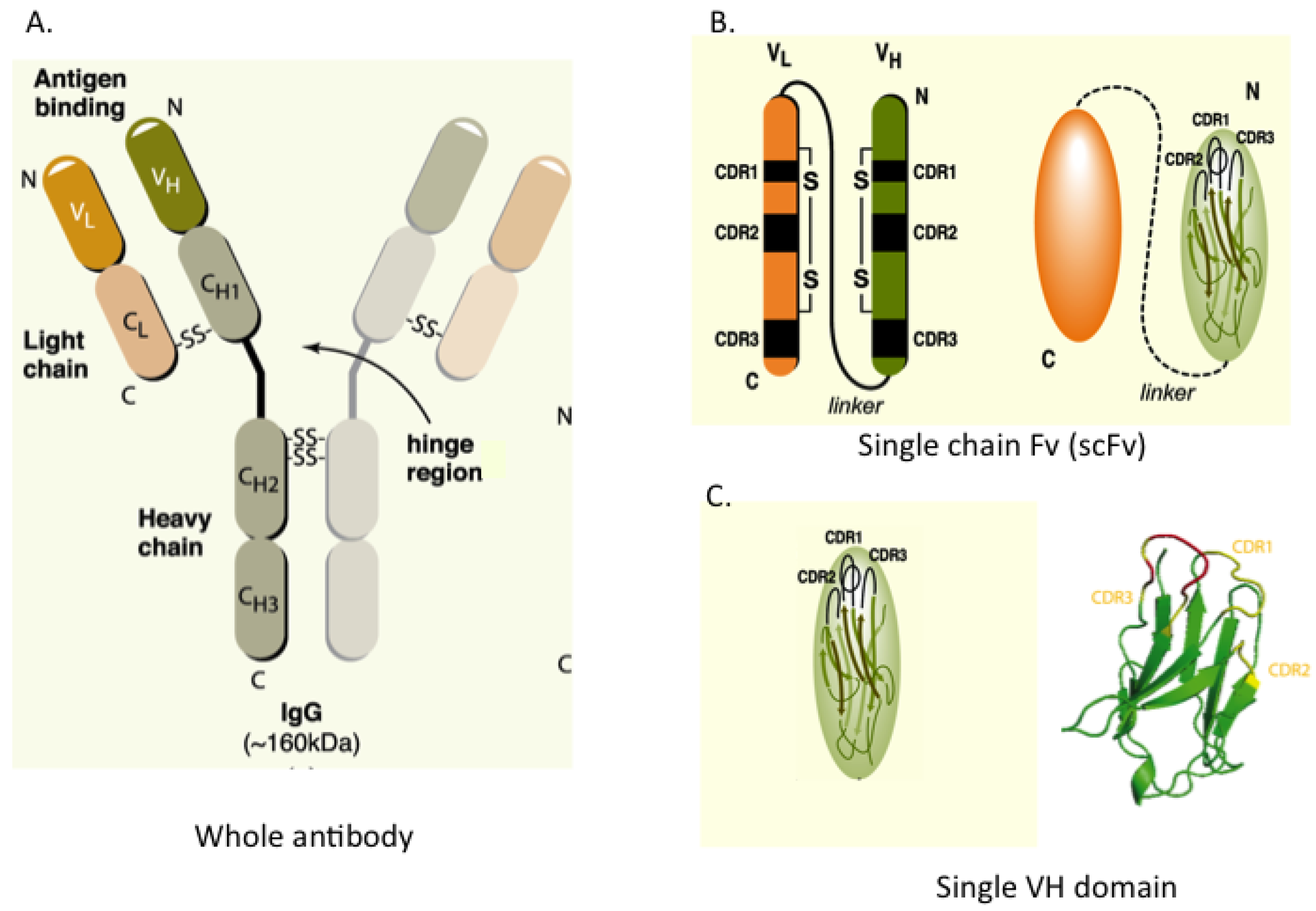
Figure 1 shows the configuration of whole IgG together with single-chain Fv (scFv), which consists of heavy (H) and light (L) chain variable regions (VH and VL) joined in a single polypeptide by a flexible linker to improve its assembly inside cells. A minimal format antibody fragment is the single VH or VL domain carrying three hyper-variable complementarity-determining regions (CDRs), which has proven sufficient for antigen recognition [
4]. We and others have shown that VH or VL domains can be used to bind to a variety of target proteins
in vivo [
5,
6,
7].
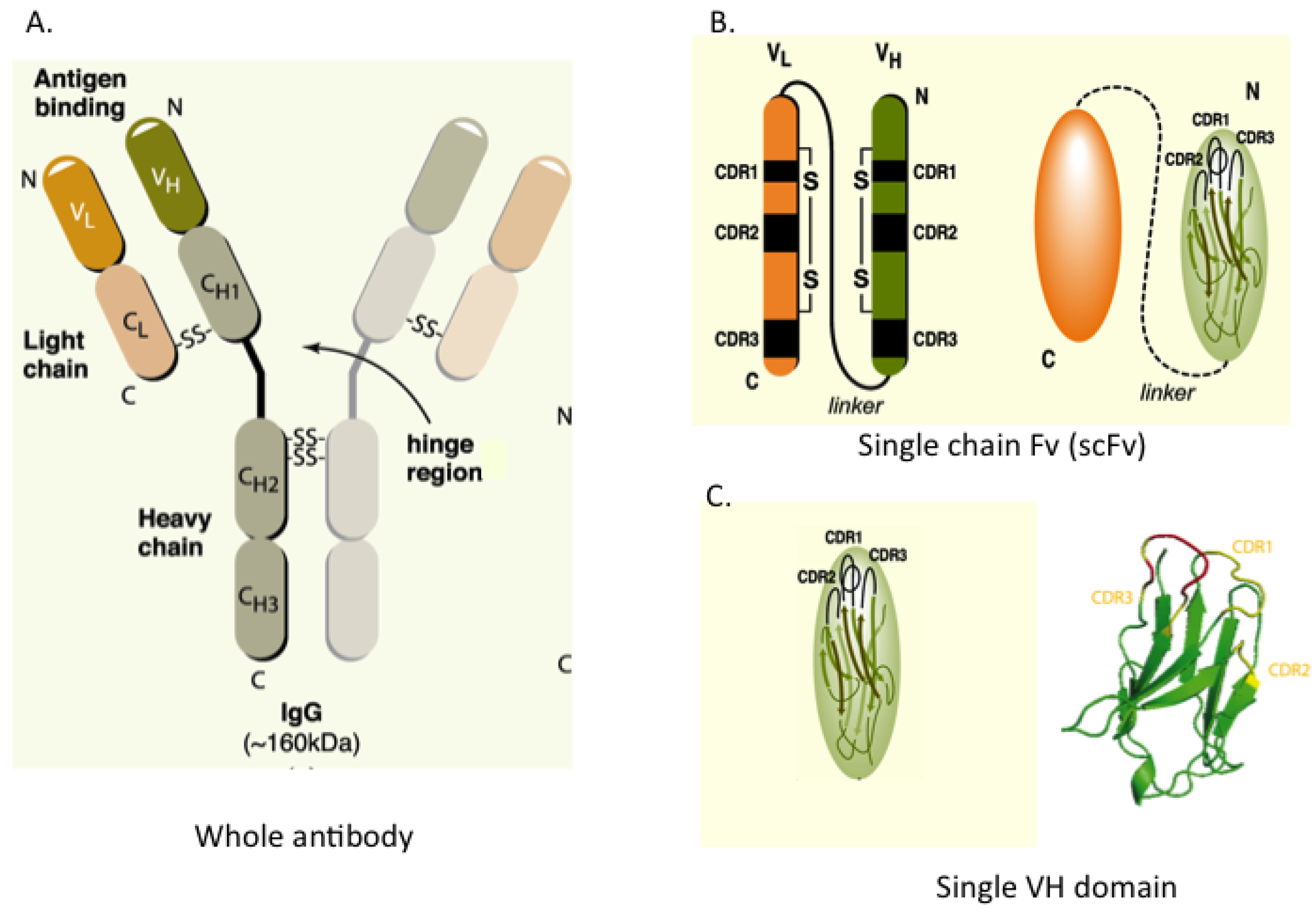
Figure 1.
Antibody and antibody fragment formats. (A) Whole antibody comprising two heavy (H) and two light (L) chains joined by inter-chain S-S bonds. The H & L chains in turn have N-terminal variable (V) regions (VH and VL) and C-terminal constant (C) regions; (B) The single chain Fv (scFv) format comprises the VH and VL segments in a single polypeptide chain joined by a short flexible linker. Each V-region has three complementarity determining regions (CDRs) in direct contact with antigen; (C) The single domain, shown here as VH, is the variable region only. The RHS shows the structure of the single VH domain with anti-parallel β-sheets and the three external complementarity determining (CDR1, 2 and 3) loops.
Figure 1.
Antibody and antibody fragment formats. (A) Whole antibody comprising two heavy (H) and two light (L) chains joined by inter-chain S-S bonds. The H & L chains in turn have N-terminal variable (V) regions (VH and VL) and C-terminal constant (C) regions; (B) The single chain Fv (scFv) format comprises the VH and VL segments in a single polypeptide chain joined by a short flexible linker. Each V-region has three complementarity determining regions (CDRs) in direct contact with antigen; (C) The single domain, shown here as VH, is the variable region only. The RHS shows the structure of the single VH domain with anti-parallel β-sheets and the three external complementarity determining (CDR1, 2 and 3) loops.
Many human cancers are caused by acquisition of somatic mutations, or when chromosomal rearrangements occur, by formation of fusion proteins [
8], which deregulate various cellular processes mainly through protein–protein interactions (PPIs). Although these aberrant protein–protein interactions are high value cancer targets, it is very difficult to target these interactions with small molecule drugs, mainly due to the relatively large and featureless protein‑protein interfaces [
9] as this means the binding affinity of small molecules may be too low. Recently, intracellular antibody fragments have been shown as promising reagents to target such protein interaction interfaces inside cells. The inherent capacity of binding protein surface with high affinity and specificity makes using antibody fragments a good option for development of macromolecule drugs (herein referred to as macrodrugs to distinguish them from conventional small molecule drugs [
10] or drug surrogates interfering with specific PPIs).
Herein we will review our results with designer VH antibody fragments as drug surrogates and suggest some prospectives for future development of the single domain molecules and potential small molecule emulators.
2. Methods for Intracellular Antibody Selection
Initially, the scFv formats were derived from V regions of high-affinity monoclonal antibodies [
11]. Despite these derived scFv being able to bind specific antigens with high binding affinity
in vitro, relatively few function effectively inside cell cytoplasm or nuclei. Similarly, scFv derived from phage antibody library screens, frequently did not function as intracellular antibodies since the selections were carried out
in vitro and their function demanded an intracellular environment. These findings pointed us towards an
in vivo selection process that would allow direct isolation of antibody fragments that work in the cellular environment. We developed
Intracellular
Antibody
Capture (IAC) for this purpose [
12,
13]. First generation IAC started with antibody selection through an initial screen
in vitro with phage scFv libraries and the selected scFv were subsequently subjected to a second screen through an antigen–antibody interaction assay inside yeast cells with a two-hybrid system [
14].
IAC technology has allowed successful isolation of intracellular antibodies against a wide variety of antigens [
5,
6,
13,
15,
16,
17]. More importantly, by comparing the recurrent residues at each position in a number of intracellular antibodies, this method has helped to define stable frameworks of VH and VL that were tolerant of the intra-cellular reducing environment, making it possible to design a consensus framework (designer VH and VL) for the human intracellular antibody fragment VH and VL [
18]. We also found that single V-region domains were more efficacious as intracellular antibodies than scFv [
19]. A comparison was made of the interaction of scFv, or the separate component VH or VL in intracellular binding to antigen using a mammalian luciferase reporter assay [
19].
Figure 2A shows these data for two independent anti-RAS scFv and their VH and VL demonstrating that the VH in these cases bind to antigen more robustly than scFv (as judged by luciferase activity) and that the separated VL have low binding ability. A further ramification of these findings was the observation that mutation of the cysteine residues, normally involved in the intra-chain disulphide bond of the V-domain, had marginal effects on the function of the VH domain intracellularly (
Figure 2A). This was not surprising since the disulphide bond would be unable to form in-cell and testifies to the intrinsic folding of the variable domain prior to disulphide formation. In addition, the crystal structures of VH and VL domains in the presence or absence of the intra-chain disulphide bond (
Figure 2B) were virtually identical [
20] suggesting further that the intra-chain disulphide bonds in V regions contribute to stability rather than to folding
per se.
Figure 2.
Single domains are effective intracellular antibody fragments and do not require the intra-chain S-S bond. (
A) Luciferase reporter assays (Firefly/Renilla activity × 10
−3 RU) using anti-RAS intracellular antibody fragment expression vectors (two different scFv or VH and VL derived from those scFv) in CHO cells co-transfected with HRASG12V or β-galactosidase (lacZ) baits. Luciferase signal (due to interaction of the antibody fragment prey and the HRASG12V bait protein) was detected when the scFv and the VH, but not the VL fragments, were used [
19] signifying that the minimal fragment of antibody (VH) can function within the reducing environment of the cell. In turn, mutation of either of the cysteine residues involved in the intra-chain S-S bridge to serine, that abolish intra-domain disulphide bonds, had little or no effect on binding. Mutation of C22S appears lower than mutation C92S but this may reflect experimental expression levels rather than structural differences (
B) The respective VH and VL single domains were produced in E. coli and crystal structures obtained with (native) or without (disulphide-free) the S-S bond [
20]. The panels show parts of the structures and that very little change has occurred in the two forms of the molecules.
Figure 2.
Single domains are effective intracellular antibody fragments and do not require the intra-chain S-S bond. (
A) Luciferase reporter assays (Firefly/Renilla activity × 10
−3 RU) using anti-RAS intracellular antibody fragment expression vectors (two different scFv or VH and VL derived from those scFv) in CHO cells co-transfected with HRASG12V or β-galactosidase (lacZ) baits. Luciferase signal (due to interaction of the antibody fragment prey and the HRASG12V bait protein) was detected when the scFv and the VH, but not the VL fragments, were used [
19] signifying that the minimal fragment of antibody (VH) can function within the reducing environment of the cell. In turn, mutation of either of the cysteine residues involved in the intra-chain S-S bridge to serine, that abolish intra-domain disulphide bonds, had little or no effect on binding. Mutation of C22S appears lower than mutation C92S but this may reflect experimental expression levels rather than structural differences (
B) The respective VH and VL single domains were produced in E. coli and crystal structures obtained with (native) or without (disulphide-free) the S-S bond [
20]. The panels show parts of the structures and that very little change has occurred in the two forms of the molecules.
![Antibodies 02 00306 g002]()
3. Single Domain Libraries, IAC3 and Selecting PPI Inhibiting Antibody Fragments
The single V-region domain is the minimal antibody fragment that can currently be used for intracellular function. The designer VH and VL frameworks were the basis of single domain libraries for yeast IAC screening in the third generation IAC (IAC
3) [
21] that uses single VH or VL domains instead of scFv as the antigen recognition unit. The IAC
3 method is outlined in
Figure 3 and directly uses yeast screens [
21] for selecting VH or VL (prey) binding to antigen (bait) of interest (depicted in
Figure 3A). Antibody screening is carried out by iterative affinity maturation making use of increasing concentrations of 3-amino-1,2,-triazole (3-AT, an inhibitor of the enzyme encoded by
HIS3) to sequentially enhance the sensitivity of screens [
21]. A single domain diverse library (~10
7 clones) of VH or VL is screened
in vivo in the yeast system where the library has randomized CDR3 (also see discussion below for VH CDR3 length). This yields variable numbers of hits and these are pooled and the CDR2 segments randomized to make a new diverse sub-library which is rescreened with antigen (
Figure 3B). If necessary, this interactive process can be repeated to increase the affinity maturation process with randomized CDR1 from the CDR2 sub-library hits, yielding nM affinity single domain binding to intracellular antigens.
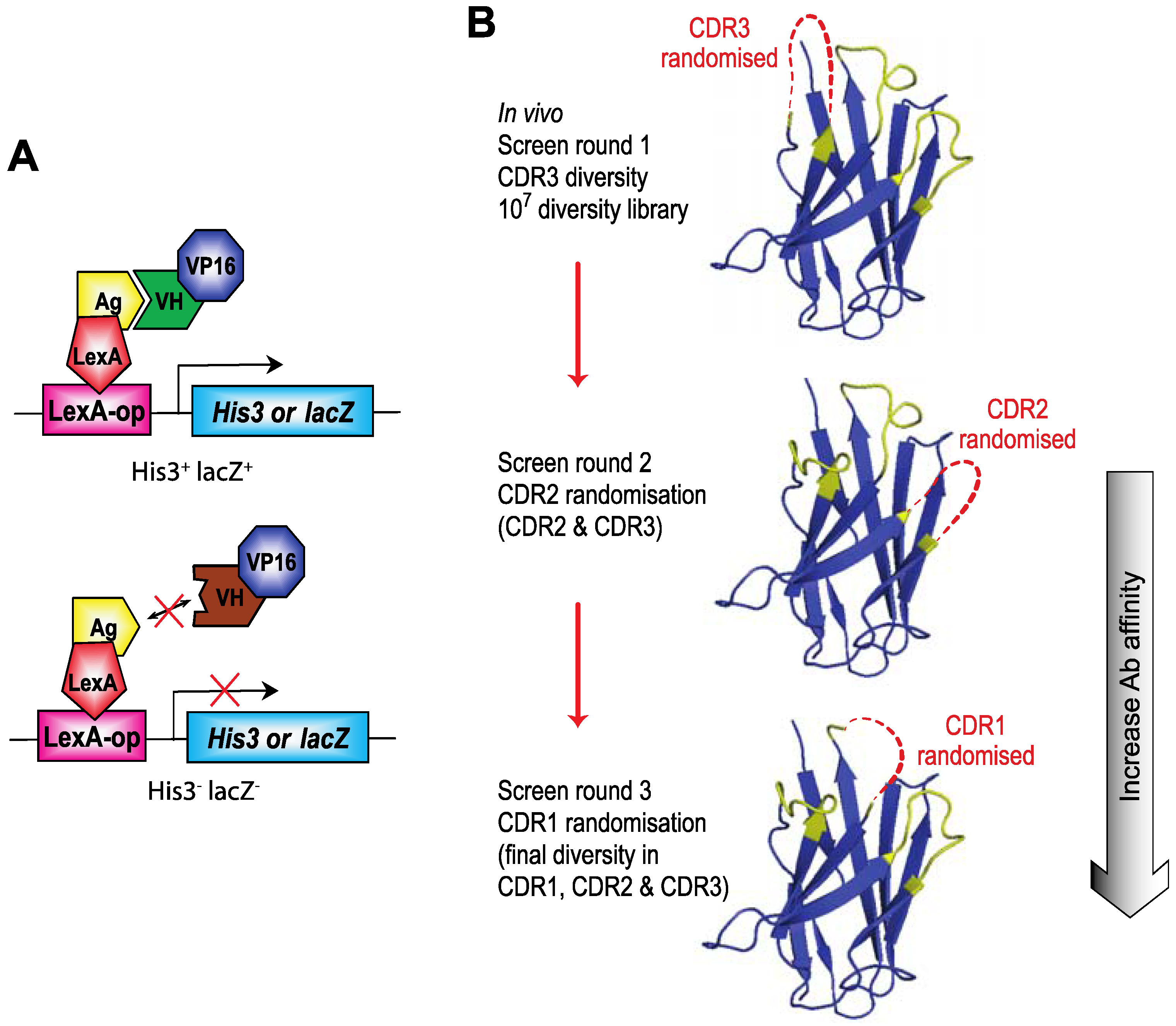
Figure 3.
Intracellular Antibody Capture (IAC). Intracellular Antibody Capture is a method based on yeast screening for interactions between, originally scFv [
12,
13], and latterly single domains, and target proteins [
19]. (
A) A diagram of the method using LexA DNA binding domain (DBD) fused to the target protein (bait, e.g., RAS) and LexA DNA binding site (DBS) in a reporter gene (either
HIS3 or
lacZ). Interaction of an antibody fragment (e.g., intracellular domain antibody, iDab), fused to a transcriptional activation domain (VP16 AD), with the target protein bait activates the reporter and allows selection of clones. (
B) The screening protocol for IAC
3 [
21] involves first using a diverse library of V genes with diversified CDR3 and subsequent affinity maturation of V regions that bind to the target protein using first CDR2 randomisation and rescreening hits from the CDR3 round of screening and, if deemed necessary, repeating the process with CDR1 randomisation and rescreening hits from the CDR2 screen. Final hits are tested for their capacity to interact with the target protein in mammalian cells.
Figure 3.
Intracellular Antibody Capture (IAC). Intracellular Antibody Capture is a method based on yeast screening for interactions between, originally scFv [
12,
13], and latterly single domains, and target proteins [
19]. (
A) A diagram of the method using LexA DNA binding domain (DBD) fused to the target protein (bait, e.g., RAS) and LexA DNA binding site (DBS) in a reporter gene (either
HIS3 or
lacZ). Interaction of an antibody fragment (e.g., intracellular domain antibody, iDab), fused to a transcriptional activation domain (VP16 AD), with the target protein bait activates the reporter and allows selection of clones. (
B) The screening protocol for IAC
3 [
21] involves first using a diverse library of V genes with diversified CDR3 and subsequent affinity maturation of V regions that bind to the target protein using first CDR2 randomisation and rescreening hits from the CDR3 round of screening and, if deemed necessary, repeating the process with CDR1 randomisation and rescreening hits from the CDR2 screen. Final hits are tested for their capacity to interact with the target protein in mammalian cells.
![Antibodies 02 00306 g003]()
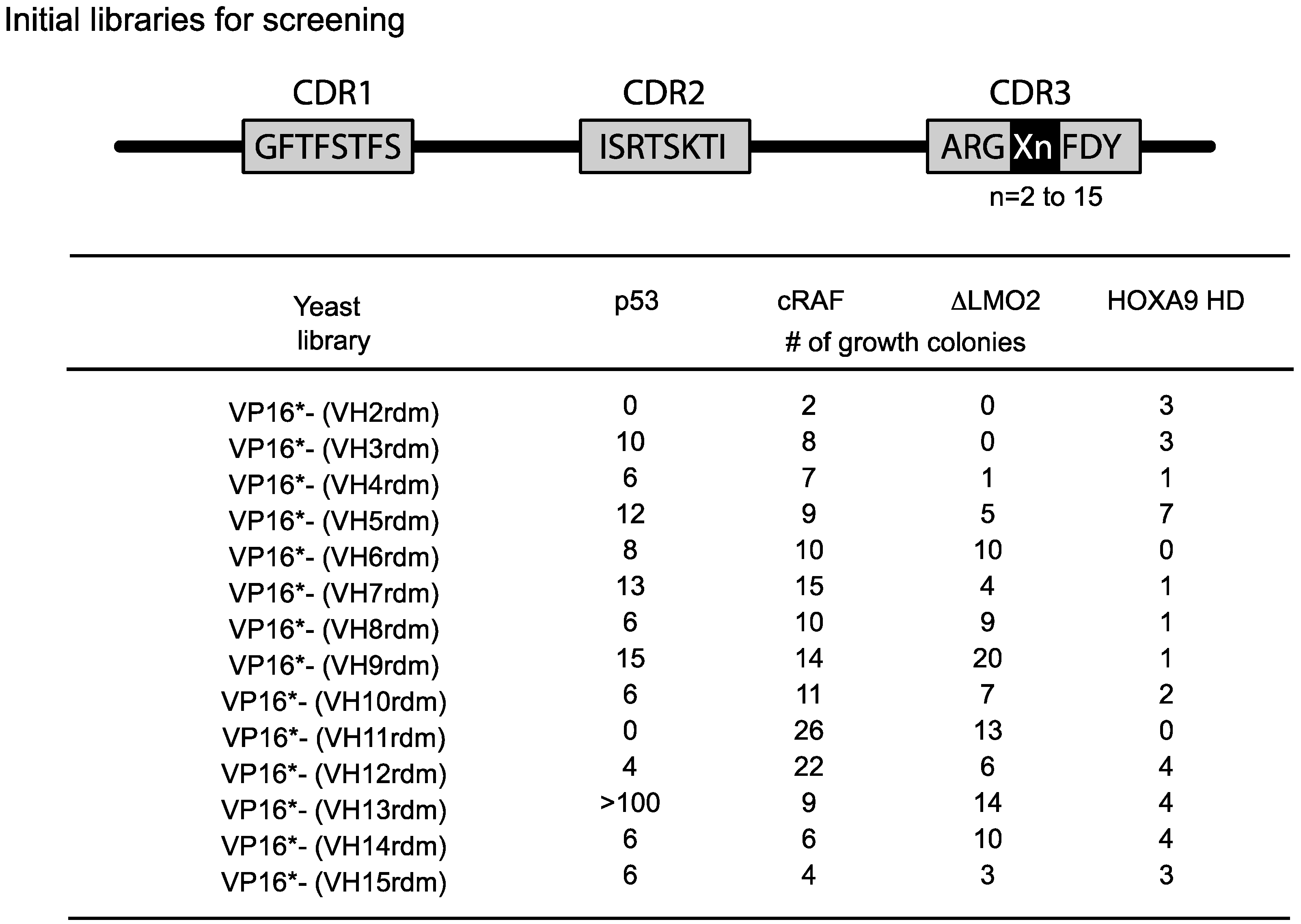
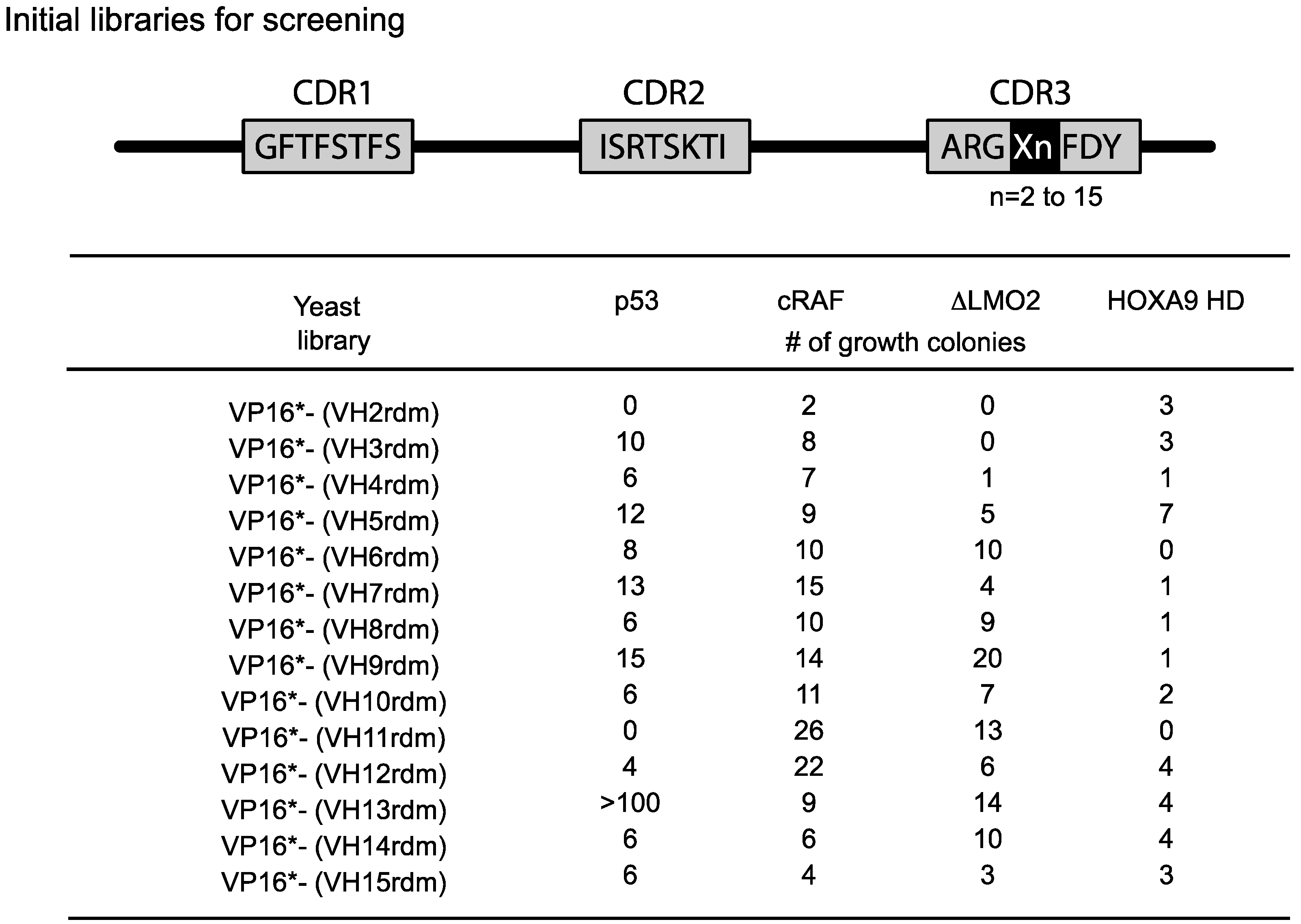
The length of the heavy chain CDR3 region naturally varies from two amino-acids to 18 or more residues due to the combinatorial joining of V-D-J genomic segments and N-region diversity. To accommodate this variability, we initially used fourteen VH diverse libraries with between 2 or 15 amino acids at the CDR3 area (
Figure 4). We obtained a variable yield of hits from screening these libraries with a range of antigens and different affinities. Recent data from deep sequencing of natural antibody repertoires have shown that human VH have CDR3 on average 13–16 in length [
22]. Accordingly, we have recently reverted to using a single diverse VH library to undertake yeast screens using 13 or 14 amino acid CDRs. Hits from these screens are affinity matured as indicated in
Figure 3 and a final examination as an indicator of “
in vivo” affinity is made by assessing the single domains in mammalian two-hybrid or functional assays in mammalian cells. Biophysical assays, such as surface plasmon resonance, are required to derive values for affinity, keeping in mind that
in vivo and
in vitro binding occur in completely dissimilar environments.
Figure 4.
Single VH domain CDR3 diverse yeast library screening data. Since the CDR3 of VH can vary from 2–15 or more residues, we prepared diversity libraries ranging from 2 amino acids to 15 randomised amino acids [
21]. These libraries have been screened in yeast reporter strains with the four target protein baits shown and various numbers of hits have been obtained. In the most recent versions of the method, we use a single library (typically VP16*-VH13rdm or 14rdm) for initial screening, prior to affinity maturation of hits. Adapted from [
6].
Figure 4.
Single VH domain CDR3 diverse yeast library screening data. Since the CDR3 of VH can vary from 2–15 or more residues, we prepared diversity libraries ranging from 2 amino acids to 15 randomised amino acids [
21]. These libraries have been screened in yeast reporter strains with the four target protein baits shown and various numbers of hits have been obtained. In the most recent versions of the method, we use a single library (typically VP16*-VH13rdm or 14rdm) for initial screening, prior to affinity maturation of hits. Adapted from [
6].
We have adopted a simple method to evaluate which of those single domains isolated by IAC3 have the binding properties to antigen and interfering with PPI. This involves genetic engineering of a vector for co-expression of target protein and its interacting partner, together with Renilla luciferase for transfection normalization (the Triplex vector [
23]), into a line of CHO cells in which a
Firefly luciferase reporter gene, with a
GAL4 DNA binding site (DBS), has been permanently integrated (CHOluc15, [
21]). The Triplex vector is illustrated in
Figure 5A. When the loaded Triplex vector is transfected into CHOluc15, interaction of protein 1 (target) with protein 2 (partner) yields a Firefly luciferase signal. A potential competitor protein (
Figure 5B) (
i.e., in this context a VH or VL single domain) can be assessed by co-transfection with the loaded Triplex vector since interaction of the competitor will diminish binding of proteins 1 and 2 and in turn reduce luciferase signal (
Figure 5C).
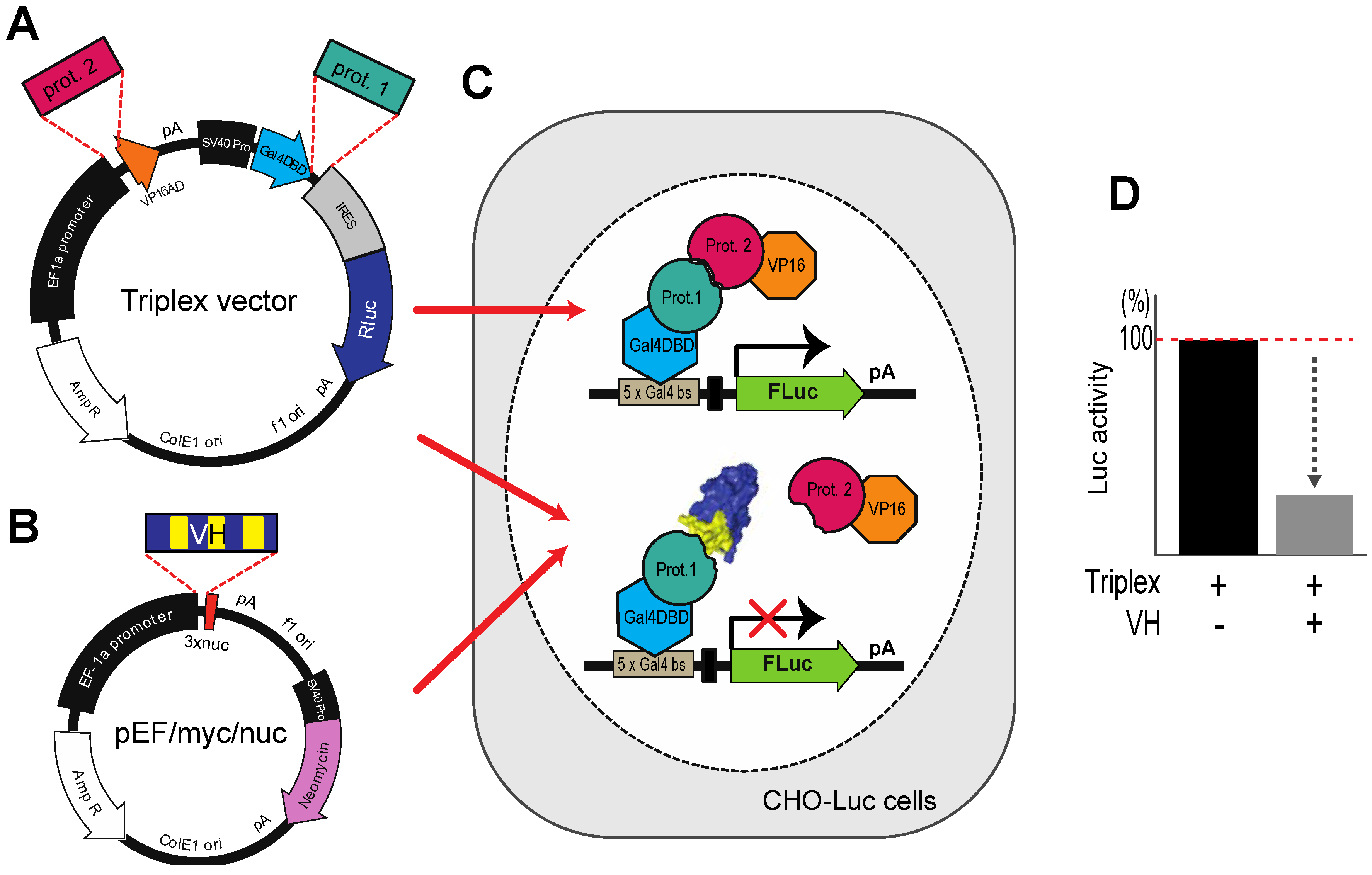
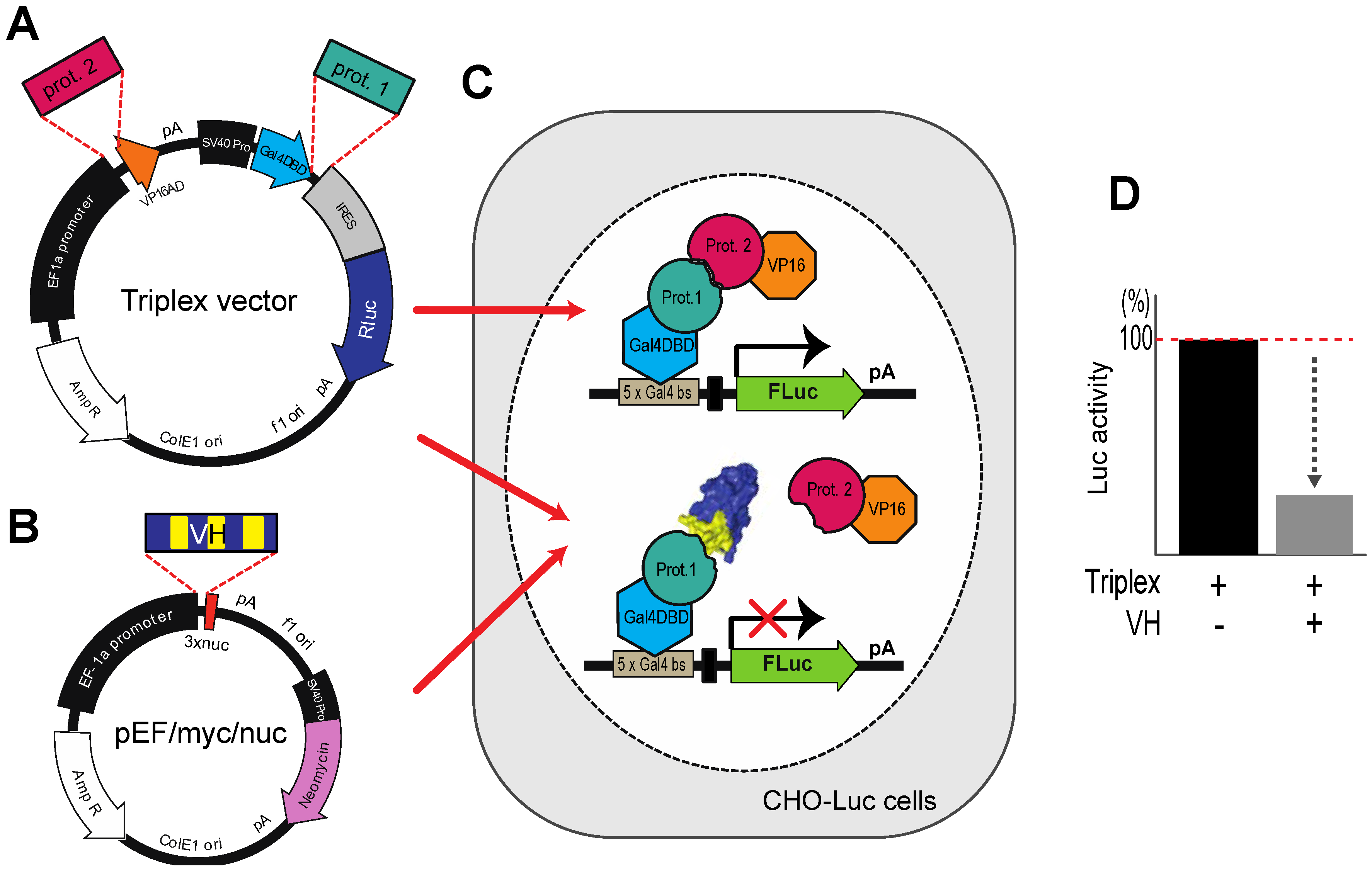
Figure 5.
The Triplex vector system for selection of antibody fragment inhibitors of PPI. (
A) The Triplex vector [
23] was engineered to co-express, on transfection in mammalian cells, two interacting proteins, one fused to a GAL4-DBD (prot. 1, e.g., target protein gene) and the other fused to VP16 AD (prot. 2, e.g. PPI partner protein gene) to bind to a
GAL4 DBS-luciferase reporter gene to express Firefly luciferase; (
B) The competitor protein (in this context VH or VL) is expressed from pEF/myc/nuc (Invitrogen) when co-transfected with the Triplex vector; (
C,
D) A stable CHO cell line (CHOluc15) has been made in which five copies of the
GAL4 DBS control the expression of Firefly luciferase. As shown in the inset panel D, transfection of a loaded Triplex vector carrying bait and prey genes will activate Firefly luciferase expression, whereas co-transfection of the loaded Triplex vector with a competitor expressing VH, diminishes luciferase signal if the VH competes with the PPI.
Figure 5.
The Triplex vector system for selection of antibody fragment inhibitors of PPI. (
A) The Triplex vector [
23] was engineered to co-express, on transfection in mammalian cells, two interacting proteins, one fused to a GAL4-DBD (prot. 1, e.g., target protein gene) and the other fused to VP16 AD (prot. 2, e.g. PPI partner protein gene) to bind to a
GAL4 DBS-luciferase reporter gene to express Firefly luciferase; (
B) The competitor protein (in this context VH or VL) is expressed from pEF/myc/nuc (Invitrogen) when co-transfected with the Triplex vector; (
C,
D) A stable CHO cell line (CHOluc15) has been made in which five copies of the
GAL4 DBS control the expression of Firefly luciferase. As shown in the inset panel D, transfection of a loaded Triplex vector carrying bait and prey genes will activate Firefly luciferase expression, whereas co-transfection of the loaded Triplex vector with a competitor expressing VH, diminishes luciferase signal if the VH competes with the PPI.
4. VH targeting Intracellular Protein-Protein Interactions as Drug Surrogates
The technologies described above have allowed us to identify VH single domains that interfere with the natural binding of RAS or LMO2 proteins [
5,
6] and we have also selected potential inhibitory VH single domains that binds to the bHLH-zipper portion of CMYC or the integrase binding domain of LEDGF (a component of the MLL complex) (unpublished).
The RAS proteins comprise a family of small GTPases that play a key role in signal transduction acting as molecular switches for a variety of important cellular processes like cell growth or survival [
24,
25]. Activating mutations of
RAS (mainly
KRAS,
NRAS or
HRAS) are involved in a very high percentage of human cancer [
26] making mutant RAS a very important potential therapeutic cancer target. The activated form of RAS transmits signals through PPI with a range of effector proteins (e.g., PI3K, RAF), most of which interact with the switch I region of GTP-bound RAS [
24] and mutated RAS is a constitutively signaling molecule. However, until recently it had not been formally demonstrated that blockade of RAS signaling could be therapeutically useful since there were no reagents that specifically target the activated form of RAS. Recently, we reported isolation of a single domain VH fragment that binds specifically to mutant RAS with high affinity [
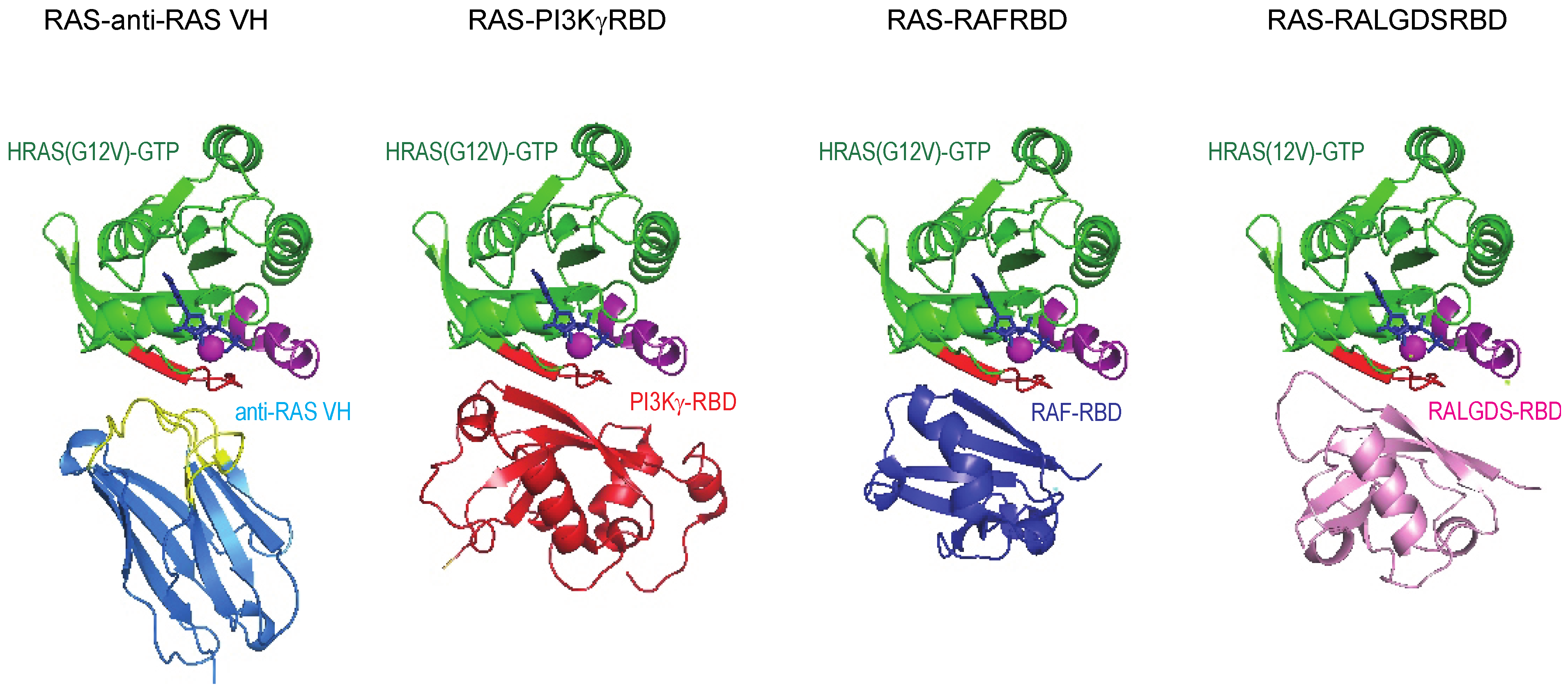
5]. The structural study showed that anti-RAS VH interacts with the switch I region of all three major RAS family members (K, H and NRAS) in an analogous fashion to the natural effector proteins and binds with affinity up to ~500 times higher than natural RAS-effector interaction (
Figure 6). Further, the anti-RAS VH does not bind to GDP-bound, inactive RAS. The massive difference in the interaction strength of the VH with RAS compared to the RAS-effectors, is one of the powerful parameters that makes working with VH as surrogates of natural interactions advantageous.
Figure 6.
Structural relationship of anti-RAS VH binding to RAS and natural effector–RAS interactions. The crystal structure of RAS complexed with anti-RAS VH (PDB, 2UZI) compared to that of RAS complexed with PI3Kγ RBD and other effectors.
Figure 6.
Structural relationship of anti-RAS VH binding to RAS and natural effector–RAS interactions. The crystal structure of RAS complexed with anti-RAS VH (PDB, 2UZI) compared to that of RAS complexed with PI3Kγ RBD and other effectors.
We have used the anti-RAS VH to demonstrate that constitutive RAS–effector interaction is necessary for neoplasia when mutant RAS is present [
27]. First by co-expression of mutant KRASG12V and anti-RAS VH, we have found complete suppression of RAS-dependent tumours in transgenic mice [
27]. Second, using human cell lines that carry mutant
RAS together with other gene mutations and an inducible form (TET-ON tetracycline inducible) of the anti-RAS
VH gene, we can show cessation of tumour growth in xenograft models.
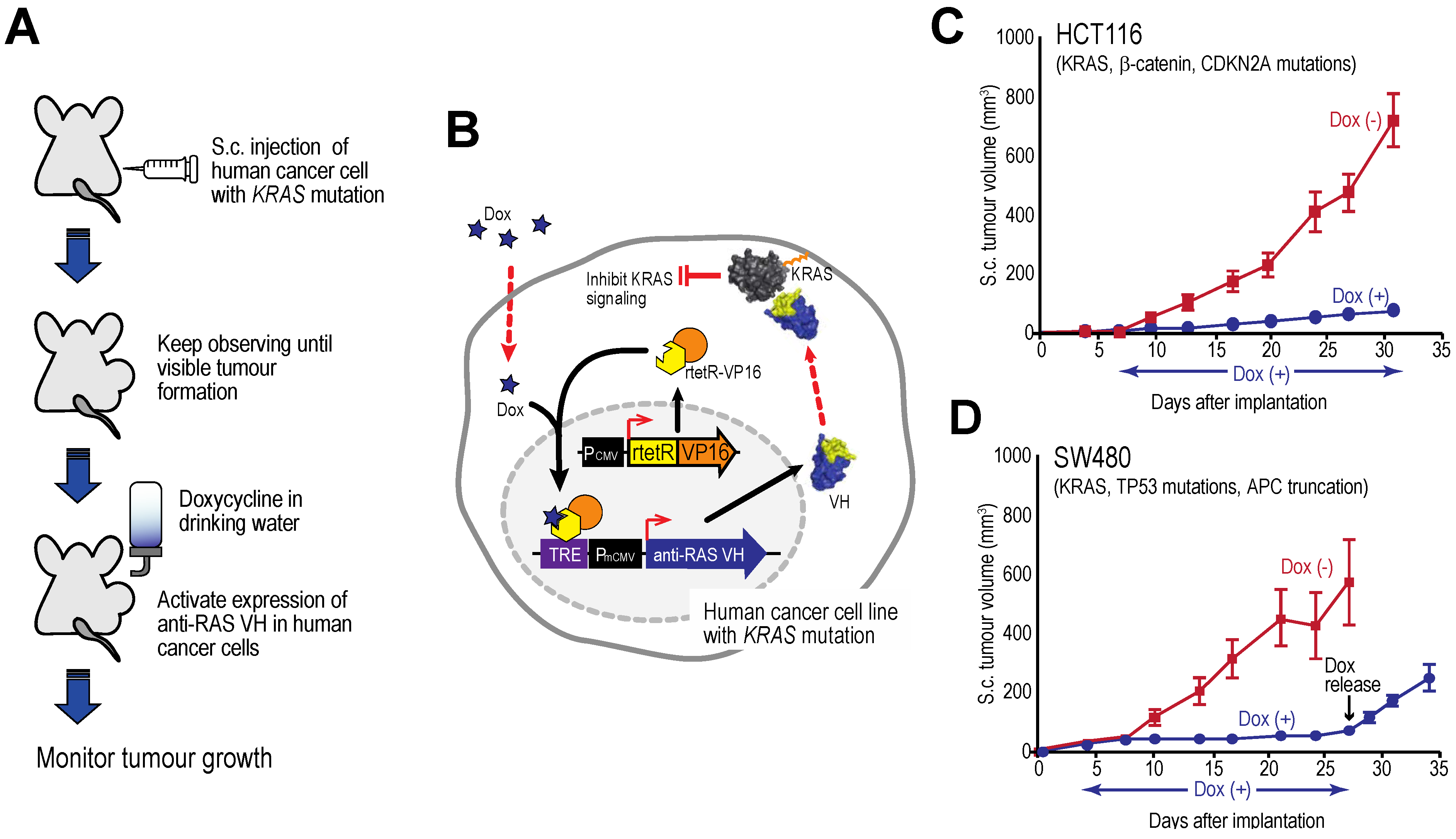
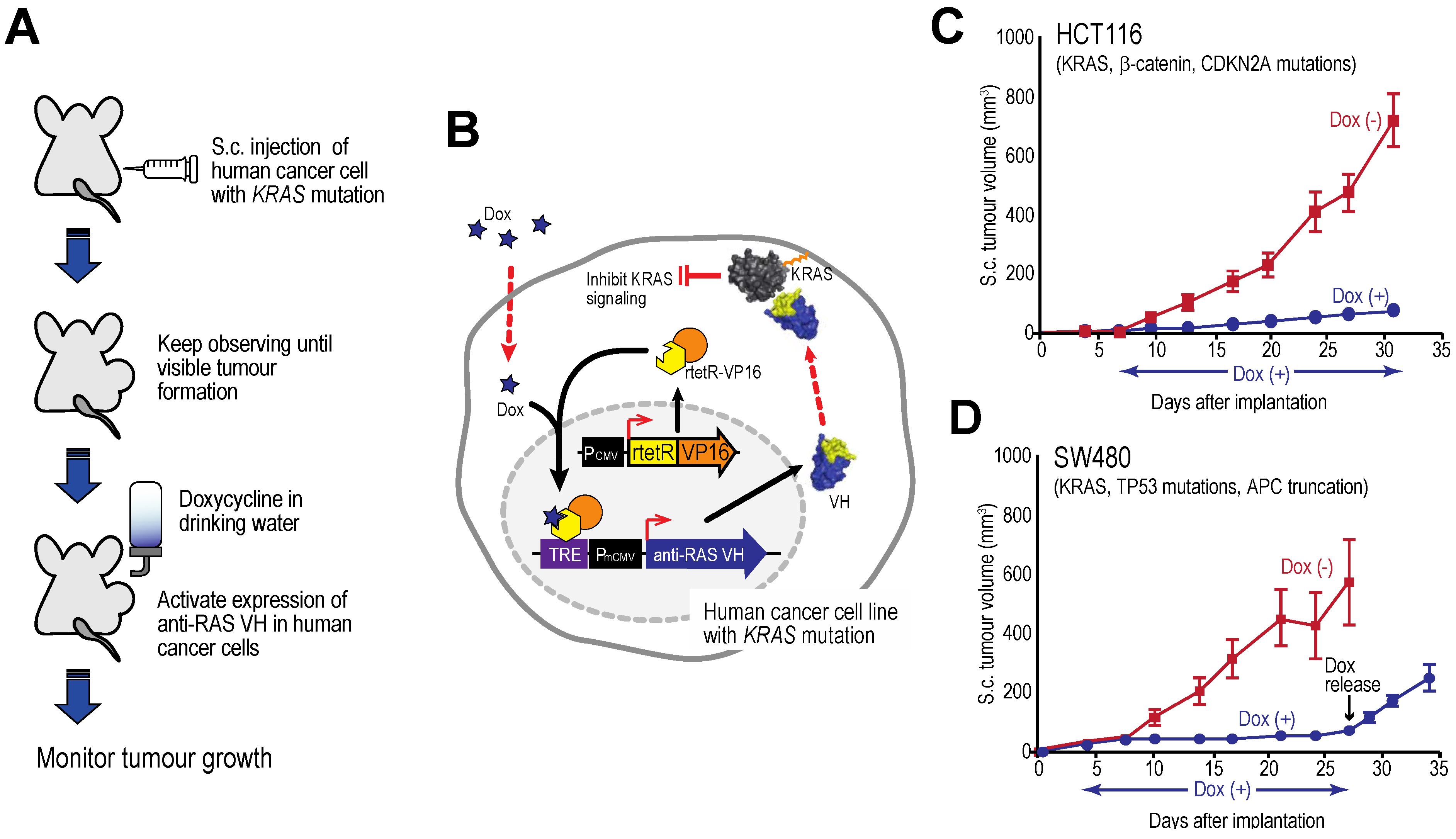
Figure 7 shows tumour growth curves for two human cell lines xenografts in immune-deficient mice [
27] showing that tumour growth is inhibited by intracellular expression of the anti-RAS VH and this cessation persists as long as the inducer is given. However, when tetracycline is withdrawn and VH expression induction is stopped, the tumours begin to grow again. The effects on the tumours are cytostatic rather than cytotoxic. A similar finding was established with an anti-LMO2 single VH domain [
6]. LMO2 is a member of the LIM-domain only protein family and is often activated in T cell acute leukemia. In a T cell transplantation assay in mice, we showed that LMO2 is necessary for overt T cell neoplasia, thereby validating this oncogenic protein as a therapeutic target. This type of experiment illustrates the potential of using single VH domains as a drug surrogate and future possible potential as anti-cancer therapeutics (see below).
Figure 7.
Tumour inhibition by anti-RAS VH. Xenograft tumour models of human cancer cells carrying KRAS mutations and a tetracycline inducible gene expressing anti-RAS VH. (A, B) The protocol comprised generating tumours in nude mice by sub-cutaneous injection of cells; when palpable tumour growth was observed (about 5 days), doxycycline was added to the drinking water to activate anti-RAS VH expression. Tumour volume was assessed over time; (C) Growth curves of HCT116 cells; (D) Growth curves of SW480 cells. In the latter case, doxycycline administration was stopped at 27 days and growth of the tumours monitored for a further 8 days. Red lines indicate doxycycline was not added; black lines indicate added doxycycline.
Figure 7.
Tumour inhibition by anti-RAS VH. Xenograft tumour models of human cancer cells carrying KRAS mutations and a tetracycline inducible gene expressing anti-RAS VH. (A, B) The protocol comprised generating tumours in nude mice by sub-cutaneous injection of cells; when palpable tumour growth was observed (about 5 days), doxycycline was added to the drinking water to activate anti-RAS VH expression. Tumour volume was assessed over time; (C) Growth curves of HCT116 cells; (D) Growth curves of SW480 cells. In the latter case, doxycycline administration was stopped at 27 days and growth of the tumours monitored for a further 8 days. Red lines indicate doxycycline was not added; black lines indicate added doxycycline.
5. Future Prospectives: Single VH Domain Macrodrug Delivery Options
Our experiments with single VH domains as drug surrogates show not only that target validation can be achieved using these high affinity reagents, but also that specific properties of target proteins involving PPI are viable drug targets. However, the single domain VH molecules are macromolecules and, as such, cannot simply penetrate cells and behave like small molecule drugs. For this reason, we call these protein ‘drugs’ macrodrugs [
10]. Although the protein drugs show promising results
in vitro, using macrodrugs is, however, likely to be restricted
in vivo for various reasons, including fast elimination due to clearance and enzymatic degradation, generation of immune response, poor efficiency and accuracy of uptake by target cells [
28]. There are, therefore, major challenges to confront in the use of macrodrugs in disease therapy. Three possible approaches for macrodrug delivery are illustrated in
Figure 8A.
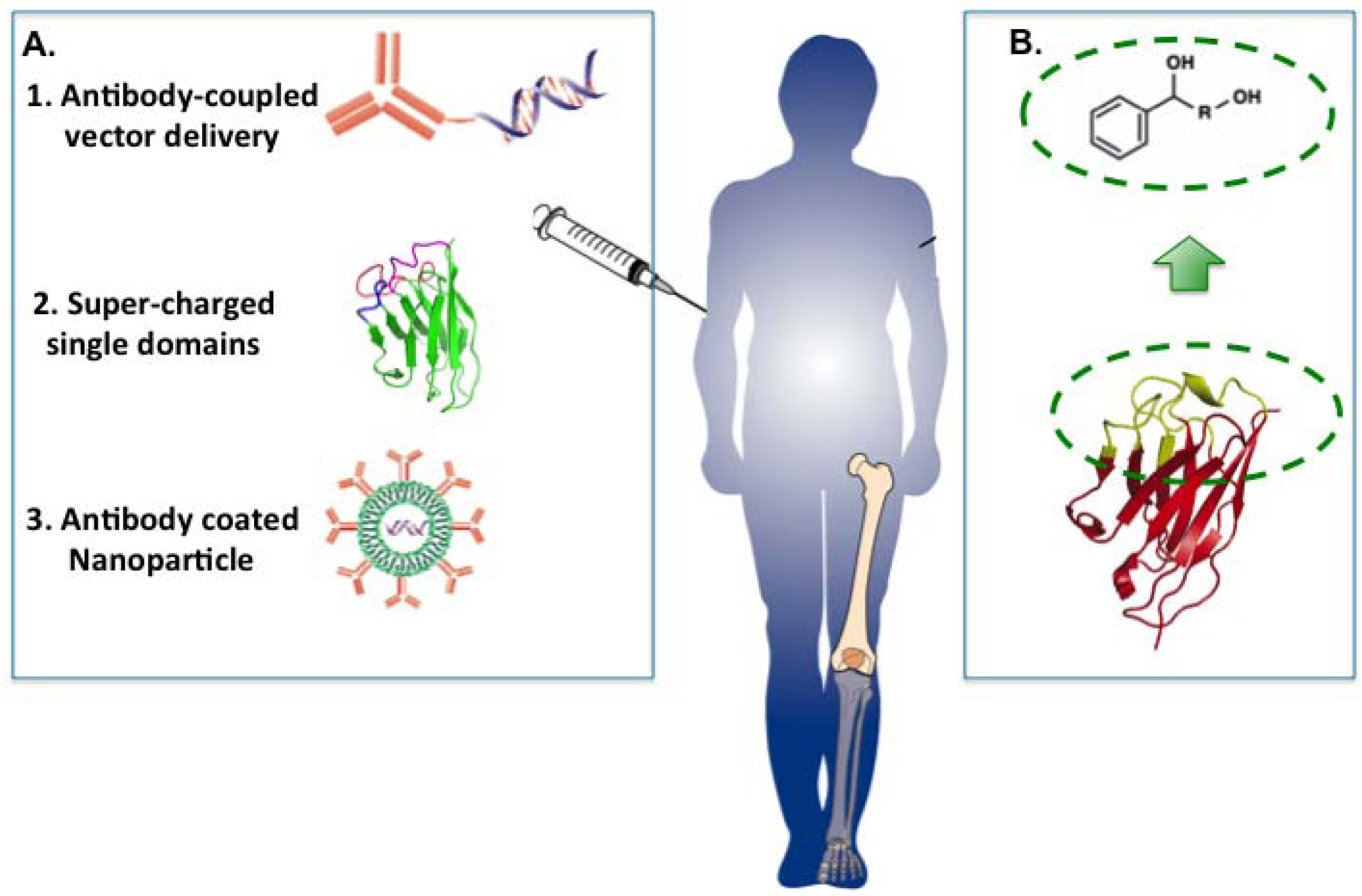
Figure 8.
Possible options for delivery of antibody fragments. There are various possible ways in which protein drugs (macrodrugs) could be introduced into cells to act on processes such as PPI inside the cell. This would currently have to involve systemic delivery into patients and localisation to affect tissue sites (e.g., joints in rheumatoid arthritis or tumours in cancer). (A) The three keys options illustrated are antibody-coupled delivery of single domain expression vectors, super-charged antibody fragments and antibody-coated nanoparticles; (B) The CDR region of the single antibody domain has a shape, and the interaction surface involved in binding can occupy up to 1,000 Å2. When anchor residues are established, it reduces this surface to a suitable size for a small molecule emulator.
Figure 8.
Possible options for delivery of antibody fragments. There are various possible ways in which protein drugs (macrodrugs) could be introduced into cells to act on processes such as PPI inside the cell. This would currently have to involve systemic delivery into patients and localisation to affect tissue sites (e.g., joints in rheumatoid arthritis or tumours in cancer). (A) The three keys options illustrated are antibody-coupled delivery of single domain expression vectors, super-charged antibody fragments and antibody-coated nanoparticles; (B) The CDR region of the single antibody domain has a shape, and the interaction surface involved in binding can occupy up to 1,000 Å2. When anchor residues are established, it reduces this surface to a suitable size for a small molecule emulator.
5.1. Antibody-Coupled Vector Delivery
Antibody-mediated delivery provides a potential method for macrodrug delivery to target cells. By using antibody that recognises a specific tumour surface antigen, the molecule carried by the antibody could be delivered to target cells specifically. This method has been tested by delivery of small interfering RNAs (siRNAs). The siRNA was delivered by protamine-antibody fusion protein of which the nucleic acid-binding protein protamine was used for loading siRNA molecules. The results indicated that siRNA was delivered by HIV envelope antibody or anti-ErbB2 single-chain antibody specifically to HIV infected cells or ErbB2-expressing cancer cells [
29,
30]. The success of antibody mediated siRNA delivery revealed the potential of cell-specific protein-expression vector delivery. Plasmid vector DNA carrying the macrodrug encoding gene and appropriate promoter could potentially be delivered and the protein could subsequently be expressed in the target cell. An antibody-mediated plasmid DNA delivery system has probable advantages in avoiding immune response and enzymatic degradation, increasing efficiency in the uptake ability and enabling the cell-specific macrodrug delivery.
5.2. Super-Charged Single Domains
Supercharged proteins are a class of protein with high net positive or negative charge [
31]. Bioactive macromolecule delivery has recently been demonstrated using supercharged proteins. These proteins could be either naturally occurring or engineered, whereby the solvent-exposed residues on the protein surface are mutated to either acidic or basic amino acids. Supercharged GFP that retains the original function performed remarkable ability as a cell-penetrating agent and enabled the functional delivery of macromolecules such as protein [
32], siRNA [
33] and plasmid DNA [
33]. Compared to commonly used protein transduction domains (such as Tat, oligo-arginine, penetratin [
34]), the supercharged protein showed enhanced efficiency in protein delivery in mammalian cells [
32,
35]. Supercharging is a possible way of creating a cell-penetrating characteristic of the antibody single domains for macrodrug delivery.
5.3. Antibody Coated Nanoparticles
Nanoparticles are particles of 100 nanometers or less in diameter and are able to overcome limitations of conventional drugs such as low solubility, stability metabolism [
36]. Nanoparticle delivery systems have been developed over the past few decades with the aim of enhancing the
in vivo properties of various anti-cancer drugs. The drug of interest can be carried by nanoparticles that are prepared with different methods to pose different characteristics for stabilizing drugs from degradation, controlling drug release and targeting for delivery.
Liposomes were the first nanoparticle formulations that applied in medicine [
37,
38]. In the past 15 years the use of liposomal nanoparticle has been extensively developed for drug delivery. Since the first clinical approved liposomal-drug formulation (Liposomal amphotericin B, AmBisome) in 1990, there are now more than 17 liposomal anticancer drugs available in clinics, with many more in various stages of development [
39,
40]. Numerous methods have been developed for coupling liposomes with antibodies or antibody fragments, which are specific to cell surface-characteristic antigens or structures [
41,
42]. Encapsulation of single domains or vectors able to express single domains would be a possible way to use nanoparticles to deliver these macrodrugs.
6. Future Prospectives: Antibody Single Domain CDRs as Templates for Drug Discovery
There are considerable difficulties with the macrodrug delivery options discussed above, not least of which being specific cell uptake and quantitative amounts of macrodrug payload delivered to elicit a therapeutic response. An alternative approach in the future could be to develop small chemical compounds or peptides that emulate the 3D structure and binding affinities of the anchor residues of the variable region CDRs. In this scenario, the single domain is critical as it provides a high affinity interaction site whose structure in contact with the target can be established to high resolution. The 3-D shape can provide the “pharmacophore” for a target and potentially be replaced by small molecules or peptides (either by library screening or by
in silico design) to identify hits and these subjected to hit to lead development by medicinal chemistry (
Figure 8B).
It should be emphasized that in the use of antibody fragments as inhibitors of PPI, there is no prior need for structural information about the PPI to be targeted. The only need is a functional interaction assay between the target and its partner protein that can be used for determination of the interference with the single domain. Further, since the determination of target structures with VH is achievable, a natural interacting partner protein need not be required at any stage. Thus this approach is generally applicable. Indeed, in the interactome studies identifying networks of interactions, often the structural basis is not known. Furthermore, the high affinity achievable with IAC
3-derived single domains is a great practical advantage allowing for mutagenesis studies to identify anchor residues [
43] and competition assays for compounds that bind to target proteins.
The scheme illustrated in
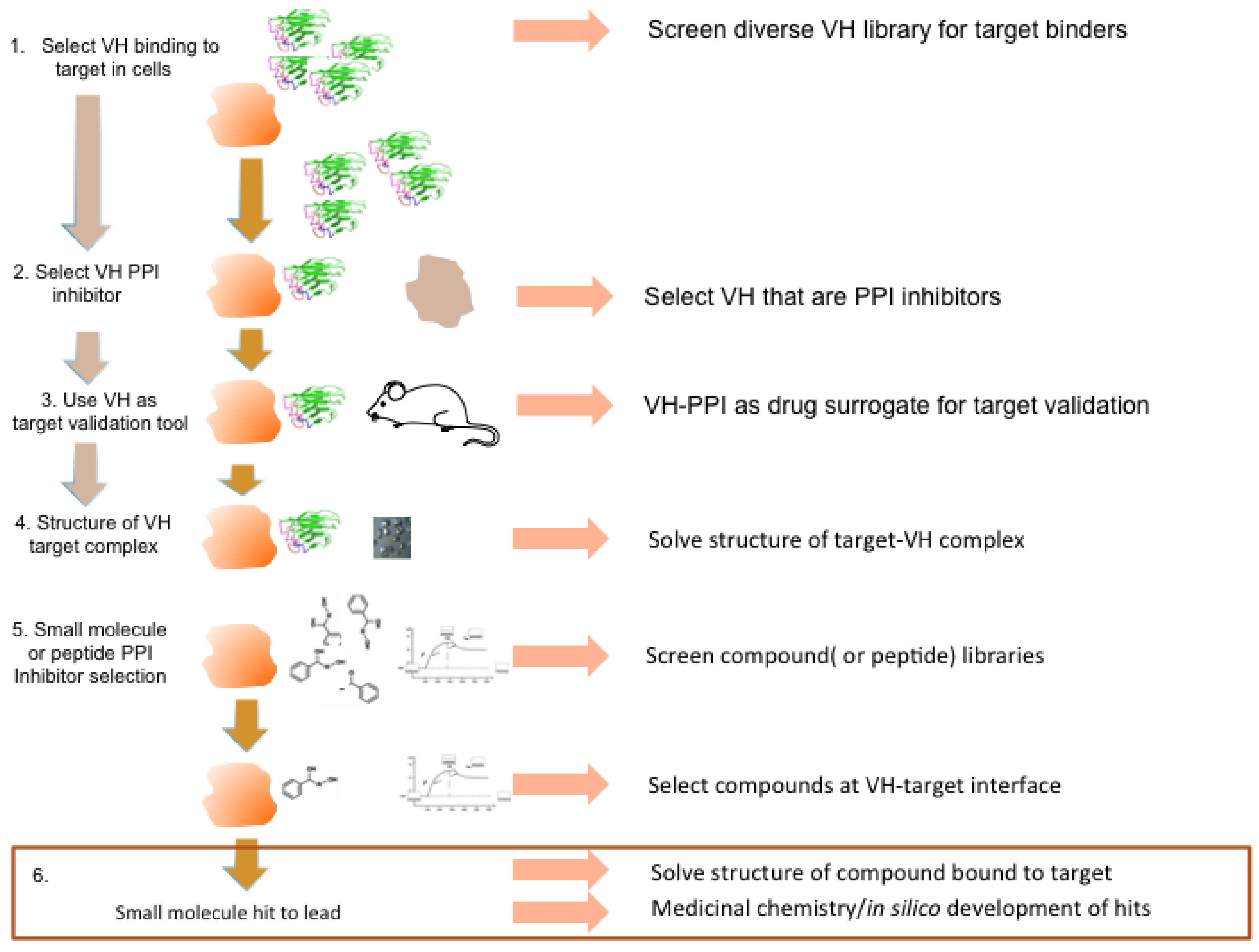
Figure 9 represents a general strategic approach to development of small molecules that can have therapeutic consequences. Finally, the structure of antibody fragments bound to targets, rather than the structure of target–natural binding partner interactions, gives a unique topological starting point for drug design. This can be exploited for chemical shapes, or peptides can be designed using the antibody fragment CDR shape to produce inhibitors of PPI in many intracellular contexts.
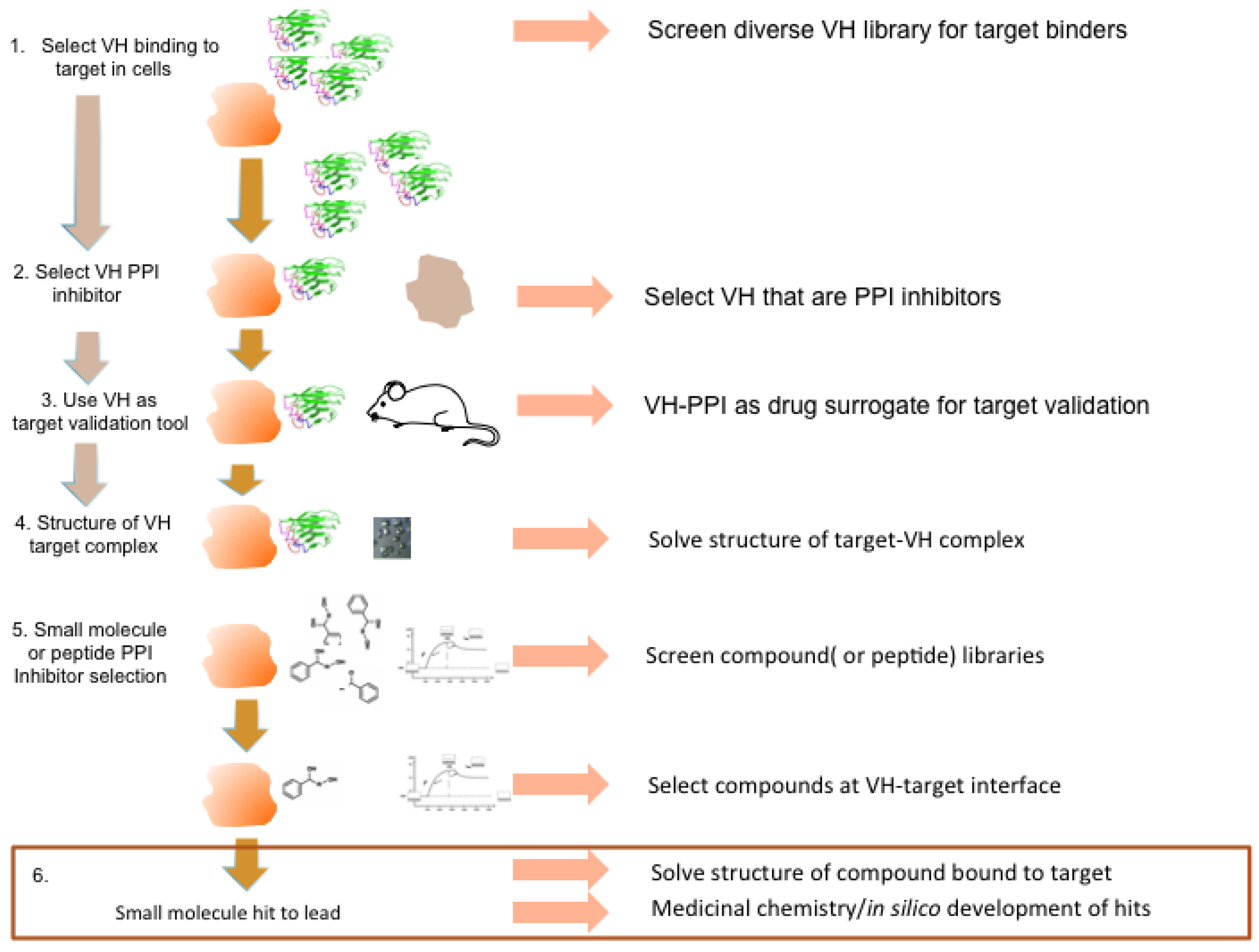
Figure 9.
Potential antibody fragment pipeline for transporting PPI inside cells. The strategy encompassed in our review to produce macrodrugs or small molecule drugs to target particular PPIs essentially has two phases and requires only the knowledge that two proteins interact rather than the use of any structural data. The first phase (steps 1–3) involves isolating and characterizing a single domain capable of inhibiting the PPI of interest and using this in preclinical models to validate the target in the specified clinical indication. The latter also validates the location as a drug target. The second phase (steps 4–6) involves structural determination of the target-VH complex and isolating chemical compounds (or peptides) that bind to the target in the target validated region where the V-region CDRs bind. The structure of the V-region CDR anchor residues can be used to define hits.
Figure 9.
Potential antibody fragment pipeline for transporting PPI inside cells. The strategy encompassed in our review to produce macrodrugs or small molecule drugs to target particular PPIs essentially has two phases and requires only the knowledge that two proteins interact rather than the use of any structural data. The first phase (steps 1–3) involves isolating and characterizing a single domain capable of inhibiting the PPI of interest and using this in preclinical models to validate the target in the specified clinical indication. The latter also validates the location as a drug target. The second phase (steps 4–6) involves structural determination of the target-VH complex and isolating chemical compounds (or peptides) that bind to the target in the target validated region where the V-region CDRs bind. The structure of the V-region CDR anchor residues can be used to define hits.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}