Improving the Therapeutic Potential of Human Granzyme B for Targeted Cancer Therapy

Abstract
:1. Introduction
2. Generation of hCFPs
2.1. Humanization of the Tumor-Targeting Component
2.2. Humanization of the Cell-Death Inducing Component
3. Granzyme B in Targeted Cancer Therapy: Advantages and Challenges
3.1. Resistance of Tumor Cells to the Induction of Apoptosis
3.2. Cell Death Pathways Induced by Granzyme B

{kind=link}
{kind=link}
| CFP (expression system) | Indication | Target receptor / antigen | Target cells | IC50 values | PI-9 expression reported from others | Structural hallmarks | Reference | |
|---|---|---|---|---|---|---|---|---|
| Gb-H22 (scFv) (HEK293T) | AML, CD64+ malignancies | CD64 | U937 | 1.7–17 nM | no * | In vitro activation via EK | [78,79] | |
| AML cells (ex vivo) | -- | |||||||
| HL60 | ~ 4–7 nM | no * | ||||||
| GrB-scFvMEL (E.coli) | Melanoma | gp240 | A375-M | ~ 20 nM | n.d. | In vitro activation via EK | [80] | |
| GrB-scFvMEL (E.coli) | Melanoma | gp240 | A375-M (in vivo) | ~30 nM | n.d. | In vitro activation via EK | [81] | |
| MEL-526 | ~50 nM | n.d. | ||||||
| TXM-18L | ~150 nM | n.d. | ||||||
| GrB-TGFα | Breast carcinoma | EGFR, | MDA-MB468 | 0.25 nM | n.d. | In vivo activation by Kex2 | [82] | |
| GrB-scFv (FRP5) | ErbB2 (Her2) | 0.29 nM | ||||||
| (P.pastoris) | (+chloroquin for both) | |||||||
| B3-GzmB*, GzmB-CD8* | Breast carcinoma | Lewis Y antigen | SK-BR3 | 98 nM, 1595 nM | Yes [83] | Polyionic adapters between GrB and dsFv-B3 | [85] | |
| (E.coli) | Yes, after induction by estrogens [84] | |||||||
| MCF-7 (MCF-7casp3) | 140 nM (35 nM), 198 nM (394 nM) | |||||||
| GrB-VEGF121 | Tumor vascular endothelial cells | Vascular endothelial growth factor | PAE/FLK-1 | ~10 nM | n.d. | In vitro activation via EK | [86] | |
| (E.coli) | PAE/FLT-1 | -- | n.d. | |||||
| ImmunoGrB-PEAII | Breast and ovarian carcinoma | Her2 | SK-BR3 (in vivo) | Not applicable since supernatant was used | Yes [83] | PEAII translocation motif to allow endogenous furin activation: e23sFv-PEAII-GrB | [87] | |
| (HeLa, Jurkat) | n.d. | |||||||
| SKOV-3 | ||||||||
| ImmunoGrB- Fpe/Fdt/R9 # | Breast, gastric and hepatocellular carcinoma | Her2 | SK-BR3 | Not applicable since supernatant was used | Yes [83] | Introduction of novel furin cleavable sites between e23Fv and GrB (compare above) | [88] | |
| n.d. | ||||||||
| (HeLa, Jurkat) | SGC-7901 (in vivo) | n.d. | ||||||
| AGS Hep G2 | n.d. | |||||||
| GrB-YCG | Ovarian, breast, endometrial and prostate carcinoma | hLHR | MA-10 (murine) | 0.16 µM | No PI-9 expected in murine cell line | In vitro activation via EK prior to use | [89] | |
| (insect Sf9 cells) | MCF-7 | -- | ||||||
| PC-3 | -- | |||||||
3.3. Heterologous Expression of Active Granzyme B
3.4. Clinical Limitations of Granzyme B
| Cell line | Cell type | PI-9 expression | Reference |
|---|---|---|---|
| L3.6pl | Pancreatic carcinoma | + | [79] |
| L428 | Hodgkin lymphoma | + | [79] |
| L1236 | Hodgkin lymphoma | + | [79] |
| L540 | Hodgkin lymphoma | - | [79] |
| K562 | Chronic myeloid leukemia in blast crisis | + | [110] |
| A431 | Epidermoid carcinoma | - | [79] |
| Jurkat | T cell leukemia | - | [110] |
| PT45 | Pancreatic adenocarcinoma | - | [79] |
| Kasumi 1 | Acute myeloid leukemia | (+) | [79] |
| Karpas 299 | T cell lymphoma | - | [79] |
| HL60 | Acute myeloid leukemia | - | [79] |
| U937 | Histiocytic lymphoma | - | [79] |
| Cancer type | Detection | Hallmarks | Reference |
|---|---|---|---|
| Lung cancer | In vitro (cell lines) | PI-9 highly expressed in lung cancer cell lines | [111] |
| In vivo (primary cancer cells) | PI-9 expression was increased in primary lung cancer cells and significantly correlated with cancer stage (dependent on granzyme B expression of CTLs) | ||
| Prostate cancer | In vitro (cell lines → qPCR and flow cytometry) | PI-9 expression in cell lines PC3 and DU-145 | [112] |
| In vivo (prostate tumor tissue → qPCR and immunohistochemistry) | PI-9 expression is up-regulated in pre-cancerous states, which is dysregulated in later stages whereas it remains in some tumors (pilot study) | ||
| Non-small cell lung carcinoma cells (NSCLCs) and tissues | In vitro (cell lines → RT-PCR and western blot analysis) | Strong PI-9 expression in 6 and low in 4 of 10 cell lines on mRNA level correlating with detection on protein level | [113] |
| In vivo (lung tissue samples from biopsies → RT-PCR) | PI-9 mRNA and protein expression in all of 150 patients at variable levels in NSCLC cells and tumors, the less differentiated lung adenocarcinomas showed significantly higher expression of PI-9 mRNA as compared to the well-differentiated tumors | ||
| Breast cancer | In vitro (cell line MCF-7 → western blot and QRT-PCR) | PI-9 expression is induced by estrogens and depends on an interplay between estrogens, estrogen receptor and EGF/EGFR | [84] |
| In vivo (MCF-7 xenograft mouse model) | Induction of PI-9 by the estrogen “genistein” | [114] | |
| Stage III and IV melanoma | In vitro (cell lines → western blot of cell lysates) | PI-9 expression in 6 of 14 melanoma cell lines | [115] |
| In vivo (paraffin embedded tissue of patients → immunohistochemistry) | PI-9 expression in 21 of 26 cases of primary melanoma and in 22 of 28 metastases | ||
| After categorizing in respect to percentage of PI-9 expressing tumor cells (+ if > 50% of cells expressed PI-9, −if < 50% of cells expressed PI-9) 15 of 26 primary tumors and 12 of 28 metastases were + | |||
| PI-9 expression in melanoma metastases correlates with poor clinical outcome following active specific immunotherapy (ASI) therapy of stage III and IV melanoma patients | |||
| Nasopharyngeal carcinoma | In vivo (formalin-fixed, paraffin-embedded tumor biopsies → immunohisto-chemistry) | PI-9 expression in 3 of 43 cases | [116] |
| Presence of many tumor infiltrating activate CTLs within patient biopsies is related to bad clinical outcome | |||
| Melanoma, breast, cervical and colon carcinoma | In vitro (cell lines → PCR) | PI-9 expression in a subset of determined tumor lines (e.g., MCF-7, SK-BR3) | [83] |
| In vivo (primary colon carcinoma cells → PCR) | PI-9 expression in 2 of 4 primary surgical specimens |
4. Bioengineering of Granzyme B
4.1. Granzyme B Variants Insensitive to PI-9—Motivation and Strategies
4.1.1. Expression of PI-9 in Different Cell Types
| Cancer type | Detection | Hallmarks | Reference |
|---|---|---|---|
| Classical Hodgkin lymphoma | In vivo (lymph node sections → gene expression via microarrays) | PI-9 expression determined in 6 EBV+ and 10 EBV- cases: EBV+ are PI-9+, EBV- PI-9- | [136] |
| Leukemia | In vitro (cell lines → western blot analysis) | PI-9 expression in 4 of 6 cell lines | [137] |
| Ex vivo (monocytes from peripheral blood) | PI-9 expression in 0 of 2 ALL cases, 3 of 4 AML cases, 2 of 3 CLL cases | ||
| NK/ T cell lymphoma | In vivo (deparaffinized tissue sections of patient biopsies → immunohisto-chemistry) | PI-9 expression in 26 of 39 cases | [138] |
| PI-9 expression level was heterogeneous from case to case with clusters of negative cells suggesting the emergence of PI-9 down-regulated subclones associated with aggressiveness and invasive potential | |||
| High levels of PI-9 associated with favorable clinical outcome | |||
| Lymphomas | In vitro (cell lines → flow cytometry) | PI-9 expression in 10 of 18 lymphoma cell lines (e.g., K562) | [110] |
| Ex vivo (isolation of monocytes from peripheral blood → flow cytometry) | PI-9 expression in 9 of 14 primary lymphomas | ||
| Using highly activated CL in vitro no inhibition of the perforin-dependent cytotoxic pathway has been observed. | |||
| ALCL (anaplastic large cell lymphoma) | In vivo (biopsies of systemic ALCL patients → mmunohistochemistry) | PI-9 expression in 6 of 45 cases (percentages of PI-9+ cells ranged from 5% to 75%), primarily found in tumors harboring many Gb+ infiltrating CTLs | [135] |
| High numbers of PI-9+ tumor cells predict resistance to chemotherapy-induced apoptosis and unfavorable outcome | |||
| Different non-Hodgkin and Hodgkin lymphomas | In vivo (formalin-fixed, paraffin-embedded tumor biopsies → immunohisto-chemistry) | Sub-division into 5 categories depending on percentage of PI-9 positive cells | [139] |
| PI-9 expression in neoplastic cells in 36 of 92 cases of T-cell lymphoma, 20 of 75 of B-cell lymphoma and 6 of 57 of Hodgkin lymphoma (expression varied between 5 and 100 %) | |||
| For further differentiation of PI-9 expression and lymphoma type see reference. |
4.1.2. Downregulation of PI-9 Expression and Activity
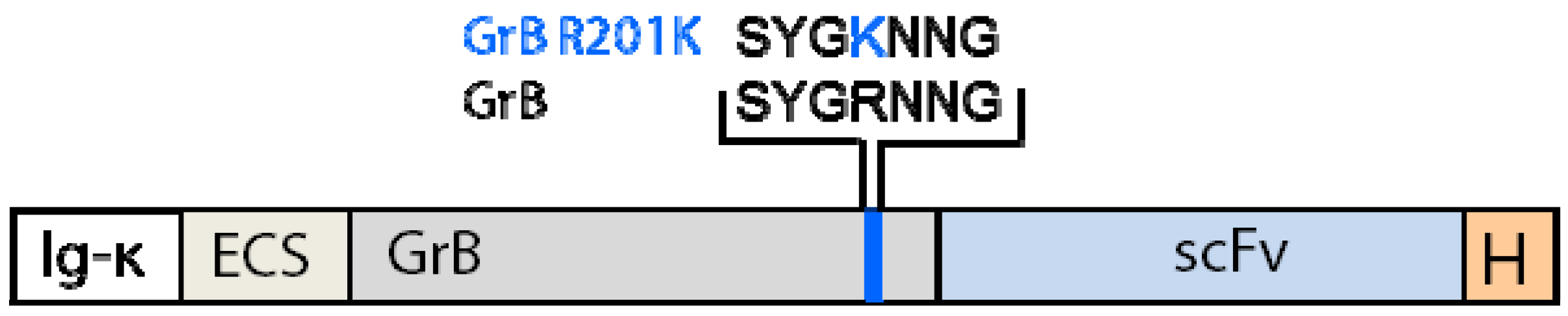
4.1.3. Generation of Granzyme B Variants Insensitive to PI-9
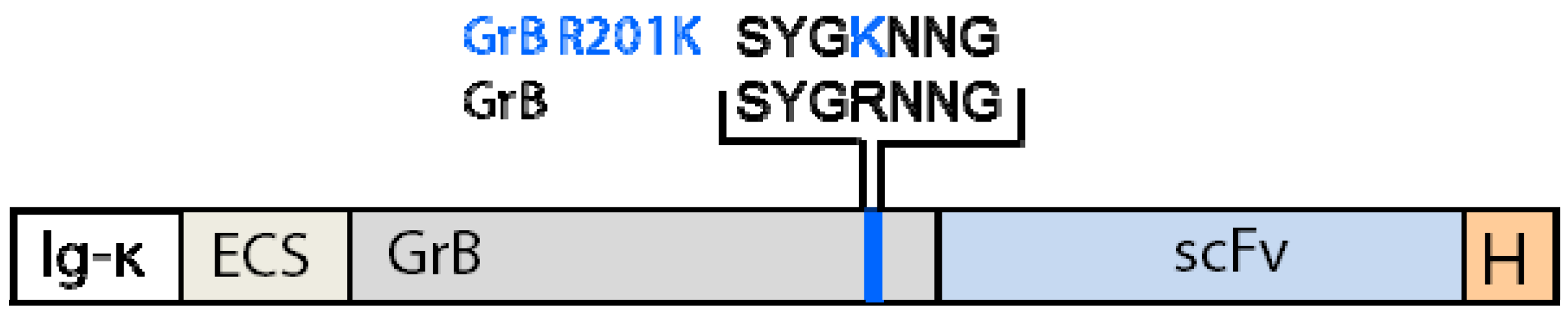
4.2. Reduction of Off-Target Effects
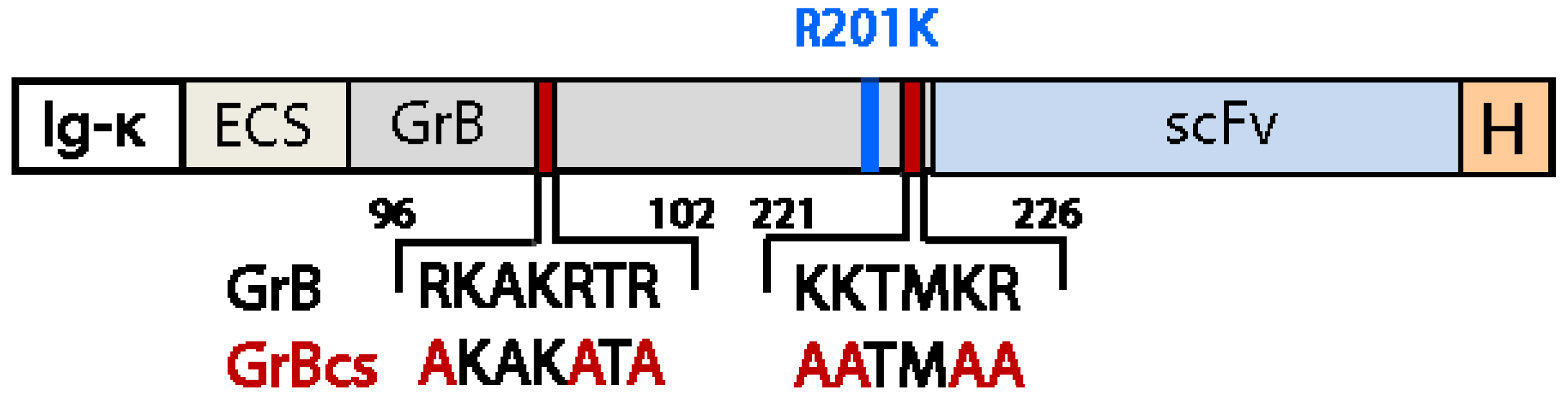
5. Effective Endosomal Release is Facilitated by Synthetic, Multifunctional Adapters
- The ECP is recognized by an endogenous endosomal enzyme, which processes the CFP following receptor-mediated endocytosis, thus exposing the MTP.
- Only following the endosomal cleavage of the ECP, the MTP promotes translocation of the effector molecule across the membrane. Since the activation of the MTP is restricted to the endosomes, non-specific uptake based on the interaction between the MTP and the membranes of bystander cells is significantly reduced.
- Once in the cytosol, cleavage of the CCP releases the active enzyme (potentially facilitating its activity by avoiding steric hindrance or conformational changes).In addition, removing the MTP prevents further undesirable membrane translocation and therefore the potential non-specific impact of the effector molecule on surrounding, healthy cells.
6. Conclusions
Acknowledgments
References and Notes
- Klebanoff, C.A.; Acquavella, N.; Yu, Z.Y.; Restifo, N.P. Therapeutic cancer vaccines: Are we there yet? Immunol. Rev. 2011, 239, 27–44. [Google Scholar] [CrossRef]
- Kreitman, R.J. Immunotoxins for targeted cancer therapy. AAPS J. 2006, 8, E532–E551. [Google Scholar] [CrossRef]
- Vanneman, M.; Dranoff, G. Combining immunotherapy and targeted therapies in cancer treatment. Nat. Rev. Cancer 2012, 12, 237–251. [Google Scholar] [CrossRef]
- Waldmann, T.A. Immunotherapy: Past, present and future. Nat. Med. 2003, 9, 269–277. [Google Scholar] [CrossRef]
- Scott, A.M.; Allison, J.P.; Wolchok, J.D. Monoclonal antibodies in cancer therapy. Cancer Immun. 2012, 12, 14. [Google Scholar]
- Reichert, J.M. Antibody-based therapeutics to watch in 2011. MAbs 2011, 3, 76–99. [Google Scholar] [CrossRef]
- Schrama, D.; Reisfeld, R.A.; Becker, J.C. Antibody targeted drugs as cancer therapeutics. Nat. Rev. Drug Discov. 2006, 5, 147–59. [Google Scholar] [CrossRef]
- Carter, P. Improving the efficacy of antibody-based cancer therapies. Nat. Rev. Cancer 2001, 1, 118–129. [Google Scholar] [CrossRef]
- Rybak, S.M.; Hoogenboom, H.R.; Meade, H.M.; Raus, J.C.; Schwartz, D.; Youle, R.J. Humanization of immunotoxins. Proc. Natl. Acad. Sci. USA 1992, 89, 3165–3169. [Google Scholar]
- Natsume, A.; Niwa, R.; Satoh, M. Improving effector functions of antibodies for cancer treatment: Enhancing ADCC and CDC. Drug Des. Devel. Ther. 2009, 3, 7–16. [Google Scholar]
- Carter, P.J.; Senter, P.D. Antibody-drug conjugates for cancer therapy. Cancer J. 2008, 14, 154–169. [Google Scholar] [CrossRef]
- Lambert, J.M. Drug-conjugated monoclonal antibodies for the treatment of cancer. Curr. Opin. Pharmacol. 2005, 5, 543–549. [Google Scholar] [CrossRef]
- Younes, A.; Yasothan, U.; Kirkpatrick, P. Brentuximab vedotin. Nat. Rev. Drug Discov. 2012, 11, 19–20. [Google Scholar] [CrossRef]
- Bross, P.F.; Beitz, J.; Chen, G.; Chen, X.H.; Duffy, E.; Kieffer, L.; Roy, S.; Sridhara, R.; Rahman, A.; Williams, G.; et al. Approval summary: Gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin. Cancer Res. 2001, 7, 1490–1496. [Google Scholar]
- Kreitman, R.J. Recombinant toxins for the treatment of cancer. Curr. Opin. Mol. Ther. 2003, 5, 44–51. [Google Scholar]
- Allen, T.M. Ligand-targeted therapeutics in anticancer therapy. Nat. Rev. Cancer 2002, 2, 750–763. [Google Scholar] [CrossRef]
- Pavlinkova, G.; Beresford, G.W.; Booth, B.J.; Batra, S.K.; Colcher, D. Pharmacokinetics and biodistribution of engineered single-chain antibody constructs of MAb CC49 in colon carcinoma xenografts. J. Nucl. Med. 1999, 40, 1536–1546. [Google Scholar]
- Pirker, R.; FitzGerald, D.J.; Willingham, M.C.; Pastan, I. Enhancement of the activity of immunotoxins made with either ricin A chain or Pseudomonas exotoxin in human ovarian and epidermoid carcinoma cell lines. Cancer Res. 1988, 48, 3919–3923. [Google Scholar]
- Barth, S.; Huhn, M.; Matthey, B.; Schnell, R.; Tawadros, S.; Schinkothe, T.; Lorenzen, J.; Diehl, V.; Engert, A. Recombinant anti-CD25 immunotoxin RFT5(SCFV)-ETA' demonstrates successful elimination of disseminated human Hodgkin lymphoma in SCID mice. Int. J. Cancer 2000, 86, 718–724. [Google Scholar] [CrossRef]
- Barth, S.; Huhn, M.; Matthey, B.; Tawadros, S.; Schnell, R.; Schinkothe, T.; Diehl, V.; Engert, A. Ki-4(scFv)-ETA', a new recombinant anti-CD30 immunotoxin with highly specific cytotoxic activity against disseminated Hodgkin tumors in SCID mice. Blood 2000, 95, 3909–3914. [Google Scholar]
- Schnell, R.; Vitetta, E.; Schindler, J.; Borchmann, P.; Barth, S.; Ghetie, V.; Hell, K.; Drillich, S.; Diehl, V.; Engert, A. Treatment of refractory Hodgkin's lymphoma patients with an anti-CD25 ricin A-chain immunotoxin. Leukemia 2000, 14, 129–135. [Google Scholar] [CrossRef]
- Kreitman, R.J. Recombinant immunotoxins for the treatment of chemoresistant hematologic malignancies. Curr. Pharm. Des. 2009, 15, 2652–2664. [Google Scholar] [CrossRef]
- Kreitman, R.J.; Wilson, W.H.; Bergeron, K.; Raggio, M.; Stetler-Stevenson, M.; FitzGerald, D.J.; Pastan, I. Efficacy of the anti-CD22 recombinant immunotoxin BL22 in chemotherapy-resistant hairy-cell leukemia. N. Engl. J. Med. 2001, 345, 241–247. [Google Scholar] [CrossRef]
- Frankel, A.E.; Powell, B.L.; Hall, P.D.; Case, L.D.; Kreitman, R.J. Phase I trial of a novel diphtheria toxin/granulocyte macrophage colony-stimulating factor fusion protein (DT388GMCSF) for refractory or relapsed acute myeloid leukemia. Clin. Cancer Res. 2002, 8, 1004–1013. [Google Scholar]
- Szatrowski, T.P.; Dodge, R.K.; Reynolds, C.; Westbrook, C.A.; Frankel, S.R.; Sklar, J.; Stewart, C.C.; Hurd, D.D.; Kolitz, J.E.; Velez-Garcia, E.; et al. Lineage specific treatment of adult patients with acute lymphoblastic leukemia in first remission with anti-B4-blocked ricin or high-dose cytarabine: Cancer and Leukemia Group B Study 9311. Cancer 2003, 97, 1471–1480. [Google Scholar] [CrossRef]
- Pai, L.H.; Pastan, I. Clinical trials with Pseudomonas exotoxin immunotoxins. Curr. Top. Microbiol. Immunol. 1998, 234, 83–96. [Google Scholar]
- Pastan, I.; Hassan, R.; Fitzgerald, D.J.; Kreitman, R.J. Immunotoxin therapy of cancer. Nat. Rev. Cancer 2006, 6, 559–565. [Google Scholar] [CrossRef]
- Pastan, I.; Hassan, R.; FitzGerald, D.J.; Kreitman, R.J. Immunotoxin treatment of cancer. Annu. Rev. Med. 2007, 58, 221–237. [Google Scholar] [CrossRef]
- Madhumathi, J.; Verma, R.S. Therapeutic targets and recent advances in protein immunotoxins. Curr. Opin. Microbiol. 2012, 15, 300–309. [Google Scholar] [CrossRef]
- Becker, N.; Benhar, I. Antibody-Based Immunotoxins for the Treatment of Cancer. Antibodies 2012, 1, 39–69. [Google Scholar] [CrossRef]
- Dang, N.H.; Pro, B.; Hagemeister, F.B.; Samaniego, F.; Jones, D.; Samuels, B.I.; Rodriguez, M.A.; Goy, A.; Romaguera, J.E.; McLaughlin, P.; et al. Phase II trial of denileukin diftitox for relapsed/refractory T-cell non-Hodgkin lymphoma. Br. J. Haematol. 2007, 136, 439–447. [Google Scholar] [CrossRef]
- Mathew, M.; Verma, R.S. Humanized immunotoxins: A new generation of immunotoxins for targeted cancer therapy. Cancer Sci. 2009, 100, 1359–1365. [Google Scholar] [CrossRef]
- Tur, M.K.; Neef, I.; Jager, G.; Teubner, A.; Stocker, M.; Melmer, G.; Barth, S. Immunokinases, a novel class of immunotherapeutics for targeted cancer therapy. Curr. Pharm. Des. 2009, 15, 2693–2699. [Google Scholar] [CrossRef]
- Huhn, M.; Sasse, S.; Tur, M.K.; Matthey, B.; Schinkothe, T.; Rybak, S.M.; Barth, S.; Engert, A. Human angiogenin fused to human CD30 ligand (Ang-CD30L) exhibits specific cytotoxicity against CD30-positive lymphoma. Cancer Res. 2001, 61, 8737–8742. [Google Scholar]
- Rosenblum, M.G.; Barth, S. Development of novel, highly cytotoxic fusion constructs containing granzyme B: Unique mechanisms and functions. Curr. Pharm. Des. 2009, 15, 2676–2692. [Google Scholar] [CrossRef]
- von Minckwitz, G.; Harder, S.; Hovelmann, S.; Jager, E.; Al-Batran, S.E.; Loibl, S.; Atmaca, A.; Cimpoiasu, C.; Neumann, A.; Abera, A.; et al. Phase I clinical study of the recombinant antibody toxin scFv(FRP5)-ETA specific for the ErbB2/HER2 receptor in patients with advanced solid malignomas. Breast Cancer Res. 2005, 7, R617–R626. [Google Scholar] [CrossRef] [Green Version]
- Engert, A.; Diehl, V.; Schnell, R.; Radszuhn, A.; Hatwig, M.T.; Drillich, S.; Schon, G.; Bohlen, H.; Tesch, H.; Hansmann, M.L.; et al. A phase-I study of an anti-CD25 ricin A-chain immunotoxin (RFT5-SMPT-dgA) in patients with refractory Hodgkin's lymphoma. Blood 1997, 89, 403–410. [Google Scholar]
- Hall, P.D.; Virella, G.; Willoughby, T.; Atchley, D.H.; Kreitman, R.J.; Frankel, A.E. Antibody response to DT-GM, a novel fusion toxin consisting of a truncated diphtheria toxin (DT) linked to human granulocyte-macrophage colony stimulating factor (GM), during a phase I trial of patients with relapsed or refractory acute myeloid leukemia. Clin. Immunol. 2001, 100, 191–17. [Google Scholar] [CrossRef]
- Posey, J.A.; Khazaeli, M.B.; Bookman, M.A.; Nowrouzi, A.; Grizzle, W.E.; Thornton, J.; Carey, D.E.; Lorenz, J.M.; Sing, A.P.; Siegall, C.B.; et al. A phase I trial of the single-chain immunotoxin SGN-10 (BR96 sFv-PE40) in patients with advanced solid tumors. Clin. Cancer Res. 2002, 8, 3092–3099. [Google Scholar]
- Scadden, D.T.; Schenkein, D.P.; Bernstein, Z.; Luskey, B.; Doweiko, J.; Tulpule, A.; Levine, A.M. Immunotoxin combined with chemotherapy for patients with AIDS-related non-Hodgkin's lymphoma. Cancer 1998, 83, 2580–2587. [Google Scholar] [CrossRef]
- Mischak, R.P.; Foxall, C.; Rosendorf, L.L.; Knebel, K.; Scannon, P.J.; Spitler, L.E. Human antibody responses to components of the monoclonal antimelanoma antibody ricin A chain immunotoxin XomaZyme-MEL. Mol. Biother. 1990, 2, 104–109. [Google Scholar]
- Tur, M.K.; Neef, I.; Jost, E.; Galm, O.; Jager, G.; Stocker, M.; Ribbert, M.; Osieka, R.; Klinge, U.; Barth, S. Targeted restoration of down-regulated DAPK2 tumor suppressor activity induces apoptosis in Hodgkin lymphoma cells. J. Immunother. 2009, 32, 431–441. [Google Scholar] [CrossRef]
- Hwang, W.Y.; Foote, J. Immunogenicity of engineered antibodies. Methods 2005, 36, 3–10. [Google Scholar] [CrossRef]
- Boulianne, G.L.; Hozumi, N.; Shulman, M.J. Production of functional chimaeric mouse/human antibody. Nature 1984, 312, 643–646. [Google Scholar] [CrossRef]
- Morrison, S.L.; Johnson, M.J.; Herzenberg, L.A.; Oi, V.T. Chimeric human antibody molecules: Mouse antigen-binding domains with human constant region domains. Proc. Natl. Acad. Sci. USA 1984, 81, 6851–6855. [Google Scholar]
- Jones, P.T.; Dear, P.H.; Foote, J.; Neuberger, M.S.; Winter, G. Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature 1986, 321, 522–525. [Google Scholar] [CrossRef]
- Verhoeyen, M.; Milstein, C.; Winter, G. Reshaping human antibodies: Grafting an antilysozyme activity. Science 1988, 239, 1534–1536. [Google Scholar]
- Hu, W.G.; Yin, J.; Chau, D.; Negrych, L.M.; Cherwonogrodzky, J.W. Humanization and characterization of an anti-ricin neutralization monoclonal antibody. PLoS One 2012, 7, e45595. [Google Scholar]
- Lonberg, N. Fully human antibodies from transgenic mouse and phage display platforms. Curr. Opin. Immunol. 2008, 20, 450–459. [Google Scholar] [CrossRef]
- Menoret, S.; Iscache, A.L.; Tesson, L.; Remy, S.; Usal, C.; Osborn, M.J.; Cost, G.J.; Bruggemann, M.; Buelow, R.; Anegon, I. Characterization of immunoglobulin heavy chain knockout rats. Eur. J. Immunol. 2010, 40, 2932–2941. [Google Scholar] [CrossRef]
- Hansen, J.K.; Weldon, J.E.; Xiang, L.; Beers, R.; Onda, M.; Pastan, I. A recombinant immunotoxin targeting CD22 with low immunogenicity, low nonspecific toxicity, and high antitumor activity in mice. J. Immunother. 2010, 33, 297–304. [Google Scholar] [CrossRef]
- Liu, W.; Onda, M.; Lee, B.; Kreitman, R.J.; Hassan, R.; Xiang, L.; Pastan, I. Recombinant immunotoxin engineered for low immunogenicity and antigenicity by identifying and silencing human B-cell epitopes. Proc. Natl. Acad. Sci. USA 2012, 109, 11782–11787. [Google Scholar]
- Tsutsumi, Y.; Onda, M.; Nagata, S.; Lee, B.; Kreitman, R.J.; Pastan, I. Site-specific chemical modification with polyethylene glycol of recombinant immunotoxin anti-Tac(Fv)-PE38 (LMB-2) improves antitumor activity and reduces animal toxicity and immunogenicity. Proc. Natl. Acad. Sci. USA 2000, 97, 8548–8553. [Google Scholar]
- Oratz, R.; Speyer, J.L.; Wernz, J.C.; Hochster, H.; Meyers, M.; Mischak, R.; Spitler, L.E. Antimelanoma monoclonal antibody-ricin A chain immunoconjugate (XMMME-001-RTA) plus cyclophosphamide in the treatment of metastatic malignant melanoma: Results of a phase II trial. J. Biol. Response Mod. 1990, 9, 345–354. [Google Scholar]
- Siegall, C.B.; Haggerty, H.G.; Warner, G.L.; Chace, D.; Mixan, B.; Linsley, P.S.; Davidson, T. Prevention of immunotoxin-induced immunogenicity by coadministration with CTLA4Ig enhances antitumor efficacy. J. Immunol. 1997, 159, 5168–5173. [Google Scholar]
- Igney, F.H.; Krammer, P.H. Immune escape of tumors: Apoptosis resistance and tumor counterattack. J. Leuk. Biol. 2002, 71, 907–920. [Google Scholar]
- Vesely, M.D.; Kershaw, M.H.; Schreiber, R.D.; Smyth, M.J. Natural innate and adaptive immunity to cancer. Annu. Rev. Immunol. 2011, 29, 235–271. [Google Scholar] [CrossRef]
- Griffith, T.S.; Chin, W.A.; Jackson, G.C.; Lynch, D.H.; Kubin, M.Z. Intracellular regulation of TRAIL-induced apoptosis in human melanoma cells. J. Immunol. 1998, 161, 2833–2840. [Google Scholar]
- Sutton, V.R.; Wowk, M.E.; Cancilla, M.; Trapani, J.A. Caspase activation by granzyme B is indirect, and caspase autoprocessing requires the release of proapoptotic mitochondrial factors. Immunity 2003, 18, 319–329. [Google Scholar] [CrossRef]
- Campos, L.; Rouault, J.P.; Sabido, O.; Oriol, P.; Roubi, N.; Vasselon, C.; Archimbaud, E.; Magaud, J.P.; Guyotat, D. High expression of bcl-2 protein in acute myeloid leukemia cells is associated with poor response to chemotherapy. Blood 1993, 81, 3091–3096. [Google Scholar]
- Ambrosini, G.; Adida, C.; Altieri, D.C. A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat. Med. 1997, 3, 917–921. [Google Scholar] [CrossRef]
- Russell, J.H.; Ley, T.J. Lymphocyte-mediated cytotoxicity. Annu. Rev. Immunol. 2002, 20, 323–370. [Google Scholar] [CrossRef]
- Boivin, W.A.; Cooper, D.M.; Hiebert, P.R.; Granville, D.J. Intracellular versus extracellular granzyme B in immunity and disease: Challenging the dogma. Lab. Invest. 2009, 89, 1195–1220. [Google Scholar] [CrossRef]
- Raja, S.M.; Wang, B.; Dantuluri, M.; Desai, U.R.; Demeler, B.; Spiegel, K.; Metkar, S.S.; Froelich, C.J. Cytotoxic cell granule-mediated apoptosis. Characterization of the macromolecular complex of granzyme B with serglycin. J. Biol. Chem. 2002, 277, 49523–49530. [Google Scholar]
- Grossman, W.J.; Revell, P.A.; Lu, Z.H.; Johnson, H.; Bredemeyer, A.J.; Ley, T.J. The orphan granzymes of humans and mice. Curr. Opin. Immunol. 2003, 15, 544–552. [Google Scholar] [CrossRef]
- Bots, M.; Medema, J.P. Granzymes at a glance. J. Cell Sci. 2006, 119, 5011–5014. [Google Scholar] [CrossRef]
- Galvin, J.P.; Spaeny-Dekking, L.H.; Wang, B.; Seth, P.; Hack, C.E.; Froelich, C.J. Apoptosis induced by granzyme B-glycosaminoglycan complexes: Implications for granule-mediated apoptosis in vivo. J. Immunol. 1999, 162, 5345–5350. [Google Scholar]
- Grujic, M.; Braga, T.; Lukinius, A.; Eloranta, M.L.; Knight, S.D.; Pejler, G.; Abrink, M. Serglycin-deficient cytotoxic T lymphocytes display defective secretory granule maturation and granzyme B storage. J. Biol. Chem. 2005, 280, 33411–33418. [Google Scholar]
- Lanier, L.L. NK cell recognition. Annu. Rev. Immunol. 2005, 23, 225–274. [Google Scholar] [CrossRef]
- Kurschus, F.C.; Jenne, D.E. Delivery and therapeutic potential of human granzyme B. Immunol. Rev. 2010, 235, 159–171. [Google Scholar]
- Andrade, F.; Casciola-Rosen, L.A.; Rosen, A. Granzyme B-induced cell death. Acta Haematol. 2004, 111, 28–41. [Google Scholar] [CrossRef]
- Heibein, J.A.; Goping, I.S.; Barry, M.; Pinkoski, M.J.; Shore, G.C.; Green, D.R.; Bleackley, R.C. Granzyme B-mediated cytochrome c release is regulated by the Bcl-2 family members bid and Bax. J. Exp. Med. 2000, 192, 1391–1402. [Google Scholar] [CrossRef]
- Pinkoski, M.J.; Waterhouse, N.J.; Heibein, J.A.; Wolf, B.B.; Kuwana, T.; Goldstein, J.C.; Newmeyer, D.D.; Bleackley, R.C.; Green, D.R. Granzyme B-mediated apoptosis proceeds predominantly through a Bcl-2-inhibitable mitochondrial pathway. J. Biol. Chem. 2001, 276, 12060–12067. [Google Scholar]
- Metkar, S.S.; Wang, B.; Ebbs, M.L.; Kim, J.H.; Lee, Y.J.; Raja, S.M.; Froelich, C.J. Granzyme B activates procaspase-3 which signals a mitochondrial amplification loop for maximal apoptosis. J. Cell Biol. 2003, 160, 875–885. [Google Scholar] [CrossRef]
- Talanian, R.V.; Yang, X.; Turbov, J.; Seth, P.; Ghayur, T.; Casiano, C.A.; Orth, K.; Froelich, C.J. Granule-mediated killing: Pathways for granzyme B-initiated apoptosis. J. Exp. Med. 1997, 186, 1323–1331. [Google Scholar] [CrossRef]
- Waterhouse, N.J.; Sedelies, K.A.; Trapani, J.A. Role of Bid-induced mitochondrial outer membrane permeabilization in granzyme B-induced apoptosis. Immunol. Cell Biol. 2006, 84, 72–78. [Google Scholar] [CrossRef]
- Hengartner, M.O. The biochemistry of apoptosis. Nature 2000, 407, 770–776. [Google Scholar] [CrossRef]
- Stahnke, B.; Thepen, T.; Stocker, M.; Rosinke, R.; Jost, E.; Fischer, R.; Tur, M.K.; Barth, S. Granzyme B-H22(scFv), a human immunotoxin targeting CD64 in acute myeloid leukemia of monocytic subtypes. Mol. Cancer Ther. 2008, 7, 2924–2932. [Google Scholar] [CrossRef]
- Schiffer, S. Fraunhofer Institute for Molecular Biology and Applied Ecology IME. unpublished work, Aachen: Germany, 2012. [Google Scholar]
- Liu, Y.; Cheung, L.H.; Hittelman, W.N.; Rosenblum, M.G. Targeted delivery of human pro-apoptotic enzymes to tumor cells: In vitro studies describing a novel class of recombinant highly cytotoxic agents. Mol. Cancer Ther. 2003, 2, 1341–1350. [Google Scholar]
- Liu, Y.; Zhang, W.; Niu, T.; Cheung, L.H.; Munshi, A.; Meyn, R.E., Jr.; Rosenblum, M.G. Targeted apoptosis activation with GrB/scFvMEL modulates melanoma growth, metastatic spread, chemosensitivity, and radiosensitivity. Neoplasia 2006, 8, 125–135. [Google Scholar] [CrossRef]
- Dalken, B.; Giesubel, U.; Knauer, S.K.; Wels, W.S. Targeted induction of apoptosis by chimeric granzyme B fusion proteins carrying antibody and growth factor domains for cell recognition. Cell Death Differ. 2006, 13, 576–585. [Google Scholar] [CrossRef]
- Medema, J.P.; de Jong, J.; Peltenburg, L.T.; Verdegaal, E.M.; Gorter, A.; Bres, S.A.; Franken, K.L.; Hahne, M.; Albar, J.P.; Melief, C.J.; et al. Blockade of the granzyme B/perforin pathway through overexpression of the serine protease inhibitor PI-9/SPI-6 constitutes a mechanism for immune escape by tumors. Proc. Natl. Acad. Sci. USA 2001, 98, 11515–11520. [Google Scholar]
- Jiang, X.; Ellison, S.J.; Alarid, E.T.; Shapiro, D.J. Interplay between the levels of estrogen and estrogen receptor controls the level of the granzyme inhibitor, proteinase inhibitor 9 and susceptibility to immune surveillance by natural killer cells. Oncogene 2007, 26, 4106–4114. [Google Scholar] [CrossRef]
- Kurschus, F.C.; Kleinschmidt, M.; Fellows, E.; Dornmair, K.; Rudolph, R.; Lilie, H.; Jenne, D.E. Killing of target cells by redirected granzyme B in the absence of perforin. FEBS Lett. 2004, 562, 87–92. [Google Scholar] [CrossRef]
- Liu, Y.; Cheung, L.H.; Thorpe, P.; Rosenblum, M.G. Mechanistic studies of a novel human fusion toxin composed of vascular endothelial growth factor (VEGF)121 and the serine protease granzyme B: Directed apoptotic events in vascular endothelial cells. Mol. Cancer Ther. 2003, 2, 949–959. [Google Scholar]
- Zhang, L.; Zhao, J.; Wang, T.; Yu, C.J.; Jia, L.T.; Duan, Y.Y.; Yao, L.B.; Chen, S.Y.; Yang, A.G. HER2-targeting recombinant protein with truncated pseudomonas exotoxin A translocation domain efficiently kills breast cancer cells. Cancer Biol. Ther. 2008, 7, 1226–1231. [Google Scholar]
- Wang, T.; Zhao, J.; Ren, J.L.; Zhang, L.; Wen, W.H.; Zhang, R.; Qin, W.W.; Jia, L.T.; Yao, L.B.; Zhang, Y.Q.; et al. Recombinant immunoproapoptotic proteins with furin site can translocate and kill HER2-positive cancer cells. Cancer Res. 2007, 67, 11830–11839. [Google Scholar]
- Kanatani, I.; Lin, X.; Yuan, X.; Manorek, G.; Shang, X.; Cheung, L.H.; Rosenblum, M.G.; Howell, S.B. Targeting granzyme B to tumor cells using a yoked human chorionic gonadotropin. Cancer Chemother. Pharmacol. 2011, 68, 979–990. [Google Scholar] [CrossRef]
- Hegde, R.; Srinivasula, S.M.; Zhang, Z.; Wassell, R.; Mukattash, R.; Cilenti, L.; DuBois, G.; Lazebnik, Y.; Zervos, A.S.; Fernandes-Alnemri, T.; et al. Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts inhibitor of apoptosis protein-caspase interaction. J. Biol. Chem. 2002, 277, 432–438. [Google Scholar]
- Liu, X.; Kim, C.N.; Yang, J.; Jemmerson, R.; Wang, X. Induction of apoptotic program in cell-free extracts: Requirement for dATP and cytochrome c. Cell 1996, 86, 147–157. [Google Scholar] [CrossRef]
- Han, J.; Goldstein, L.A.; Gastman, B.R.; Froelich, C.J.; Yin, X.M.; Rabinowich, H. Degradation of Mcl-1 by granzyme B: Implications for Bim-mediated mitochondrial apoptotic events. J. Biol. Chem. 2004, 279, 22020–22029. [Google Scholar]
- Alimonti, J.B.; Shi, L.; Baijal, P.K.; Greenberg, A.H. Granzyme B induces BID-mediated cytochrome c release and mitochondrial permeability transition. J. Biol. Chem. 2001, 276, 6974–6982. [Google Scholar] [CrossRef]
- Waterhouse, N.J.; Sedelies, K.A.; Browne, K.A.; Wowk, M.E.; Newbold, A.; Sutton, V.R.; Clarke, C.J.; Oliaro, J.; Lindemann, R.K.; Bird, P.I.; et al. A central role for Bid in granzyme B-induced apoptosis. J. Biol. Chem. 2005, 280, 4476–4482. [Google Scholar]
- Cullen, S.P.; Adrain, C.; Luthi, A.U.; Duriez, P.J.; Martin, S.J. Human and murine granzyme B exhibit divergent substrate preferences. J. Cell Biol. 2007, 176, 435–444. [Google Scholar] [CrossRef]
- Chowdhury, D.; Lieberman, J. Death by a thousand cuts: Granzyme pathways of programmed cell death. Annu. Rev. Immunol. 2008, 26, 389–420. [Google Scholar] [CrossRef]
- Trapani, J.A.; Sutton, V.R. Granzyme B: Pro-apoptotic, antiviral and antitumor functions. Curr. Opin. Immunol. 2003, 15, 533–543. [Google Scholar] [CrossRef]
- Bleackley, R.C. A molecular view of cytotoxic T lymphocyte induced killing. Biochem. Cell Biol. 2005, 83, 747–51. [Google Scholar] [CrossRef]
- Sun, J.; Bird, C.H.; Sutton, V.; McDonald, L.; Coughlin, P.B.; De Jong, T.A.; Trapani, J.A.; Bird, P.I. A cytosolic granzyme B inhibitor related to the viral apoptotic regulator cytokine response modifier A is present in cytotoxic lymphocytes. J. Biol. Chem. 1996, 271, 27802–27809. [Google Scholar]
- Poe, M.; Blake, J.T.; Boulton, D.A.; Gammon, M.; Sigal, N.H.; Wu, J.K.; Zweerink, H.J. Human cytotoxic lymphocyte granzyme B. Its purification from granules and the characterization of substrate and inhibitor specificity. J. Biol. Chem. 1991, 266, 98–103. [Google Scholar]
- Zhao, J.; Zhang, L.H.; Jia, L.T.; Zhang, L.; Xu, Y.M.; Wang, Z.; Yu, C.J.; Peng, W.D.; Wen, W.H.; Wang, C.J.; et al. Secreted antibody/granzyme B fusion protein stimulates selective killing of HER2-overexpressing tumor cells. J. Biol. Chem. 2004, 279, 21343–21348. [Google Scholar]
- Caputo, A.; Garner, R.S.; Winkler, U.; Hudig, D.; Bleackley, R.C. Activation of recombinant murine cytotoxic cell proteinase-1 requires deletion of an amino-terminal dipeptide. J. Biol. Chem. 1993, 268, 17672–17675. [Google Scholar]
- Dalken, B.; Jabulowsky, R.A.; Oberoi, P.; Benhar, I.; Wels, W.S. Maltose-binding protein enhances secretion of recombinant human granzyme B accompanied by in vivo processing of a precursor MBP fusion protein. PLoS One 2010, 5, e14404. [Google Scholar]
- Kam, C.M.; Hudig, D.; Powers, J.C. Granzymes (lymphocyte serine proteases): Characterization with natural and synthetic substrates and inhibitors. Biochim. Biophys. Acta 2000, 1477, 307–323. [Google Scholar] [CrossRef]
- Griffiths, G.M.; Isaaz, S. Granzymes A and B are targeted to the lytic granules of lymphocytes by the mannose-6-phosphate receptor. J. Cell Biol. 1993, 120, 885–896. [Google Scholar] [CrossRef]
- Edwards, K.M.; Davis, J.E.; Browne, K.A.; Sutton, V.R.; Trapani, J.A. Anti-viral strategies of cytotoxic T lymphocytes are manifested through a variety of granule-bound pathways of apoptosis induction. Immunol. Cell Biol. 1999, 77, 76–89. [Google Scholar] [CrossRef]
- Giesubel, U.; Dalken, B.; Mahmud, F.; Wels, W.S. Cell binding, internalization and cytotoxic activity of human granzyme B expressed in the yeast Pichia pastoris. Biochem. J. 2006, 394, 563–573. [Google Scholar] [CrossRef]
- Xia, Z.; Kam, C.M.; Huang, C.; Powers, J.C.; Mandle, R.J.; Stevens, R.L.; Lieberman, J. Expression and purification of enzymatically active recombinant granzyme B in a baculovirus system. Biochem. Biophys. Res. Commun. 1998, 243, 384–389. [Google Scholar] [CrossRef]
- Smyth, M.J.; McGuire, M.J.; Thia, K.Y. Expression of recombinant human granzyme B. A processing and activation role for dipeptidyl peptidase I. J. Immunol. 1995, 154, 6299–6305. [Google Scholar]
- Godal, R.; Keilholz, U.; Uharek, L.; Letsch, A.; Asemissen, A.M.; Busse, A.; Na, I.K.; Thiel, E.; Scheibenbogen, C. Lymphomas are sensitive to perforin-dependent cytotoxic pathways despite expression of PI-9 and overexpression of bcl-2. Blood 2006, 107, 3205–3211. [Google Scholar] [CrossRef]
- Soriano, C.; Mukaro, V.; Hodge, G.; Ahern, J.; Holmes, M.; Jersmann, H.; Moffat, D.; Meredith, D.; Jurisevic, C.; Reynolds, P.N.; et al. Increased proteinase inhibitor-9 (PI-9) and reduced granzyme B in lung cancer: Mechanism for immune evasion? Lung Cancer 2012, 77, 38–45. [Google Scholar] [CrossRef]
- Ray, M.; Hostetter, D.R.; Loeb, C.R.; Simko, J.; Craik, C.S. Inhibition of Granzyme B by PI-9 protects prostate cancer cells from apoptosis. Prostate 2012, 72, 846–855. [Google Scholar] [CrossRef]
- Rousalova, I.; Krepela, E.; Prochazka, J.; Cermak, J.; Benkova, K. Expression of proteinase inhibitor-9/serpinB9 in non-small cell lung carcinoma cells and tissues. Int. J. Oncol. 2010, 36, 275–283. [Google Scholar]
- Jiang, X.G.; Patterson, N.M.; Ling, Y.; Xie, J.W.; Helferich, W.G.; Shapiro, D.J. Low Concentrations of the Soy Phytoestrogen Genistein Induce Proteinase Inhibitor 9 and Block Killing of Breast Cancer Cells by Immune Cells. Endocrinology 2008, 149, 5366–5373. [Google Scholar] [CrossRef]
- van Houdt, I.S.; Oudejans, J.J.; van den Eertwegh, A.J.; Baars, A.; Vos, W.; Bladergroen, B.A.; Rimoldi, D.; Muris, J.J.; Hooijberg, E.; Gundy, C.M.; et al. Expression of the apoptosis inhibitor protease inhibitor 9 predicts clinical outcome in vaccinated patients with stage III and IV melanoma. Clin. Cancer Res. 2005, 11, 6400–6407. [Google Scholar]
- Oudejans, J.J.; Harijadi, H.; Kummer, J.A.; Tan, I.B.; Bloemena, E.; Middeldorp, J.M.; Bladergroen, B.; Dukers, D.F.; Vos, W.; Meijer, C.J. High numbers of granzyme B/CD8-positive tumour-infiltrating lymphocytes in nasopharyngeal carcinoma biopsies predict rapid fatal outcome in patients treated with curative intent. J. Pathol. 2002, 198, 468–475. [Google Scholar] [CrossRef]
- Bots, M.; Liesbeth, V.A.N.B.; Rademaker, M.T.; Offringa, R.; Medema, J.P. Serpins prevent granzyme-induced death in a species-specific manner. Immunol. Cell Biol. 2006, 84, 79–86. [Google Scholar] [CrossRef]
- Huntington, J.A.; Read, R.J.; Carrell, R.W. Structure of a serpin-protease complex shows inhibition by deformation. Nature 2000, 407, 923–926. [Google Scholar] [CrossRef]
- Bladergroen, B.A.; Strik, M.C.; Bovenschen, N.; van Berkum, O.; Scheffer, G.L.; Meijer, C.J.; Hack, C.E.; Kummer, J.A. The granzyme B inhibitor, protease inhibitor 9, is mainly expressed by dendritic cells and at immune-privileged sites. J. Immunol. 2001, 166, 3218–3225. [Google Scholar]
- Buzza, M.S.; Hosking, P.; Bird, P.I. The granzyme B inhibitor, PI-9, is differentially expressed during placental development and up-regulated in hydatidiform moles. Placenta 2006, 27, 62–69. [Google Scholar] [CrossRef]
- Kannan-Thulasiraman, P.; Shapiro, D.J. Modulators of inflammation use nuclear factor-kappa B and activator protein-1 sites to induce the caspase-1 and granzyme B inhibitor, proteinase inhibitor 9. J. Biol. Chem. 2002, 277, 41230–41239. [Google Scholar] [CrossRef]
- El Haddad, N.; Moore, R.; Heathcote, D.; Mounayar, M.; Azzi, J.; Mfarrej, B.; Batal, I.; Ting, C.; Atkinson, M.; Sayegh, M.H.; et al. The novel role of SERPINB9 in cytotoxic protection of human mesenchymal stem cells. J. Immunol. 2011, 187, 2252–2260. [Google Scholar] [CrossRef]
- Heutinck, K.M.; Kassies, J.; Florquin, S.; Ten Berge, I.J.; Hamann, J.; Rowshani, A.T. SerpinB9 expression in human renal tubular epithelial cells is induced by triggering of the viral dsRNA sensors TLR3, MDA5 and RIG-I. Nephrol. Dial. Transplant. 2012, 27, 2746–2754. [Google Scholar] [CrossRef]
- Rowshani, A.T.; Strik, M.C.; Molenaar, R.; Yong, S.L.; Wolbink, A.M.; Bemelman, F.J.; Hack, C.E.; Ten Berge, I.J. The granzyme B inhibitor SERPINB9 (protease inhibitor 9) circulates in blood and increases on primary cytomegalovirus infection after renal transplantation. J. Infect. Dis. 2005, 192, 1908–1911. [Google Scholar] [CrossRef]
- Classen, C.F.; Bird, P.I.; Debatin, K.M. Modulation of the granzyme B inhibitor proteinase inhibitor 9 (PI-9) by activation of lymphocytes and monocytes in vitro and by Epstein-Barr virus and bacterial infection. Clin. Exp. Immunol. 2006, 143, 534–542. [Google Scholar] [CrossRef]
- Buzza, M.S.; Hirst, C.E.; Bird, C.H.; Hosking, P.; McKendrick, J.; Bird, P.I. The granzyme B inhibitor, PI-9, is present in endothelial and mesothelial cells, suggesting that it protects bystander cells during immune respons. Cell. Immunol. 2001, 210, 21–29. [Google Scholar] [CrossRef]
- Barrie, M.B.; Stout, H.W.; Abougergi, M.S.; Miller, B.C.; Thiele, D.L. Antiviral cytokines induce hepatic expression of the granzyme B inhibitors, proteinase inhibitor 9 and serine proteinase inhibitor 6. J. Immunol. 2004, 172, 6453–6459. [Google Scholar]
- Horie, O.; Saigo, K.; Murayama, T.; Ryo, R. Differential expression of proteinase inhibitor-9 and granzyme B mRNAs in activated immunocompetent cells. Tohoku J. Exp. Med. 2005, 205, 103–113. [Google Scholar] [CrossRef]
- Hirst, C.E.; Buzza, M.S.; Bird, C.H.; Warren, H.S.; Cameron, P.U.; Zhang, M.; Ashton-Rickardt, P.G.; Bird, P.I. The intracellular granzyme B inhibitor, proteinase inhibitor 9, is up-regulated during accessory cell maturation and effector cell degranulation, and its overexpression enhances CTL potency. J. Immunol. 2003, 170, 805–815. [Google Scholar]
- Rowshani, A.T.; Florquin, S.; Bemelman, F.; Kummer, J.A.; Hack, C.E.; Ten Berge, I.J. Hyperexpression of the granzyme B inhibitor PI-9 in human renal allografts: A potential mechanism for stable renal function in patients with subclinical rejection. Kidney Int. 2004, 66, 1417–1422. [Google Scholar] [CrossRef]
- Walker, P.R.; Saas, P.; Dietrich, P.Y. Role of Fas ligand (CD95L) in immune escape: The tumor cell strikes back. J. Immunol. 1997, 158, 4521–4524. [Google Scholar]
- Buehring, G.C.; Eby, E.A.; Eby, M.J. Cell line cross-contamination: How aware are Mammalian cell culturists of the problem and how to monitor it? In Vitro Cell. Dev. Biol. Anim. 2004, 40, 211–215. [Google Scholar] [CrossRef]
- Bird, C.H.; Sutton, V.R.; Sun, J.; Hirst, C.E.; Novak, A.; Kumar, S.; Trapani, J.A.; Bird, P.I. Selective regulation of apoptosis: The cytotoxic lymphocyte serpin proteinase inhibitor 9 protects against granzyme B-mediated apoptosis without perturbing the Fas cell death pathway. Mol. Cell. Biol. 1998, 18, 6387–6398. [Google Scholar]
- Schiffer, S. Efficacy of an adapted Granzyme B-based anti-CD30 cytolytic fusion protein against PI-9-positive classical Hodgkin lymphoma cells in a murine model. Blood Cancer J. 2012. submitted for publication. [Google Scholar]
- ten Berge, R.L.; Meijer, C.J.; Dukers, D.F.; Kummer, J.A.; Bladergroen, B.A.; Vos, W.; Hack, C.E.; Ossenkoppele, G.J.; Oudejans, J.J. Expression levels of apoptosis-related proteins predict clinical outcome in anaplastic large cell lymphoma. Blood 2002, 99, 4540–4546. [Google Scholar] [CrossRef]
- Tiacci, E.; Doring, C.; Brune, V.; van Noesel, C.J.; Klapper, W.; Mechtersheimer, G.; Falini, B.; Kuppers, R.; Hansmann, M.L. Analyzing primary Hodgkin and Reed-Sternberg cells to capture the molecular and cellular pathogenesis of classical Hodgkin lymphoma. Blood 2012, 120, 4609–4620. [Google Scholar] [CrossRef]
- Fritsch, K. Die Bedeutung des Proteinase Inhibitor 9 für die Apoptoseresistenz in leukämischen Zelllinien. Ph.D. Thesis, Albert-Ludwigs-University, Freiburg im Breisgau.
- Bossard, C.; Belhadj, K.; Reyes, F.; Martin-Garcia, N.; Berger, F.; Kummer, J.A.; Briere, J.; Baglin, A.C.; Cheze, S.; Bosq, J.; et al. Expression of the granzyme B inhibitor PI9 predicts outcome in nasal NK/T-cell lymphoma: Results of a Western series of 48 patients treated with first-line polychemotherapy within the Groupe d'Etude des Lymphomes de l'Adulte (GELA) trials. Blood 2007, 109, 2183–2189. [Google Scholar] [CrossRef]
- Bladergroen, B.A.; Meijer, C.J.; ten Berge, R.L.; Hack, C.E.; Muris, J.J.; Dukers, D.F.; Chott, A.; Kazama, Y.; Oudejans, J.J.; van Berkum, O.; et al. Expression of the granzyme B inhibitor, protease inhibitor 9, by tumor cells in patients with non-Hodgkin and Hodgkin lymphoma: A novel protective mechanism for tumor cells to circumvent the immune system? Blood 2002, 99, 232–237. [Google Scholar] [CrossRef]
- Mahrus, S.; Kisiel, W.; Craik, C.S. Granzyme M is a regulatory protease that inactivates proteinase inhibitor 9, an endogenous inhibitor of granzyme B. J. Biol. Chem. 2004, 279, 54275–54282. [Google Scholar]
- Madison, E.L.; Goldsmith, E.J.; Gerard, R.D.; Gething, M.J.; Sambrook, J.F. Serpin-resistant mutants of human tissue-type plasminogen activator. Nature 1989, 339, 721–724. [Google Scholar] [CrossRef]
- Potempa, J.; Korzus, E.; Travis, J. The serpin superfamily of proteinase inhibitors: Structure, function, and regulation. J. Biol. Chem. 1994, 269, 15957–15960. [Google Scholar]
- Marszal, E.; Shrake, A. Serpin crystal structure and serpin polymer structure. Arch. Biochem. Biophys. 2006, 453, 123–129. [Google Scholar] [CrossRef]
- Silverman, G.A.; Bird, P.I.; Carrell, R.W.; Church, F.C.; Coughlin, P.B.; Gettins, P.G.; Irving, J.A.; Lomas, D.A.; Luke, C.J.; Moyer, R.W.; et al. The serpins are an expanding superfamily of structurally similar but functionally diverse proteins. Evolution, mechanism of inhibition, novel functions, and a revised nomenclature. J. Biol. Chem. 2001, 276, 33293–33296. [Google Scholar]
- Sun, J.; Whisstock, J.C.; Harriott, P.; Walker, B.; Novak, A.; Thompson, P.E.; Smith, A.I.; Bird, P.I. Importance of the P4' residue in human granzyme B inhibitors and substrates revealed by scanning mutagenesis of the proteinase inhibitor 9 reactive center loop. J. Biol. Chem. 2001, 276, 15177–15184. [Google Scholar]
- Hedstrom, L. Serine protease mechanism and specificity. Chem. Rev. 2002, 102, 4501–4524. [Google Scholar] [CrossRef]
- Ye, S.; Cech, A.L.; Belmares, R.; Bergstrom, R.C.; Tong, Y.; Corey, D.R.; Kanost, M.R.; Goldsmith, E.J. The structure of a Michaelis serpin-protease complex. Nat. Struct. Biol. 2001, 8, 979–983. [Google Scholar]
- Rotonda, J.; Garcia-Calvo, M.; Bull, H.G.; Geissler, W.M.; McKeever, B.M.; Willoughby, C.A.; Thornberry, N.A.; Becker, J.W. The three-dimensional structure of human granzyme B compared to caspase-3, key mediators of cell death with cleavage specificity for aspartic acid in P1. Chem. Biol. 2001, 8, 357–368. [Google Scholar]
- Kortemme, T.; Baker, D. A simple physical model for binding energy hot spots in protein-protein complexes. Proc. Natl. Acad. Sci. USA 2002, 99, 14116–14121. [Google Scholar] [CrossRef]
- Losasso, V.; Schiffer, S.; Barth, S.; Carloni, P. Design of human granzyme B variants resistant to serpin B9. Proteins 2012, 80, 2514–2522. [Google Scholar]
- Jabulowsky, R.A.; Oberoi, P.; Bahr-Mahmud, H.; Dalken, B.; Wels, W.S. Surface Charge-Modification Prevents Sequestration and Enhances Tumor-Cell Specificity of a Recombinant Granzyme B-TGF alpha Fusion Protein. Bioconjug. Chem. 2012, 23, 1567–1576. [Google Scholar]
- Motyka, B.; Korbutt, G.; Pinkoski, M.J.; Heibein, J.A.; Caputo, A.; Hobman, M.; Barry, M.; Shostak, I.; Sawchuk, T.; Holmes, C.F.; et al. Mannose 6-phosphate/insulin-like growth factor II receptor is a death receptor for granzyme B during cytotoxic T cell-induced apoptosis. Cell 2000, 103, 491–500. [Google Scholar] [CrossRef]
- Veugelers, K.; Motyka, B.; Goping, I.S.; Shostak, I.; Sawchuk, T.; Bleackley, R.C. Granule-mediated killing by granzyme B and perforin requires a mannose 6-phosphate receptor and is augmented by cell surface heparan sulfate. Mol. Biol. Cell 2006, 17, 623–633. [Google Scholar]
- Kurschus, F.C.; Bruno, R.; Fellows, E.; Falk, C.S.; Jenne, D.E. Membrane receptors are not required to deliver granzyme B during killer cell attack. Blood 2005, 105, 2049–2058. [Google Scholar] [CrossRef]
- Trapani, J.A.; Sutton, V.R.; Thia, K.Y.; Li, Y.Q.; Froelich, C.J.; Jans, D.A.; Sandrin, M.S.; Browne, K.A. A clathrin/dynamin- and mannose-6-phosphate receptor-independent pathway for granzyme B-induced cell death. J. Cell Biol. 2003, 160, 223–233. [Google Scholar] [CrossRef]
- Bretthauer, R.K.; Castellino, F.J. Glycosylation of Pichia pastoris-derived proteins. Biotechnol. Appl. Biochem. 1999, 30, 193–200. [Google Scholar]
- Raja, S.M.; Metkar, S.S.; Froelich, C.J. Cytotoxic granule-mediated apoptosis: Unraveling the complex mechanism. Curr. Opin. Immunol. 2003, 15, 528–532. [Google Scholar] [CrossRef]
- Gross, C.; Koelch, W.; DeMaio, A.; Arispe, N.; Multhoff, G. Cell surface-bound heat shock protein 70 (Hsp70) mediates perforin-independent apoptosis by specific binding and uptake of granzyme B. J. Biol. Chem. 2003, 278, 41173–41181. [Google Scholar]
- Bird, C.H.; Sun, J.; Ung, K.; Karambalis, D.; Whisstock, J.C.; Trapani, J.A.; Bird, P.I. Cationic sites on granzyme B contribute to cytotoxicity by promoting its uptake into target cells. Mol. Cell. Biol. 2005, 25, 7854–7867. [Google Scholar]
- Shi, L.; Keefe, D.; Durand, E.; Feng, H.; Zhang, D.; Lieberman, J. Granzyme B binds to target cells mostly by charge and must be added at the same time as perforin to trigger apoptosis. J. Immunol. 2005, 174, 5456–5461. [Google Scholar]
- Metkar, S.S.; Wang, B.; Aguilar-Santelises, M.; Raja, S.M.; Uhlin-Hansen, L.; Podack, E.; Trapani, J.A.; Froelich, C.J. Cytotoxic cell granule-mediated apoptosis: Perforin delivers granzyme B-serglycin complexes into target cells without plasma membrane pore formation. Immunity 2002, 16, 417–428. [Google Scholar] [CrossRef]
- Onda, M.; Nagata, S.; Tsutsumi, Y.; Vincent, J.J.; Wang, Q.; Kreitman, R.J.; Lee, B.; Pastan, I. Lowering the isoelectric point of the Fv portion of recombinant immunotoxins leads to decreased nonspecific animal toxicity without affecting antitumor activity. Cancer Res. 2001, 61, 5070–5077. [Google Scholar]
- Buzza, M.S.; Zamurs, L.; Sun, J.; Bird, C.H.; Smith, A.I.; Trapani, J.A.; Froelich, C.J.; Nice, E.C.; Bird, P.I. Extracellular matrix remodeling by human granzyme B via cleavage of vitronectin, fibronectin, and laminin. J. Biol. Chem. 2005, 280, 23549–23558. [Google Scholar]
- Ronday, H.K.; van der Laan, W.H.; Tak, P.P.; de Roos, J.A.; Bank, R.A.; TeKoppele, J.M.; Froelich, C.J.; Hack, C.E.; Hogendoorn, P.C.; Breedveld, F.C.; et al. Human granzyme B mediates cartilage proteoglycan degradation and is expressed at the invasive front of the synovium in rheumatoid arthritis. Rheumatology (Oxford) 2001, 40, 55–61. [Google Scholar] [CrossRef]
- Tak, P.P.; Spaeny-Dekking, L.; Kraan, M.C.; Breedveld, F.C.; Froelich, C.J.; Hack, C.E. The levels of soluble granzyme A and B are elevated in plasma and synovial fluid of patients with rheumatoid arthritis (RA). Clin. Exp. Immunol. 1999, 116, 366–370. [Google Scholar] [CrossRef]
- Goldbach-Mansky, R.; Suson, S.; Wesley, R.; Hack, C.E.; El-Gabalawy, H.S.; Tak, P.P. Raised granzyme B levels are associated with erosions in patients with early rheumatoid factor positive rheumatoid arthritis. Ann. Rheum. Dis. 2005, 64, 715–721. [Google Scholar] [CrossRef]
- Chamberlain, C.M.; Ang, L.S.; Boivin, W.A.; Cooper, D.M.; Williams, S.J.; Zhao, H.; Hendel, A.; Folkesson, M.; Swedenborg, J.; Allard, M.F.; et al. Perforin-independent extracellular granzyme B activity contributes to abdominal aortic aneurysm. Am. J. Pathol. 2010, 176, 1038–1049. [Google Scholar] [CrossRef]
- Saito, Y.; Kondo, H.; Hojo, Y. Granzyme B as a novel factor involved in cardiovascular diseases. J. Cardiol. 2011, 57, 141–147. [Google Scholar] [CrossRef]
- Kurschus, F.C.; Fellows, E.; Stegmann, E.; Jenne, D.E. Granzyme B delivery via perforin is restricted by size, but not by heparan sulfate-dependent endocytosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13799–13804. [Google Scholar] [CrossRef]
- Thomas, G. Furin at the cutting edge: From protein traffic to embryogenesis and disease. Nat. Rev. Mol. Cell Biol. 2002, 3, 753–766. [Google Scholar] [CrossRef]
- Molloy, S.S.; Anderson, E.D.; Jean, F.; Thomas, G. Bi-cycling the furin pathway: From TGN localization to pathogen activation and embryogenesis. Trends Cell Biol. 1999, 9, 28–35. [Google Scholar] [CrossRef]
- Ogata, M.; Fryling, C.M.; Pastan, I.; FitzGerald, D.J. Cell-mediated cleavage of Pseudomonas exotoxin between Arg279 and Gly280 generates the enzymatically active fragment which translocates to the cytosol. J. Biol. Chem. 1992, 267, 25396–25401. [Google Scholar]
- Theuer, C.P.; FitzGerald, D.; Pastan, I. A recombinant form of Pseudomonas exotoxin directed at the epidermal growth factor receptor that is cytotoxic without requiring proteolytic processing. J. Biol. Chem. 1992, 267, 16872–16877. [Google Scholar]
- Wolf, P.; Elsasser-Beile, U. Pseudomonas exotoxin A: From virulence factor to anti-cancer agent. Int. J. Med. Microbiol. 2009, 299, 161–176. [Google Scholar] [CrossRef]
- Collier, R.J. Understanding the mode of action of diphtheria toxin: A perspective on progress during the 20th century. Toxicon 2001, 39, 1793–1803. [Google Scholar] [CrossRef]
- Zhan, H.; Choe, S.; Huynh, P.D.; Finkelstein, A.; Eisenberg, D.; Collier, R.J. Dynamic transitions of the transmembrane domain of diphtheria toxin: Disulfide trapping and fluorescence proximity studies. Biochemistry 1994, 33, 11254–11263. [Google Scholar]
- Plank, C.; Oberhauser, B.; Mechtler, K.; Koch, C.; Wagner, E. The influence of endosome-disruptive peptides on gene transfer using synthetic virus-like gene transfer systems. J. Biol. Chem. 1994, 269, 12918–12924. [Google Scholar]
- Vives, E.; Brodin, P.; Lebleu, B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J. Biol. Chem. 1997, 272, 16010–16017. [Google Scholar] [CrossRef]
- Gius, D.R.; Ezhevsky, S.A.; Becker-Hapak, M.; Nagahara, H.; Wei, M.C.; Dowdy, S.F. Transduced p16INK4a peptides inhibit hypophosphorylation of the retinoblastoma protein and cell cycle progression prior to activation of Cdk2 complexes in late G1. Cancer Res. 1999, 59, 2577–2580. [Google Scholar]
- Vocero-Akbani, A.M.; Heyden, N.V.; Lissy, N.A.; Ratner, L.; Dowdy, S.F. Killing HIV-infected cells by transduction with an HIV protease-activated caspase-3 protein. Nat. Med. 1999, 5, 29–33. [Google Scholar] [CrossRef]
- Falnes, P.O.; Wesche, J.; Olsnes, S. Ability of the Tat basic domain and VP22 to mediate cell binding, but not membrane translocation of the diphtheria toxin A-fragment. Biochemistry 2001, 40, 4349–4358. [Google Scholar] [CrossRef]
- Ziegler, A.; Seelig, J. Interaction of the protein transduction domain of HIV-1 TAT with heparan sulfate: Binding mechanism and thermodynamic parameters. Biophys. J. 2004, 86, 254–263. [Google Scholar] [CrossRef]
- Noguchi, H.; Ueda, M.; Matsumoto, S.; Kobayashi, N.; Hayashi, S. BETA2/NeuroD protein transduction requires cell surface heparan sulfate proteoglycans. Hum. Gene Ther. 2007, 18, 10–17. [Google Scholar] [CrossRef]
- Richard, J.P.; Melikov, K.; Brooks, H.; Prevot, P.; Lebleu, B.; Chernomordik, L.V. Cellular uptake of unconjugated TAT peptide involves clathrin-dependent endocytosis and heparan sulfate receptors. J. Biol. Chem. 2005, 280, 15300–15306. [Google Scholar]
- Tilstra, J.; Rehman, K.K.; Hennon, T.; Plevy, S.E.; Clemens, P.; Robbins, P.D. Protein transduction: Identification, characterization and optimization. Biochem. Soc. Trans. 2007, 35, 811–815. [Google Scholar] [CrossRef]
- Zahid, M.; Lu, X.; Mi, Z.; Robbins, P.D. Cationic and tissue-specific protein transduction domains identification, characterization, and therapeutic application. Adv. Genet. 2010, 69, 83–95. [Google Scholar] [CrossRef]
- Fuchs, H.; Bachran, C.; Heisler, I.; Sutherland, M. A Closer Look at Protein Transduction Domains as a Tool in Drug Delivery. Curr. Nanosci. 2005, 1, 117–124. [Google Scholar] [CrossRef]
- Kabouridis, P.S. Biological applications of protein transduction technology. Trends Biotechnol. 2003, 21, 498–503. [Google Scholar] [CrossRef]
- van den Berg, A.; Dowdy, S.F. Protein transduction domain delivery of therapeutic macromolecules. Curr. Opin. Biotechnol. 2011, 22, 888–893. [Google Scholar] [CrossRef]
- Snyder, E.L.; Meade, B.R.; Dowdy, S.F. Anti-cancer protein transduction strategies: Reconstitution of p27 tumor suppressor function. J. Control Release 2003, 91, 45–51. [Google Scholar] [CrossRef]
- Plescia, J.; Salz, W.; Xia, F.; Pennati, M.; Zaffaroni, N.; Daidone, M.G.; Meli, M.; Dohi, T.; Fortugno, P.; Nefedova, Y.; et al. Rational design of shepherdin, a novel anticancer agent. Cancer Cell 2005, 7, 457–468. [Google Scholar] [CrossRef]
- Hong, F.D.; Clayman, G.L. Isolation of a peptide for targeted drug delivery into human head and neck solid tumors. Cancer Res. 2000, 60, 6551–6556. [Google Scholar]
- Wang, J.L.; Zhang, Z.J.; Choksi, S.; Shan, S.; Lu, Z.; Croce, C.M.; Alnemri, E.S.; Korngold, R.; Huang, Z. Cell permeable Bcl-2 binding peptides: A chemical approach to apoptosis induction in tumor cells. Cancer Res. 2000, 60, 1498–1502. [Google Scholar]
- Curnis, F.; Arrigoni, G.; Sacchi, A.; Fischetti, L.; Arap, W.; Pasqualini, R.; Corti, A. Differential binding of drugs containing the NGR motif to CD13 isoforms in tumor vessels, epithelia, and myeloid cells. Cancer Res. 2002, 62, 867–874. [Google Scholar]
- Snyder, E.L.; Saenz, C.C.; Denicourt, C.; Meade, B.R.; Cui, X.S.; Kaplan, I.M.; Dowdy, S.F. Enhanced targeting and killing of tumor cells expressing the CXC chemokine receptor 4 by transducible anticancer peptides. Cancer Res. 2005, 65, 10646–10650. [Google Scholar] [CrossRef]
- Keller, J.; Heisler, I.; Tauber, R.; Fuchs, H. Development of a novel molecular adapter for the optimization of immunotoxins. J. Control Release 2001, 74, 259–261. [Google Scholar] [CrossRef]
- Heisler, I.; Keller, J.; Tauber, R.; Sutherland, M.; Fuchs, H. A cleavable adapter to reduce nonspecific cytotoxicity of recombinant immunotoxins. Int. J. Cancer 2003, 103, 277–282. [Google Scholar] [CrossRef]
- Hetzel, C.; Bachran, C.; Tur, M.K.; Fuchs, H.; Stocker, M. Improved immunotoxins with novel functional elements. Curr. Pharm Des. 2009, 15, 2700–2711. [Google Scholar] [CrossRef]
- Hetzel, C.; Bachran, C.; Fischer, R.; Fuchs, H.; Barth, S.; Stocker, M. Small cleavable adapters enhance the specific cytotoxicity of a humanized immunotoxin directed against CD64-positive cells. J. Immunother. 2008, 31, 370–376. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hehmann-Titt, G.; Schiffer, S.; Berges, N.; Melmer, G.; Barth, S. Improving the Therapeutic Potential of Human Granzyme B for Targeted Cancer Therapy. Antibodies 2013, 2, 19-49. https://doi.org/10.3390/antib2010019
Hehmann-Titt G, Schiffer S, Berges N, Melmer G, Barth S. Improving the Therapeutic Potential of Human Granzyme B for Targeted Cancer Therapy. Antibodies. 2013; 2(1):19-49. https://doi.org/10.3390/antib2010019
Chicago/Turabian StyleHehmann-Titt, Grit, Sonja Schiffer, Nina Berges, Georg Melmer, and Stefan Barth. 2013. "Improving the Therapeutic Potential of Human Granzyme B for Targeted Cancer Therapy" Antibodies 2, no. 1: 19-49. https://doi.org/10.3390/antib2010019