Antibody-Based Immunotoxins for the Treatment of Cancer

Department of Molecular Microbiology and Biotechnology, The George S. Wise Faculty of Life Sciences, Tel-Aviv University, Ramat Aviv 69978, Israel
*
Author to whom correspondence should be addressed.
Antibodies 2012, 1(1), 39-69; https://doi.org/10.3390/antib1010039
Submission received: 28 March 2012
/
Revised: 1 May 2012
/
Accepted: 8 May 2012
/
Published: 15 May 2012
(This article belongs to the Special Issue Modes of Antibody Action for Cancer Therapy)
Abstract
:Antibody-based immunotoxins comprise an important group in targeted cancer therapeutics. These chimeric proteins are a form of biological guided missiles that combine a targeting moiety with a potent effector molecule. The targeting moiety is mostly a monoclonal antibody (MAb) or a recombinant antibody-based fragment that confers target specificity to the immunotoxin. The effector domain is a potent protein toxin of bacterial or plant origin, which, following binding to the target cells, undergoes internalization and causes cell death. Over time and following research progression, immunotoxins become better fitted to their purpose, losing immunogenic fragments and non-specific targeting moieties. Many immunotoxins have gone through clinical evaluation. Some of these have been shown to be active and work is progressing with them in the form of further clinical trials. Others, mostly developed in the previous century, failed to generate a response in patients, or even caused undesired side effects. This article reviews the antibody and protein-toxin based immunotoxins that were clinically evaluated up to the present day.
1. Introduction
Cancer is becoming the most frequent cause of death in most developed countries. In the US, the estimated number of new cancer cases was above 1.5 million in 2010, with a mortality rate accounting for 23% of total deaths [1]. In order to use highly potent agents as a cancer therapy, it is necessary to target the killing agent selectively to the cancer cells. This strategy in not new and many directions have been taken in the development of such therapies. In this way it is possible to use more potent drugs on the one hand, and on the other, reduce systematic toxicity [2].
Antibody-based therapeutics are of growing significance in cancer therapy, and so far 28 such drugs have been approved for cancer therapy by the US Food and Drug Administration (FDA) for marketing in the USA, with combined global revenues that exceed US $50 billion [3]. The market for therapeutic antibodies is the fastest growing sector in the pharmaceutical industry. Currently, hundreds of MAbs for oncology use are in clinical development and progress in the development of antibody-based therapeutics is dramatically increasing [4,5]. A big incentive to this field was given by the ability to genetically design the antibody-based moiety using recombinant DNA technologies. Chimerization and humanization of MAbs have enhanced the clinical efficiency of murine MAbs and accelerated the rate of approvals for MAbs and classic monovalent antibody fragment (Fab) molecules for the treatment of cancer, and promoted the use of other small MAb fragments in the development of new treatments [6,7]. Despite the clinical success of therapeutic MAbs, naked antibodies, targeting cell surface tumor antigens expressed on carcinomas, are rarely potent enough by themselves, and thus are usually administered in combination with chemotherapy [8]. Several approaches were developed to solve this problem. One approach is to enhance the potency of MAbs and antibody fragments by linking them to toxic moieties. Plant and bacterial toxins that cause cellular cytotoxicity through the inhibition of protein synthesis after internalization were found to be good candidates. Toxins like Pseudomonas exotoxin A (PE), ricin toxin (RT) and others are used. These are highly potent and active in very small quantities, such that killing a cell is possible even by a single molecule in the cytoplasm [9]. Another new approach in therapeutic antibody development is to bind the antibody to non-protein therapeutic payloads, such as radioactive isotopes or chemotherapeutic drugs to target cells [3]. The field of antibody drug conjugates (ADCs) in which the toxic moiety is not a protein has been reviewed recently [10,11] and will not be discussed here.
This review provides an update of this fast-moving field, describes the toxins that are used as a part of the immunotoxins, the antibody and antibody-fragment based immunotoxins that were clinically tested up to now for cancer therapy, and evaluates their advantages and disadvantages.
2. General Features of Immunotoxins
Immunotoxins are chimeric proteins composed of an antibody or antibody fragment derived from the immune system (conferring target specificity) that is fused or conjugated to a toxic protein. Immunotoxins have evolved with time and technology, and can be generally divided into three generations. The first generation immunotoxins were produced by chemically coupling native toxins to antibodies using crosslinking reagents that form disulfide bonds connecting the toxin to the antibody. Second generation immunotoxins, like the first generation, were made by chemical coupling methods. The first immunotoxins contained full length toxins, including their targeting domains that are not cancer cell-specific. As research progressed, toxin autonomic cell-binding domains were recognized and removed. The resulting toxin fragment, that could no longer bind normal cells, was coupled to an antibody [12,13,14,15]. Although full length immunoglobulin G (IgG) has good in vivo half-life and effector functions, its large size limits antibody tissue penetration, especially in solid tumors, and complicates the manufacturing process [6]. Moreover, most of the MAbs that were used in the first two generations were murine MAbs. The usage of non-human MAbs has many therapeutic disadvantages (discussed later). Third generation immunotoxins are made by recombinant DNA techniques and combine variable fragments of an antibody (Fv) and toxins without their cell binding domains on the same protein [16]. More than 1,000 third generation immunotoxins have been developed since the first report of Fv production in 1988, in which the variable domains of the heavy chain and light chain of the antibody were connected by a peptide linker [17,18]. The majority of these immunotoxins targeted antigens selectively expressed on cancer cells. It has been hoped (and in some cases successfully demonstrated) that these agents could cause regression of malignant disease in patients [19]. Being of bacterial or plant origin, toxins and toxin domains are highly immunogenic to humans and the immune reaction to the immunotoxin restricts the treatment of each patient to a few doses. Today, efforts are being made to eliminate immunogenic epitopes from toxin surfaces to reduce the immunogenicity of the native toxins [20,21].
3. Immunotoxins in Clinical Evaluation
Clinically evaluated immunotoxins can be easily differentiated according to the immunotoxins’ periods of development. Immunotoxins developed as part of early first generation of immunotoxins were found more often to cause severe side effects in patients, and in many cases, late clinical phases were not perused (like OVB3-PE, 260F9-rRTA and others) [22,23]. Some of these immunotoxins returned to be clinically evaluated after insertion of improvements, such as mutations, as in the case of BL22 that was mutated into HA22, or like shorter antibody fragments, as in the case of LMB-1 that was changed to create LMB-7 and LMB-9 [15]. In other cases, immunotoxins were combined with different chemicals to reduce their immunogenicity (like Xomazyme-Mel), or, in order to increase their potency as in the case of Combotox, immunotoxins were even given together. Some immunotoxins were tested in a few trials, including several different cancer types or diseases other than cancer (like IMTOX-25 and BL22) [24,25]. To conclude, until today, none of the antibody-based immunotoxins has received the FDA approval for use, although a few immunotoxins are in final stages of clinical evaluation.
This chapter describes the toxins that are used as the killing agents in the immunotoxins that are under clinical evaluation and their native mechanism of action. For each toxin, the immunotoxins that are derived from it are described, and clinical trials that were conducted are detailed mainly in terms of patient responses.
3.1. Pseudomonas Exotoxin A-Based Immunotoxins
Pseudomonas exotoxin A (PE) is a bacterial toxin originally produced by the bacterium Pseudomonas aeruginosa as one of its virulence factors [26]. PE is one of the most common toxins used as toxin moiety in targeted cancer therapy. The reasons for that are that PE is one of the most investigated toxins and is easy to produce in E. coli [15,27,28,29,30,31,32,33]. Another important feature of PE is that it withstands many mutations without harming its toxicity. This characteristic enables raising PE stability and dramatically lowering immunogenicity, making it an even better candidate for cancer therapy [20,21,34,35,36,37,38]. When attached to an antibody-based targeting moiety, PE is usually shortened to a 38kDa or 40kDa fragment (named PE38 and PE40 respectively), eliminating its natural non-selective targeting moiety. Table 1 and Table 2 respectively list the clinically evaluated/under evaluation PE-based immunotoxins against hematologic malignancies and solid tumors.

{kind=link}
{kind=link}
Table 1.
Clinically evaluated/under evaluation PE-based immunotoxins against hematologic malignancies.
| Immunotoxin | Target antigen | Toxic moiety | Target malignancy | Clinical trial phase (Year the trial ended) | References |
|---|---|---|---|---|---|
| LMB-2 * | CD25 | PE38 | Leukemia, lymphoma | II (ongoing) | [39,40] |
| RFB4(dsFv)-PE38 (BL22/CAT3888) | CD22 | PE38 | NHL, CLL, HCL, ALL | I,II (2008) | [25,41,42,43] |
| Mutated RFB4(dsFv)-PE38 (HA22/CAT-8015) | CD22 | PE38 | HCL, ALL, NHL, CLL, PLL, SLL | I (2012 **) | [44,45] |
** Year of publication (Year of trial unknown); Abbreviations: CLL: chronic lymphocytic leukemia; dsFv: disulfide-stabilized variable fragment of an antibody; HCL: hairy cell leukemia; NHL: non-Hodgkin’s lymphoma; PLL: prolymphocytic leukemia; SLL: small lymphocytic leukemia.
| Immunotoxin | Target antigen | Toxic moiety | Target malignancy | Clinical trial phase (Year the trial ended) | References |
|---|---|---|---|---|---|
| LMB-2 * | CD25 | PE38 | Metastatic melanoma | I (2008) | [50] |
| OVB3-PE | Ovarian Antigen | Full length PE | Ovarian cancer | I (1991 **) | [22] |
| ERB-38 | erbB2/HER2 | PE38 | Breast, esophageal cancers | I (1999 **) | [51] |
| SS1(dsFv)PE38 (SS1P) | Mesothelin | PE38 | Mesothelioma, ovarian, pancreatic cancers | I (Ongoing) | [52,53] |
| LMB-1 | Lewis Y | PE38 | Adenoca | I (1996) | [54] |
| B3(Fv)-PE38 (LMB-7) | Lewis Y | PE38 | Adenoca | I (2011) | [15] |
| B3(dsFv)-PE38 (LMB-9) | Lewis Y | PE38 | Adenoca | I (2009) | [15] |
| BR96sFv-PE40 (SGN-10) | Lewis Y | PE40 | Adenoca | I (2002 **) | [55] |
| scFv(FRP5)-ETA | erbB2/HER2 | PE40 | Melanoma, breast, colon cancers | I (2005 **) | [56,57] |
* Immunotoxin that was tested both for hematologic malignancies and solid tumor therapy; ** Year of publication (Year of trial unknown); Abbreviations: Adenoca: adenocarcinoma; scFv: single-chain variable fragment of an antibody.
3.1.1. Pseudomonas Exotoxin A—Mechanism of Action
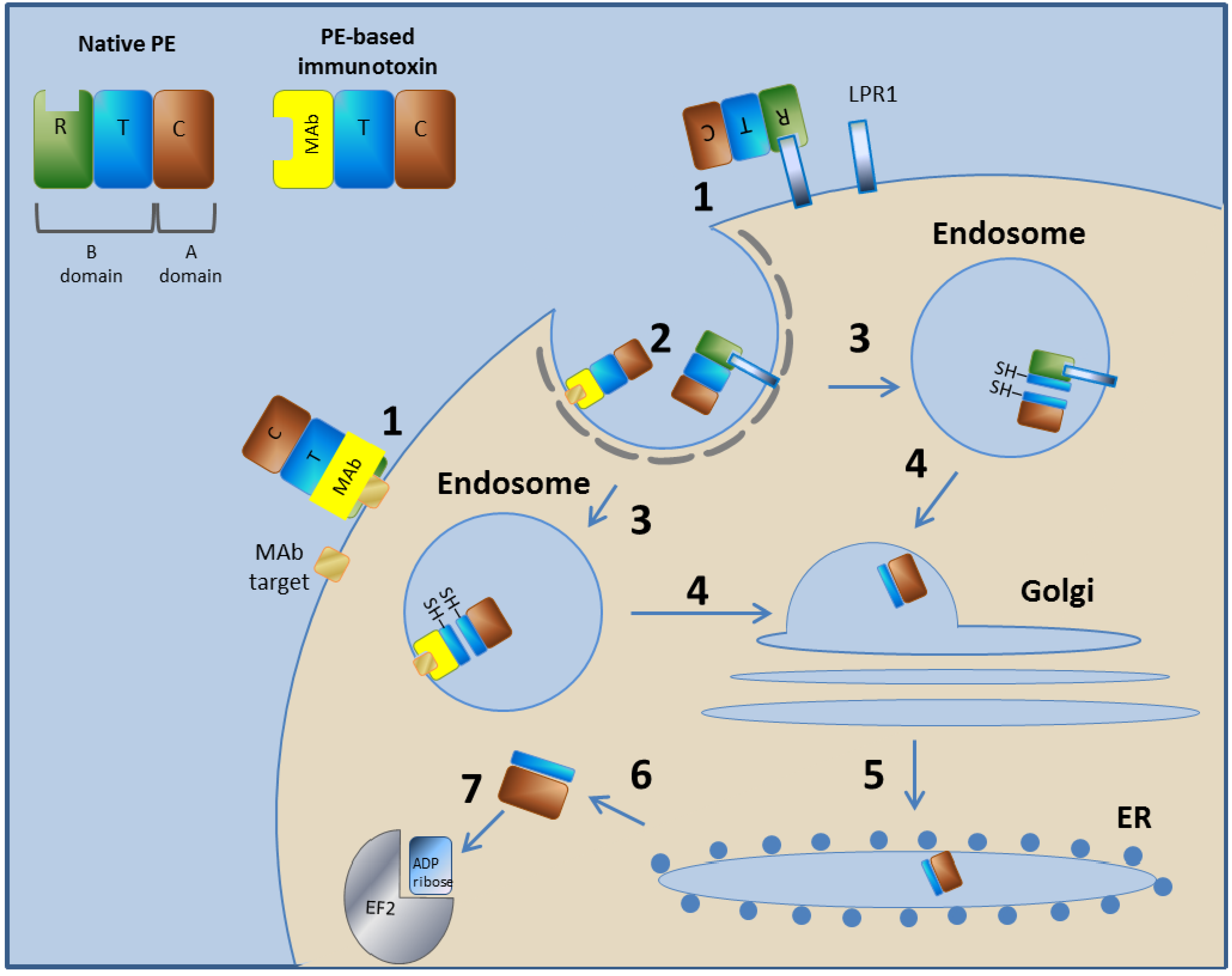
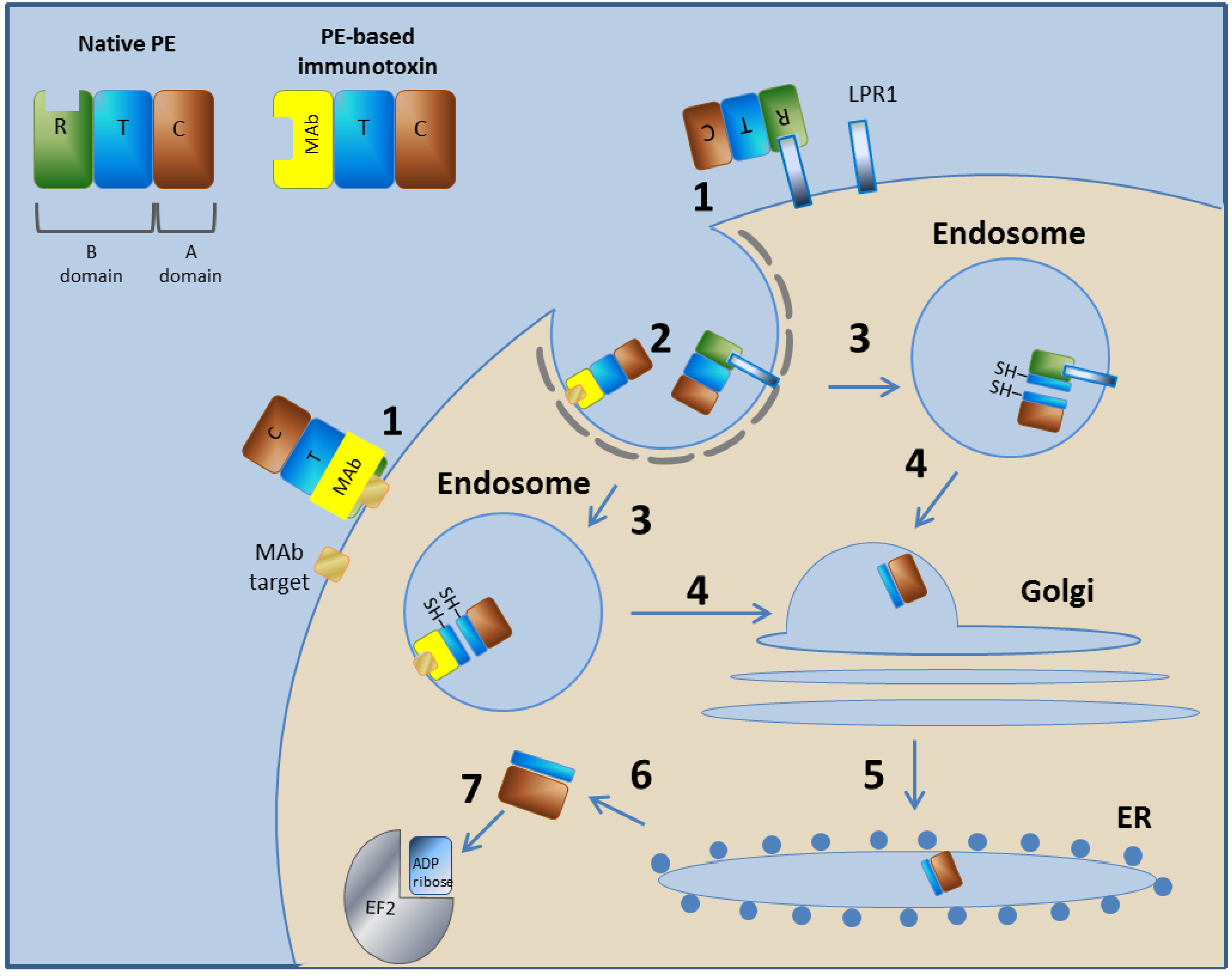
PE is a 613-amino acid protein with AB toxin structure-function properties, meaning both the A and B domains are encoded on a single protein. The A domain encodes the catalytic (C) domain, and the B domain comprises a translocation (T) domain that facilitates translocation of the A domain into the cytoplasm of the cells into which it is internalized and a receptor binding (R) domain that facilitates the binding of the toxin to cell surface receptors [26]. The N terminus encodes the R domain, the internal domain encodes the T domain, and the C terminus possesses the adenosine diphosphate (ADP)-ribosyltransferase A domain. PE binds to the low-density lipoprotein (LDL) Receptor Related Protein 1 [46,47], enters cells via receptor-mediated endocytosis [48], retrograde traffics within the cell to the endoplasmic reticulum (ER), where the A domain is translocated across the ER membrane and delivered into the host cytoplasm [49], and finally the catalytic ADP ribosylation of elongation factor II occurs, leading to apoptotic cell death [40] (Figure 1).
Figure 1.
Mechanism of action of PE and PE-based immunotoxins. In step 1, the toxin binds to its cell-surface receptor (LPR1). The immunotoxin binds to the target molecule specified by its targeting moiety. Steps 2–7 are common to PE and to the PE-based immunotoxins. In step 2, the toxin/immunotoxin undergoes receptor-mediated endocytosis into clathrin-coated pits. In step 3, in the endosome, the T subunit is cleaved by the Furin protease and then reduced to create two separated domains. In step 4, the C subunit, along with most of the T subunit (C+T), traffics in a retrograde manner within the cell to the Golgi system. In step 5, the C+T subunit is further retrograde transported to the ER. In step 6, the C+T subunit translocate from the ER to the cytoplasm, where, in step 7, the C subunit inactivates translation elongation factor II by catalytic ADP ribosylation. Diphtheria toxin (DT) and DT-based immunotoxins use a different mechanism as they escape from the endosome directly to the cytoplasm.
Figure 1.
Mechanism of action of PE and PE-based immunotoxins. In step 1, the toxin binds to its cell-surface receptor (LPR1). The immunotoxin binds to the target molecule specified by its targeting moiety. Steps 2–7 are common to PE and to the PE-based immunotoxins. In step 2, the toxin/immunotoxin undergoes receptor-mediated endocytosis into clathrin-coated pits. In step 3, in the endosome, the T subunit is cleaved by the Furin protease and then reduced to create two separated domains. In step 4, the C subunit, along with most of the T subunit (C+T), traffics in a retrograde manner within the cell to the Golgi system. In step 5, the C+T subunit is further retrograde transported to the ER. In step 6, the C+T subunit translocate from the ER to the cytoplasm, where, in step 7, the C subunit inactivates translation elongation factor II by catalytic ADP ribosylation. Diphtheria toxin (DT) and DT-based immunotoxins use a different mechanism as they escape from the endosome directly to the cytoplasm.
3.1.2. LMB-2
LMB-2 is a third generation anti-CD25 immunotoxin. It is composed from the variable domains VH and VL of the murine anti-CD25 MAb anti T-cell activation antigen (Tac), fused in a scFv form via a peptide linker (G4S)3, and the VL is further fused at its C-terminus to PE38 via a short connector ASGGPE [40]. CD25 is the α chain of the interleukin-2 receptor (IL-2R) [58]. It is not expressed on resting lymphocytes and stem cells but is efficiently induced after T-cell activation. IL-2Rs have been detected on cells at high levels in autoimmune disorders and on malignant cells in different hematopoietic malignancies [59]. LMB-2 was evaluated in a phase I study that was completed in 2011. In this trial, conducted in patients with hematologic malignancies, out of 35 patients, 1 showed complete response (CR) and 7 showed partial response (PR), a total response rate of 24% [19,39,60]. It is important to mention that 4 out of 4 of the HCL patients responded (1 CR and 3 PRs). The activity has also been observed in a follow-up phase II trial in HCL [40]. Another phase I trial was conducted on 8 patients with metastatic melanoma in order to induce antitumor response. This trial ended in 2008. In this trial, partial reduction in regulatory T-cell (Treg) frequency in the peripheral blood and at the tumor site was detected, although there was no tumor regression [50]. A phase II trial for HCL and a phase II trial of LMB-2, fludarabine and cyclophosphamide for adult T-cell leukemia are still ongoing [61,62].
Among HCL patients, 10% are CD25 negative. Although it is a minority, since they do not respond to cladribine, their percent figures among relapsed/refractory HCL patients are more significant. This fact promoted the development of anti-CD22 immunotoxins, since all HCL patients are CD22 positive [40].
3.1.3. RFB4(dsFv)-PE38 (BL22/CAT3888)
The recombinant immunotoxin, RFB4(dsFv)-PE38 (BL22, CAT-3888), contains the variable domains of the anti-CD22 murine MAb RFB4, in the form of a dsFv fused to PE38 [15,63]. CD22 is an adhesion molecule that is expressed exclusively on B cells and common on tumor cells of B-cell lymphomas [64,65,66,67,68,69]. In a phase I trial conducted on 16 HCL patients, 11 showed complete remissions and one showed partial remission. Three of the complete remission patients relapsed a year or less after treatment. This trial ended in 2001 [41]. In a phase I trial conducted on 46 patients with B-cell malignancies, 19 patients showed CR, all of which were HCL patients, and 7 showed PR: 6 HCL patients and 1 CLL patient. All 3 CD25 negative patients were among the CR patients. This trial was completed in 2009 [42]. In a phase II trial conducted on 36 HCL patients, 17 patients showed CR and 4 patients showed PR. This trial was completed in 2008 [43]. In a pediatric phase I trial that was conducted on 23 CD22-Positive hematologic malignancies of children patients, no CR or PR were observed, although activity was achieved. This trial also ended in 2008 [25].
3.1.4. Mutated RFB4(dsFv)-PE38 (HA22/CAT-8015)
Since BL22 showed a low response rate in CLL patients, with an intention to increase the affinity of the antibody, mutations were introduced to complementarity-determining regions in the antibody variable domain (CDRs) of the RFB4(dsFv) fragment, producing mutated RFB4(dsFv)-PE38 (HA22/CAT-8015) [70]. In a phase I trial reported this year (2012), in which 28 HCL patients were treated with the immunotoxin, 13 patients showed CR and 11 showed PR [40,44].
3.1.5. OVB3-PE
OVB3-PE is an immunotoxin consisting of the murine IgG2b MAb OVB3 that was coupled with the whole native PE by a thioether bond. OVB3 reacts with a variety of ovarian cancer cell lines, fresh ovarian cancer tissue, and ascites specimens with minimal reactivity with normal tissues [22]. In a phase I trial that was reported in 1991, 23 ovarian cancer patients were treated with OVB3-PE. No partial or complete responses were observed [22]. Further clinical trials were not conducted since OVB3 MAb was found to be not selective enough to cancer cells.
3.1.6. ERB-38
ERB-38 is composed of the Fv portion of the murine MAb e23 fused to PE38 [51,71]. MAb e23 was found to be very active as a PE38 linked immunotoxin, and therefore was chosen from a variety of erbB2 binding MAbs [72]. ErbB2 (also named Her2/neu) is highly expressed in many breast cancers and several other cancers [15,51]. In a phase I study that was reported in 1999, 5 breast cancer patients and 1 esophageal cancer patient were treated with ERB-38. In this trial, no responses were observed, however, hepatotoxicity was observed in all patients [51].
3.1.7. SS1(dsFv)PE38 (SS1P)
SS1(dsFv)PE38 (SS1P) is composed of the dsFv fragment of the SS1 murine MAb fused to PE38 [73]. SS1 MAb binds with high affinity to mesothelin, a differentiation antigen expressed on normal mesothelial cells and highly expressed in mesothelioma, as well as in ovarian and pancreatic cancers [74,75]. Two phase I trials were conducted on patients with mesothelin-expressing cancers, such as ovarian cancer, malignant mesothelioma and pancreatic adenocarcinoma. Both trials were carried out between the years 2000–2009 [52]. The trials differ in the dosing method that was used. In one trial, SS1P was given by 30-min infusion every 2 days for 3–6 doses to 34 patients. Only 4 patients showed minor responses [52]. In the other trial, SS1P was given by continuous infusion over 10 days, in order to saturate the tumor, to 24 patients. Only 1 patient showed a PR [53]. The responses were minor but significant, therefore, phase II evaluation of SS1P for mesothelin-expressing malignancies might be pursued, and current clinical trials are evaluating it in combination with chemotherapy [15,76,77].
3.1.8. LMB-1
LMB-1 (also known as B3-LysPE38) is composed of the murine MAb B3 fused to PE38 [78]. LMB-1 was the first PE-based immunotoxin to show evidence of clinical activity in a phase I clinical trial [15]. B3 reacts with the carbohydrate antigen Lewis Y and with several closely related carbohydrate antigens found in carcinomas of the breast, colon, stomach, esophagus, ovary and lung [79]. In a phase I study carried out between 1993–1996 and conducted on 38 patients with malignant solid tumors expressing Lewis Y antigen, 1 patient showed CR and 1 patient showed PR [54]. Further development of LMB-1 was not pursued; instead, other recombinant immunotoxins (LMB-7, LMB-9, BR96-sFv-PE40) that targeted the Lewis Y antigen were produced and evaluated in clinical trials of patients with Lewis Y-expressing malignancies [15,55].
3.1.9. B3(Fv)-PE38 (LMB-7)
LMB-7 is composed of the scFv of the murine MAb B3 fused to PE38. LMB-7 was tested in a phase I trial conducted on patients with leptomeningeal metastases. In this trial that began in 1997, no significant antitumor activity was observed [15]. No further data were published.
3.1.10. LMB-9
LMB-9 is composed of the dsFv of murine MAb B3 fused to PE38. LMB-9 was tested in 2 phase I trials conducted on patients with advanced carcinomas that express the Lewis Y antigen. In these trials that began in 1998 and 2000, as in the case of LMB-7, no significant antitumor activity was observed [15], and no further data were published.
3.1.11. BR96sFv-PE40 (SGN-10)
BR96sFv-PE40 (SGN-10) is composed of the VH and VL fragments of the BR96 murine Mab, linked together in a scFv format by a 15-amino-acid flexible linker, fused via a 7-amino-acid linker to PE40 [80,81]. In the phase I trial, reported in 2002 and conducted on 46 patients with Lewis Y-positive metastatic carcinoma, no objective responses were observed [55].
3.1.12. scFv(FRP5)-ETA
ScFv(FRP5)-ETA was constructed from the scFv of the humanized FRP5 MAb fused to PE38. FRP5 binds the erbB2 antigen [82,83]. A phase I trial, reported in 2003, was conducted on 11 patients with metastatic breast and colorectal cancers and malignant melanoma. The treatment was given by intra-tumoral injection. Out of 11 patients, 4 showed complete remission and 3 showed partial remissions [56]. Another phase I trial was reported in 2005. This trial was conducted on 18 patients suffering from erbB2-expressing metastatic breast cancers, prostate cancers, head and neck cancer, non-small cell lung cancer (SCLC), or transitional cell carcinoma. In this trial, no objective responses were observed [57].
3.2. Ricin Toxin-Based Immunotoxins
RT is a 60 kDa type II ribosome inactivating protein (RIP) produced in the seeds of the castor oil plant (Ricinus communis). Type II RIPs, such as ricin, are composed of two domains connected by a disulfide bond. The A chain is the catalytic domain of the RIP and the B chain is a galactose binding lectin. The lectin chain can bind to galactosyl moieties of glycoproteins and/or glycolipids found on the surface of eukaryotic cells [84]. Ricin was selected as a candidate for therapy because of its high potency, and raised the interest in other RIPs as well [85,86]. The development of MAbs enabled the coupling of ricin to a specific targeting agent [84]. Because the ricin toxin A chain (RTA) by itself has an unwanted affinity to hepatic cells [87,88,89], several strategies have been developed to prevent this binding. Deglycosylated A chain (dgA) is one strategy that has been taken, since the glycosylated side residues of RTA are responsible for the non-specific binding to hepatic cells [87,88,90]. Since the B chain is known to reduce the cytotoxicity of the A chain [91], another strategy, named blocked-ricin (bR), was developed. In this strategy, the whole ricin toxin is used and its oligosaccharide-binding sites are blocked [13,14]. Unfortunately, RIP-derived immunotoxins were found to have some clinical disadvantages. For example, they are very immunogenic and many times caused vascular leak syndrome (VLS), which limited clinical efficacy as a cancer therapy [92]. Attempts to refine the toxins in order to solve these problems are being made [93,94]. Table 3 and Table 4 list the clinically evaluated/under evaluation RT-based immunotoxins against hematologic malignancies and solid tumors, respectively.
Table 3.
Clinically evaluated/under evaluation RT-based immunotoxins against hematologic malignancies.
| Immunotoxin | Target antigen | Toxic moiety | Target malignancy | Clinical trial phase (Year the trial ended) | References |
|---|---|---|---|---|---|
| RFB4-Fab'-dgA | CD22 | dgA | B-NHL | I (1991 **) | [64] |
| RFB4-dgA (IMTOX-22) | CD22 | dgA | B-NHL, CLL | I (2001) | [104,105] |
| HD37-dgA (IMTOX-19) | CD19 | dgA | NHL | I (1996 **) | [106,107] |
| RFB4-dgA + HD37-dgA (Combotox) | CD22, CD19 | dgA | NHL, ALL | I (Ongoing) | [108,109] |
| RFT5-dgA (IMTOX-25) * | CD25 | dgA | HD, CTCL, GVHD, haploidentical SCT | I,II (Ongoing) | [24,110,111,112,113] |
| Ki-4.dgA | CD30 | dgA | HD, NHL | I (2002 **) | [112,114] |
| Anti-B4-bR | CD19 | Blocked ricin | B-NHL | II,III (1992-2011 **) | [115,116,117,118,119,120] |
| Anti-CD7-dgA (DA7) | CD7 | dgA | T-NHL | I (1997 **) | [121] |
| H65-RTA | CD5 | RTA | CTCL, GVHD | I, I/II (1991 **) | [122,123,124] |
| T101-RTA | CD5 | RTA | CLL | I (1989 **) | [125,126,127] |
** Year of publication (Years of trial unknown); Abbreviations: ALL: acute lymphoblastic leukemia; B-NHL: B-cell non-Hodgkin’s lymphoma; CTCL: cutaneous T cell lymphomas; GVHD: graft-versus-host disease; SCT: stem cell transplantation; T-NHL: T-cell non-Hodgkin’s lymphoma.
| Immunotoxin | Target antigen | Toxic moiety | Target malignancy | Clinical trial phase (Year the trial ended) | References |
|---|---|---|---|---|---|
| RFT5-dgA (IMTOX-25) * | CD25 | dgA | Melanoma | I,II (Ongoing) | [128] |
| Anti-CEA-bR | CEA | Blocked ricin | Colorectal cancer | I/II (1994 **) | [129] |
| N901-bR | CD56 | Blocked ricin | SCLC | I,II (2002 **) | [130,131,132,133] |
| XomaZyme-Mel (XMMME-001-RTA) | Melanoma antigen | RTA | Melanoma | I
II with CTL I/II with cyclosporine (1993 **) | [134,135,136,137,138,139] |
| XomaZyme-791 (79IT/36-RTA) | 72 kDa TAA | RTA | Colorectal cancer | I (1995 **) | [140,141,142] |
| 454A12-rRA | TfR | RTA | Leptomeningeal neoplasia | I (1997 **) | [143] |
| 260F9-rRTA | 55 kDa Breast cancer antigen | RTA | Breast cancer | I (1989) | [23,144] |
* Immunotoxin that was tested both for hematologic malignancies and solid tumor therapy; ** Year of publication (Years of trial unknown); Abbreviations: CEA: carcinoembryonic antigen; rRA: recombinant ricin A chain.
3.2.1. Ricin Toxin—Mechanism of Action
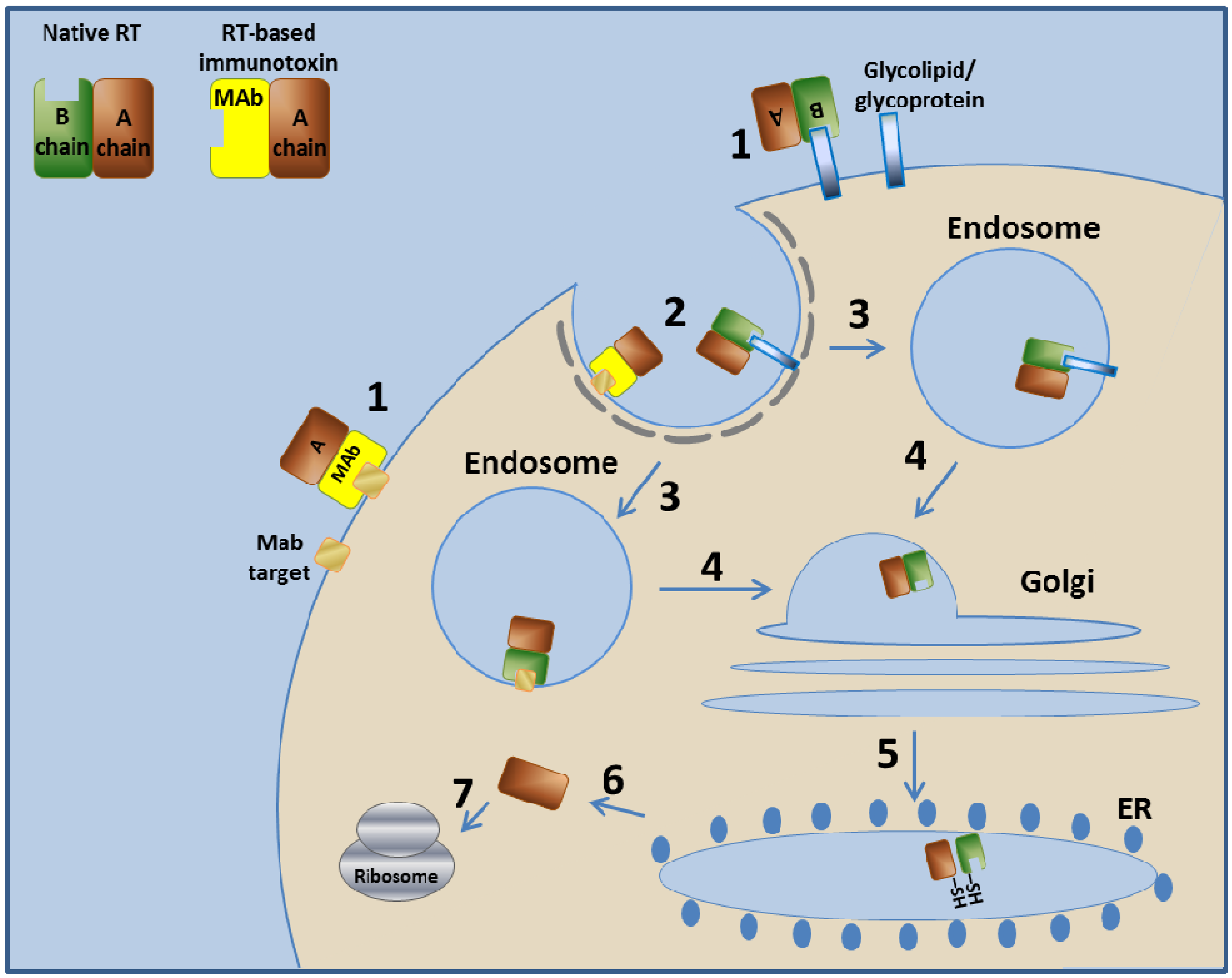
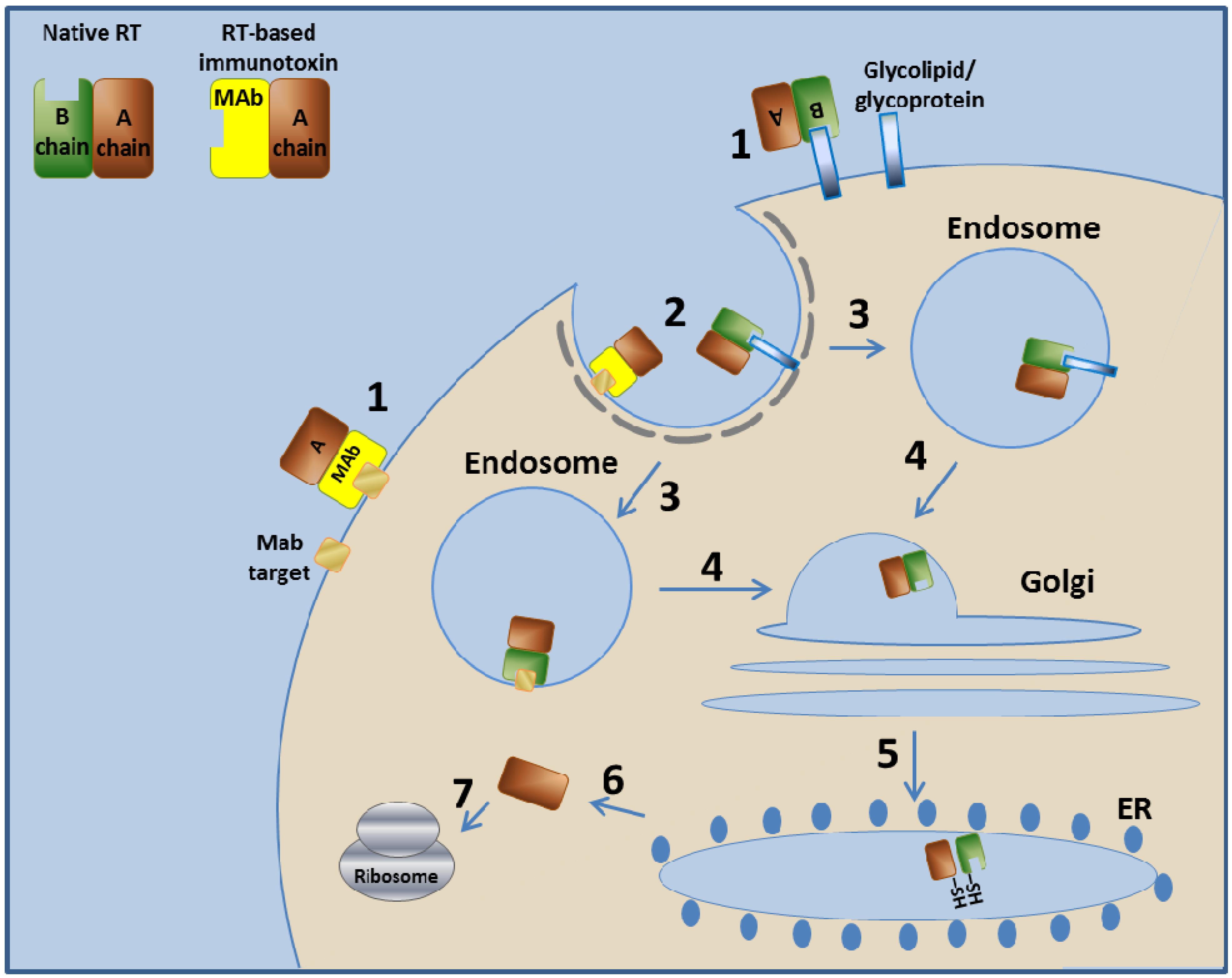
The native RT binds the target cell via glycolipids or glycoproteins found on the cell surface. The ricin enters the cell by endocytosis and is next transported to the endosome. The endosome unites with the Golgi system and then with the ER, where the A and B chains of ricin are separated by disulfide isomerase that reduces the disulfide bond between them [95,96]. From the ER, the A chain is retrograde-translocated to the cytosol, where the RTA, a glycosidase, inactivates ribosomes by removing an adenine from position 4324 of the 28S ribosomal RNA of the 60S ribosomal subunit [97,98,99]. This action eventually stops protein production in the cell and leads to cell death [100,101,102,103] (Figure 2).
Figure 2.
Mechanism of action of RT and RT-based immunotoxins. In step 1, the toxin binds to its cell-surface receptor (Glycolipid/ glycoprotein). The immunotoxin binds to the target molecule specified by its targeting moiety. Steps 2–7 are common to RT and to the RT-based immunotoxins. In step 2, the toxin/immunotoxin undergoes receptor-mediated endocytosis into clathrin-coated pits. In step 3, the toxin/immunotoxin enters the endosome. In step 4, the toxin/immunotoxin retrograde traffics within the cell to the Golgi system. In step 5, the toxin/immunotoxin further travels to the ER where the disulfide bond between the A chain and the B chain/MAb is enzymatically reduced. In step 6, the A chain translocates to the cytoplasm, where, in step 7, it inactivates the ribosome. Immunotoxins based on other type II RIPs use a similar mechanism.
Figure 2.
Mechanism of action of RT and RT-based immunotoxins. In step 1, the toxin binds to its cell-surface receptor (Glycolipid/ glycoprotein). The immunotoxin binds to the target molecule specified by its targeting moiety. Steps 2–7 are common to RT and to the RT-based immunotoxins. In step 2, the toxin/immunotoxin undergoes receptor-mediated endocytosis into clathrin-coated pits. In step 3, the toxin/immunotoxin enters the endosome. In step 4, the toxin/immunotoxin retrograde traffics within the cell to the Golgi system. In step 5, the toxin/immunotoxin further travels to the ER where the disulfide bond between the A chain and the B chain/MAb is enzymatically reduced. In step 6, the A chain translocates to the cytoplasm, where, in step 7, it inactivates the ribosome. Immunotoxins based on other type II RIPs use a similar mechanism.
3.2.2. RFB4-Fab'-dgA
RFB4-Fab'-dgA immunotoxin is composed of the Fab moiety of the murine IgGl-κ anti-CD22 antibody RFB4 [145], coupled to chemically dgA [146]. In a phase I trial, reported in 1991, 15 patients of B-cell lymphomas were treated with the immunotoxin. In this trial, 5 patients have shown PR that lasted up to 4 months, out of the 14 patients that were evaluated [64].
3.2.3. RFB4-dgA (IMTOX-22)
RFB4-dgA (also known as IgG-RFB4-SMPT-dgA) was prepared by conjugating the IgG, murine monoclonal anti-CD22 antibody RFB4 to dgA using the heterobifunctional hindered disulfide linker, N-succinimidyl-oxycarbonyl-cu-methyl-a-(2-pyridyldithio) toluene (SMPT) [146,147]. RFB4-dgA was tested in 2 phase I studies. The first trial was a dose escalation study on 26 patients of B-cell lymphomas that were found to be resistant to conventional therapy. In this trial, reported in 1993, one patient showed CR and five patients showed PR [104]. The second trial was a continuous infusion study in 18 patients with B-cell lymphomas, carried out between the years 1991–2001. In this trial, four patients showed PR [105]. It is important to mention that as a contrast to the common development of immunotoxins, in which the antibody-based fragment gets smaller as research develops, in the case of RFB4-dgA, the Fab fragment was tested in clinical trials prior to the full IgG. The two were compared and no significant advantage was found in one over the other [104]. The RFB4 antibody was also tested later as a dsFv fragment fused to PE38, as detailed earlier in this chapter.
3.2.4. HD37-dgA (IMTOX-19)
HD37-dgA (also known as IgG-HD37-SMPT-dgA) was prepared by conjugating the murine MAb anti-CD19, HD37, to dgA using the disulfide linker SMPT. CD19 is expressed on all normal B cells from the pre-B cell stage to the plasma cell [69,145,148]. HD37-dgA was tested in two concomitant phase I trials in which two regimens for the administration of HD37-dgA to patients with NHL were used. One trial, which examined four intermittent bolus infusions administered at 48-hour intervals, was reported on 1995. In this trial, conducted in eight patients with NHL, one patient showed near CR but then died on day 8 of the experiment. No other responses were observed [107]. The other phase I trial, reported in 1996, studied a continuous infusion administered over the same 8-day period. This phase I trial was performed in order to support the notion of using HD37-dgA in a cocktail with RFB4-dgA to treat NHL (as discussed later). In this phase I trial conducted on 23 patients of B-cell lymphoma, two patients showed partial remission of up to two months and one patient showed CR [106].
3.2.5. RFB4-dgA + HD37-dgA (Combotox)
RFB4-dgA + HD37-dgA (Combotox) is a drug combination of a 1:1 ratio of the RFB4-dgA and HD37-dgA immunotoxins. CD19 and CD22 are expressed on greater than 90% and 60–80% of B lymphomas, respectively [149]. The rationale for using Combotox in this setting is to avoid the emergence of antigen-negative variants [109], in addition to the additive activity of the two immunotoxins to each other, as was detected in the preclinical studies [150]. In a phase I trial, reported in 2000 and conducted on 22 patients with refractory CD19+- CD22+ B cell lymphoma, 2 patients showed partial remission [109]. Another phase I study using combotox was conducted on 17 pediatric patients with refractory B-lineage ALL. In this trial, reported in 2009, 3 patients experienced complete remission. Six additional patients experienced a significant hematological improvement (HI): a decrease of 75–100% in their peripheral blood blast counts [108]. A third phase I trial of combotox with cytarabine in relapsed/refractory B-lineage is now ongoing [151].
3.2.6. RFT5-dgA (IMTOX-25)
RFT5-dgA contains the anti-CD25 RFT5 murine Mab, chemically conjugated to dgA using the disulfide linker SMPT [110]. In a phase I study, reported in 1997 and conducted on 15 patients with refractory Hodgkin’s Lymphoma (HL), two patients showed partial remission [110]. The immunotoxin in this trial was administered intravenously over four hours, specifically on days 1, 3, 5 and 7. Since this administration method seemed to cause side effects, an extension to this trial was conducted as five more patients were treated at the maximal tolerated dose (MTD). No objective responses were observed [111]. Later on, 13 additional patients were added to this trial, two of which showed partial remission. This trial was last reported in 2000 [113]. RFT5-dgA was also used in an attempt to prevent GVHD in patients undergoing allogeneic SCT but unexpectedly produced a higher incidence of grade III/IV GVHD than historical controls, as reported in 2004 [24]. Phase I trials for depleted alloreactive cells, prior to and after haploidentical SCT, were also conducted between the years 2000–2009, but no data regarding those trials were published. RFT5-dgA is currently evaluated in phase I/II trials for metastatic melanoma and in a phase II trial for Refractory/Relapsed CD25 Positive Adult T Cell Leukemia/Lymphoma [152].
3.2.7. Ki-4.dgA
Ki-4.dgA is an anti-CD30 immunotoxin that was constructed by coupling the murine MAb Ki-4 via SMPT linker to dgA. The lymphocyte activation marker CD30 is expressed consistently on HL cells, while normal human organs revealed no major cross-reactivity to the anti-CD30 MAbs [153]. In a phase I trial in 17 patients with refractory CD30 positive HL and NHL, reported in 2002, one partial remission was observed out of 15 patients that have been evaluated [114].
3.2.8. Anti-B4-bR
Anti-B4-bR immunotoxin was constructed from the anti-CD19 murine MAb B4, chemically linked to bR using succinimidyl 4-(N-maleimidomethyl) cyclohexane carbovlate (SMCC) linker [120]. A phase I trial of intermittent bolus administration in 25 patients of B-cell neoplasms was conducted. In this trial, reported in 1992, one complete remission and two partial remissions were observed [120]. Another phase I trial of continuous infusion administration was conducted on 43 patients of B-cell neoplasms. In this trial, reported in 1993, two complete remissions and two partial remissions were observed [117]. Unfortunately, the following phase II trial, conducted on 16 NHL patients did not show any objective response. This trial was reported in 1998 [115]. Subsequent trials reported between the years 1998–2003, used anti-B4-bR in combination with chemotherapy, and no activity was observed [154,155,156]. Anti-B4-bR was tested also in phase I, II and III as adjuvant therapy to post-autologous bone marrow transplant in B-cell lymphoma patients. Although the phase I and II trials suggested a possible role for anti-B4-bR as consolidation after high-dose chemotherapy and autologous SCT, the phase III trial results failed to show any connection between anti-B4-bR administration and even free survival of patients. These trials were reported between the years 1993–2011 [116,118,119].
3.2.9. Anti-CD7-dgA (DA7)
Anti-CD7-dgA was constructed from the 3Ale murine MAb that selectively binds CD7, linked via N-succinimidyl 3-(2-pyridyl-dithioproprionic acid) (SPDP) linker to dgA [121]. CD7 is a 40 kDa transmembrane glycoprotein antigen that is known to appear on the majority of malignant T cells [157,158]. In a phase I trial for T-NHL patients, two out of 11 patients showed PR. No other activity was observed [121]. This trial was reported in 1997 and no further clinical trials were conducted.
3.2.10. H65-RTA
H65-RTA comprises a murine MAb that recognizes the CD5 lymphocyte differentiation antigen coupled to RTA. CD5, a 65 kDa glycoprotein is among the T-cell surface antigens expressed by CTCLs. The expression of CD5 by CTCL, while heterogeneous, is often present in higher densities than on normal lymphocytes, is identical between malignant lymphocyte infiltrating different sites, and appears to be consistent over time [159,160]. In a phase I trial for CTCL patients, four out of 14 showed a PR that was not clearly related to the dose of H65-RTA [124]. This trial was reported in 1991 and phase II trials were not pursued. H65-RTA was also tested in a phase I trial for GVHD as reported in 1990, where no apparent relationship was observed between the dose level and the response rate [123].
3.2.11. T101-RTA
T101-RTA is a conjugate of the RTA via disulfide bond to the T101 murine MAb that also targets CD5. T101 does not bind to other hematopoietic cells or to non-hematopoietic tissue [161]. In a phase I trial conducted on 4 leukemia patients, no objective reaction was detected. This trial was last reported in 1988 [125,127]. In another phase I trial, T101-RTA was administered in the presence of monensin, covalently linked to human serum albumin (HAS) for cytotoxicity enhancement. In this trial, conducted on 5 B-cell chronic lymphocytic leukemia (B-CLL) patients, no long term clinical effect was observed [126]. These trials were reported in 1989 and no further clinical trials were pursued.
3.2.12. Anti-CEA-bR
Anti-CEA-bR is composed of the murine MAb I-1 that targets anti-CEA conjugated to blocked ricin via disulfide bridges [162,163]. CEA is a glycoprotein normally expressed on fetal cells. The presence of CEA on adult cells has been connected to colon carcinoma where it serves as an adhesion agent [164]. A phase I/II study of the intralesional administration of Anti-CEA-bR that was conducted on 27 patients with hepatic metastases of gastrointestinal origin was conducted. In this trial, no major objective responses or changes in the growth rate of injected lesions were observed. This trial was reported in 1994 and overall, the therapeutic approach of intralesional administration was determined as unsuccessful [129].
3.2.13. N901-bR
N901-bR is an immunotoxin comprising the N901 murine MAb and bR. N901 recognizes the natural killer cell-associated antigen, a neural cell adhesion molecule isoform that is expressed on SCLC cell lines and fresh human tumors [14]. In a phase I study in 21 patients with relapsed or refractory SCLC, only one patient showed PR out of the 20 patients that have been evaluated. This trial was last reported in 1997 [130,131,132]. The phase II trial in nine SCLC patients was unsuccessful, since no objective response to N901-bR was observed. Moreover, it was associated with a fatal incident of capillary leak syndrome (CLS) and a nearly universal development of human anti-mouse antibodies (HAMA) and human anti-ricin antibodies (HARA). After this phase II that was reported in 2002, no other trials were conducted using N901-bR [133].
3.2.14. XomaZyme-Mel (XMMME-001-RTA)
XomaZyme-Mel is a conjugate between a murine anti-melanoma IgG2a MAb (XMMME-001) and RTA [135]. XMMME-001 targets melanoma-associated antigens having molecular weights of 220,000 and over 500,000 [134]. XomaZyme-Mel was tested in several clinical trials for metastatic melanoma patients, as a single treatment and together with cyclophosphamide or cyclosporine. In a phase I study of XomaZyme-Mel as a single treatment in 20 patients, one CR of more than 12 months’ duration was observed. This trial was reported in 1991 [138]. In a phase II study with cyclophosphamide as an antibody response blocker in 20 patients with metastatic malignant melanoma, an overall response rate of 20% was observed, but no decrease in HAMA or HARA was detected. This study was reported in 1990 [137]. In a phase I/II study with cyclosporine as an antibody response blocker in nine patients with metastatic melanoma, one patient showed partial remission [139]. This last trial was reported in 1993 and further trials were not pursued.
3.2.15. XomaZyme-791 (79IT/36-RTA)
XomaZyme-791 is composed of the murine IgG2b MAb XMMCO-791, conjugated via a disulfide bond to RTA. XMMCO-791 recognizes a 72 kD glycoprotein on tumor cells [165,166]. A Phase I dose escalation study was carried out in 17 patients with metastatic colorectal cancer. In this trial, reported in 1989, two patients showed decreasing sizes of large metastases and the disappearance of smaller lesions [142]. Another phase I trial in seven patients with metastatic colorectal cancer was conducted to test humoral immune responses. In this trial, reported in 1989 and 1995, all patients showed very high levels of HAMA and HARA [140]. Overall, no antitumor activity was detected [141]. No further trials were conducted.
3.2.16. 454A12-rRA
454A12-rRA immunotoxin is a chemically-linked conjugate of a murine MAb targeting the human transferrin receptor (454A12) and rRA via disulfide linkage [143]. Transferrin receptors transport iron-transferrin complexes into cells and are overexpressed on rapidly dividing cells, including cancer cells, reflecting increased iron requirements [167,168,169,170,171,172,173,174]. A pilot phase I trial of intraventricular therapy for eight leptomeningeal neoplasia patients was conducted. In this trial, only one patient experienced clinical improvement. However, no patient had their cerebrospinal fluid (CSF) cleared of tumor, and evidence of tumor progression was demonstrated in seven of the eight patients after treatment [143]. This trial was reported in 1997 and no further trials were conducted.
3.2.17. 260F9-rRTA
260F9-rRTA was constructed from the IgGl murine MAb 260F9, conjugated to rRA via a disulfide bond [175]. 260F9 is specific for a 55 kDa antigen expressed by approximately 50% of breast cancer cells [176]. Two phase I trials were conducted using 2 different administration schedules: by daily injection on four patients [23], and by continuous infusion on five patients [144]: all patients with metastatic breast carcinoma. In these trials it was found that 260F9-rRTA binds to human cells that do not express the antigen identified by 260F9 via uptake by cellular constant fragment of an antibody heavy chain (Fc)-γ receptors, which caused severe toxic effects, including marked fluid overload and debilitating sensorimotor neuropathies [23,144]. Because of the severe toxicity of 260F9-rRTA, trials were suspended in 1989, and no further trials were conducted using this immunotoxin.
3.3. Additional Toxin-Based Immunotoxins Evaluated/Under Clinical Evaluation
Besides PE and RT, other protein toxins are in use for the construction of immunotoxins. The most common is the diphtheria toxin (DT). It is in extensive use, mostly in non-antibody-based targeted toxins. In fact, the only targeted toxin that was already approved by the FDA is the IL2-DAB389 fusion protein, also known as denileukin diftitox or Ontak (Ontak®; Eisai Inc., Woodcliff Lake, NJ, USA). The targeting moiety of these targeted toxins is interleukin-2 (IL-2) that binds IL-2R on T cells. It was approved in 1999 for use in CTCL. DT, a 535 amino acid protein, is the prototype protein of the ADP-ribosylating family. It is the only virulence factor secreted by the bacterium Corynebacterium diphtheriae, which is the pathogen that causes the disease diphtheria [26]. Like PE, DT has an AB organization (detailed in the PE section) but its subunits are organized in the reverse order of PE: The N terminal encodes the A domain and the C terminal encodes the B domain [26]. Functionally, DT is very similar to PE. Other DT-based targeted toxins included DTGM that was evaluated for acute myelogenous leukemia (AML) [177], DAB486IL2 that was evaluated for lymphoma/leukemia [178,179,180,181,182] and DAB389EGF that was evaluated for EGFR-expressing carcinoma [12].
Pokeweed antiviral protein (PAP), saporin (from soapwort, Saponaria officinalis L.) and gelonin (from Gelonium multiflorum) are all type I RIPs. Like RT, these proteins are N-glycosidases (detailed in the ricin section), but unlike RT, which is a type II RIP, these proteins do not contain a lectin-binding domain, or any other specific targeting domain [84]. The use of these toxins in an immunotoxin context is not common, and no other immunotoxin for cancer treatment was clinically evaluated other than the ones that appear in this section. Table 5 lists the clinically evaluated/under evaluation immunotoxins against hematologic malignancies and solid tumors that are based on protein toxins besides PE and RT.
Table 5.
Clinically evaluated/under evaluation immunotoxins against hematologic malignancies and solid tumors.
| Immunotoxin | Target antigen | Toxic moiety | Target malignancy | Clinical trial phase (Year the trial ended) | References |
|---|---|---|---|---|---|
| A-dmDT390- bisFV (UCHT1) | CD3ε | DT390 | T-cell lymphoma/leukemia | I/II (2010 **) | [183,185] |
| B43-PAP | CD19 | PAP | ALL | I (1993 **) | [187] |
| BER-H2-Sap6 | CD30 | Saporin | HD | I (1992 **) | [188] |
| HUM-195/rGel | CD33 | Gelonin | AML, CML | I (2010 **) | [189] |
** Year of publication (Year of trial unknown); Abbreviations: CML: chronic myelogenous leukemia.
3.3.1. A-dmDT390- bisFv (UCHT1)
Anti-CD3 recombinant diphtheria immunotoxin, A-dmDT390-bisFv(UCHT1), consists of the catalytic and translocation domains of diphtheria toxin fused to 2 scFvs of an anti-CD3ε murine MAb UCHT1 [183,184]. To study the pharmacology of A-dmDT390-bisFv(UCHT1) in patients with CD3 positive T-cell lymphoma, a phase I clinical trial was conducted in six patients. In this trial, depletion greater than 99.0% of normal T cells in all six patients was observed for a short period of time (2–3 days). Two patients had partial remissions lasting one and over six months. This trial was reported in 2009–2010 [183,185]. Currently, a phase I/II trial on patients with T-cell lymphomas is under way) [186].
3.3.2. B43-PAP
B43-PAP immunotoxin is constructed of the anti-CD19 murine MAb B43 and the plant toxin PAP [190]. A phase I dose escalation study was conducted on 17 relapsed patients with B-lineage acute lymphoblastic leukemia (ALL). In this trial, four complete remissions and one partial remission were detected, as well as a rapid reductions in the numbers of leukemic blasts in circulation in an additional five patients [187]. This trial was reported in 1993 and no other trials were reported.
3.3.3. BER-H2-Sap6
BER-H2-Sap6 immunotoxin was prepared by conjugation of the anti-CD30 murine MAb (Ber-H2) to saporin (SO6). In a pilot clinical trial conducted on four patients with advanced refractory HL, three patients showed rapid and substantial reduction in tumor mass (50% to >75%). Clinical responses were transient (6–10 weeks). There was no growth at new sites but re-growth at all original sites except the skin of one patient [188]. This trial was reported in 1992 and no further trials were reported. Because of the similar antigen of BER-H2-Sap6 and Ki-4.dgA, their performance has been compared [191].
3.3.4. HUM-195/rGel
HuM195/rGel is composed from the humanized MAb HuM195 and the recombinant gelonin toxin. HuM195 is a humanized version of the murine M195: An anti CD-33 monoclonal antibody. CD33 is heavily expressed on the surface of myeloid leukemia cells. A phase I clinical trial of HUM-195/rGel in 28 patients with advanced myeloid malignancies was started in 1999. In this trial, as reported in 2010, no complete or partial responses were observed [189].
4. Advantages and Disadvantages of Immunotoxins
Immunotoxins, together with all antibody-based therapeutics, are a growing field in targeted cancer therapy. New and better antibodies are developed nowadays and toxins are being improved to better fit their target. As can be seen in this review, most of the immunotoxins that have already undergone clinical evaluation were constructed, based on murine antibodies developed in the 80s and 90s of the last century and therefore did not reach their full potential.
Many common disadvantages still need to be improved, as many unwanted side effects are caused due to immunotoxin use. Of course, many therapeutic drugs, especially in the field of cancer therapy, cause side effects. Some of which are relatively “minor” like diarrhea, nausea or mild fever, but some are harsh and dose-limiting. As opposed to chemotherapy, immunotoxins are considered specific to their target cells, and therefore they can deliver highly potent toxic moieties that could not be given systematically. Moreover, the dosage could be less limited by systematic poisoning. Unfortunately, other characteristics of the immunotoxins prevent them from being of benefit because of these disadvantages. One problem that should be referred to is immunogenicity. Immunotoxins, as opposed to chemotherapy, are constructed from proteins that the human immune system cannot ignore. The immune system reacts to both the murine antibody fragment and to the protein toxin, by generating HAMA and human anti-toxin antibodies (HATA). When antibody levels in the blood are high, the patient can no longer receive the treatment, as in the case of N901-bR, XomaZyme-Mel and XomaZyme-791. This problem is addressed by the new generation of immunotoxins that are being constructed from humanized or human antibody fragments and, as was mentioned in the previous chapter, efforts are also being made to minimize toxin immunogenicity [20,21,93,94]. Another major disadvantage of immunotoxins is the cause of CSL or VSL. Immunotoxins are usually given intravenously, and therefore encounter epithelial cells surrounding the blood vessels, this can cause VLS that can be severe and even cause death. Other severe side effects were observed, like hepatotoxicity, which brought about the termination of some clinical trials [15].
The antibody moiety’s size also affects the immunotoxins’ potency. Although the abnormality of angiogenesis in cancer tumors increase their leakiness and cause accumulation of high molar weight molecules in the tumor area [8], the permeability of solid tumors is size-limited. Therefore, the potency of immunotoxins may be increased by lowering its molar weight. Since immunotoxins are defined as chimera of two proteins, their minimization is limited to about 45kDa. This fact led to the development of many immunotoxins for the treatment of non solid tumors where tumor penetration is not an issue [12].
5. Conclusions
This article discusses the antibody and protein toxin-based immunotoxins that have been clinically evaluated to date. As the pioneers in the field, many did not go further than phase I trial for various reasons that have been mentioned, mostly toxicity and lack of efficacy. These immunotoxins will most probably never be approved for use, but their contribution to the understanding of patient responses to immunotoxins has led to the improvements that are currently applied in new immunotoxins. It is hoped that these improvements will eventually lead to the production of closer-to-perfection immunotoxins that will combine specificity and high potency with an absence of the disadvantages encountered today.
References
- Society, A.c. Cancer facts and figures 2010. In Atlanta: American cancer society. 2010. Available online: http://www.cancer.org/acs/groups/content/@epidemiologysurveilance/documents/document/acspc-026238.pdf (accessed on 9 May 2012).
- Choudhary, S.; Mathew, M.; Verma, R.S. Therapeutic potential of anticancer immunotoxins. Drug Discov. Today 2011, 16, 495–503. [Google Scholar]
- Li, J.; Zhu, Z. Research and development of next generation of antibody-based therapeutics. Acta Pharma. Sin. 2010, 31, 1198–1207. [Google Scholar]
- Reichert, J.M. Antibody-based therapeutics to watch in 2011. mAbs 2011, 3, 76–99. [Google Scholar]
- Reichert, J.M. Monoclonal antibodies as innovative therapeutics. Curr. Pharm. Biotechnol. 2008, 9, 423–430. [Google Scholar]
- Holliger, P.; Hudson, P.J. Engineered antibody fragments and the rise of single domains. Nat. Biotechnol. 2005, 23, 1126–1136. [Google Scholar]
- Adams, G.P.; Weiner, L.M. Monoclonal antibody therapy of cancer. Nat. Biotechnol. 2005, 23, 1147–1157. [Google Scholar]
- Dosio, F.; Brusa, P.; Cattel, L. Immunotoxins and anticancer drug conjugate assemblies: The role of the linkage between components. Toxins (Basel) 2011, 3, 848–883. [Google Scholar]
- Yamaizumi, M.; Mekada, E.; Uchida, T.; Okada, Y. One molecule of diphtheria toxin fragment A introduced into a cell can kill the cell. Cell 1978, 15, 245–250. [Google Scholar]
- Alley, S.C.; Okeley, N.M.; Senter, P.D. Antibody-drug conjugates: Targeted drug delivery for cancer. Curr. Opin. Chem. Biol. 2010, 14, 529–537. [Google Scholar]
- Govindan, S.V.; Goldenberg, D.M. New antibody conjugates in cancer therapy. TheScientificWorldJo 2010, 10, 2070–2089. [Google Scholar]
- Kreitman, R.J. Immunotoxins for targeted cancer therapy. AAPS J. 2006, 8, E532–E551. [Google Scholar]
- Lambert, J.M.; Goldmacher, V.S.; Collinson, A.R.; Nadler, L.M.; Blattler, W.A. An immunotoxin prepared with blocked ricin: A natural plant toxin adapted for therapeutic use. Cancer Res. 1991, 51, 6236–6242. [Google Scholar]
- Lambert, J.M.; McIntyre, G.; Gauthier, M.N.; Zullo, D.; Rao, V.; Steeves, R.M.; Goldmacher, V.S.; Blattler, W.A. The galactose-binding sites of the cytotoxic lectin ricin can be chemically blocked in high yield with reactive ligands prepared by chemical modification of glycopeptides containing triantennary N-linked oligosaccharides. Biochemistry 1991, 30, 3234–3247. [Google Scholar]
- Pastan, I.; Hassan, R.; FitzGerald, D.J.; Kreitman, R.J. Immunotoxin treatment of cancer. Annu. Rev. Med. 2007, 58, 221–237. [Google Scholar]
- Chaudhary, V.K.; Queen, C.; Junghans, R.P.; Waldmann, T.A.; FitzGerald, D.J.; Pastan, I. A recombinant immunotoxin consisting of two antibody variable domains fused to Pseudomonas exotoxin. Nature 1989, 339, 394–397. [Google Scholar]
- Bird, R.E.; Hardman, K.D.; Jacobson, J.W.; Johnson, S.; Kaufman, B.M.; Lee, S.M.; Lee, T.; Pope, S.H.; Riordan, G.S.; Whitlow, M. Single-chain antigen-binding proteins. Science 1988, 242, 423–426. [Google Scholar]
- Huston, J.S.; Levinson, D.; Mudgett-Hunter, M.; Tai, M.S.; Novotny, J.; Margolies, M.N.; Ridge, R.J.; Bruccoleri, R.E.; Haber, E.; Crea, R.; et al. Protein engineering of antibody binding sites: Recovery of specific activity in an anti-digoxin single-chain Fv analogue produced in Escherichia coli. Proc. Natl. Acad. Sci. USA 1988, 85, 5879–5883. [Google Scholar]
- Kreitman, R.J.; Wilson, W.H.; Robbins, D.; Margulies, I.; Stetler-Stevenson, M.; Waldmann, T.A.; Pastan, I. Responses in refractory hairy cell leukemia to a recombinant immunotoxin. Blood 1999, 94, 3340–3348. [Google Scholar]
- Liu, W.; Onda, M.; Kim, C.; Xiang, L.; Weldon, J.E.; Lee, B.; Pastan, I. A recombinant immunotoxin engineered for increased stability by adding a disulfide bond has decreased immunogenicity. Protein Eng. Des. Sel. 2012, 25, 1–6. [Google Scholar]
- Onda, M.; Beers, R.; Xiang, L.; Nagata, S.; Wang, Q.C.; Pastan, I. An immunotoxin with greatly reduced immunogenicity by identification and removal of B cell epitopes. Proc. Natl. Acad. Sci. USA 2008, 105, 11311–11316. [Google Scholar]
- Pai, L.H.; Bookman, M.A.; Ozols, R.F.; Young, R.C.; Smith, J.W., 2nd; Longo, D.L.; Gould, B.; Frankel, A.; McClay, E.F.; Howell, S.; et al. Clinical evaluation of intraperitoneal Pseudomonas exotoxin immunoconjugate OVB3-PE in patients with ovarian cancer. J. Clin Oncol. 1991, 9, 2095–2103. [Google Scholar]
- Weiner, L.M.; O'Dwyer, J.; Kitson, J.; Comis, R.L.; Frankel, A.E.; Bauer, R.J.; Konrad, M.S.; Groves, E.S. Phase I evaluation of an anti-breast carcinoma monoclonal antibody 260F9-recombinant ricin A chain immunoconjugate. Cancer Res. 1989, 49, 4062–4067. [Google Scholar]
- Martin, P.J.; Pei, J.; Gooley, T.; Anasetti, C.; Appelbaum, F.R.; Deeg, J.; Hansen, J.A.; Nash, R.A.; Petersdorf, E.W.; Storb, R.; et al. Evaluation of a CD25-specific immunotoxin for prevention of graft-versus-host disease after unrelated marrow transplantation. Biol. Blood Marrow Transplant. 2004, 10, 552–560. [Google Scholar] [CrossRef]
- Wayne, A.S.; Kreitman, R.J.; Findley, H.W.; Lew, G.; Delbrook, C.; Steinberg, S.M.; Stetler-Stevenson, M.; Fitzgerald, D.J.; Pastan, I. Anti-CD22 immunotoxin RFB4(dsFv)-PE38 (BL22) for CD22-positive hematologic malignancies of childhood: Preclinical studies and phase I clinical trial. Clin. Cancer Res. 2010, 16, 1894–1903. [Google Scholar]
- Deng, Q.; Barbieri, J.T. Molecular mechanisms of the cytotoxicity of ADP-ribosylating toxins. Annu. Rev. Microbiol. 2008, 62, 271–288. [Google Scholar]
- Saelinger, C.B.; Morris, R.E. Intracellular trafficking of Pseudomonas exotoxin A. Antibiot. Chemother. 1987, 39, 149–159. [Google Scholar]
- FitzGerald, D.J.; Willingham, M.C.; Pastan, I. Pseudomonas exotoxin—Immunotoxins. Cancer Treat. Res. 1988, 37, 161–173. [Google Scholar]
- Collier, R.J. Structure-activity relationships in diphtheria toxin and Pseudomonas aeruginosa exotoxin A. Cancer Treat. Res. 1988, 37, 25–35. [Google Scholar]
- Wick, M.J.; Frank, D.W.; Storey, D.G.; Iglewski, B.H. Structure, function, and regulation of Pseudomonas aeruginosa exotoxin A. Ann. Rev. Microbiol. 1990, 44, 335–363. [Google Scholar] [CrossRef]
- Wilson, B.A.; Collier, R.J. Diphtheria toxin and Pseudomonas aeruginosa exotoxin A: Active-site structure and enzymic mechanism. Curr. Top. Microbiol. Immunol. 1992, 175, 27–41. [Google Scholar]
- Mrsny, R.J.; Daugherty, A.L.; McKee, M.L.; FitzGerald, D.J. Bacterial toxins as tools for mucosal vaccination. Drug Discov. Today 2002, 7, 247–258. [Google Scholar]
- Wolf, P.; Elsasser-Beile, U. Pseudomonas exotoxin A: From virulence factor to anti-cancer agent. Int. J. Med. Microbiol. 2009, 299, 161–176. [Google Scholar]
- Chaudhary, V.K.; Jinno, Y.; Gallo, M.G.; FitzGerald, D.; Pastan, I. Mutagenesis of Pseudomonas exotoxin in identification of sequences responsible for the animal toxicity. J. Biol. Chem. 1990, 265, 16306–16310. [Google Scholar]
- Brinkmann, U.; Pai, L.H.; FitzGerald, D.J.; Pastan, I. Alteration of a protease-sensitive region of Pseudomonas exotoxin prolongs its survival in the circulation of mice. Proc. Natl. Acad. Sci. USA 1992, 89, 3065–3069. [Google Scholar]
- Kasturi, S.; Kihara, A.; FitzGerald, D.; Pastan, I. Alanine scanning mutagenesis identifies surface amino acids on domain II of Pseudomonas exotoxin required for cytotoxicity, proper folding, and secretion into periplasm. J. Biol. Chem. 1992, 267, 23427–23433. [Google Scholar]
- Kuan, C.T.; Wang, Q.C.; Pastan, I. Pseudomonas exotoxin A mutants. Replacement of surface exposed residues in domain II with cysteine residues that can be modified with polyethylene glycol in a site-specific manner. J. Biol. Chem. 1994, 269, 7610–7616. [Google Scholar]
- Benhar, I.; Wang, Q.C.; FitzGerald, D.; Pastan, I. Pseudomonas exotoxin A mutants. Replacement of surface-exposed residues in domain III with cysteine residues that can be modified with polyethylene glycol in a site-specific manner. J. Biol. Chem. 1994, 269, 13398–13404. [Google Scholar]
- Kreitman, R.J.; Wilson, W.H.; White, J.D.; Stetler-Stevenson, M.; Jaffe, E.S.; Giardina, S.; Waldmann, T.A.; Pastan, I. Phase I trial of recombinant immunotoxin anti-Tac(Fv)-PE38 (LMB-2) in patients with hematologic malignancies. J. Clin. Oncol. 2000, 18, 1622–1636. [Google Scholar]
- Kreitman, R.J.; Arons, E.; Stetler-Stevenson, M.; Fitzgerald, D.J.; Wilson, W.H.; Pastan, I. Recombinant immunotoxins and other therapies for relapsed/refractory hairy cell leukemia. Leuk. Lymphoma 2011, 52 Suppl. 2, 82–86. [Google Scholar] [CrossRef]
- Kreitman, R.J.; Wilson, W.H.; Bergeron, K.; Raggio, M.; Stetler-Stevenson, M.; FitzGerald, D.J.; Pastan, I. Efficacy of the anti-CD22 recombinant immunotoxin BL22 in chemotherapy-resistant hairy-cell leukemia. N. Engl. J. Med. 2001, 345, 241–247. [Google Scholar]
- Kreitman, R.J.; Squires, D.R.; Stetler-Stevenson, M.; Noel, P.; FitzGerald, D.J.; Wilson, W.H.; Pastan, I. Phase I trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with B-cell malignancies. J. Clin. Oncol. 2005, 23, 6719–6729. [Google Scholar]
- Kreitman, R.J.; Stetler-Stevenson, M.; Margulies, I.; Noel, P.; Fitzgerald, D.J.; Wilson, W.H.; Pastan, I. Phase II trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with hairy cell leukemia. J. Clin. Oncol. 2009, 27, 2983–2990. [Google Scholar]
- Kreitman, R.J.; Tallman, M.S.; Robak, T.; Coutre, S.; Wilson, W.H.; Stetler-Stevenson, M.; Fitzgerald, D.J.; Lechleider, R.; Pastan, I. Phase I trial of anti-CD22 recombinant immunotoxin moxetumomab pasudotox (CAT-8015 or HA22) in patients with hairy cell leukemia. J. Clin. Oncol. 2012. [Google Scholar] [CrossRef]
- Mussai, F.; Campana, D.; Bhojwani, D.; Stetler-Stevenson, M.; Steinberg, S.M.; Wayne, A.S.; Pastan, I. Cytotoxicity of the anti-CD22 immunotoxin HA22 (CAT-8015) against paediatric acute lymphoblastic leukaemia. Br. J. Haematol. 2010, 150, 352–358. [Google Scholar]
- Fitzgerald, D.J.; Fryling, C.M.; Zdanovsky, A.; Saelinger, C.B.; Kounnas, M.; Strickland, D.K.; Leppla, S. Selection of Pseudomonas exotoxin-resistant cells with altered expression of alpha 2MR/LRP. Ann. NY Acad. Sci. 1994, 737, 138–144. [Google Scholar]
- Kounnas, M.Z.; Morris, R.E.; Thompson, M.R.; FitzGerald, D.J.; Strickland, D.K.; Saelinger, C.B. The alpha 2-macroglobulin receptor/low density lipoprotein receptor-related protein binds and internalizes Pseudomonas exotoxin A. J. Biol. Chem. 1992, 267, 12420–12423. [Google Scholar]
- FitzGerald, D.; Morris, R.E.; Saelinger, C.B. Receptor-mediated internalization of Pseudomonas toxin by mouse fibroblasts. Cell 1980, 21, 867–873. [Google Scholar]
- Smith, D.C.; Spooner, R.A.; Watson, P.D.; Murray, J.L.; Hodge, T.W.; Amessou, M.; Johannes, L.; Lord, J.M.; Roberts, L.M. Internalized Pseudomonas exotoxin A can exploit multiple pathways to reach the endoplasmic reticulum. Traffic 2006, 7, 379–393. [Google Scholar]
- Powell, D.J., Jr.; Felipe-Silva, A.; Merino, M.J.; Ahmadzadeh, M.; Allen, T.; Levy, C.; White, D.E.; Mavroukakis, S.; Kreitman, R.J.; Rosenberg, S.A.; Pastan, I. Administration of a CD25-directed immunotoxin, LMB-2, to patients with metastatic melanoma induces a selective partial reduction in regulatory T cells in vivo. J. Immunol. 2007, 179, 4919–4928. [Google Scholar]
- Pai-Scherf, L.H.; Villa, J.; Pearson, D.; Watson, T.; Liu, E.; Willingham, M.C.; Pastan, I. Hepatotoxicity in cancer patients receiving erb-38, a recombinant immunotoxin that targets the erbB2 receptor. Clin. Cancer Res. 1999, 5, 2311–2315. [Google Scholar]
- Hassan, R.; Bullock, S.; Premkumar, A.; Kreitman, R.J.; Kindler, H.; Willingham, M.C.; Pastan, I. Phase I study of SS1P, a recombinant anti-mesothelin immunotoxin given as a bolus I.V. infusion to patients with mesothelin-expressing mesothelioma, ovarian, and pancreatic cancers. Clin. Cancer Res. 2007, 13, 5144–5149. [Google Scholar]
- Kreitman, R.J.; Hassan, R.; Fitzgerald, D.J.; Pastan, I. Phase I trial of continuous infusion anti-mesothelin recombinant immunotoxin SS1P. Clin. Cancer Res. 2009, 15, 5274–5279. [Google Scholar]
- Pai, L.H.; Wittes, R.; Setser, A.; Willingham, M.C.; Pastan, I. Treatment of advanced solid tumors with immunotoxin LMB-1: An antibody linked to Pseudomonas exotoxin. Nat. Med. 1996, 2, 350–353. [Google Scholar]
- Posey, J.A.; Khazaeli, M.B.; Bookman, M.A.; Nowrouzi, A.; Grizzle, W.E.; Thornton, J.; Carey, D.E.; Lorenz, J.M.; Sing, A.P.; Siegall, C.B.; et al. A phase I trial of the single-chain immunotoxin SGN-10 (BR96 sFv-PE40) in patients with advanced solid tumors. Clin. Cancer Res. 2002, 8, 3092–3099. [Google Scholar]
- Azemar, M.; Djahansouzi, S.; Jager, E.; Solbach, C.; Schmidt, M.; Maurer, A.B.; Mross, K.; Unger, C.; von Minckwitz, G.; Dall, P.; et al. Regression of cutaneous tumor lesions in patients intratumorally injected with a recombinant single-chain antibody-toxin targeted to ErbB2/HER2. Breast Cancer Res. Treat. 2003, 82, 155–164. [Google Scholar] [CrossRef]
- von Minckwitz, G.; Harder, S.; Hovelmann, S.; Jager, E.; Al-Batran, S.E.; Loibl, S.; Atmaca, A.; Cimpoiasu, C.; Neumann, A.; Abera, A.; et al. Phase I clinical study of the recombinant antibody toxin scFv(FRP5)-ETA specific for the ErbB2/HER2 receptor in patients with advanced solid malignomas. Breast Cancer Res. 2005, 7, R617–R626. [Google Scholar] [CrossRef] [Green Version]
- Taniguchi, T.; Minami, Y. The IL-2/IL-2 receptor system: A current overview. Cell 1993, 73, 5–8. [Google Scholar]
- Diamantstein, T.; Osawa, H. The interleukin-2 receptor, its physiology and a new approach to a selective immunosuppressive therapy by anti-interleukin-2 receptor monoclonal antibodies. Immunol. Rev. 1986, 92, 5–27. [Google Scholar]
- Anti-Tac(Fv)-PE38 (LMB-2) to Treat Chronic Lymphocytic Leukemia. Available online: http://www.clinicaltrials.gov/ct2/show/NCT00077922?term=LMB2&rank=2 (accessed on 31 March 2012).
- LMB-2 to Treat Hairy Cell Leukemia. Available online: http://clinicaltrials.gov/ct2/show/NCT00321555?term=LMB-2&rank=5 (accessed on 31 March 2012).
- Phase II Trial of LMB-2, Fludarabine and Cyclophosphamide for Adult T-Cell Leukemia. Available online: http://clinicaltrials.gov/ct2/show/NCT00924170?term=LMB-2&rank=1 (accessed on 31 March 2012).
- Li, J.L.; Shen, G.L.; Ghetie, M.A.; May, R.D.; Till, M.; Ghetie, V.; Uhr, J.W.; Janossy, G.; Thorpe, P.E.; Amlot, P.; et al. The epitope specificity and tissue reactivity of four murine monoclonal anti-CD22 antibodies. Cell. Immunol. 1989, 118, 85–99. [Google Scholar] [CrossRef]
- Vitetta, E.S.; Stone, M.; Amlot, P.; Fay, J.; May, R.; Till, M.; Newman, J.; Clark, P.; Collins, R.; Cunningham, D.; et al. Phase I immunotoxin trial in patients with B-cell lymphoma. Cancer Res. 1991, 51, 4052–4058. [Google Scholar]
- Clark, E.A. CD22, a B cell-specific receptor, mediates adhesion and signal transduction. J. Immunol. 1993, 150, 4715–4718. [Google Scholar]
- Robbins, B.A.; Ellison, D.J.; Spinosa, J.C.; Carey, C.A.; Lukes, R.J.; Poppema, S.; Saven, A.; Piro, L.D. Diagnostic application of two-color flow cytometry in 161 cases of hairy cell leukemia. Blood 1993, 82, 1277–1287. [Google Scholar]
- Cordone, I.; Annino, L.; Masi, S.; Pescarmona, E.; Rahimi, S.; Ferrari, A.; Giubilei, E.; Pignoloni, P.; Faraggiana, T.; Mandelli, F. Diagnostic relevance of peripheral blood immunocytochemistry in hairy cell leukaemia. J. Clin. Pathol. 1995, 48, 955–960. [Google Scholar]
- Robbins, D.H.; Margulies, I.; Stetler-Stevenson, M.; Kreitman, R.J. Hairy cell leukemia, a B-cell neoplasm that is particularly sensitive to the cytotoxic effect of anti-Tac(Fv)-PE38 (LMB-2). Clin. Cancer Res. 2000, 6, 693–700. [Google Scholar]
- Anderson, K.C.; Bates, M.P.; Slaughenhoupt, B.L.; Pinkus, G.S.; Schlossman, S.F.; Nadler, L.M. Expression of human B cell-associated antigens on leukemias and lymphomas: A model of human B cell differentiation. Blood 1984, 63, 1424–1433. [Google Scholar]
- Salvatore, G.; Beers, R.; Margulies, I.; Kreitman, R.J.; Pastan, I. Improved cytotoxic activity toward cell lines and fresh leukemia cells of a mutant anti-CD22 immunotoxin obtained by antibody phage display. Clin. Cancer Res. 2002, 8, 995–1002. [Google Scholar]
- Batra, J.K.; Kasprzyk, P.G.; Bird, R.E.; Pastan, I.; King, C.R. Recombinant anti-erbB2 immunotoxins containing Pseudomonas exotoxin. Proc. Natl. Acad. Sci. USA 1992, 89, 5867–5871. [Google Scholar]
- Reiter, Y.; Brinkmann, U.; Jung, S.H.; Lee, B.; Kasprzyk, P.G.; King, C.R.; Pastan, I. Improved binding and antitumor activity of a recombinant anti-erbB2 immunotoxin by disulfide stabilization of the Fv fragment. J. Biol. Chem. 1994, 269, 18327–18331. [Google Scholar]
- Chowdhury, P.S.; Pastan, I. Improving antibody affinity by mimicking somatic hypermutation in vitro. Nat. Biotechnol. 1999, 17, 568–572. [Google Scholar] [CrossRef]
- Hassan, R.; Bera, T.; Pastan, I. Mesothelin: A new target for immunotherapy. Clin. Cancer Res. 2004, 10, 3937–3942. [Google Scholar]
- Chowdhury, P.S.; Viner, J.L.; Beers, R.; Pastan, I. Isolation of a high-affinity stable single-chain Fv specific for mesothelin from DNA-immunized mice by phage display and construction of a recombinant immunotoxin with anti-tumor activity. Proc. Natl. Acad. Sci. USA 1998, 95, 669–674. [Google Scholar]
- Ricart, A.D. Immunoconjugates against solid tumors: Mind the gap. Clin. Pharmacol. Ther. 2011, 89, 513–523. [Google Scholar]
- SS1P and Pentostatin Plus Cyclophosphamide for Mesothelioma. Available online: http://clinicaltrials.gov/ct2/show/NCT01362790?term=SS1P&rank=1 (accessed on 31 March 2012).
- Bigner, D.D.; Archer, G.E.; McLendon, R.E.; Friedman, H.S.; Fuchs, H.E.; Pai, L.H.; Herndon, J.E., 2nd; Pastan, I.H. Efficacy of compartmental administration of immunotoxin LMB-1 (B3-LysPE38) in a rat model of carcinomatous meningitis. Clin. Cancer Res. 1995, 1, 1545–1555. [Google Scholar]
- Pai, L.H.; Batra, J.K.; FitzGerald, D.J.; Willingham, M.C.; Pastan, I. Anti-tumor activities of immunotoxins made of monoclonal antibody B3 and various forms of Pseudomonas exotoxin. Proc. Natl. Acad. Sci. USA 1991, 88, 3358–3362. [Google Scholar]
- Friedman, P.N.; McAndrew, S.J.; Gawlak, S.L.; Chace, D.; Trail, P.A.; Brown, J.P.; Siegall, C.B. BR96 sFv-PE40, a potent single-chain immunotoxin that selectively kills carcinoma cells. Cancer Res. 1993, 53, 334–339. [Google Scholar]
- Siegall, C.B.; Chace, D.; Mixan, B.; Garrigues, U.; Wan, H.; Paul, L.; Wolff, E.; Hellstrom, I.; Hellstrom, K.E. In vitro and in vivo characterization of BR96 sFv-PE40. A single-chain immunotoxin fusion protein that cures human breast carcinoma xenografts in athymic mice and rats. J. Immunol. 1994, 152, 2377–2384. [Google Scholar]
- Wels, W.; Harwerth, I.M.; Mueller, M.; Groner, B.; Hynes, N.E. Selective inhibition of tumor cell growth by a recombinant single-chain antibody-toxin specific for the erbB-2 receptor. Cancer Res. 1992, 52, 6310–6317. [Google Scholar]
- Wels, W.; Beerli, R.; Hellmann, P.; Schmidt, M.; Marte, B.M.; Kornilova, E.S.; Hekele, A.; Mendelsohn, J.; Groner, B.; Hynes, N.E. EGF receptor and p185erbB-2-specific single-chain antibody toxins differ in their cell-killing activity on tumor cells expressing both receptor proteins. Int. J. Cancer 1995, 60, 137–144. [Google Scholar]
- Nielsen, K.; Boston, R.S. RIBOSOME-INACTIVATING PROTEINS: A Plant Perspective. Annu. Rev. Plant Phys. 2001, 52, 785–816. [Google Scholar]
- Frankel, A.E.; FitzGerald, D.; Siegall, C.; Press, O.W. Advances in immunotoxin biology and therapy: A summary of the Fourth International Symposium on Immunotoxins. Cancer Res. 1996, 56, 926–932. [Google Scholar]
- Pastan, I.; FitzGerald, D. Recombinant toxins for cancer treatment. Science 1991, 254, 1173–1177. [Google Scholar]
- Fulton, R.J.; Uhr, J.W.; Vitetta, E.S. In vivo therapy of the BCL1 tumor: Effect of immunotoxin valency and deglycosylation of the ricin A chain. Cancer Res. 1988, 48, 2626–2631. [Google Scholar]
- Fulton, R.J.; Tucker, T.F.; Vitetta, E.S.; Uhr, J.W. Pharmacokinetics of tumor-reactive immunotoxins in tumor-bearing mice: Effect of antibody valency and deglycosylation of the ricin A chain on clearance and tumor localization. Cancer Res. 1988, 48, 2618–2625. [Google Scholar]
- Bourrie, B.J.; Casellas, P.; Blythman, H.E.; Jansen, F.K. Study of the plasma clearance of antibody—Ricin-A-chain immunotoxins. Evidence for specific recognition sites on the A chain that mediate rapid clearance of the immunotoxin. Eur. J. Biochem. 1986, 155, 1–10. [Google Scholar] [CrossRef]
- Blakey, D.C.; Watson, G.J.; Knowles, P.P.; Thorpe, P.E. Effect of chemical deglycosylation of ricin A chain on the in vivo fate and cytotoxic activity of an immunotoxin composed of ricin A chain and anti-Thy 1.1 antibody. Cancer Res. 1987, 47, 947–952. [Google Scholar]
- Ramakrishnan, S.; Bjorn, M.J.; Houston, L.L. Recombinant ricin A chain conjugated to monoclonal antibodies: Improved tumor cell inhibition in the presence of lysosomotropic compounds. Cancer Res. 1989, 49, 613–617. [Google Scholar]
- Kreitman, R.J. Immunotoxins in cancer therapy. Curr. Opin. Immunol. 1999, 11, 570–578. [Google Scholar]
- Baluna, R.; Rizo, J.; Gordon, B.E.; Ghetie, V.; Vitetta, E.S. Evidence for a structural motif in toxins and interleukin-2 that may be responsible for binding to endothelial cells and initiating vascular leak syndrome. Proc. Natl. Acad. Sci. USA 1999, 96, 3957–3962. [Google Scholar]
- Hirao, I.; Madin, K.; Endo, Y.; Yokoyama, S.; Ellington, A.D. RNA aptamers that bind to and inhibit the ribosome-inactivating protein, pepocin. J. Biol. Chem. 2000, 275, 4943–4948. [Google Scholar]
- Audi, J.; Belson, M.; Patel, M.; Schier, J.; Osterloh, J. Ricin poisoning: A comprehensive review. JAMA 2005, 294, 2342–2351. [Google Scholar]
- Shapira, A.; Benhar, I. Toxin-based therapeutic approaches. Toxins (Basel) 2010, 2, 2519–2583. [Google Scholar]
- Endo, Y.; Tsurugi, K. The RNA N-glycosidase activity of ricin A-chain. The characteristics of the enzymatic activity of ricin A-chain with ribosomes and with rRNA. J. Biol. Chem. 1988, 263, 8735–8739. [Google Scholar]
- Endo, Y.; Tsurugi, K.; Lambert, J.M. The site of action of six different ribosome-inactivating proteins from plants on eukaryotic ribosomes: The RNA N-glycosidase activity of the proteins. Biochem. Biophys. Res. Commun. 1988, 150, 1032–1036. [Google Scholar]
- Stirpe, F.; Bailey, S.; Miller, S.P.; Bodley, J.W. Modification of ribosomal RNA by ribosome-inactivating proteins from plants. Nucleic Acids Res. 1988, 16, 1349–1357. [Google Scholar]
- Montanaro, L.; Sperti, S.; Mattioli, A.; Testoni, G.; Stirpe, F. Inhibition by ricin of protein synthesis in vitro. Inhibition of the binding of elongation factor 2 and of adenosine diphosphate-ribosylated elongation factor 2 to ribosomes. Biochem. J. 1975, 146, 127–131. [Google Scholar]
- Griffiths, G.D.; Leek, M.D.; Gee, D.J. The toxic plant proteins ricin and abrin induce apoptotic changes in mammalian lymphoid tissues and intestine. J. Pathol. 1987, 151, 221–229. [Google Scholar]
- Bolognesi, A.; Tazzari, P.L.; Olivieri, F.; Polito, L.; Falini, B.; Stirpe, F. Induction of apoptosis by ribosome-inactivating proteins and related immunotoxins. Int. J. Cancer 1996, 68, 349–355. [Google Scholar]
- Narayanan, S.; Surolia, A.; Karande, A.A. Ribosome-inactivating protein and apoptosis: Abrin causes cell death via mitochondrial pathway in Jurkat cells. Biochem. J. 2004, 377, 233–240. [Google Scholar]
- Amlot, P.L.; Stone, M.J.; Cunningham, D.; Fay, J.; Newman, J.; Collins, R.; May, R.; McCarthy, M.; Richardson, J.; Ghetie, V.; et al. A phase I study of an anti-CD22-deglycosylated ricin A chain immunotoxin in the treatment of B-cell lymphomas resistant to conventional therapy. Blood 1993, 82, 2624–2633. [Google Scholar]
- Sausville, E.A.; Headlee, D.; Stetler-Stevenson, M.; Jaffe, E.S.; Solomon, D.; Figg, W.D.; Herdt, J.; Kopp, W.C.; Rager, H.; Steinberg, S.M.; et al. Continuous infusion of the anti-CD22 immunotoxin IgG-RFB4-SMPT-dgA in patients with B-cell lymphoma: A phase I study. Blood 1995, 85, 3457–3465. [Google Scholar]
- Stone, M.J.; Sausville, E.A.; Fay, J.W.; Headlee, D.; Collins, R.H.; Figg, W.D.; Stetler-Stevenson, M.; Jain, V.; Jaffe, E.S.; Solomon, D.; et al. A phase I study of bolus versus continuous infusion of the anti-CD19 immunotoxin, IgG-HD37-dgA, in patients with B-cell lymphoma. Blood 1996, 88, 1188–1197. [Google Scholar]
- Conry, R.M.; Khazaeli, M.B.; Saleh, M.N.; Ghetie, V.; Vitetta, E.S.; Liu, T.; LoBuglio, A.F. Phase I trial of an anti-CD19 deglycosylated ricin A chain immunotoxin in non-Hodgkin's lymphoma: Effect of an intensive schedule of administration. J. Immunother. Emphasis Tumor Immunol. 1995, 18, 231–241. [Google Scholar]
- Herrera, L.; Bostrom, B.; Gore, L.; Sandler, E.; Lew, G.; Schlegel, P.G.; Aquino, V.; Ghetie, V.; Vitetta, E.S.; Schindler, J. A phase 1 study of Combotox in pediatric patients with refractory B-lineage acute lymphoblastic leukemia. J. Pediatr. Hematol. Oncol. 2009, 31, 936–941. [Google Scholar]
- Messmann, R.A.; Vitetta, E.S.; Headlee, D.; Senderowicz, A.M.; Figg, W.D.; Schindler, J.; Michiel, D.F.; Creekmore, S.; Steinberg, S.M.; Kohler, D.; et al. A phase I study of combination therapy with immunotoxins IgG-HD37-deglycosylated ricin A chain (dgA) and IgG-RFB4-dgA (Combotox) in patients with refractory CD19(+), CD22(+) B cell lymphoma. Clin. Cancer Res. 2000, 6, 1302–1313. [Google Scholar]
- Engert, A.; Diehl, V.; Schnell, R.; Radszuhn, A.; Hatwig, M.T.; Drillich, S.; Schon, G.; Bohlen, H.; Tesch, H.; Hansmann, M.L.; et al. A phase-I study of an anti-CD25 ricin A-chain immunotoxin (RFT5-SMPT-dgA) in patients with refractory Hodgkin's lymphoma. Blood 1997, 89, 403–410. [Google Scholar]
- Schnell, R.; Vitetta, E.; Schindler, J.; Barth, S.; Winkler, U.; Borchmann, P.; Hansmann, M.L.; Diehl, V.; Ghetie, V.; Engert, A. Clinical trials with an anti-CD25 ricin A-chain experimental and immunotoxin (RFT5-SMPT-dgA) in Hodgkin's lymphoma. Leuk. Lymphoma 1998, 30, 525–537. [Google Scholar]
- Schnell, R.; Borchmann, P.; Staak, J.O.; Schindler, J.; Ghetie, V.; Vitetta, E.S.; Engert, A. Clinical evaluation of ricin A-chain immunotoxins in patients with Hodgkin's lymphoma. Ann. Oncol. 2003, 14, 729–736. [Google Scholar]
- Schnell, R.; Vitetta, E.; Schindler, J.; Borchmann, P.; Barth, S.; Ghetie, V.; Hell, K.; Drillich, S.; Diehl, V.; Engert, A. Treatment of refractory Hodgkin's lymphoma patients with an anti-CD25 ricin A-chain immunotoxin. Leukemia 2000, 14, 129–135. [Google Scholar]
- Schnell, R.; Staak, O.; Borchmann, P.; Schwartz, C.; Matthey, B.; Hansen, H.; Schindler, J.; Ghetie, V.; Vitetta, E.S.; Diehl, V.; Engert, A. A Phase I study with an anti-CD30 ricin A-chain immunotoxin (Ki-4.dgA) in patients with refractory CD30+ Hodgkin's and non-Hodgkin's lymphoma. Clin. Cancer Res. 2002, 8, 1779–1786. [Google Scholar]
- Multani, P.S.; O'Day, S.; Nadler, L.M.; Grossbard, M.L. Phase II clinical trial of bolus infusion anti-B4 blocked ricin immunoconjugate in patients with relapsed B-cell non-Hodgkin's lymphoma. Clin. Cancer Res. 1998, 4, 2599–2604. [Google Scholar]
- Grossbard, M.L.; Gribben, J.G.; Freedman, A.S.; Lambert, J.M.; Kinsella, J.; Rabinowe, S.N.; Eliseo, L.; Taylor, J.A.; Blattler, W.A.; Epstein, C.L.; et al. Adjuvant immunotoxin therapy with anti-B4-blocked ricin after autologous bone marrow transplantation for patients with B-cell non-Hodgkin's lymphoma. Blood 1993, 81, 2263–2271. [Google Scholar]
- Grossbard, M.L.; Lambert, J.M.; Goldmacher, V.S.; Spector, N.L.; Kinsella, J.; Eliseo, L.; Coral, F.; Taylor, J.A.; Blattler, W.A.; Epstein, C.L.; et al. Anti-B4-blocked ricin: A phase I trial of 7-day continuous infusion in patients with B-cell neoplasms. J. Clin. Oncol. 1993, 11, 726–737. [Google Scholar]
- Grossbard, M.L.; Multani, P.S.; Freedman, A.S.; O'Day, S.; Gribben, J.G.; Rhuda, C.; Neuberg, D.; Nadler, L.M. A Phase II study of adjuvant therapy with anti-B4-blocked ricin after autologous bone marrow transplantation for patients with relapsed B-cell non-Hodgkin's lymphoma. Clin. Cancer Res. 1999, 5, 2392–2398. [Google Scholar]
- Furman, R.R.; Grossbard, M.L.; Johnson, J.L.; Pecora, A.L.; Cassileth, P.A.; Jung, S.H.; Peterson, B.A.; Nadler, L.M.; Freedman, A.; Bayer, R.L.; et al. A phase III study of anti-B4-blocked ricin as adjuvant therapy post-autologous bone marrow transplant: CALGB 9254. Leuk. Lymphoma 2011, 52, 587–596. [Google Scholar]
- Grossbard, M.L.; Freedman, A.S.; Ritz, J.; Coral, F.; Goldmacher, V.S.; Eliseo, L.; Spector, N.; Dear, K.; Lambert, J.M.; Blattler, W.A.; et al. Serotherapy of B-cell neoplasms with anti-B4-blocked ricin: A phase I trial of daily bolus infusion. Blood 1992, 79, 576–585. [Google Scholar]
- Frankel, A.E.; Laver, J.H.; Willingham, M.C.; Burns, L.J.; Kersey, J.H.; Vallera, D.A. Therapy of patients with T-cell lymphomas and leukemias using an anti-CD7 monoclonal antibody-ricin A chain immunotoxin. Leuk. Lymphoma 1997, 26, 287–298. [Google Scholar]
- Kernan, N.A.; Byers, V.; Scannon, P.J.; Mischak, R.P.; Brochstein, J.; Flomenberg, N.; Dupont, B.; O'Reilly, R.J. Treatment of steroid-resistant acute graft-vs-host disease by in vivo administration of an anti-T-cell ricin A chain immunotoxin. JAMA 1988, 259, 3154–3157. [Google Scholar]
- Byers, V.S.; Henslee, P.J.; Kernan, N.A.; Blazar, B.R.; Gingrich, R.; Phillips, G.L.; LeMaistre, C.F.; Gilliland, G.; Antin, J.H.; Martin, P.; et al. Use of an anti-pan T-lymphocyte ricin a chain immunotoxin in steroid-resistant acute graft-versus-host disease. Blood 1990, 75, 1426–1432. [Google Scholar]
- LeMaistre, C.F.; Rosen, S.; Frankel, A.; Kornfeld, S.; Saria, E.; Meneghetti, C.; Drajesk, J.; Fishwild, D.; Scannon, P.; Byers, V. Phase I trial of H65-RTA immunoconjugate in patients with cutaneous T-cell lymphoma. Blood 1991, 78, 1173–1182. [Google Scholar]
- Hertler, A.A.; Schlossman, D.M.; Borowitz, M.J.; Laurent, G.; Jansen, F.K.; Schmidt, C.; Frankel, A.E. A phase I study of T101-ricin A chain immunotoxin in refractory chronic lymphocytic leukemia. J. Biol. Response Mod. 1988, 7, 97–113. [Google Scholar]
- Hertler, A.A.; Schlossman, D.M.; Borowitz, M.J.; Blythman, H.E.; Casellas, P.; Frankel, A.E. An anti-CD5 immunotoxin for chronic lymphocytic leukemia: Enhancement of cytotoxicity with human serum albumin-monensin. Int. J. Cancer 1989, 43, 215–219. [Google Scholar]
- Laurent, G.; Pris, J.; Farcet, J.P.; Carayon, P.; Blythman, H.; Casellas, P.; Poncelet, P.; Jansen, F.K. Effects of therapy with T101 ricin A-chain immunotoxin in two leukemia patients. Blood 1986, 67, 1680–1687. [Google Scholar]
- Phan, G.Q.; Yang, J.C.; Sherry, R.M.; Hwu, P.; Topalian, S.L.; Schwartzentruber, D.J.; Restifo, N.P.; Haworth, L.R.; Seipp, C.A.; Freezer, L.J.; et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc. Natl. Acad. Sci. USA 2003, 100, 8372–8377. [Google Scholar]
- Zalcberg, J.R.; Pietersz, G.; Toohey, B.; Laird, J.; Huggins, R.; Zimet, A.S.; Hennessy, O.; McKenzie, A.; McKenzie, I.F. A phase I/II study of the intralesional injection of ricin-monoclonal antibody conjugates in patients with hepatic metastases. Eur. J. Cancer 1994, 30A, 1227–1231. [Google Scholar]
- Lynch, T.J., Jr. Immunotoxin therapy of small-cell lung cancer. N901-blocked ricin for relapsed small-cell lung cancer. Chest 1993, 103, 436S–439S. [Google Scholar] [CrossRef]
- Lynch, T.J., Jr.; Lambert, J.M.; Coral, F.; Shefner, J.; Wen, P.; Blattler, W.A.; Collinson, A.R.; Ariniello, P.D.; Braman, G.; Cook, S.; et al. Immunotoxin therapy of small-cell lung cancer: A phase I study of N901-blocked ricin. J. Clin. Oncol. 1997, 15, 723–734. [Google Scholar]
- Epstein, C.; Lynch, T.; Shefner, J.; Wen, P.; Maxted, D.; Braman, V.; Ariniello, P.; Coral, F.; Ritz, J. Use of the immunotoxin N901-blocked ricin in patients with small-cell lung cancer. Int. J. Cancer Suppl. 1994, 8, 57–59. [Google Scholar]
- Fidias, P.; Grossbard, M.; Lynch, T.J., Jr. A phase II study of the immunotoxin N901-blocked ricin in small-cell lung cancer. Clin. Lung Cancer 2002, 3, 219–222. [Google Scholar]
- Hertler, A.A.; Spitler, L.E.; Frankel, A.E. Humoral immune response to a ricin A chain immunotoxin in patients with metastatic melanoma. Cancer Drug Deliv. 1987, 4, 245–253. [Google Scholar]
- Spitler, L.E.; del Rio, M.; Khentigan, A.; Wedel, N.I.; Brophy, N.A.; Miller, L.L.; Harkonen, W.S.; Rosendorf, L.L.; Lee, H.M.; Mischak, R.P.; et al. Therapy of patients with malignant melanoma using a monoclonal antimelanoma antibody-ricin A chain immunotoxin. Cancer Res. 1987, 47, 1717–1723. [Google Scholar]
- Mischak, R.P.; Foxall, C.; Rosendorf, L.L.; Knebel, K.; Scannon, P.J.; Spitler, L.E. Human antibody responses to components of the monoclonal antimelanoma antibody ricin A chain immunotoxin XomaZyme-MEL. Mol. Biother. 1990, 2, 104–109. [Google Scholar]
- Oratz, R.; Speyer, J.L.; Wernz, J.C.; Hochster, H.; Meyers, M.; Mischak, R.; Spitler, L.E. Antimelanoma monoclonal antibody-ricin A chain immunoconjugate (XMMME-001-RTA) plus cyclophosphamide in the treatment of metastatic malignant melanoma: Results of a phase II trial. J. Biol. Response Mod. 1990, 9, 345–354. [Google Scholar]
- Gonzalez, R.; Salem, P.; Bunn, P.A., Jr.; Zukiwski, A.A.; Lamb, R.; Benjamin, R.S.; Spitler, L.; Wedel, N.; Robinson, W.A. Single-dose murine monoclonal antibody ricin A chain immunotoxin in the treatment of metastatic melanoma: A phase I trial. Mol. Biother. 1991, 3, 192–196. [Google Scholar]
- Selvaggi, K.; Saria, E.A.; Schwartz, R.; Vlock, D.R.; Ackerman, S.; Wedel, N.; Kirkwood, J.M.; Jones, H.; Ernstoff, M.S. Phase I/II study of murine monoclonal antibody-ricin A chain (XOMAZYME-Mel) immunoconjugate plus cyclosporine A in patients with metastatic melanoma. J. Immunother. Emphasis Tumor Immunol. 1993, 13, 201–207. [Google Scholar]
- Durrant, L.G.; Byers, V.S.; Scannon, P.J.; Rodvien, R.; Grant, K.; Robins, R.A.; Marksman, R.A.; Baldwin, R.W. Humoral immune responses to XMMCO-791-RTA immunotoxin in colorectal cancer patients. Clin. Exp. Immunol. 1989, 75, 258–264. [Google Scholar]
- LoRusso, P.M.; Lomen, P.L.; Redman, B.G.; Poplin, E.; Bander, J.J.; Valdivieso, M. Phase I study of monoclonal antibody-ricin A chain immunoconjugate Xomazyme-791 in patients with metastatic colon cancer. Am. J. Clin. Oncol. 1995, 18, 307–312. [Google Scholar]
- Byers, V.S.; Rodvien, R.; Grant, K.; Durrant, L.G.; Hudson, K.H.; Baldwin, R.W.; Scannon, P.J. Phase I study of monoclonal antibody-ricin A chain immunotoxin XomaZyme-791 in patients with metastatic colon cancer. Cancer Res. 1989, 49, 6153–6160. [Google Scholar]
- Laske, D.W.; Muraszko, K.M.; Oldfield, E.H.; DeVroom, H.L.; Sung, C.; Dedrick, R.L.; Simon, T.R.; Colandrea, J.; Copeland, C.; Katz, D.; et al. Intraventricular immunotoxin therapy for leptomeningeal neoplasia. Neurosurgery 1997, 41, 1039–1049, discussion 1049–1051. [Google Scholar]
- Gould, B.J.; Borowitz, M.J.; Groves, E.S.; Carter, P.W.; Anthony, D.; Weiner, L.M.; Frankel, A.E. Phase I study of an anti-breast cancer immunotoxin by continuous infusion: Report of a targeted toxic effect not predicted by animal studies. J. Natl. Cancer Inst. 1989, 81, 775–781. [Google Scholar]
- Campana, D.; Janossy, G.; Bofill, M.; Trejdosiewicz, L.K.; Ma, D.; Hoffbrand, A.V.; Mason, D.Y.; Lebacq, A.M.; Forster, H.K. Human B cell development. I. Phenotypic differences of B lymphocytes in the bone marrow and peripheral lymphoid tissue. J. Immunol. 1985, 134, 1524–1530. [Google Scholar]
- Thorpe, P.E.; Detre, S.I.; Foxwell, B.M.; Brown, A.N.; Skilleter, D.N.; Wilson, G.; Forrester, J.A.; Stirpe, F. Modification of the carbohydrate in ricin with metaperiodate-cyanoborohydride mixtures. Effects on toxicity and in vivo distribution. Eur. J. Biochem. 1985, 147, 197–206. [Google Scholar] [CrossRef]
- Thorpe, P.E.; Wallace, P.M.; Knowles, P.P.; Relf, M.G.; Brown, A.N.; Watson, G.J.; Blakey, D.C.; Newell, D.R. Improved antitumor effects of immunotoxins prepared with deglycosylated ricin A-chain and hindered disulfide linkages. Cancer Res. 1988, 48, 6396–6403. [Google Scholar]
- Dorken, B.; Moldenhauer, G.; Schwartz, R.; Pez-zutto, A.; Hammerling, G. B cell differentiation antigens identified by monoclonal antibodies (HD6, HD28, HD37, HD39). Immunobiology 1983, 165, 253–254. [Google Scholar]
- Ghetie, M.A.; Ghetie, V.; Vitetta, E.S. Immunotoxins for the treatment of B-cell lymphomas. Mol. Med. 1997, 3, 420–427. [Google Scholar]
- Ghetie, M.A.; Tucker, K.; Richardson, J.; Uhr, J.W.; Vitetta, E.S. The antitumor activity of an anti-CD22 immunotoxin in SCID mice with disseminated Daudi lymphoma is enhanced by either an anti-CD19 antibody or an anti-CD19 immunotoxin. Blood 1992, 80, 2315–2320. [Google Scholar]
- Phase 1 Study of Combotox With Cytarabine in Relapsed/Refractory B-lineage Acute Lymphoblastic Leukemia (ALL). Available online: http://clinicaltrials.gov/ct2/show/NCT01408160?term=combotox&rank=1 (accessed on 31 March 2012).
- RFT5-dgA. Available online: http://clinicaltrials.gov/ct2/results?term=RFT5-dgA (accessed on 31 March 2012).
- Schwab, U.; Stein, H.; Gerdes, J.; Lemke, H.; Kirchner, H.; Schaadt, M.; Diehl, V. Production of a monoclonal antibody specific for Hodgkin and Sternberg-Reed cells of Hodgkin's disease and a subset of normal lymphoid cells. Nature 1982, 299, 65–67. [Google Scholar]
- Scadden, D.T.; Schenkein, D.P.; Bernstein, Z.; Luskey, B.; Doweiko, J.; Tulpule, A.; Levine, A.M. Immunotoxin combined with chemotherapy for patients with AIDS-related non-Hodgkin's lymphoma. Cancer 1998, 83, 2580–2587. [Google Scholar]
- Longo, D.L.; Duffey, P.L.; Gribben, J.G.; Jaffe, E.S.; Curti, B.D.; Gause, B.L.; Janik, J.E.; Braman, V.M.; Esseltine, D.; Wilson, W.H.; et al. Combination chemotherapy followed by an immunotoxin (anti-B4-blocked ricin) in patients with indolent lymphoma: Results of a phase II study. Cancer J. 2000, 6, 146–150. [Google Scholar]
- Szatrowski, T.P.; Dodge, R.K.; Reynolds, C.; Westbrook, C.A.; Frankel, S.R.; Sklar, J.; Stewart, C.C.; Hurd, D.D.; Kolitz, J.E.; Velez-Garcia, E.; et al. Lineage specific treatment of adult patients with acute lymphoblastic leukemia in first remission with anti-B4-blocked ricin or high-dose cytarabine: Cancer and Leukemia Group B Study 9311. Cancer 2003, 97, 1471–1480. [Google Scholar]
- Aruffo, A.; Seed, B. Molecular cloning of two CD7 (T-cell leukemia antigen) cDNAs by a COS cell expression system. EMBO J. 1987, 6, 3313–3316. [Google Scholar]
- Haynes, B.F.; Eisenbarth, G.S.; Fauci, A.S. Human lymphocyte antigens: Production of a monoclonal antibody that defines functional thymus-derived lymphocyte subsets. Proc. Natl. Acad. Sci. USA 1979, 76, 5829–5833. [Google Scholar]
- Wood, G.S.; Deneau, D.G.; Miller, R.A.; Levy, R.; Hoppe, R.T.; Warnke, R.A. Subtypes of cutaneous T-cell lymphoma defined by expression of leu-1 and Ia. Blood 1982, 59, 876–882. [Google Scholar]
- Holden, C.A.; Staughton, R.C.; Campbell, M.A.; MacDonald, D.M. Differential loss of T lymphocyte markers in advanced cutaneous T cell lymphoma. J. Am. Acad. Dermatol. 1982, 6, 507–513. [Google Scholar]
- Royston, I.; Majda, J.A.; Baird, S.M.; Meserve, B.L.; Griffiths, J.C. Human T cell antigens defined by monoclonal antibodies: The 65,000-dalton antigen of T cells (T65) is also found on chronic lymphocytic leukemia cells bearing surface immunoglobulin. J. Immunol. 1980, 125, 725–731. [Google Scholar]
- Pietersz, G.A.; Kanellos, J.; McKenzie, I.F. Novel synthesis and in vitro characterization of disulfide-linked ricin-monoclonal antibody conjugates devoid of galactose binding activity. Cancer Res. 1988, 48, 4469–4476. [Google Scholar]
- Teh, J.G.; Thompson, C.H.; McKenzie, I.F. Production of monoclonal antibodies to serum antigens in colorectal carcinoma. J. Immunol. Methods 1988, 110, 101–109. [Google Scholar]
- Thomas, S.N.; Zhu, F.; Schnaar, R.L.; Alves, C.S.; Konstantopoulos, K. Carcinoembryonic antigen and CD44 variant isoforms cooperate to mediate colon carcinoma cell adhesion to E- and L-selectin in shear flow. J. Biol. Chem. 2008, 283, 15647–15655. [Google Scholar]
- Campbell, D.G.; Price, M.R.; Baldwin, R.W. Analysis of a human osteogenic sarcoma antigen and its expression on various human tumour cell lines. Int. J. Cancer 1984, 34, 31–37. [Google Scholar]
- Price, M.R.; Campbell, D.G.; Robins, R.A.; Baldwin, R.W. Characteristics of a cell surface antigen defined by an anti-human osteogenic sarcoma monoclonal antibody. Eur. J. Cancer Clin. Oncol. 1983, 19, 81–90. [Google Scholar]
- Faulk, W.P.; Hsi, B.L.; Stevens, P.J. Transferrin and transferrin receptors in carcinoma of the breast. Lancet 1980, 2, 390–392. [Google Scholar]
- Galbraith, G.M.; Galbraith, R.M.; Faulk, W.P. Transferrin binding by human lymphoblastoid cell lines and other transformed cells. Cell. Immunol. 1980, 49, 215–222. [Google Scholar]
- Gatter, K.C.; Brown, G.; Trowbridge, I.S.; Woolston, R.E.; Mason, D.Y. Transferrin receptors in human tissues: Their distribution and possible clinical relevance. J. Clin. Pathol. 1983, 36, 539–545. [Google Scholar]
- Larrick, J.W.; Cresswell, P. Modulation of cell surface iron transferrin receptors by cellular density and state of activation. J. Supramol. Struct. 1979, 11, 579–586. [Google Scholar]
- Shindelman, J.E.; Ortmeyer, A.E.; Sussman, H.H. Demonstration of the transferrin receptor in human breast cancer tissue. Potential marker for identifying dividing cells. Int. J. Cancer 1981, 27, 329–334. [Google Scholar] [CrossRef]
- Trowbridge, I.S.; Domingo, D.L. Anti-transferrin receptor monoclonal antibody and toxin-antibody conjugates affect growth of human tumour cells. Nature 1981, 294, 171–173. [Google Scholar]
- Trowbridge, I.S.; Omary, M.B. Human cell surface glycoprotein related to cell proliferation is the receptor for transferrin. Proc. Natl. Acad. Sci. USA 1981, 78, 3039–3043. [Google Scholar]
- Trowbridge, I.S.; Newman, R.A.; Domingo, D.L.; Sauvage, C. Transferrin receptors: Structure and function. Biochem. Pharmacol. 1984, 33, 925–932. [Google Scholar]
- Bjorn, M.J.; Ring, D.; Frankel, A. Evaluation of monoclonal antibodies for the development of breast cancer immunotoxins. Cancer Res. 1985, 45, 1214–1221. [Google Scholar]
- Frankel, A.E.; Ring, D.B.; Tringale, F.; Hsieh-Ma, S.T. Tissue distribution of breast cancer-associated antigens defined by monoclonal antibodies. J. Biol. Response Mod. 1985, 4, 273–286. [Google Scholar]
- Frankel, A.E.; Powell, B.L.; Hall, P.D.; Case, L.D.; Kreitman, R.J. Phase I trial of a novel diphtheria toxin/granulocyte macrophage colony-stimulating factor fusion protein (DT388GMCSF) for refractory or relapsed acute myeloid leukemia. Clin. Cancer Res. 2002, 8, 1004–1013. [Google Scholar]
- LeMaistre, C.F.; Meneghetti, C.; Rosenblum, M.; Reuben, J.; Parker, K.; Shaw, J.; Deisseroth, A.; Woodworth, T.; Parkinson, D.R. Phase I trial of an interleukin-2 (IL-2) fusion toxin (DAB486IL-2) in hematologic malignancies expressing the IL-2 receptor. Blood 1992, 79, 2547–2554. [Google Scholar]
- Kuzel, T.M.; Rosen, S.T.; Gordon, L.I.; Winter, J.; Samuelson, E.; Kaul, K.; Roenigk, H.H.; Nylen, P.; Woodworth, T. Phase I trial of the diphtheria toxin/interleukin-2 fusion protein DAB486IL-2: Efficacy in mycosis fungoides and other non-Hodgkin's lymphomas. Leuk. Lymphoma 1993, 11, 369–377. [Google Scholar]
- LeMaistre, C.F.; Craig, F.E.; Meneghetti, C.; McMullin, B.; Parker, K.; Reuben, J.; Boldt, D.H.; Rosenblum, M.; Woodworth, T. Phase I trial of a 90-minute infusion of the fusion toxin DAB486IL-2 in hematological cancers. Cancer Res. 1993, 53, 3930–3934. [Google Scholar]
- Platanias, L.C.; Ratain, M.J.; O'Brien, S.; Larson, R.A.; Vardiman, J.W.; Shaw, J.P.; Williams, S.F.; Baron, J.M.; Parker, K.; Woodworth, T.G. Phase I trial of a genetically engineered interleukin-2 fusion toxin (DAB486IL-2) as a 6 hour intravenous infusion in patients with hematologic malignancies. Leuk. Lymphoma 1994, 14, 257–262. [Google Scholar]
- Tepler, I.; Schwartz, G.; Parker, K.; Charette, J.; Kadin, M.E.; Woodworth, T.G.; Schnipper, L.E. Phase I trial of an interleukin-2 fusion toxin (DAB486IL-2) in hematologic malignancies: Complete response in a patient with Hodgkin's disease refractory to chemotherapy. Cancer 1994, 73, 1276–1285. [Google Scholar]
- Woo, J.H.; Lee, Y.J.; Neville, D.M.; Frankel, A.E. Pharmacology of anti-CD3 diphtheria immunotoxin in CD3 positive T-cell lymphoma trials. Methods Mol. Biol. 2010, 651, 157–175. [Google Scholar]
- Thompson, J.; Hu, H.; Scharff, J.; Neville, D.M., Jr. An anti-CD3 single-chain immunotoxin with a truncated diphtheria toxin avoids inhibition by pre-existing antibodies in human blood. J. Biol. Chem. 1995, 270, 28037–28041. [Google Scholar]
- Frankel, A.E.; Zuckero, S.L.; Mankin, A.A.; Grable, M.; Mitchell, K.; Lee, Y.J.; Neville, D.M.; Woo, J.H. Anti-CD3 recombinant diphtheria immunotoxin therapy of cutaneous T cell lymphoma. Curr. Drug Targets 2009, 10, 104–109. [Google Scholar]
- A-dmDT390-bisFv (UCHT1) Immunotoxin Therapy for Patients With T-cell Diseases. Available online: http://clinicaltrials.gov/ct2/show/NCT00611208?term=UCHT1&rank=1 (accessed on 31 March 2012).
- Uckun, F.M. Immunotoxins for the treatment of leukaemia. Br. J. Haematol. 1993, 85, 435–438. [Google Scholar]
- Falini, B.; Bolognesi, A.; Flenghi, L.; Tazzari, P.L.; Broe, M.K.; Stein, H.; Durkop, H.; Aversa, F.; Corneli, P.; Pizzolo, G.; et al. Response of refractory Hodgkin's disease to monoclonal anti-CD30 immunotoxin. Lancet 1992, 339, 1195–1196. [Google Scholar]
- Dean, A.; Talpaz, M.; Kantarjian, H.; Faderl, S.; Jabbour, E.; Ravandi Kashani, F.; O'Brien, S.M.; Rosenblum, M.; Cortes, J.E. Phase I clinical trial of the anti-CD33 immunotoxin HuM195/rgel in patients (pts) with advanced myeloid malignancies. ASCO Meeting Abstracts 2010, 28, 6549. [Google Scholar]
- Uckun, F.M.; Chelstrom, L.M.; Irvin, J.D.; Finnegan, D.; Gunther, R.; Young, J.; Kuebelbeck, V.; Myers, D.E.; Houston, L.L. In vivo efficacy of B43 (anti-CD19)-pokeweed antiviral protein immunotoxin against BCL-1 murine B-cell leukemia. Blood 1992, 79, 2649–2661. [Google Scholar]
- Foyil, K.V.; Bartlett, N.L. Anti-CD30 Antibodies for Hodgkin lymphoma. Curr. Hematol. Malign. Rep. 2010, 5, 140–147. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Becker, N.; Benhar, I. Antibody-Based Immunotoxins for the Treatment of Cancer. Antibodies 2012, 1, 39-69. https://doi.org/10.3390/antib1010039
AMA Style
Becker N, Benhar I. Antibody-Based Immunotoxins for the Treatment of Cancer. Antibodies. 2012; 1(1):39-69. https://doi.org/10.3390/antib1010039
Chicago/Turabian StyleBecker, Nurit, and Itai Benhar. 2012. "Antibody-Based Immunotoxins for the Treatment of Cancer" Antibodies 1, no. 1: 39-69. https://doi.org/10.3390/antib1010039