
Evaluation and Inter-Comparison of Oxygen-Based OC-EC Separation Methods for Radiocarbon Analysis of Ambient Aerosol Particle Samples

 , ,
, , 
Abstract
:1. Introduction
2. Methods
2.1. Sampling
2.2. Filter Handling and Pretreatment at CIO
2.3. 2stepCIO Separation Method and Radiocarbon Analysis of EC at CIO
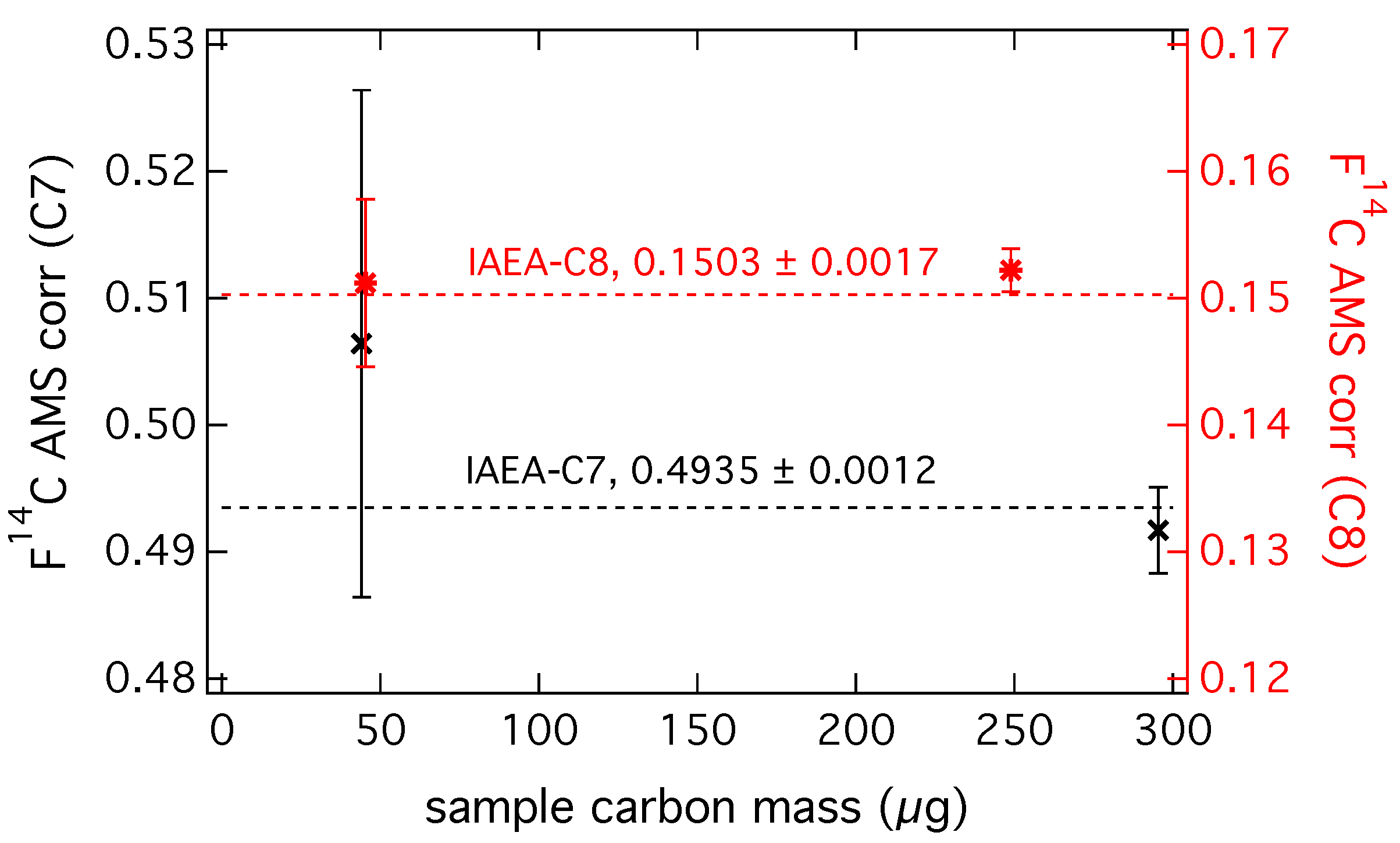
2.4. Radiocarbon Analysis of EC at LARA
2.5. Radiocarbon Analysis of EC at LABEC
2.6. Thermo-Optical Analysis with a Commercial OC-EC Analyzer
3. Results and Discussion
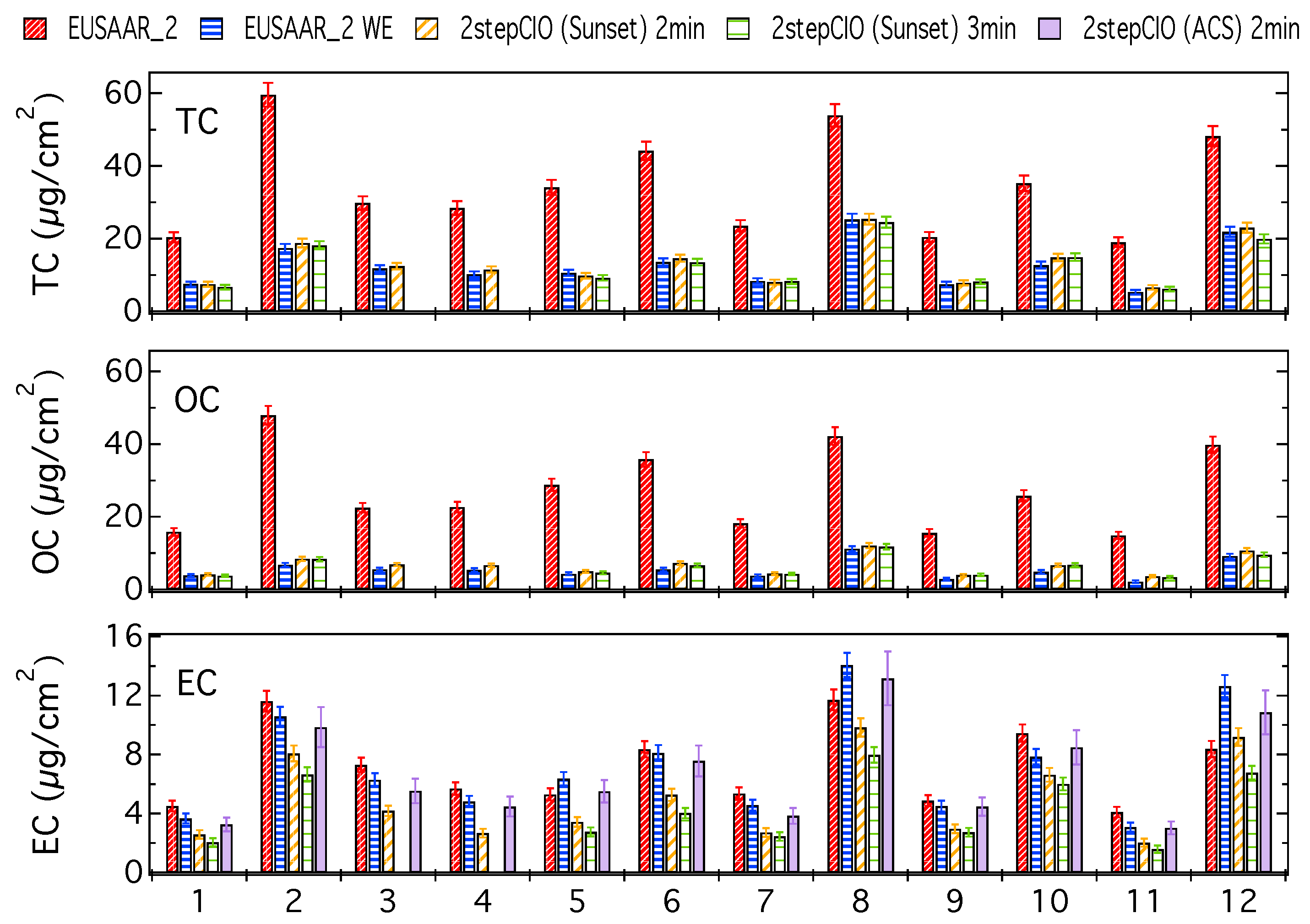
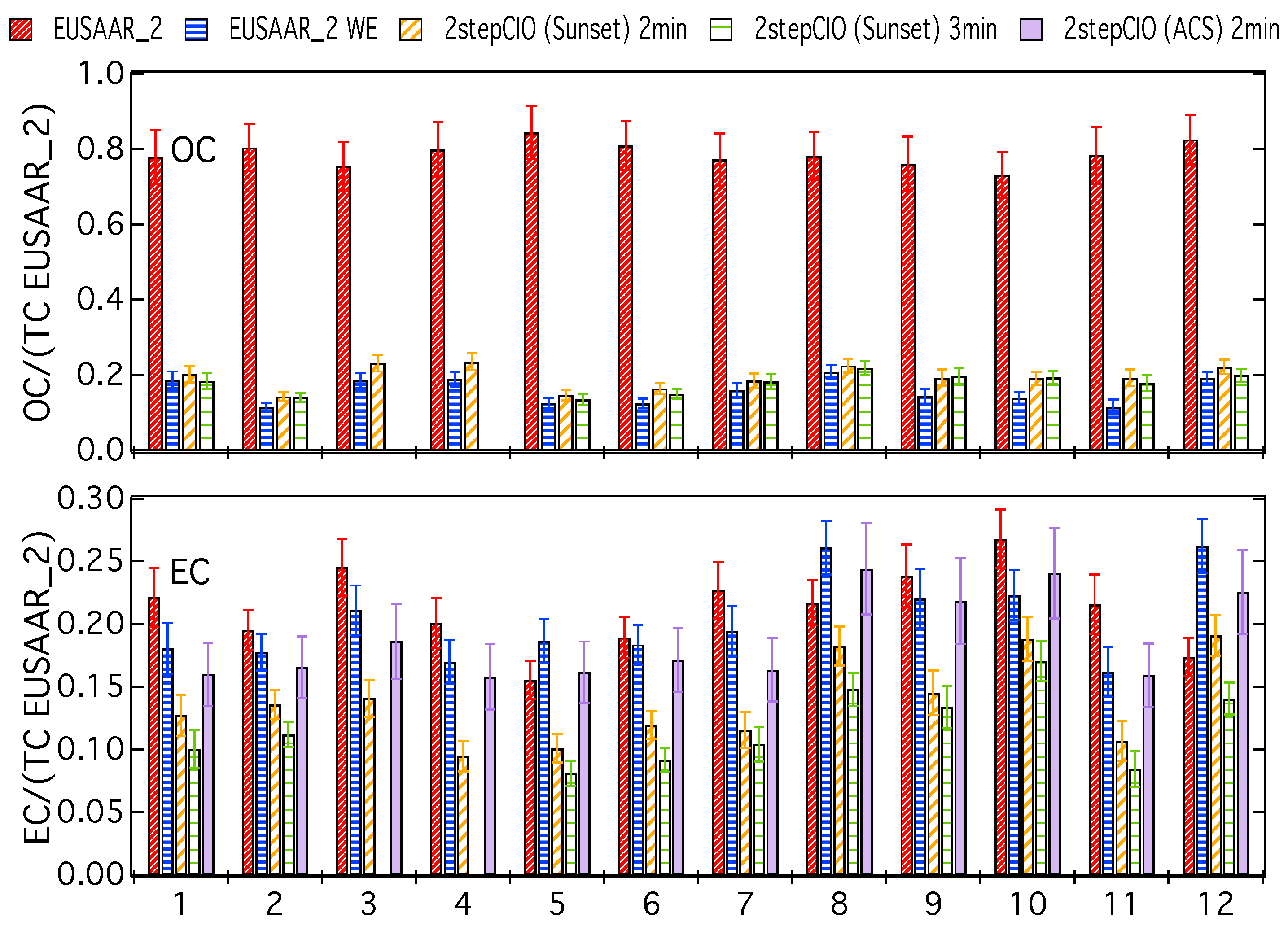
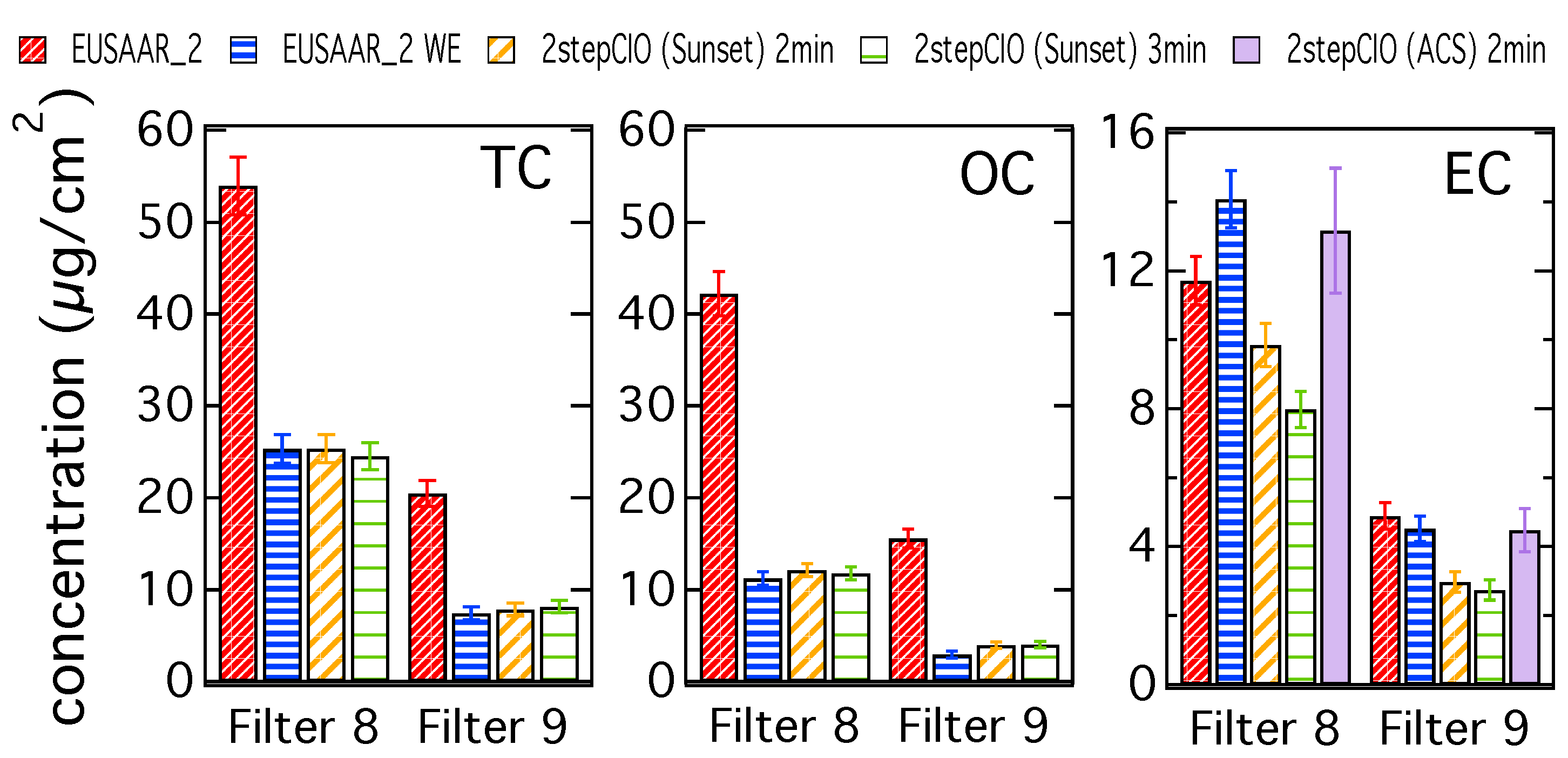
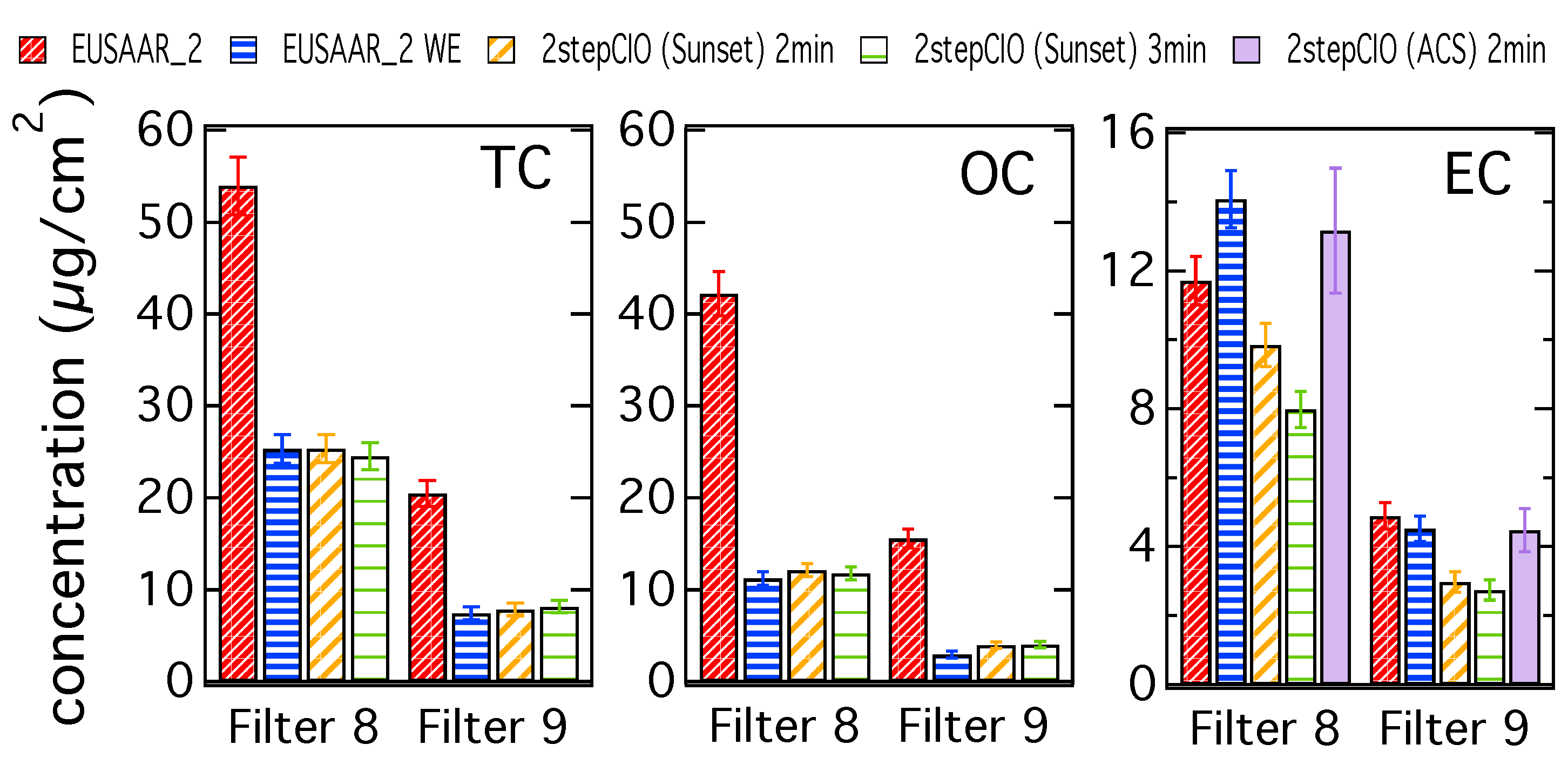
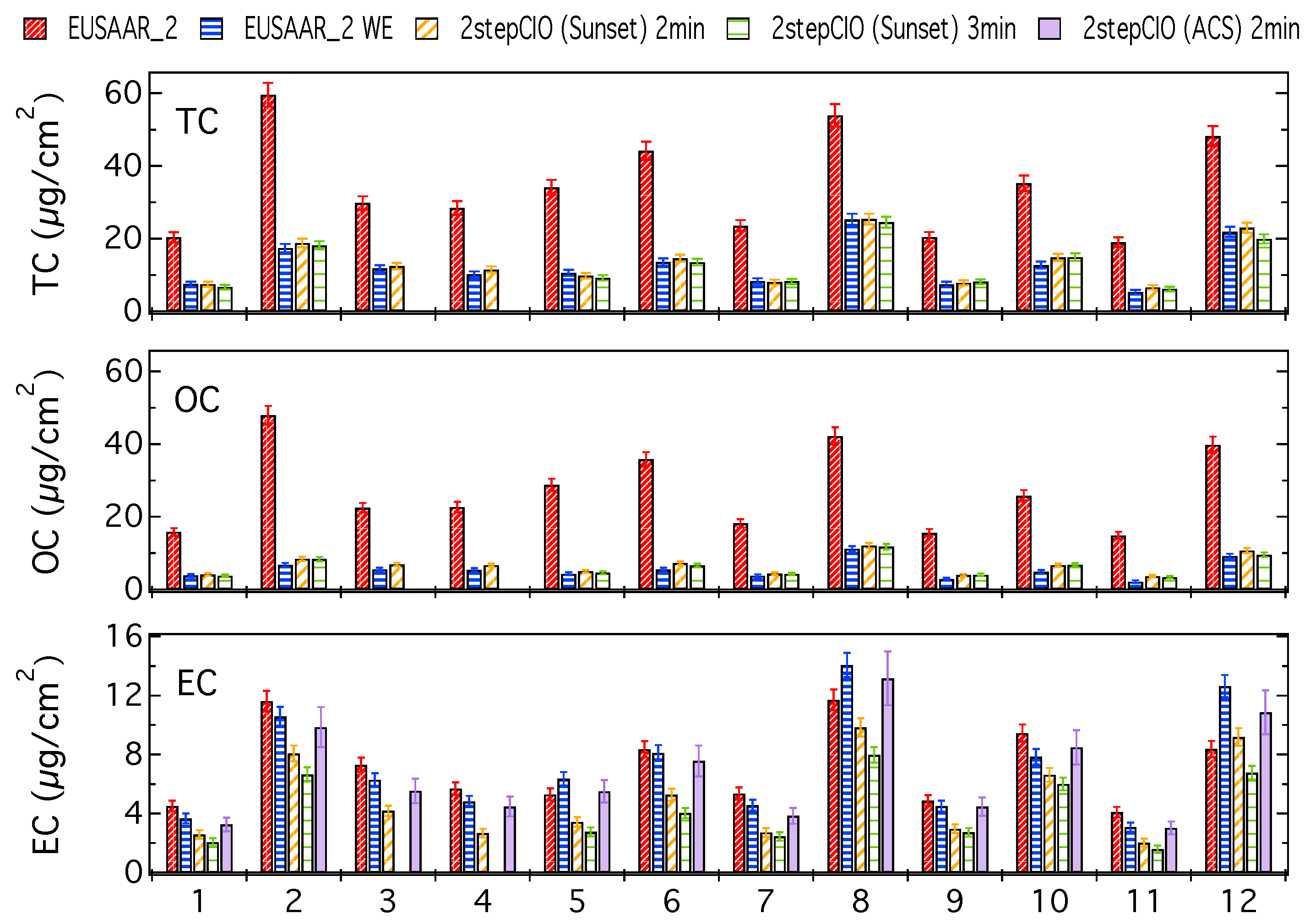
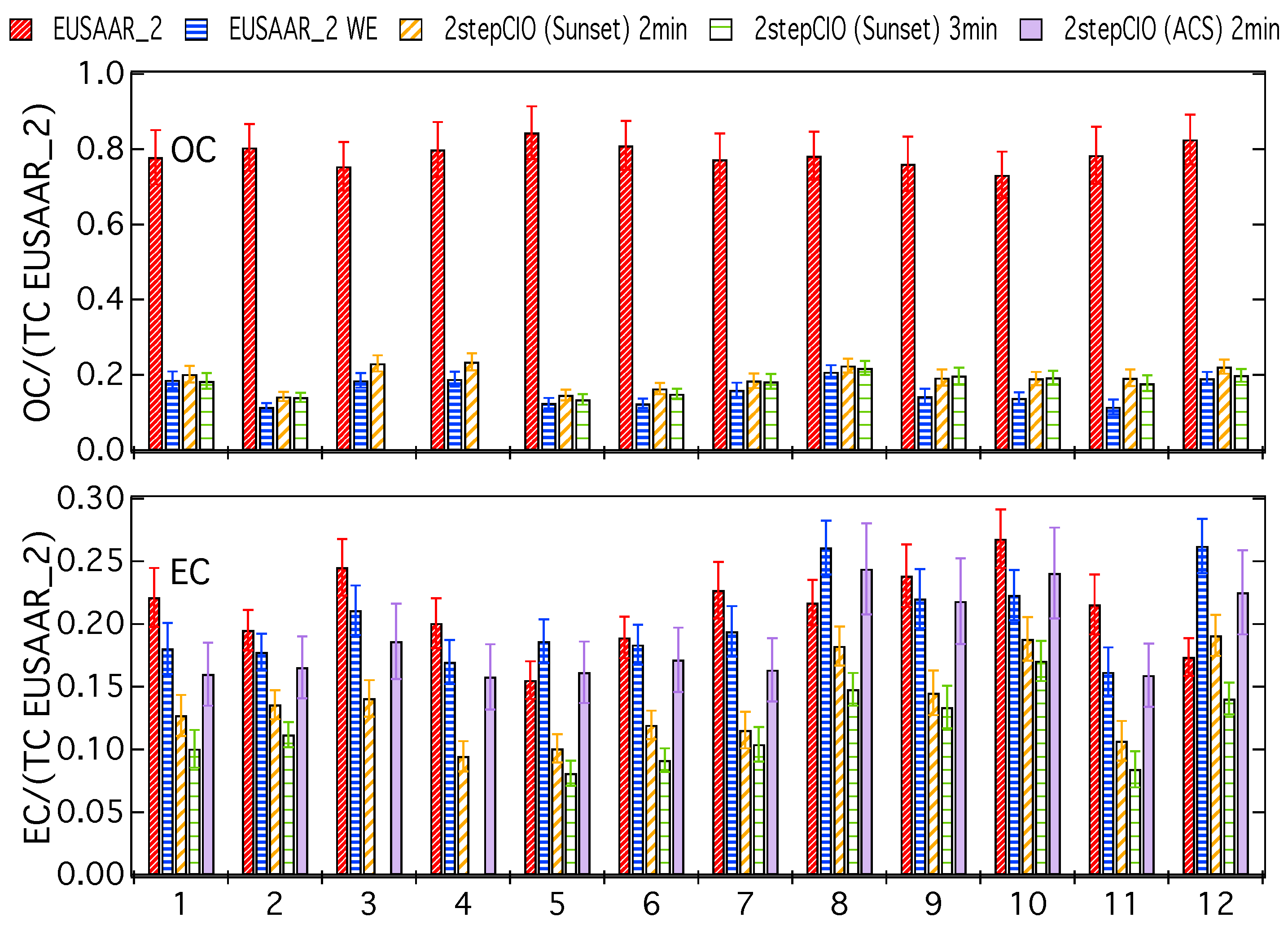
3.1. Analysis of OC-EC Concentrations (CIO)
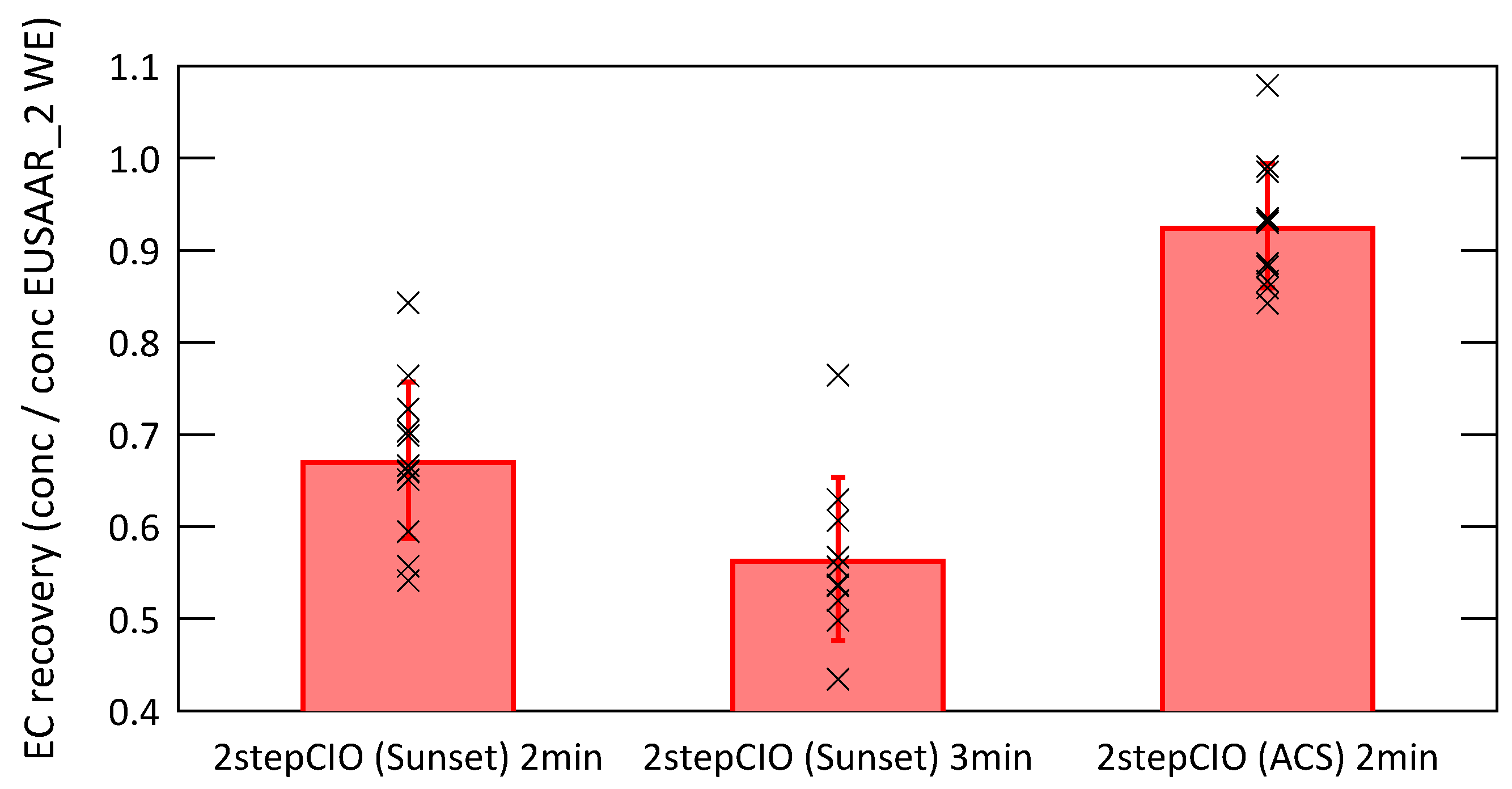
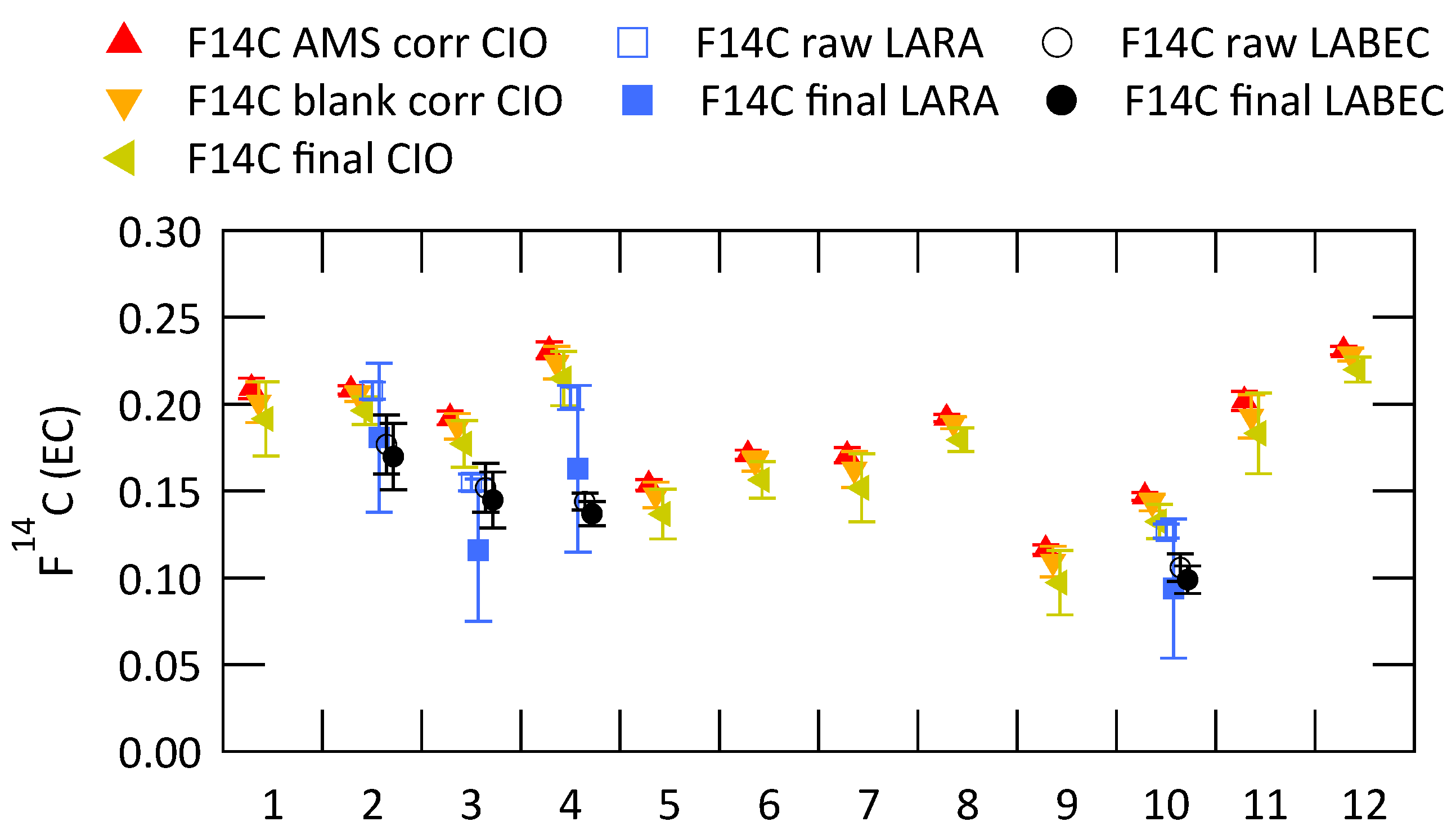
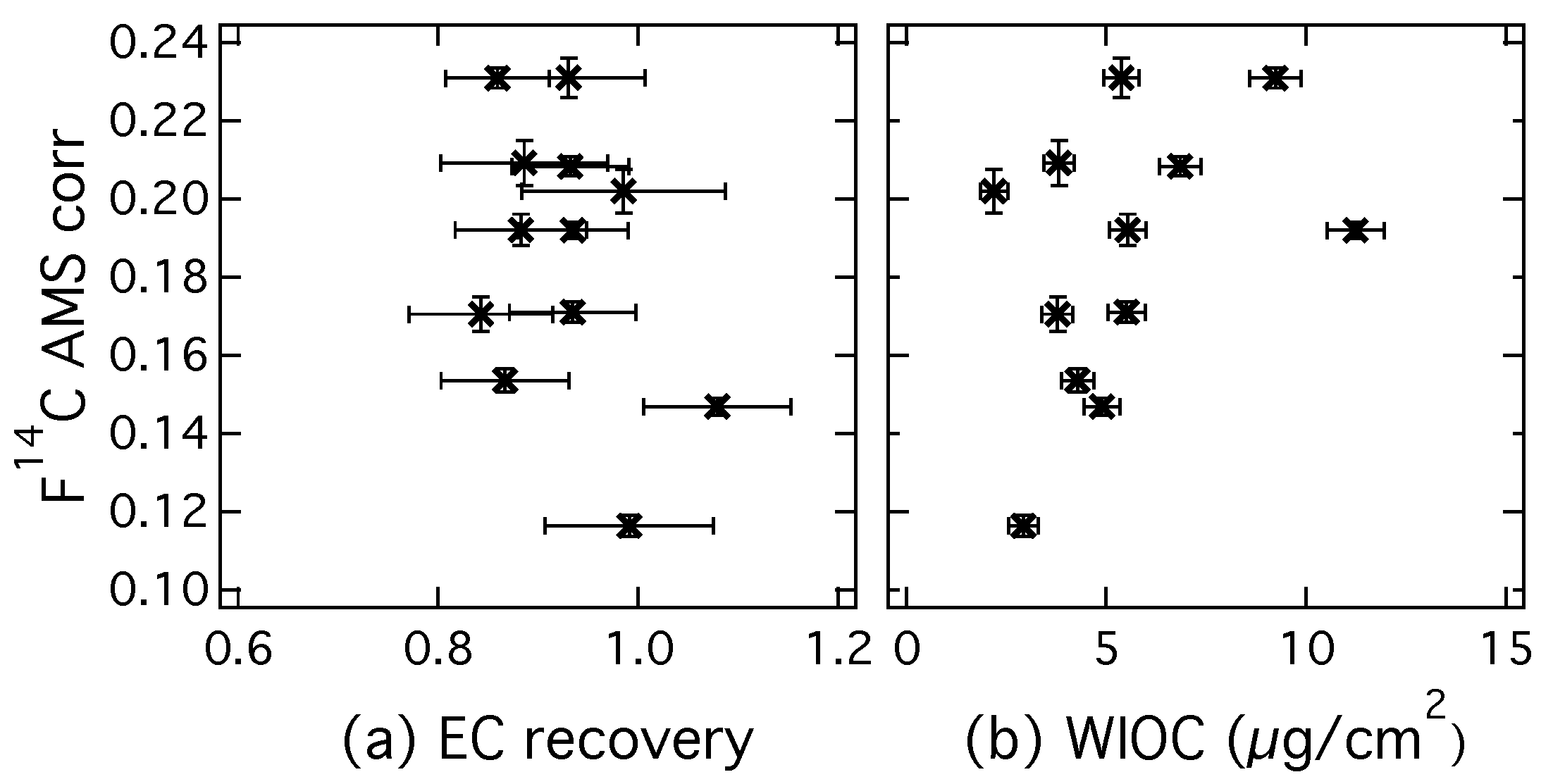
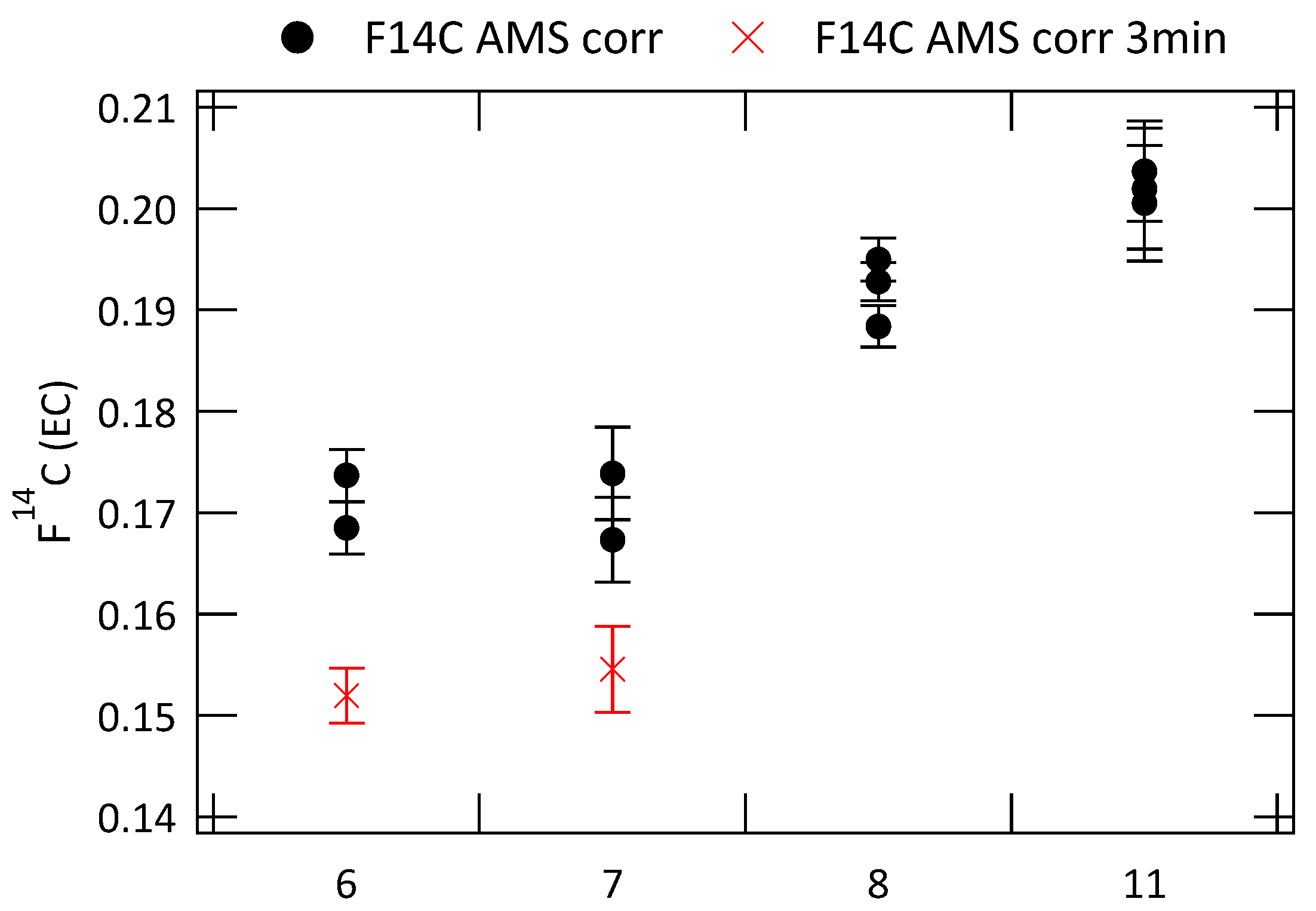
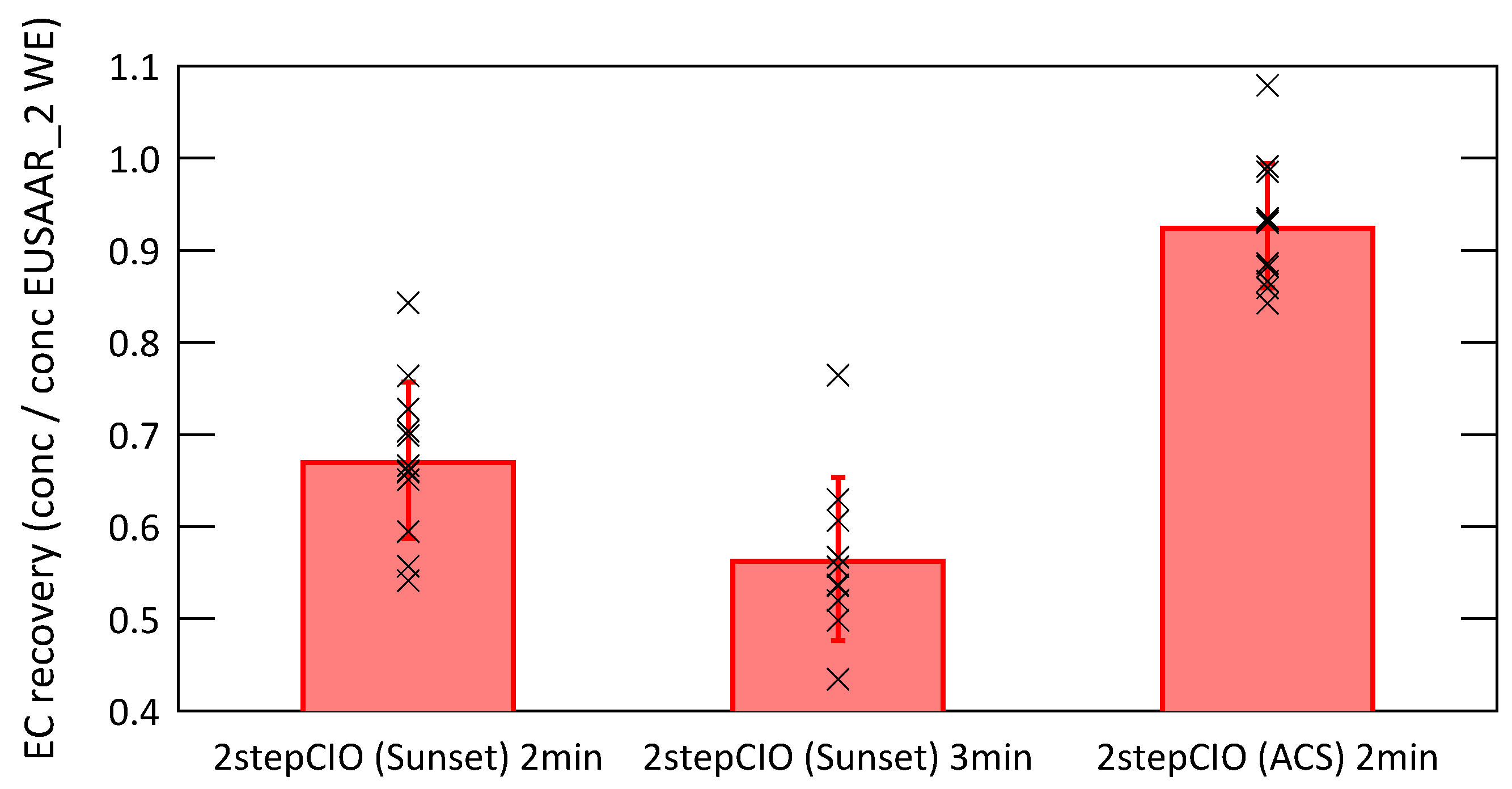
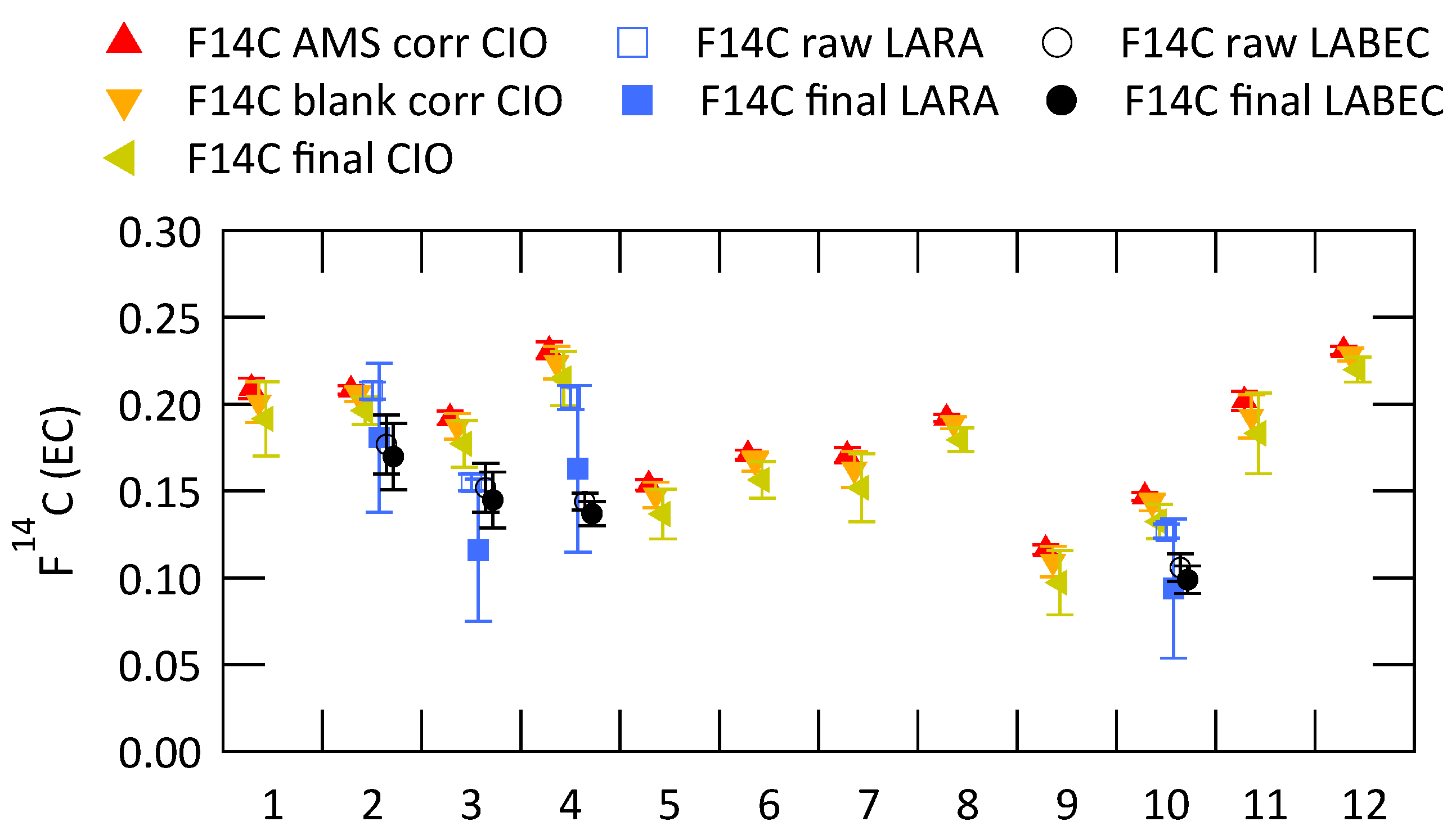
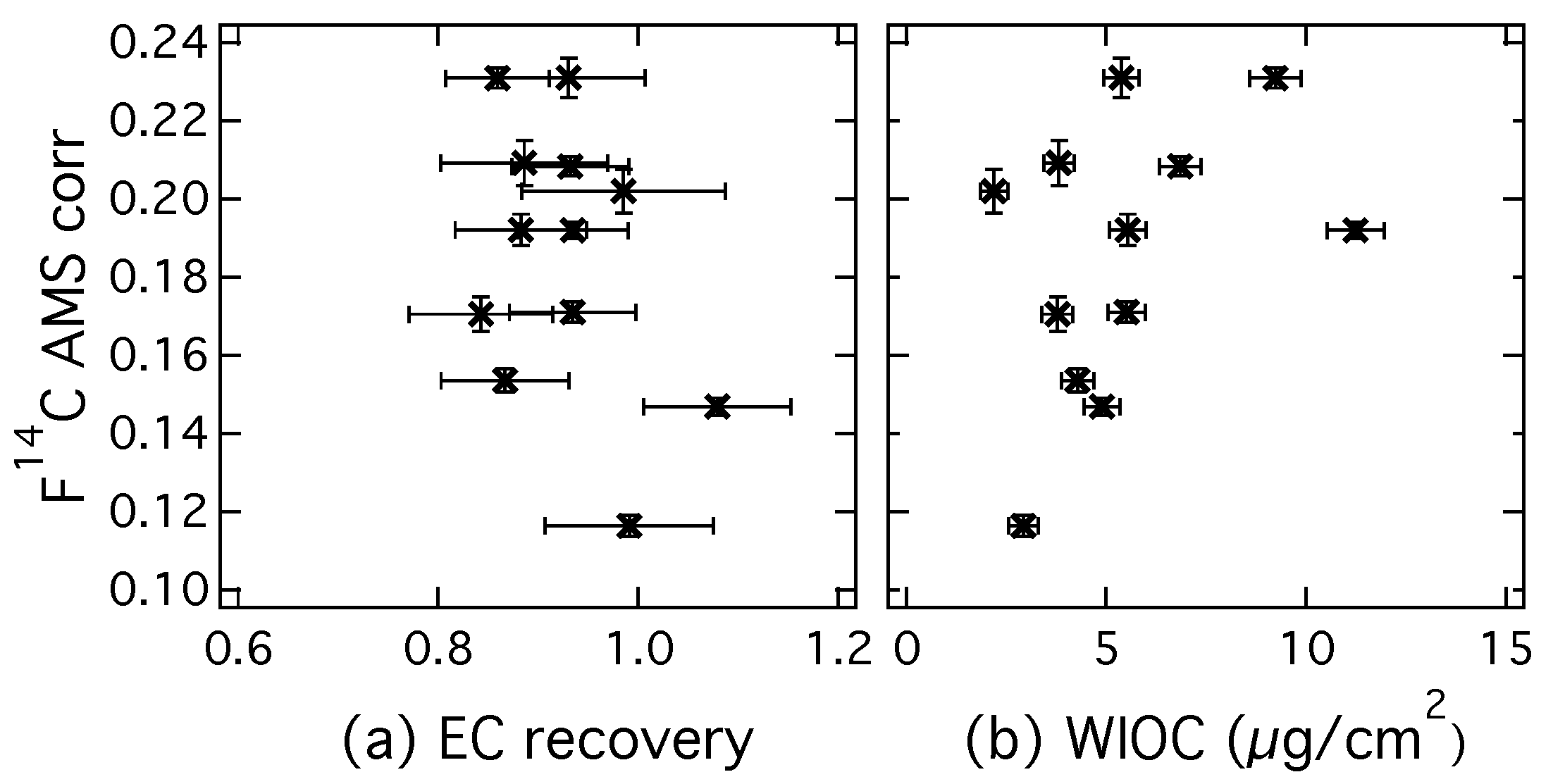
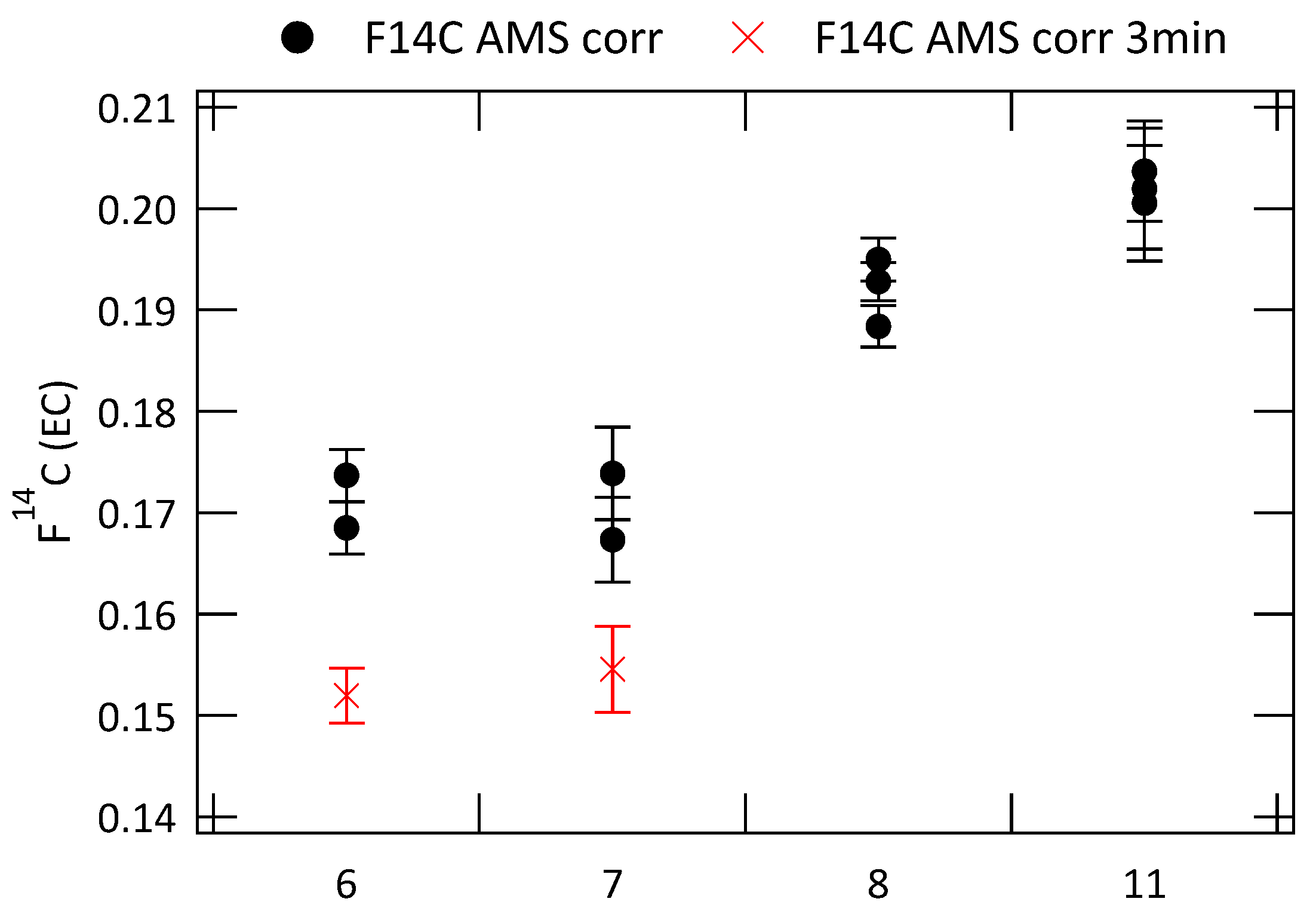
3.2. Analysis of Radiocarbon in EC: Results of an Inter-Comparison Study
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix A

References
- Pöschl, U. Atmospheric Aerosols: Composition, transformation, climate and health effects. Angew. Chem. Int. Ed. 2005, 44, 7520–7540. [Google Scholar] [CrossRef] [PubMed]
- Petzold, A.; Ogren, J.A.; Fiebig, M.; Laj, P.; Li, S.M.; Baltensperger, U.; Holzer-Popp, T.; Kinne, S.; Pappalardo, G.; Sugimoto, N.; et al. Recommendations for reporting “black carbon” measurements. Atmos. Chem. Phys. 2013, 13, 8365–8379. [Google Scholar] [CrossRef] [Green Version]
- Boucher, O.; Randall, D.; Artaxo, P.; Bretherton, C.; Feingold, G.; Forster, P.; Kerminen, V.M.; Kondo, Y.; Liao, H.; Lohmann, U.; et al. Clouds and aerosols. In Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; Stocker, T., Qin, D., Plattner, G.K., Tignor, M., Allen, S., Boschung, J., Nauels, A., Xia, Y., Bex, V., Midgley, P., Eds.; Book Section 7; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2013; pp. 571–658. [Google Scholar]
- Myhre, G.; Shindell, D.; Bréon, F.M.; Collins, W.; Fuglestvedt, J.; Huang, J.; Koch, D.; Lamarque, J.F.; Lee, D.; Mendoza, B.; et al. Anthropogenic and natural radiative forcing. In Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; Stocker, T., Qin, D., Plattner, G.K., Tignor, M., Allen, S., Boschung, J., Nauels, A., Xia, Y., Bex, V., Midgley, P., Eds.; Book Section 8; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2013; pp. 659–740. [Google Scholar]
- Andersson, A.; Sheesley, R.J.; Kruså, M.; Johansson, C.; Gustafsson, Ö. 14C-Based source assessment of soot aerosols in Stockholm and the Swedish EMEP-Aspvreten regional background site. Atmos. Environ. 2011, 45, 215–222. [Google Scholar] [CrossRef]
- Bernardoni, V.; Calzolai, G.; Chiari, M.; Fedi, M.; Lucarelli, F.; Nava, S.; Piazzalunga, A.; Riccobono, F.; Taccetti, F.; Valli, G.; et al. Radiocarbon analysis on organic and elemental carbon in aerosol samples and source apportionment at an urban site in Northern Italy. J. Aerosol Sci. 2013, 56, 88–99. [Google Scholar] [CrossRef]
- Chen, B.; Andersson, A.; Lee, M.; Kirillova, E.N.; Xiao, Q.; Kruså, M.; Shi, M.; Hu, K.; Lu, Z.; Streets, D.G.; et al. Source forensics of black carbon aerosols from China. Environ. Sci. Technol. 2013, 47, 9102–9108. [Google Scholar] [CrossRef] [PubMed]
- Dusek, U.; ten Brink, H.M.; Meijer, H.A.J.; Kos, G.; Mrozek, D.; Röckmann, T.; Holzinger, R.; Weijers, E.P. The contribution of fossil sources to the organic aerosol in The Netherlands. Atmos. Environ. 2013, 74, 169–176. [Google Scholar] [CrossRef]
- Dusek, U.; Hitzenberger, R.; Kasper-Giebl, A.; Kistler, M.; Meijer, H.A.J.; Szidat, S.; Wacker, L.; Holzinger, R.; Röckmann, T. Sources and formation mechanisms of carbonaceous aerosol at a regional background site in the Netherlands: Insights from a year-long radiocarbon study. Atmos. Chem. Phys. 2017, 17, 3233–3251. [Google Scholar] [CrossRef]
- Fushimi, A.; Wagai, R.; Uchida, M.; Hasegawa, S.; Takahashi, K.; Kondo, M.; Hirabayashi, M.; Morino, Y.; Shibata, Y.; Ohara, T.; et al. Radiocarbon (14C) diurnal variations in fine particles at Sites Downwind from Tokyo, Japan in Summer. Environ. Sci. Technol. 2011, 45, 6784–6792. [Google Scholar] [CrossRef] [PubMed]
- Heal, M.R.; Naysmith, P.; Cook, G.T.; Xu, S.; Duran, T.R.; Harrison, R.M. Application of 14C analyses to source apportionment of carbonaceous PM2.5 in the UK. Atmos. Environ. 2011, 45, 2341–2348. [Google Scholar] [CrossRef]
- Minguillón, M.C.; Perron, N.; Querol, X.; Szidat, S.; Fahrni, S.M.; Alastuey, A.; Jimenez, J.L.; Mohr, C.; Ortega, A.M.; Day, D.A.; et al. Fossil versus contemporary sources of fine elemental and organic carbonaceous particulate matter during the DAURE campaign in Northeast Spain. Atmos. Chem. Phys. 2011, 11, 12067–12084. [Google Scholar] [CrossRef] [Green Version]
- Morino, Y.; Takahashi, K.; Fushimi, A.; Tanabe, K.; Ohara, T.; Hasegawa, S.; Uchida, M.; Takami, A.; Yokouchi, Y.; Kobayashi, S. Contrasting diurnal variations in fossil and nonfossil secondary organic aerosol in Urban Outflow, Japan. Environ. Sci. Technol. 2010, 44, 8581–8586. [Google Scholar] [CrossRef] [PubMed]
- Szidat, S.; Jenk, T.M.; Synal, H.A.; Kalberer, M.; Wacker, L.; Hajdas, I.; Kasper-Giebl, A.; Baltensperger, U. Contributions of fossil fuel, biomass-burning, and biogenic emissions to carbonaceous aerosols in Zurich as traced by 14C. J. Geophys. Res. Atmos. 2006, 111, D07206. [Google Scholar] [CrossRef]
- Uchida, M.; Kumata, H.; Koike, Y.; Tsuzuki, M.; Uchida, T.; Fujiwara, K.; Shibata, Y. Radiocarbon-based source apportionment of black carbon (BC) in PM10 aerosols from residential area of suburban Tokyo. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 2010, 268, 1120–1124. [Google Scholar] [CrossRef]
- Zencak, Z.; Elmquist, M.; Gustafsson, Ö. Quantification and radiocarbon source apportionment of black carbon in atmospheric aerosols using the CTO-375 method. Atmos. Environ. 2007, 41, 7895–7906. [Google Scholar] [CrossRef]
- Cavalli, F.; Viana, M.; Yttri, K.E.; Genberg, J.; Putaud, J.P. Toward a standardised thermal-optical protocol for measuring atmospheric organic and elemental carbon: the EUSAAR protocol. Atmos. Meas. Tech. 2010, 3, 79–89. [Google Scholar] [CrossRef] [Green Version]
- Chow, J.C.; Watson, J.G.; Chen, L.W.A.; Chang, M.C.O.; Robinson, N.F.; Trimble, D.; Kohl, S. The IMPROVE-A temperature protocol for thermal/optical carbon analysis: Maintaining consistency with a long-term database. J. Air Waste Manag. Assoc. 2007, 57, 1014–1023. [Google Scholar] [CrossRef] [PubMed]
- Chow, J.C.; Watson, J.G.; Crow, D.; Lowenthal, D.H.; Merrifield, T. Comparison of IMPROVE and NIOSH Carbon Measurements. Aerosol Sci. Technol. 2001, 34, 23–34. [Google Scholar] [CrossRef]
- Chiappini, L.; Verlhac, S.; Aujay, R.; Maenhaut, W.; Putaud, J.P.; Sciare, J.; Jaffrezo, J.L.; Liousse, C.; Galy-Lacaux, C.; Alleman, L.Y.; et al. Clues for a standardised thermal-optical protocol for the assessment of organic and elemental carbon within ambient air particulate matter. Atmos. Meas. Tech. 2014, 7, 1649–1661. [Google Scholar] [CrossRef] [Green Version]
- Panteliadis, P.; Hafkenscheid, T.; Cary, B.; Diapouli, E.; Fischer, A.; Favez, O.; Quincey, P.; Viana, M.; Hitzenberger, R.; Vecchi, R.; et al. ECOC comparison exercise with identical thermal protocols after temperature offset correction—Instrument diagnostics by in-depth evaluation of operational parameters. Atmos. Meas. Tech. 2015, 8, 779–792. [Google Scholar] [CrossRef] [Green Version]
- Cachier, H.; Bremond, M.P.; Buat-Ménard, P. Determination of atmospheric soot carbon with a simple thermal method. Tellus B 1989, 41B, 379–390. [Google Scholar] [CrossRef]
- Dusek, U.; Monaco, M.; Prokopiou, M.; Gongriep, F.; Hitzenberger, R.; Meijer, H.A.J.; Röckmann, T. Evaluation of a two-step thermal method for separating organic and elemental carbon for radiocarbon analysis. Atmos. Meas. Tech. 2014, 7, 1943–1955. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.L.; Perron, N.; Ciobanu, V.G.; Zotter, P.; Minguillón, M.C.; Wacker, L.; Prévôt, A.S.H.; Baltensperger, U.; Szidat, S. On the isolation of OC and EC and the optimal strategy of radiocarbon-based source apportionment of carbonaceous aerosols. Atmos. Chem. Phys. 2012, 12, 10841–10856. [Google Scholar] [CrossRef] [Green Version]
- Elmquist, M.; Cornelissen, G.; Kukulska, Z.; Gustafsson, Ö. Distinct oxidative stabilities of char versus soot black carbon: Implications for quantification and environmental recalcitrance. Glob. Biogeochem. Cycles 2006, 20, GB2009. [Google Scholar] [CrossRef]
- Yu, J.Z.; Xu, J.; Yang, H. Charring characteristics of atmospheric organic particulate matter in thermal analysis. Environ. Sci. Technol. 2002, 36, 754–761. [Google Scholar] [CrossRef] [PubMed]
- Szidat, S.; Bench, G.; Bernardoni, V.; Calzolai, G.; Czimczik, C.I.; Derendorp, L.; Dusek, U.; Elder, K.; Fedi, M.E.; Genberg, J.; et al. Intercomparison of 14C analysis of carbonaceous aerosols: Exercise 2009. Radiocarbon 2013, 55, 1496–1509. [Google Scholar] [CrossRef]
- Chow, J.C.; Watson, J.G. PM2.5 carbonate concentrations at regionally representative Interagency Monitoring of Protected Visual Environment sites. J. Geophys. Res. Atmos. 2002, 107, 8344. [Google Scholar] [CrossRef]
- de Rooij, M.; van der Plicht, J.; Meijer, H.A.J. Porous iron pellets for AMS 14C analysis of small samples down to ultra-microscale size (10–25 µgC). Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 2010, 268, 947–951. [Google Scholar] [CrossRef]
- van der Plicht, J.; Wijma, S.; Aerts, A.T.; Pertuisot, M.H.; Meijer, H.A.J. Status report: The Groningen AMS facility. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 2000, 172, 58–65. [Google Scholar] [CrossRef]
- Reimer, P.J.; Brown, T.A.; Reimer, R.W. Discussion: Reporting and Calibration of post-bomb 14C Data. Radiocarbon 2004, 46, 1299–1304. [Google Scholar] [CrossRef]
- Santos, G.M.; Southon, J.R.; Griffin, S.; Beaupre, S.R.; Druffel, E.R.M. Ultra small-mass AMS 14C sample preparation and analyses at KCCAMS/UCI Facility. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 2007, 259, 293–302. [Google Scholar] [CrossRef]
- Ruff, M.; Wacker, L.; Gäggeler, H.W.; Suter, M.; Synal, H.A.; Szidat, S. A gas ion source for radiocarbon measurements at 200 kV. Radiocarbon 2007, 49, 307–314. [Google Scholar] [CrossRef]
- Salazar, G.; Zhang, Y.L.; Agrios, K.; Szidat, S. Development of a method for fast and automatic radiocarbon measurement of aerosol samples by online coupling of an elemental analyzer with a MICADAS AMS. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 2015, 361, 163–167. [Google Scholar] [CrossRef]
- Synal, H.A.; Stocker, M.; Suter, M. MICADAS: A new compact radiocarbon AMS system. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 2007, 259, 7–13. [Google Scholar] [CrossRef]
- Zotter, P.; El Haddad, I.; Zhang, Y.; Hayes, P.L.; Zhang, X.; Lin, Y.H.; Wacker, L.; Schnelle-Kreis, J.; Abbaszade, G.; Zimmermann, R.; et al. Diurnal cycle of fossil and nonfossil carbon using radiocarbon analyses during CalNex. J. Geophys. Res. Atmos. 2014, 119, 6818–6835. [Google Scholar] [CrossRef] [Green Version]
- Calzolai, G.; Bernardoni, V.; Chiari, M.; Fedi, M.; Lucarelli, F.; Nava, S.; Riccobono, F.; Taccetti, F.; Valli, G.; Vecchi, R. The new sample preparation line for radiocarbon measurements on atmospheric aerosol at LABEC. Nucl. Instrum. Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 2011, 269, 203–208. [Google Scholar] [CrossRef]
- Piazzalunga, A.; Bernardoni, V.; Fermo, P.; Valli, G.; Vecchi, R. Technical Note: On the effect of water-soluble compounds removal on EC quantification by TOT analysis in urban aerosol samples. Atmos. Chem. Phys. 2011, 11, 10193–10203. [Google Scholar] [CrossRef]
- Subramanian, R.; Khlystov, A.Y.; Robinson, A.L. Effect of peak inert-mode temperature on elemental carbon measured using thermal-optical analysis. Aerosol Sci. Technol. 2006, 40, 763–780. [Google Scholar] [CrossRef]
- Weingartner, E.; Saathoff, H.; Schnaiter, M.; Streit, N.; Bitnar, B.; Baltensperger, U. Absorption of light by soot particles: Determination of the absorption coefficient by means of aethalometers. J. Aerosol Sci. 2003, 34, 1445–1463. [Google Scholar] [CrossRef]
- Birch, M.E.; Cary, R.A. Elemental carbon-based method for monitoring occupational exposures to particulate diesel exhaust. Aerosol Sci. Technol. 1996, 25, 221–241. [Google Scholar] [CrossRef]
- Cheng, Y.; He, K.B. Measurement of carbonaceous aerosol with different sampling configurations and frequencies. Atmos. Meas. Tech. 2015, 8, 2639–2648. [Google Scholar] [CrossRef]
- Szidat, S.; Ruff, M.; Perron, N.; Wacker, L.; Synal, H.A.; Hallquist, M.; Shannigrahi, A.S.; Yttri, K.E.; Dye, C.; Simpson, D. Fossil and non-fossil sources of organic carbon (OC) and elemental carbon (EC) in Göteborg, Sweden. Atmos. Chem. Phys. 2009, 9, 1521–1535. [Google Scholar] [CrossRef] [Green Version]
- Zotter, P.; Ciobanu, V.G.; Zhang, Y.L.; El-Haddad, I.; Macchia, M.; Daellenbach, K.R.; Salazar, G.A.; Huang, R.J.; Wacker, L.; Hueglin, C.; et al. Radiocarbon analysis of elemental and organic carbon in Switzerland during winter-smog episodes from 2008 to 2012—Part 1: Source apportionment and spatial variability. Atmos. Chem. Phys. 2014, 14, 13551–13570. [Google Scholar] [CrossRef]
- Gustafsson, Ö.; Gschwend, P.M. Soot as a strong partition medium for polycyclic aromatic hydrocarbons in aquatic systems. In Molecular Markers in Environmental Geochemistry; ACS Publication: Washington, DC, USA, 1997; pp. 365–381. [Google Scholar] [CrossRef]
- Gustafsson, O.; Bucheli, T.D.; Kukulska, Z.; Andersson, M.; Largeau, C.; Rouzaud, J.N.; Reddy, C.M.; Eglinton, T.I. Evaluation of a protocol for the quantification of black carbon in sediments. Glob. Biogeochem. Cycles 2001, 15, 881–890. [Google Scholar] [CrossRef]
- Reddy, C.M.; Pearson, A.; Xu, L.; McNichol, A.P.; Benner, B.A.; Wise, S.A.; Klouda, G.A.; Currie, L.A.; Eglinton, T.I. Radiocarbon as a tool to apportion the sources of polycyclic aromatic hydrocarbons and black carbon in environmental samples. Environ. Sci. Technol. 2002, 36, 1774–1782. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Hu, M.; Guo, S.; Liu, K.; Zhou, L. 14C-Based source assessment of carbonaceous aerosols at a rural site. Atmos. Environ. 2012, 50, 36–40. [Google Scholar] [CrossRef]
- Winiger, P.; Andersson, A.; Yttri, K.E.; Tunved, P.; Gustafsson, Ö. Isotope-based source apportionment of EC aerosol particles during winter high-pollution events at the Zeppelin Observatory, Svalbard. Environ. Sci. Technol. 2015, 49, 11959–11966. [Google Scholar] [CrossRef] [PubMed]
- Minoura, H.; Morikawa, T.; Mizohata, A.; Sakamoto, K. Carbonaceous aerosol and its characteristics observed in Tokyo and south Kanto region. Atmos. Environ. 2012, 61, 605–613. [Google Scholar] [CrossRef]
- Liu, J.; Li, J.; Ding, P.; Zhang, Y.; Liu, D.; Shen, C.; Zhang, G. Optimizing isolation protocol of organic carbon and elemental carbon for 14C analysis using fine particulate samples. Atmos. Environ. 2017, 154, 9–19. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Sampling | Sampling | Sampling | Main Air | |
|---|---|---|---|---|---|
| Identity | Start | End | Duration (days) | Mass | |
| 1 | 2 September 2014 | 4 September 2014 | 2 | mixed | |
| 2 | 4 September 2014 | 7 September 2014 | 3 | continental | x |
| 3 | 7 September 2014 | 12 September 2014 | 5 | regional | x |
| 4 | 12 September 2014 | 16 September 2014 | 4 | mixed | x |
| 5 | 16 September 2014 | 18 September 2014 | 2 | continental | o |
| 6 | 18 September 2014 | 21 September 2014 | 3 | continental | o |
| 7 | 21 September 2014 | 25 September 2014 | 4 | regional | o |
| 8 | 25 September 2014 | 30 September 2014 | 5 | mixed | o |
| 9 | 30 September 2014 | 2 October 2014 | 2 | mixed | |
| 10 | 2 October 2014 | 5 October 2014 | 3 | mixed | x |
| 11 | 5 October 2014 | 9 October 2014 | 4 | mixed | |
| 12 | 9 October 2014 | 14 October 2014 | 5.39 | mixed |
| Laboratory | LARA | LABEC | CIO | |
|---|---|---|---|---|
| Pretreatment | water extraction | water extraction | water extraction | |
| Type Oven | commercial OC-EC analyzer | self-built | self-built | |
| Thermal Protocol | Step 1 | 180 °C, 50 s+ 375 °C, 150 s, O2 | 375 °C, 40 min, O2 | 375 °C, 10 min, O2 |
| Step 2 | 475 °C, 120 s, O2 | 750 °C, flash heating, He | 450 °C, 2 min/3 min, O2 | |
| Step 3 | 450 °C, 180 s+ 650 °C, 180 s, He | 800 °C, 20 min, O2 | 650 °C, 5 min, O2 | |
| Step 4 | 500 °C, 120 s+ 760 °C, 150 s, O2 | |||
| AMS Measurements | direct CO2 injection MICADAS | graphitization on iron powder 3 MV Tandetron accelerator (HVEE) | graphitization on iron pellets 3 MV Tandetron accelerator (HVEE) | |
| Corrections | •background contamination | •background contamination | •background contamination | |
| • cross contamination | • blank correction | |||
| • EC yield correction | • OC charring correction | |||
| • OC charring correction | ||||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zenker, K.; Vonwiller, M.; Szidat, S.; Calzolai, G.; Giannoni, M.; Bernardoni, V.; Jedynska, A.D.; Henzing, B.; Meijer, H.A.J.; Dusek, U. Evaluation and Inter-Comparison of Oxygen-Based OC-EC Separation Methods for Radiocarbon Analysis of Ambient Aerosol Particle Samples. Atmosphere 2017, 8, 226. https://doi.org/10.3390/atmos8110226
Zenker K, Vonwiller M, Szidat S, Calzolai G, Giannoni M, Bernardoni V, Jedynska AD, Henzing B, Meijer HAJ, Dusek U. Evaluation and Inter-Comparison of Oxygen-Based OC-EC Separation Methods for Radiocarbon Analysis of Ambient Aerosol Particle Samples. Atmosphere. 2017; 8(11):226. https://doi.org/10.3390/atmos8110226
Chicago/Turabian StyleZenker, Katrin, Matthias Vonwiller, Sönke Szidat, Giulia Calzolai, Martina Giannoni, Vera Bernardoni, Aleksandra D. Jedynska, Bas Henzing, Harro A. J. Meijer, and Ulrike Dusek. 2017. "Evaluation and Inter-Comparison of Oxygen-Based OC-EC Separation Methods for Radiocarbon Analysis of Ambient Aerosol Particle Samples" Atmosphere 8, no. 11: 226. https://doi.org/10.3390/atmos8110226