Contribution from Selected Organic Species to PM2.5 Aerosol during a Summer Field Campaign at K-Puszta, Hungary

 ,
, 
Abstract
:1. Introduction
2. Experiments
3. Results and Discussion
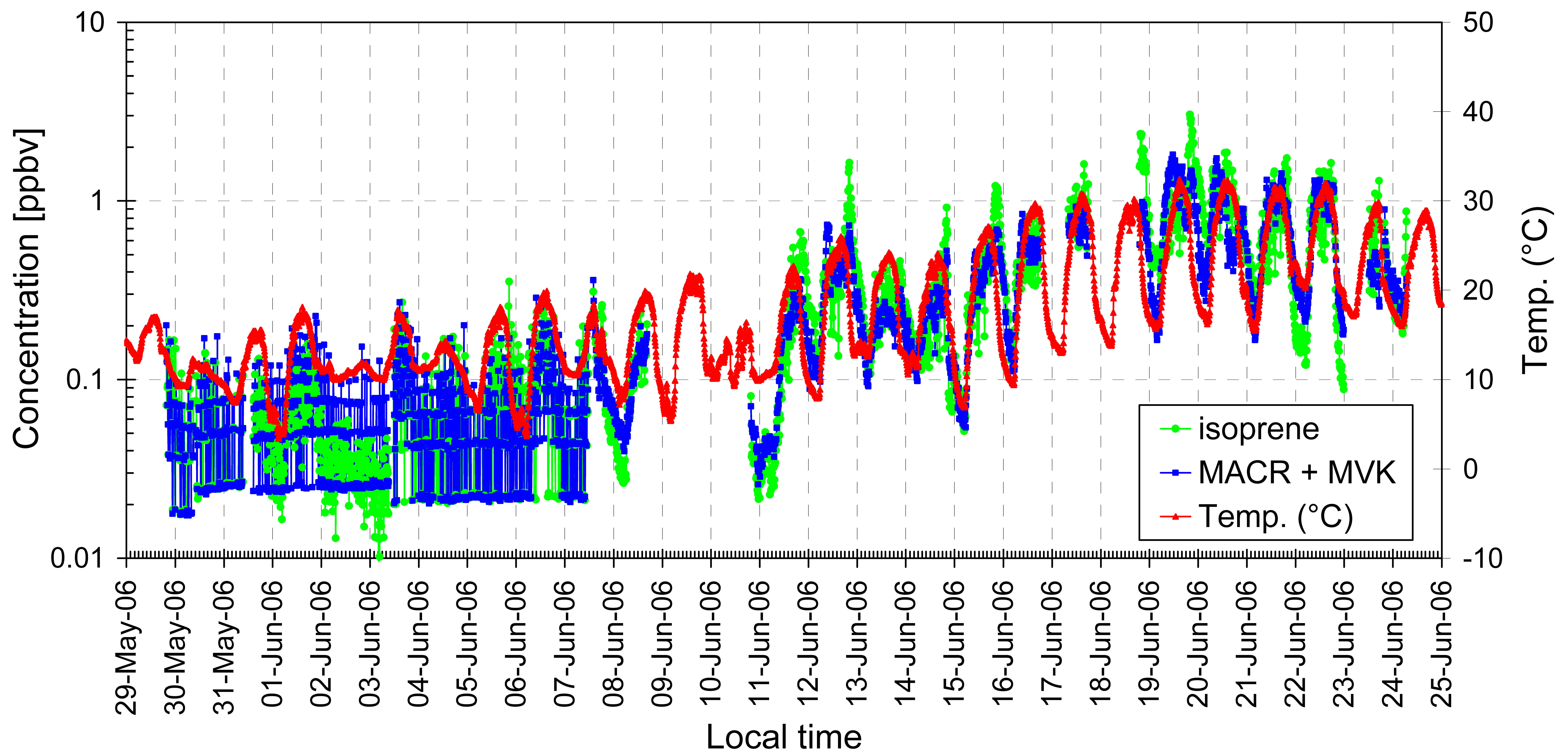
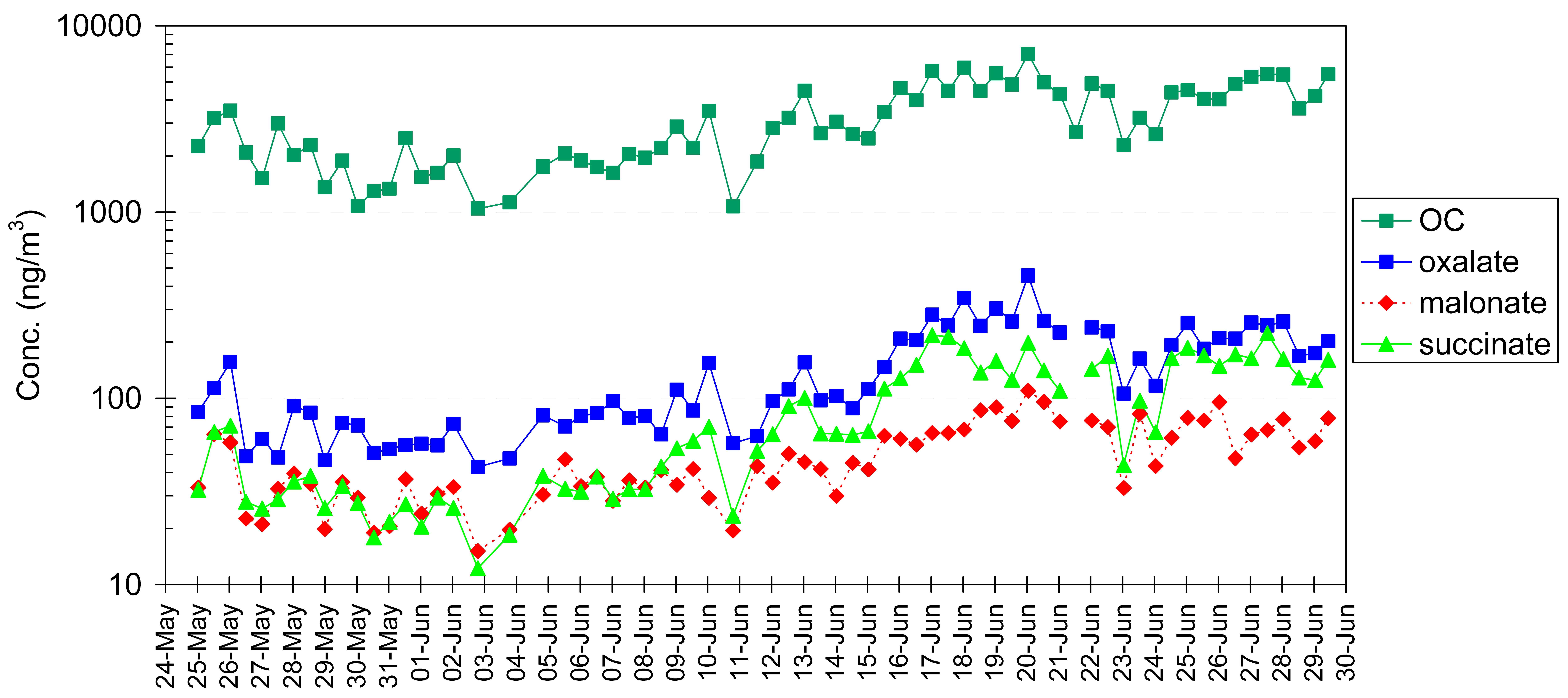
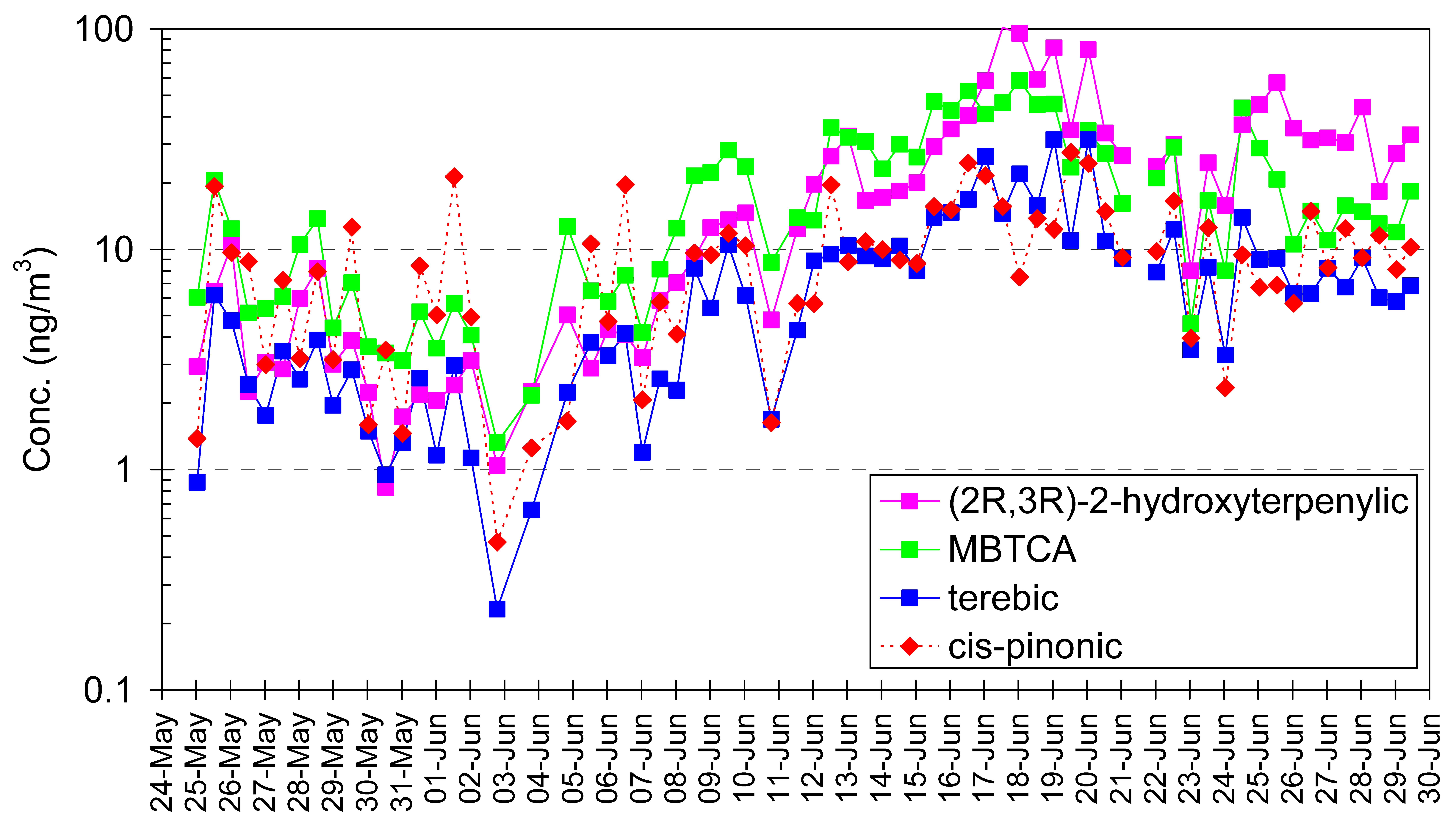
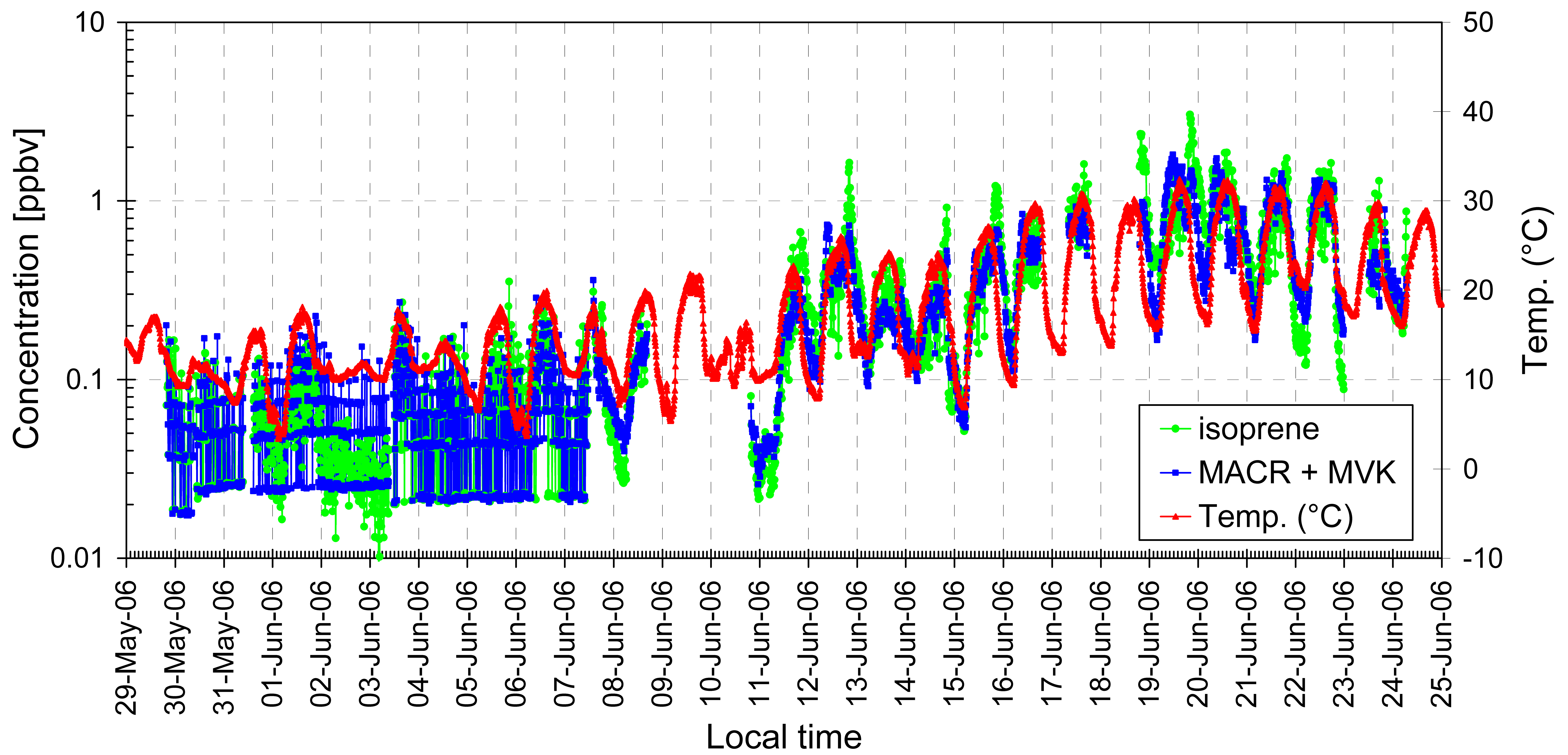
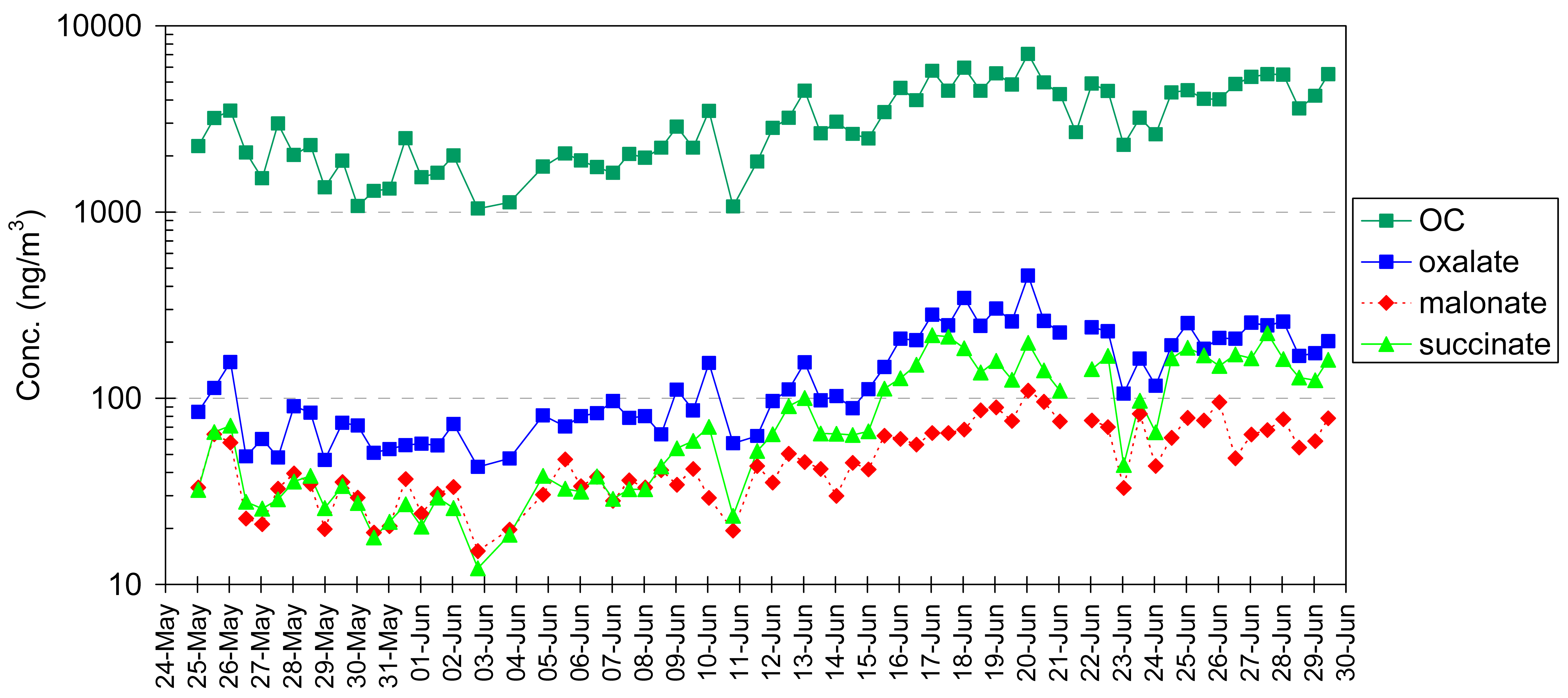
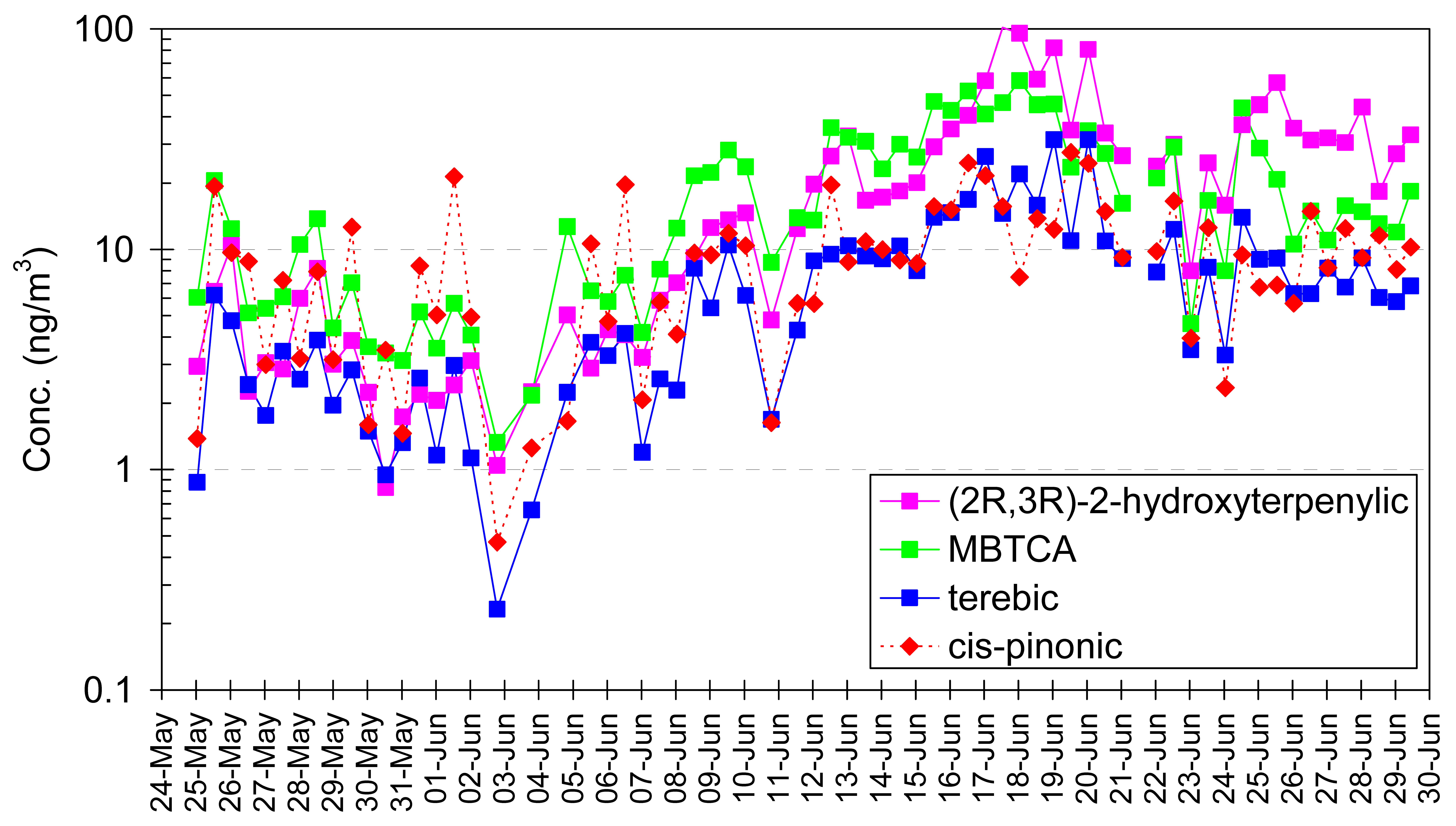
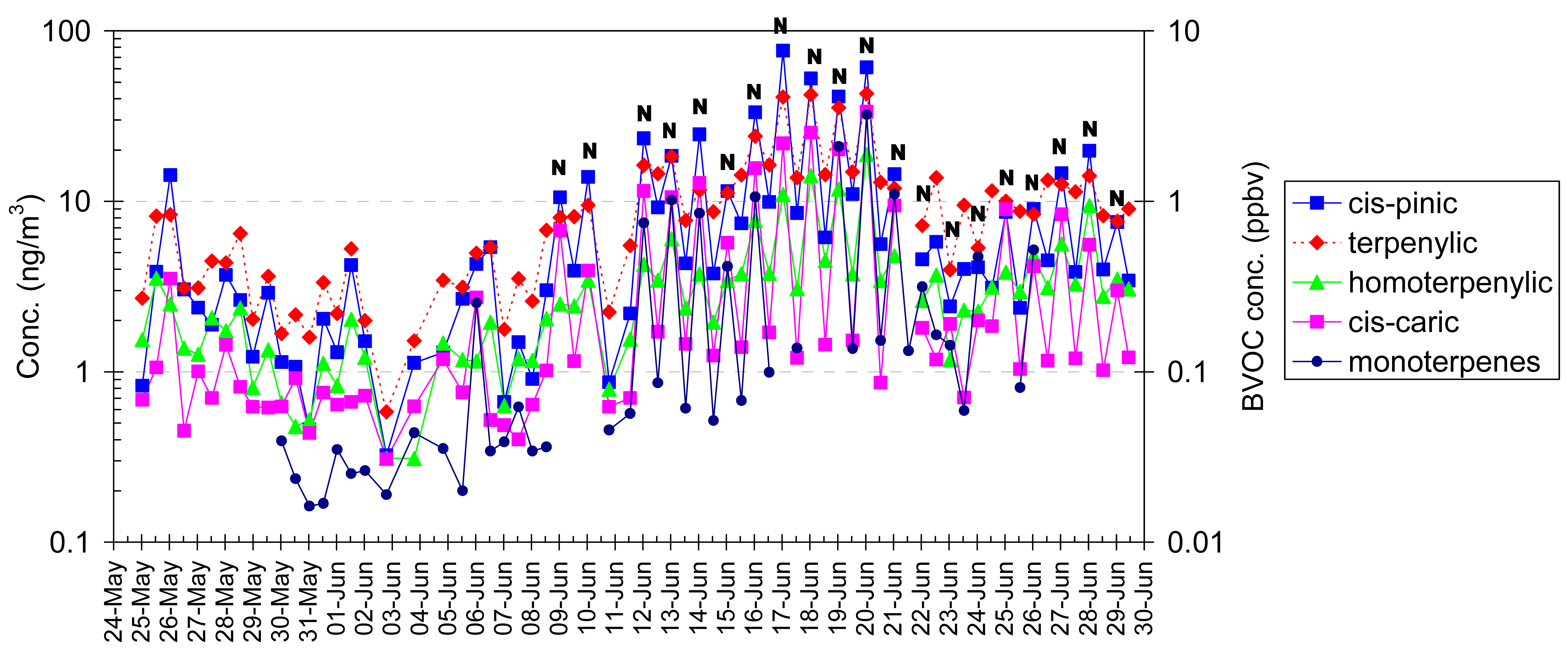
3.1. Concentrations, Time Series, and Correlations
3.2. Contribution of the Measured Organic Compounds and α-Pinene Secondary Organic Carbon to the OC
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gelencsér, A.; May, B.; Simpson, D.; Sanchez-Ochoa, A.; Kasper-Giebl, A.; Puxbaum, H.; Caseiro, A.; Pio, C.; Legrand, M. Source apportionment of PM2.5 organic aerosol over Europe: Primary/secondary, natural/anthropogenic, and fossil/biogenic origin. J. Geophys. Res. Atmos. 2007, 112, D23S04. [Google Scholar] [CrossRef]
- Ion, A.C.; Vermeylen, R.; Kourtchev, I.; Cafmeyer, J.; Chi, X.; Gelencsér, A.; Maenhaut, W.; Claeys, M. Polar organic compounds in rural PM2.5 aerosols from K-puszta, Hungary, during a 2003 summer field campaign: Sources and diel variations. Atmos. Chem. Phys. 2005, 5, 1805–1814. [Google Scholar] [CrossRef]
- Kourtchev, I.; Copolovici, L.; Claeys, M.; Maenhaut, W. Characterization of atmospheric aerosols at a forested site in Central Europe. Environ. Sci. Technol. 2009, 43, 4665–4671. [Google Scholar] [CrossRef] [PubMed]
- Maenhaut, W.; Wang, W.; Chi, X. Semivolatile behaviour and filter sampling artifacts for dicarboxylic acids during summer campaigns at three forested sites in Europe. Boreal Environ. Res. 2011, 16, 273–287. [Google Scholar]
- Claeys, M.; Iinuma, Y.; Szmigielski, R.; Surratt, J.D.; Blockhuys, F.; Van Alsenoy, C.; Böge, O.; Sierau, B.; Gómez-González, Y.; Vermeylen, R.; et al. Terpenylic acid and related compounds from the oxidation of alpha-pinene: Implications for new particle formation and growth above forests. Environ. Sci. Technol. 2009, 43, 6976–6982. [Google Scholar] [CrossRef] [PubMed]
- Kahnt, A.; Iinuma, Y.; Blockhuys, F.; Mutzel, A.; Vermeylen, R.; Kleindienst, T.E.; Jaoui, M.; Offenberg, J.H.; Lewandowski, M.; Böge, O.; et al. 2-Hydroxyterpenylic acid: An oxygenated marker compound for alpha-pinene secondary organic aerosol in ambient fine aerosol. Environ. Sci. Technol. 2014, 48, 4901–4908. [Google Scholar] [CrossRef] [PubMed]
- Kubátová, A.; Vermeylen, R.; Claeys, M.; Cafmeyer, J.; Maenhaut, W. Organic compounds in urban aerosols from Gent, Belgium: Characterization, sources, and seasonal differences. J. Geophys. Res. 2002, 107, 8343. [Google Scholar] [CrossRef]
- Oliveira, T.S.; Pio, C.A.; Alves, C.A.; Silvestre, A.J.D.; Evtyugina, M.; Afonso, J.V.; Fialho, P.; Legrand, M.; Puxbaum, H.; Gelencsér, A. Seasonal variation of particulate lipophilic organic compounds at nonurban sites in Europe. J. Geophys. Res. 2007, 112, D23S09. [Google Scholar] [CrossRef]
- Kourtchev, I.; Warnke, J.; Maenhaut, W.; Hoffmann, T.; Claeys, M. Polar organic marker compounds in PM2.5 aerosol from a mixed forest site in western Germany. Chemosphere 2008, 73, 1308–1314. [Google Scholar] [CrossRef] [PubMed]
- Kourtchev, I.; Ruuskanen, T.; Maenhaut, W.; Kulmala, M.; Claeys, M. Observation of 2-methyltetrols and related photo-oxidation products of isoprene in boreal forest aerosols from Hyytiälä, Finland. Atmos. Chem. Phys. 2005, 5, 2761–2770. [Google Scholar] [CrossRef]
- Kourtchev, I.; Ruuskanen, T.M.; Keronen, P.; Sogacheva, L.; Dal Maso, M.; Reissell, A.; Chi, X.; Vermeylen, R.; Kulmala, M.; Maenhaut, W.; et al. Determination of isoprene and alpha-/beta-pinene oxidation products in boreal forest aerosols from Hyytiälä, Finland: Diel variations and possible link with particle formation events. Plant Biol. 2008, 10, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Plewka, A.; Gnauk, T.; Brüggemann, E.; Herrmann, H. Biogenic contributions to the chemical composition of airborne particles in a coniferous forest in Germany. Atmos. Environ. 2006, 40, 103–115. [Google Scholar] [CrossRef]
- Maenhaut, W.; Claeys, M.; Janssens, I.; Kulmala, M. Formation Mechanisms, Marker Compounds and Source Apportionment for BIOgenic Atmospheric aeroSOLs (BIOSOL); Research Programme Science for a Sustainable Development (SSD); Research Project SD/AT/02; Final Report; (D2011/1191/15); Belgian Science Policy: Brussels, Belgium, 2011; p. 121. Available online: http://www.belspo.be/belspo/SSD/science/Reports/BIOSOL_FinalRepor_ML.pdf (accessed on 7 November 2017).
- Ocskay, R.; Salma, I.; Wang, W.; Maenhaut, W. Characterization and diurnal variation of size-resolved inorganic water-soluble ions at a rural background site. J. Environ. Monit. 2006, 8, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Pio, C.; Legrand, M.; Oliveira, T.; Afonso, J.; Santos, C.; Caseiro, A.; Fialho, P.; Barata, F.; Puxbaum, H.; Sanchez-Ochoa, A.; et al. Climatology of aerosol composition (organic versus inorganic) at non-urban areas on a west-east transect across Europe. J. Geophys. Res. 2007, 112, D23S02. [Google Scholar] [CrossRef]
- Solomon, P.A.; Moyers, J.L.; Fletcher, R.A. High-volume dichotomous sampler virtual impactor for the fractionation and collection of particles according to aerodynamic size. Aerosol Sci. Technol. 1983, 2, 455–464. [Google Scholar] [CrossRef]
- Lindinger, W.; Hansel, A.; Jordan, A. On-line monitoring of volatile organic compounds at pptv levels by means of proton-transfer-reaction mass spectrometry (PTR-MS)—Medical applications, food control and environmental research. Int. J. Mass Spectrom. Ion Process. 1998, 173, 191–241. [Google Scholar] [CrossRef]
- Yli-Juuti, T.; Riipinen, I.; Aalto, P.P.; Nieminen, T.; Maenhaut, W.; Janssens, I.A.; Claeys, M.; Salma, I.; Ocskay, R.; Hoffer, A.; et al. Characteristics of aerosol particle formation events at K-puszta, Hungary. Boreal Environ. Res. 2009, 14, 683–698. [Google Scholar]
- Birch, M.E.; Cary, R.A. Elemental carbon-based method for monitoring occupational exposures to particulate diesel exhaust. Aerosol Sci. Technol. 1996, 25, 221–241. [Google Scholar] [CrossRef]
- Viana, M.; Chi, X.; Maenhaut, W.; Querol, X.; Alastuey, A.; Mikuška, P.; Večeřa, Z. Organic and elemental carbon concentrations in carbonaceous aerosols during summer and winter sampling campaigns in Barcelona, Spain. Atmos. Environ. 2006, 40, 2180–2193. [Google Scholar] [CrossRef]
- Gómez-González, Y.; Wang, W.; Vermeylen, R.; Chi, X.; Neirynck, J.; Janssens, I.A.; Maenhaut, W.; Claeys, M. Chemical characterisation of atmospheric aerosols during a 2007 summer field campaign at Brasschaat, Belgium: Sources and source processes of biogenic secondary organic aerosol. Atmos. Chem. Phys. 2012, 12, 125–138. [Google Scholar] [CrossRef] [Green Version]
- Maenhaut, W.; Raes, N.; Chi, X.; Cafmeyer, J.; Wang, W. Chemical composition and mass closure for PM2.5 and PM10 aerosols at K-puszta, Hungary, in summer 2006. X-ray Spectrom. 2008, 37, 193–197. [Google Scholar] [CrossRef]
- Kesselmeier, J.; Staudt, M. Biogenic volatile organic compounds (VOC): An overview on emission, physiology and ecology. J. Atmos. Chem. 1999, 33, 23–88. [Google Scholar] [CrossRef]
- Atkinson, R. Gas-phase tropospheric chemistry of volatile organic compounds. I. Alkanes and alkenes. J. Phys. Chem. Ref. Data 1997, 26, 215–290. [Google Scholar] [CrossRef]
- Atkinson, R.; Arey, J. Atmospheric chemistry of biogenic organic compounds. Acc. Chem. Res. 1998, 31, 574–583. [Google Scholar] [CrossRef]
- Calogirou, A.; Larsen, B.R.; Kotzias, D. Gas-phase terpene oxidation products: A review. Atmos. Environ. 1999, 33, 1423–1439. [Google Scholar] [CrossRef]
- Hallquist, M.; Wenger, J.C.; Baltensperger, U.; Rudich, Y.; Simpson, D.; Claeys, M.; Dommen, J.; Donahue, N.M.; George, C.; Goldstein, A.H.; et al. The formation, properties and impact of secondary organic aerosol: Current and emerging issues. Atmos. Chem. Phys. 2009, 9, 5155–5236. [Google Scholar] [CrossRef] [Green Version]
- Jokinen, T.; Berndt, T.; Makkonen, R.; Kerminen, V.-M.; Junninen, H.; Paasonen, P.; Stratmann, F.; Herrmann, H.; Guenther, A.B.; Worsnop, D.R.; et al. Production of extremely low volatile organic compounds from biogenic emissions: Measured yields and atmospheric implications. Proc. Natl. Acad. Sci. USA 2015, 112, 7123–7128. [Google Scholar] [CrossRef] [PubMed]
- Nozière, B.; Kalberer, M.; Claeys, M.; Allan, J.; D’Anna, B.; Decesari, S.; Finessi, E.; Glasius, M.; Grgić, I.; Hamilton, J.; et al. The molecular identification of organic compounds in the atmosphere: State of the art and challenges. Chem. Rev. 2015, 115, 3919–3983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surratt, J.D.; Gómez-González, Y.; Chan, A.W.H.; Vermeylen, R.; Shahgholi, M.; Kleindienst, T.E.; Edney, E.O.; Offenberg, J.H.; Lewandowski, M.; Jaoui, M.; et al. Organosulfate formation in biogenic secondary organic aerosol. J. Phys. Chem. A 2008, 112, 8345–8378. [Google Scholar] [CrossRef] [PubMed]
- Bond, T.C.; Doherty, S.J.; Fahey, D.W.; Forster, P.M.; Berntsen, T.; DeAngelo, B.J.; Flanner, M.G.; Ghan, S.; Karcher, B.; Koch, D.; et al. Bounding the role of black carbon in the climate system: A scientific assessment. J. Geophys. Res. 2013, 118, 5380–5552. [Google Scholar] [CrossRef] [Green Version]
- Ayers, G.P.; Ivey, J.P.; Gillett, R.W. Coherence between seasonal cycles of dimethyl sulfide, methanesulfonate and sulfate in marine air. Nature 1991, 349, 404–406. [Google Scholar] [CrossRef]
- Camredon, M.; Hamilton, J.F.; Alam, M.S.; Wyche, K.P.; Carr, T.; White, I.R.; Monks, P.S.; Rickard, A.R.; Bloss, W.J. Distribution of gaseous and particulate organic composition during dark alpha-pinene ozonolysis. Atmos. Chem. Phys. 2010, 10, 2893–2917. [Google Scholar] [CrossRef] [Green Version]
- Legrand, M.; Preunkert, S.; Oliveira, T.; Hammer, S.; Gelencsér, A.; Kasper-Giebl, A.; Laj, P. Origin of C2–C5 dicarboxylic acids in the European atmosphere inferred from year-round aerosol study conducted at a west-east transect. J. Geophys. Res. 2007, 112, D23S07. [Google Scholar] [CrossRef]
- Mochizuki, T.; Kawamura, K.; Miyazaki, Y.; Wada, R.; Takahashi, Y.; Saigusa, N.; Tani, A. Secondary formation of oxalic acid and related organic species from biogenic sources in a larch forest at the northern slope of Mt. Fuji. Atmos. Environ. 2017, 166, 255–262. [Google Scholar] [CrossRef]
- Chebbi, A.; Carlier, P. Carboxylic acids in the troposphere, occurrence, sources, and sinks: A review. Atmos. Environ. 1996, 30, 4233–4249. [Google Scholar] [CrossRef]
- Wang, W. Inorganic and Organic Speciation of Atmospheric Aerosols by Ion Chromatography and Aerosol Chemical Mass Closure. Ph.D. Thesis, Ghent University, Ghent, Belgium, 2010; p. 363. Available online: http://biblio.ugent.be/record/1048356 (accessed on 7 November 2017).
- Cahill, T.M.; Seaman, V.Y.; Charles, M.J.; Holzinger, R.; Goldstein, A.H. Secondary organic aerosols formed from oxidation of biogenic volatile organic compounds in the Sierra Nevada Mountains of California. J. Geophys. Res. 2006, 111, D16312. [Google Scholar] [CrossRef]
- Kleindienst, T.E.; Jaoui, M.; Lewandowski, M.; Offenberg, J.H.; Lewis, C.W.; Bhave, P.V.; Edney, E.O. Estimates of the contributions of biogenic and anthropogenic hydrocarbons to secondary organic aerosol at a southeastern US location. Atmos. Environ. 2007, 41, 8288–8300. [Google Scholar] [CrossRef]
- Szmigielski, R.; Surratt, J.D.; Gómez-González, Y.; Van der Veken, P.; Kourtchev, I.; Vermeylen, R.; Blockhuys, F.; Jaoui, M.; Kleindienst, T.E.; Lewandowski, M.; et al. 3-Methyl-1,2,3-butanetricarboxylic acid: An atmospheric tracer for terpene secondary organic aerosol. Geophys. Res. Lett. 2007, 34, L24811. [Google Scholar] [CrossRef]
- Guenther, A.B.; Jiang, X.; Heald, C.L.; Sakulyanontvittaya, T.; Duhl, T.; Emmons, L.K.; Wang, X. The Model of Emissions of Gases and Aerosols from Nature version 2.1 (MEGAN2.1): An extended and updated framework for modeling biogenic emissions. Geosci. Model Dev. 2012, 5, 1471–1492. [Google Scholar] [CrossRef] [Green Version]
- Lewandowski, M.; Jaoui, M.; Offenberg, J.H.; Kleindienst, T.E.; Edney, E.O.; Sheesley, R.J.; Schauer, J.J. Primary and secondary contributions to ambient PM in the Midwestern United States. Environ. Sci. Technol. 2008, 42, 3303–3309. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Bian, Q.; Li, T.W.Y.; Lau, A.K.H.; Yu, J.Z. Contributions of isoprene, monoterpenes, β-caryophyllene, and toluene to secondary organic aerosol in Hong Kong during the summer of 2006. J. Geophys. Res. 2008, 113, D22206. [Google Scholar] [CrossRef]
- El Haddad, I.; Marchand, N.; Temime-Roussel, B.; Wortham, H.; Piot, C.; Besombes, J.L.; Baduel, C.; Voisin, D.; Armengaud, A.; Jaffrezo, J.L. Insights into the secondary fraction of the organic aerosol in a Mediterranean urban area: Marseille. Atmos. Chem. Phys. 2011, 11, 2059–2079. [Google Scholar] [CrossRef] [Green Version]
- Lewandowski, M.; Piletic, I.R.; Kleindienst, T.E.; Offenberg, J.H.; Beaver, M.R.; Jaoui, M.; Docherty, K.S.; Edney, E.O. Secondary organic aerosol characterisation at field sites across the United States during the spring-summer period. Int. J. Environ. Anal. Chem. 2013, 93, 1084–1103. [Google Scholar] [CrossRef]
- Ding, X.; Zhang, Y.Q.; He, Q.F.; Yu, Q.Q.; Shen, R.Q.; Zhang, Y.L.; Zhang, Z.; Lyu, S.J.; Hu, Q.H.; Wang, Y.S.; et al. Spatial and seasonal variations of secondary organic aerosol from terpenoids over China. J. Geophys. Res. 2016, 121, 14661–14678. [Google Scholar] [CrossRef]
- Li, X.R.; Liu, Y.S.; Li, D.; Wang, G.A.; Bai, Y.; Diao, H.L.; Shen, R.R.; Hu, B.; Xin, J.Y.; Liu, Z.R.; et al. Molecular composition of organic aerosol over an agricultural site in North China Plain: Contribution of biogenic sources to PM2.5. Atmos. Environ. 2017, 164, 448–457. [Google Scholar] [CrossRef]
- Glasius, M.; Goldstein, A.H. Recent discoveries and future challenges in atmospheric organic chemistry. Environ. Sci. Technol. 2016, 50, 2754–2764. [Google Scholar] [CrossRef] [PubMed]
- Ehn, M.; Thornton, J.A.; Kleist, E.; Sipilä, M.; Junninen, H.; Pullinen, I.; Springer, M.; Rubach, F.; Tillmann, R.; Lee, B.; et al. A large source of low-volatility secondary organic aerosol. Nature 2014, 506, 476–479. [Google Scholar] [CrossRef] [PubMed]
- Kirkby, J.; Duplissy, J.; Sengupta, K.; Frege, C.; Gordon, H.; Williamson, C.; Heinritzi, M.; Simon, M.; Yan, C.; Trostl, J.; et al. Ion-induced nucleation of pure biogenic particles. Nature 2016, 533, 521–526. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cold Period—Day | Cold Period—Night | Warm Period—Day | Warm Period—Night | |
|---|---|---|---|---|
| Species | Median (Interq. Range) | Median (Interq. Range) | Median (Interq. Range) | Median (Interq. Range) |
| isoprene (m/z 69) | 0.074 (0.042–0.125) | 0.041 (0.025–0.065) | 0.58 (0.34–0.90) | 0.33 (0.179–0.69) |
| MACR and MVK (m/z 71) | 0.072 (0.042–0.123) | 0.047 (0.025–0.075) | 0.60 (0.36–0.91) | 0.29 (0.168–0.50) |
| monoterpenes (m/z 137) | 0.028 (0.0190–0.041) | 0.030 (0.0192–0.047) | 0.095 (0.062–0.140) | 0.66 (0.29–1.35) |
| nopinone (m/z 139) | 0.0057 (0.0040–0.0085) | 0.0054 (0.0038–0.0075) | 0.036 (0.022–0.052) | 0.044 (0.033–0.077) |
| pinonaldehyde (m/z 151) | 0.0039 (0.0025–0.0053) | 0.0049 (0.0034–0.0072) | 0.0143 (0.0077–0.022) | 0.035 (0.022–0.065) |
| MBO (m/z 87) | 0.062 (0.049–0.078) | 0.056 (0.044–0.072) | 0.172 (0.131–0.21) | 0.150 (0.110–0.20) |
| benzene (m/z 79) | 0.046 (0.034–0.059) | 0.055 (0.042–0.072) | 0.056 (0.043–0.073) | 0.075 (0.058–0.097) |
| Compound (MW) | Day (N = 14) Median (Interq. Range) | Night (N = 15) Median (Interq. Range) |
|---|---|---|
| EC | 144 (129–180) | 165 (127–230) |
| OC | 2100 (1870–2300) | 1960 (1530–2500) |
| WSOC | 960 (860–1150) | 1050 (740–1330) |
| MSA− | 37 (15.1–53) | 23 (15.2–36) |
| oxalate | 67 (56–82) | 80 (66–97) |
| malonate | 36 (33–42) | 33 (26–34) |
| succinate | 33 (29–42) | 32 (26–45) |
| MBTCA (204) | 7.3 (5.8–13.9) | 5.8 (4.1–12.4) |
| cis-pinonic acid (184) | 9.2 (7.4–12.4) | 4.1 (2.5–5.4) |
| cis-pinic acid (186) | 2.8 (2.1–3.7) | 1.51 (1.02–7.4) |
| terebic acid (158) | 3.6 (2.7–4.3) | 1.96 (1.26–4.0) |
| terpenylic acid (172) | 4.9 (3.4–6.2) | 2.7 (2.0–6.5) |
| diaterpenylic acid acetate (232) | 0.82 (0.72–1.51) | 0.74 (0.55–1.34) |
| cis-caronic acid (184) | 1.11 (0.72–2.3) | 0.27 (0.21–0.52) |
| cis-caric acid (186) | 0.73 (0.63–0.89) | 0.72 (0.63–3.1) |
| ketolimononic acid (186) | 0.92 (0.71–1.19) | 0.54 (0.41–0.69) |
| limonic acid (186) | 1.31 (1.02–1.62) | 1.71 (1.36–2.5) |
| homoterpenylic acid (186) | 1.75 (1.23–2.1) | 1.22 (0.82–2.1) |
| 4-hydroxyterpenylic acid (188) | 1.15 (0.72–2.1) | 1.19 (0.88–2.5) |
| (2R,3R)-2-hydroxyterpenylic (188) | 4.0 (2.5–7.7) | 3.2 (3.0–8.7) |
| (2S,3R)-2-hydroxyterpenylic (188) | 1.97 (1.53–4.0) | 2.3 (1.48–4.7) |
| unknown (188) | 2.0 (1.39–3.3) | 1.23 (1.15–2.9) |
| pinanediol mono-nitrate OSs [∑5 isomers] (295) | 3.6 (2.3–4.4) | 13.7 (9.3–25) |
| Compound (MW) | Day (N = 17) Median (Interq. Range) | Night (N = 17) Median (Interq. Range) |
|---|---|---|
| EC | 162 (131–250) | 179 (122–230) |
| OC | 4200 (3300–4800) | 4500 (4000–5500) |
| WSOC | 2900 (2100–3200) | 3000 (2400–3100) |
| MSA− | 32 (25–35) | 24 (19.4–37) |
| oxalate | 200 (163–240) | 230 (156–260) |
| malonate | 65 (54–76) | 65 (45–77) |
| succinate | 141 (113–169) | 143 (100–164) |
| MBTCA (204) | 29 (18.3–44) | 23 (12.0–34) |
| cis-pinonic acid (184) | 13.8 (10.8–15.6) | 8.8 (7.5–10.0) |
| cis-pinic acid (186) | 4.5 (3.9–7.4) | 14.6 (8.6–33) |
| terebic acid (158) | 10.3 (8.3–13.9) | 9.0 (7.9–14.7) |
| terpenylic acid (172) | 12.9 (9.0–14.3) | 11.9 (8.4–24) |
| diaterpenylic acid acetate (232) | 4.4 (3.9–6.5) | 3.8 (2.9–6.0) |
| cis-caronic acid (184) | 1.70 (1.42–2.5) | 1.24 (1.06–1.87) |
| cis-caric acid (186) | 1.21 (1.16–1.46) | 9.0 (4.2–15.7) |
| ketolimononic acid (186) | 1.88 (1.58–2.1) | 1.26 (1.09–1.84) |
| limonic acid (186) | 1.87 (1.70–2.5) | 3.1 (2.1–3.9)) |
| homoterpenylic acid (186) | 3.1 (3.0–3.7) | 4.8 (3.5–9.4) |
| 4-hydroxyterpenylic acid (188) | 5.4 (3.8–5.9) | 6.1 (4.6–13.5) |
| (2R,3R)-2-hydroxyterpenylic (188) | 31 (26–37) | 33 (24–34) |
| (2S,3R)-2-hydroxyterpenylic (188) | 7.1 (4.8–12.1) | 11.5 (7.5–23) |
| unknown (188) | 8.0 (6.5–9.2) | 5.5 (4.7–8.0) |
| pinanediol mono-nitrate OSs [∑5 isomers] (295) | 1.23 (0.57–1.93) | 2.7 (1.83–3.3) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maenhaut, W.; Chi, X.; Wang, W.; Cafmeyer, J.; Yasmeen, F.; Vermeylen, R.; Szmigielska, K.; Janssens, I.A.; Claeys, M. Contribution from Selected Organic Species to PM2.5 Aerosol during a Summer Field Campaign at K-Puszta, Hungary. Atmosphere 2017, 8, 221. https://doi.org/10.3390/atmos8110221
Maenhaut W, Chi X, Wang W, Cafmeyer J, Yasmeen F, Vermeylen R, Szmigielska K, Janssens IA, Claeys M. Contribution from Selected Organic Species to PM2.5 Aerosol during a Summer Field Campaign at K-Puszta, Hungary. Atmosphere. 2017; 8(11):221. https://doi.org/10.3390/atmos8110221
Chicago/Turabian StyleMaenhaut, Willy, Xuguang Chi, Wan Wang, Jan Cafmeyer, Farhat Yasmeen, Reinhilde Vermeylen, Katarzyna Szmigielska, Ivan A. Janssens, and Magda Claeys. 2017. "Contribution from Selected Organic Species to PM2.5 Aerosol during a Summer Field Campaign at K-Puszta, Hungary" Atmosphere 8, no. 11: 221. https://doi.org/10.3390/atmos8110221