
Inland Concentrations of Cl2 and ClNO2 in Southeast Texas Suggest Chlorine Chemistry Significantly Contributes to Atmospheric Reactivity

Abstract
:1. Introduction
2. Results and Discussion
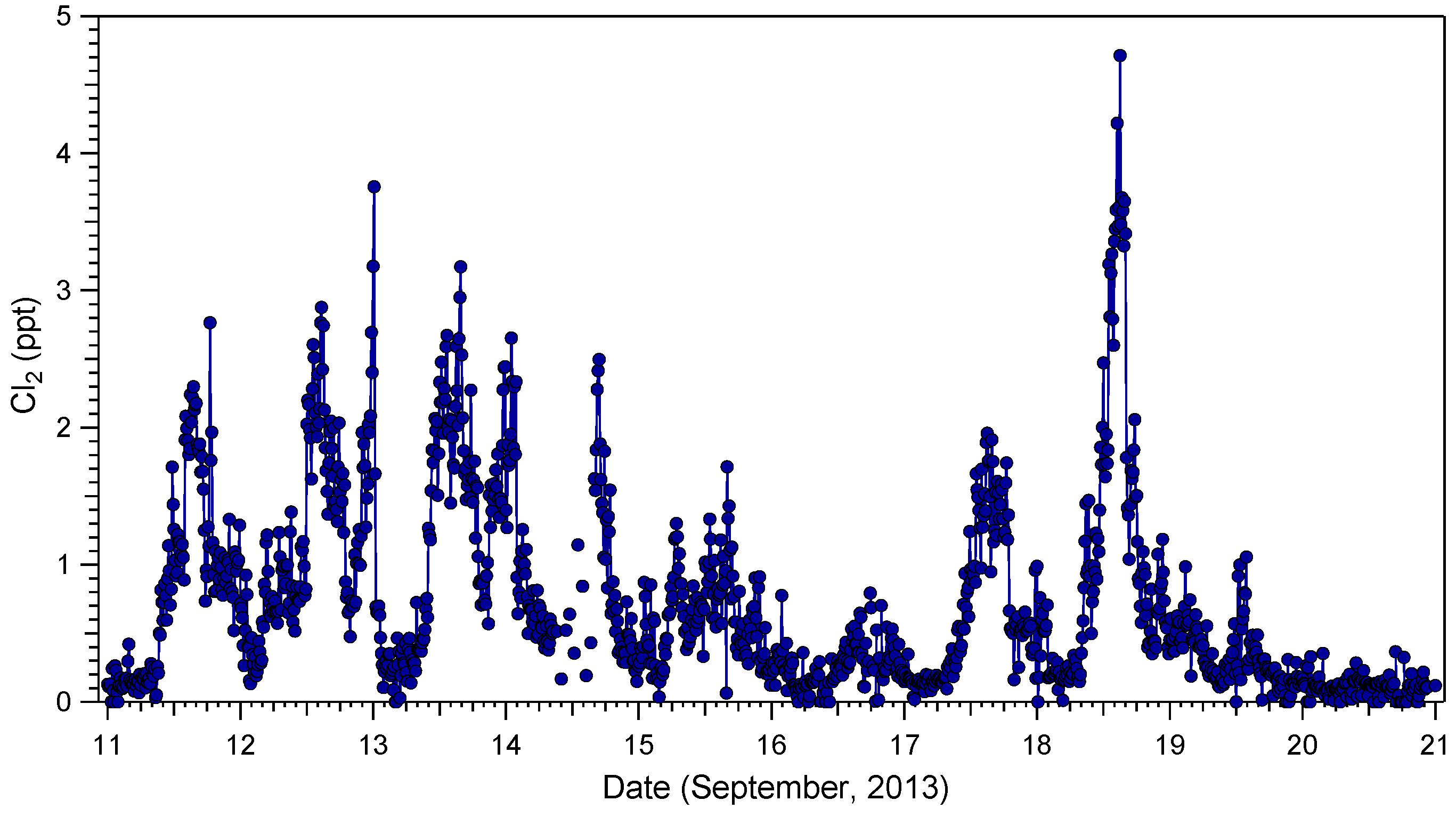
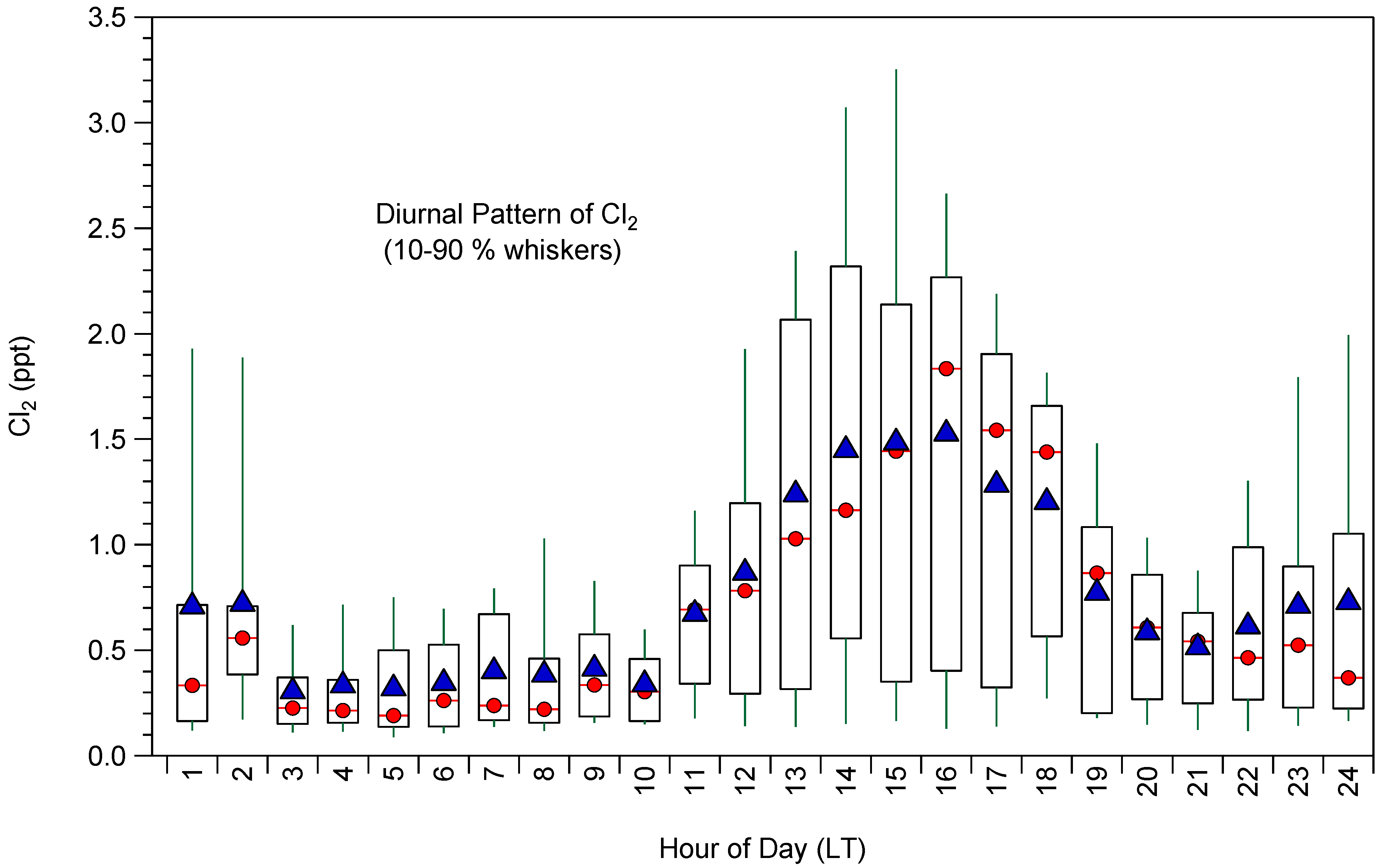
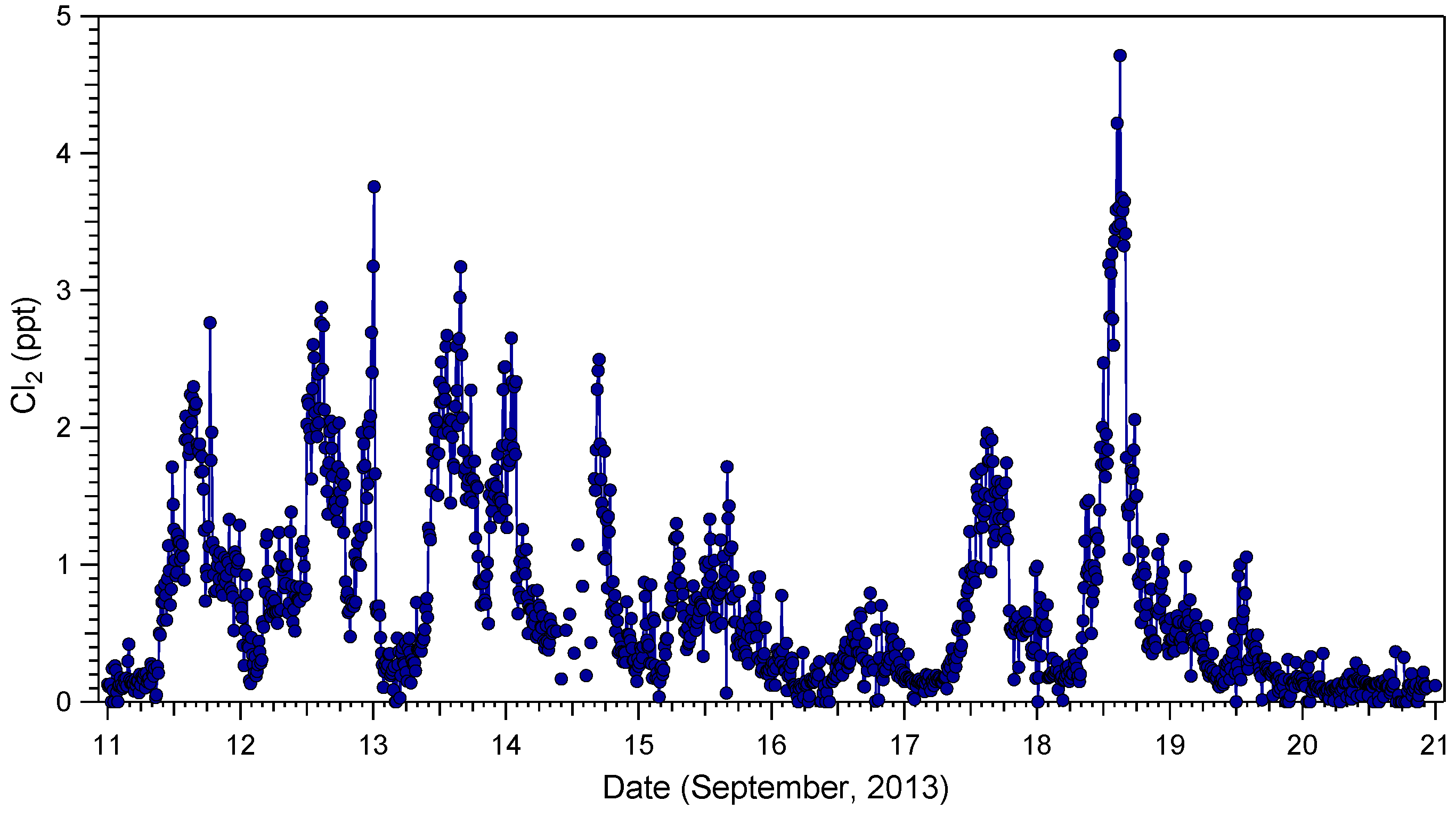
2.1. Cl2 Measurements
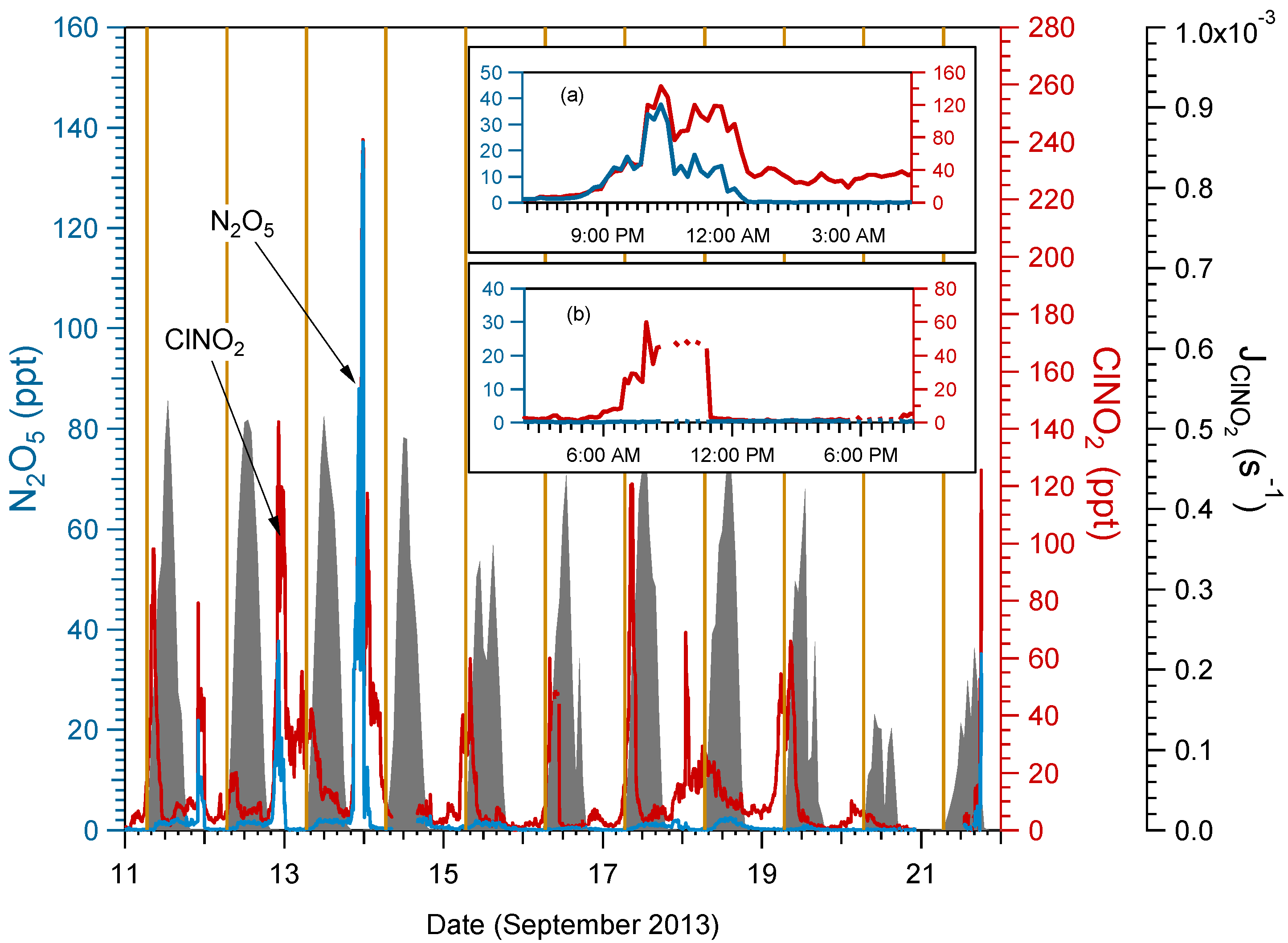
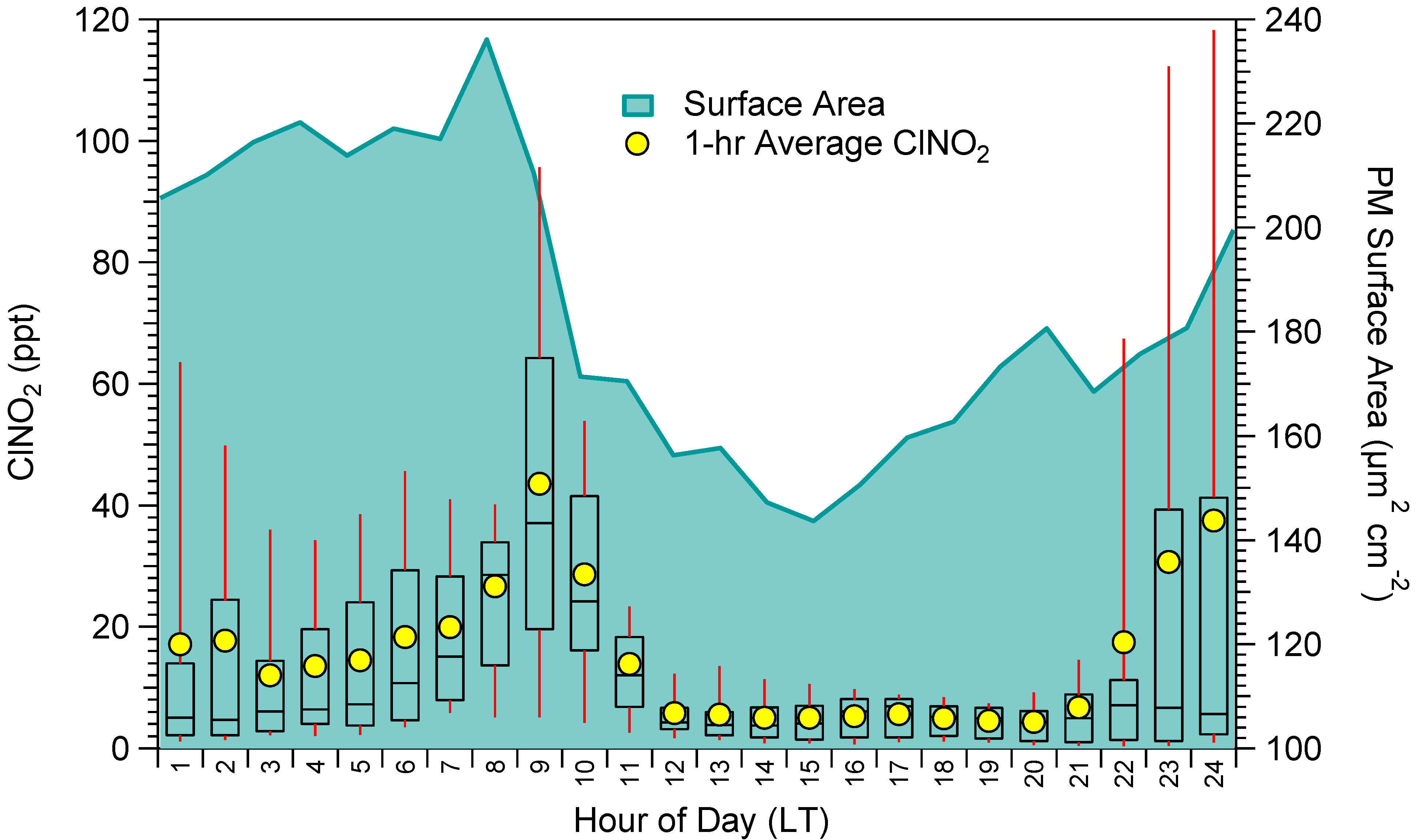
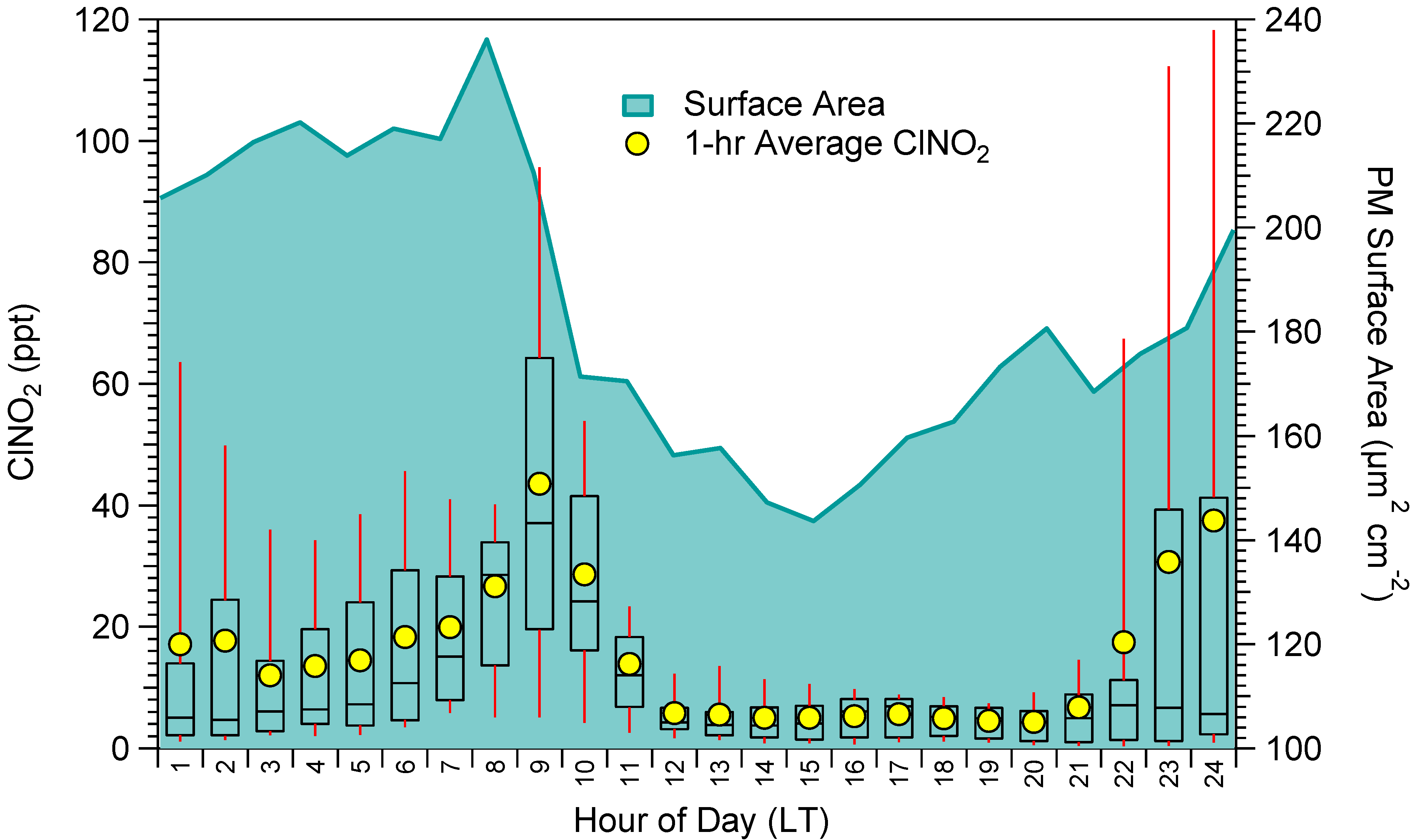
2.2. ClNO2 and N2O5 Measurements
2.3. Air Source Regions and Back Trajectories

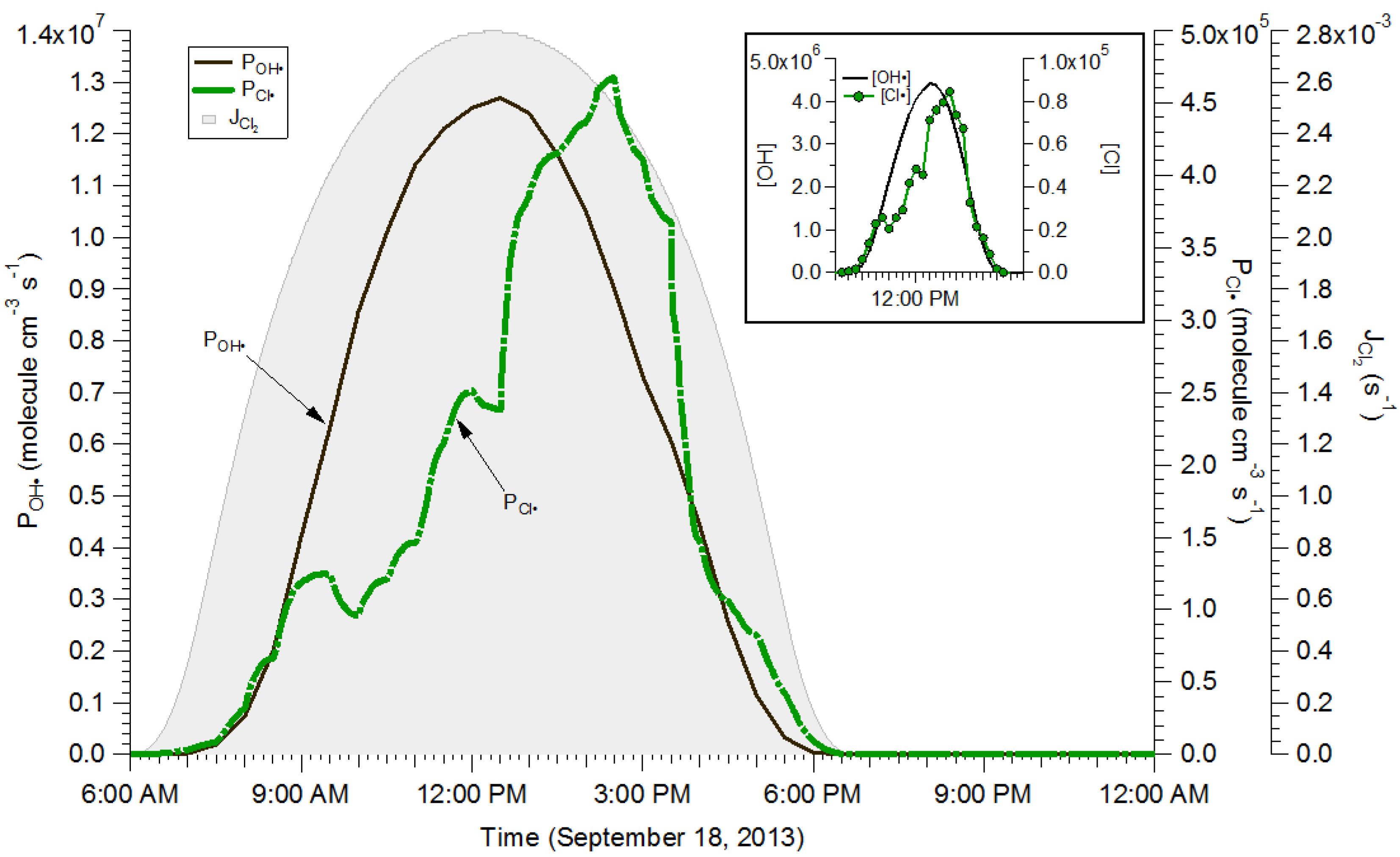
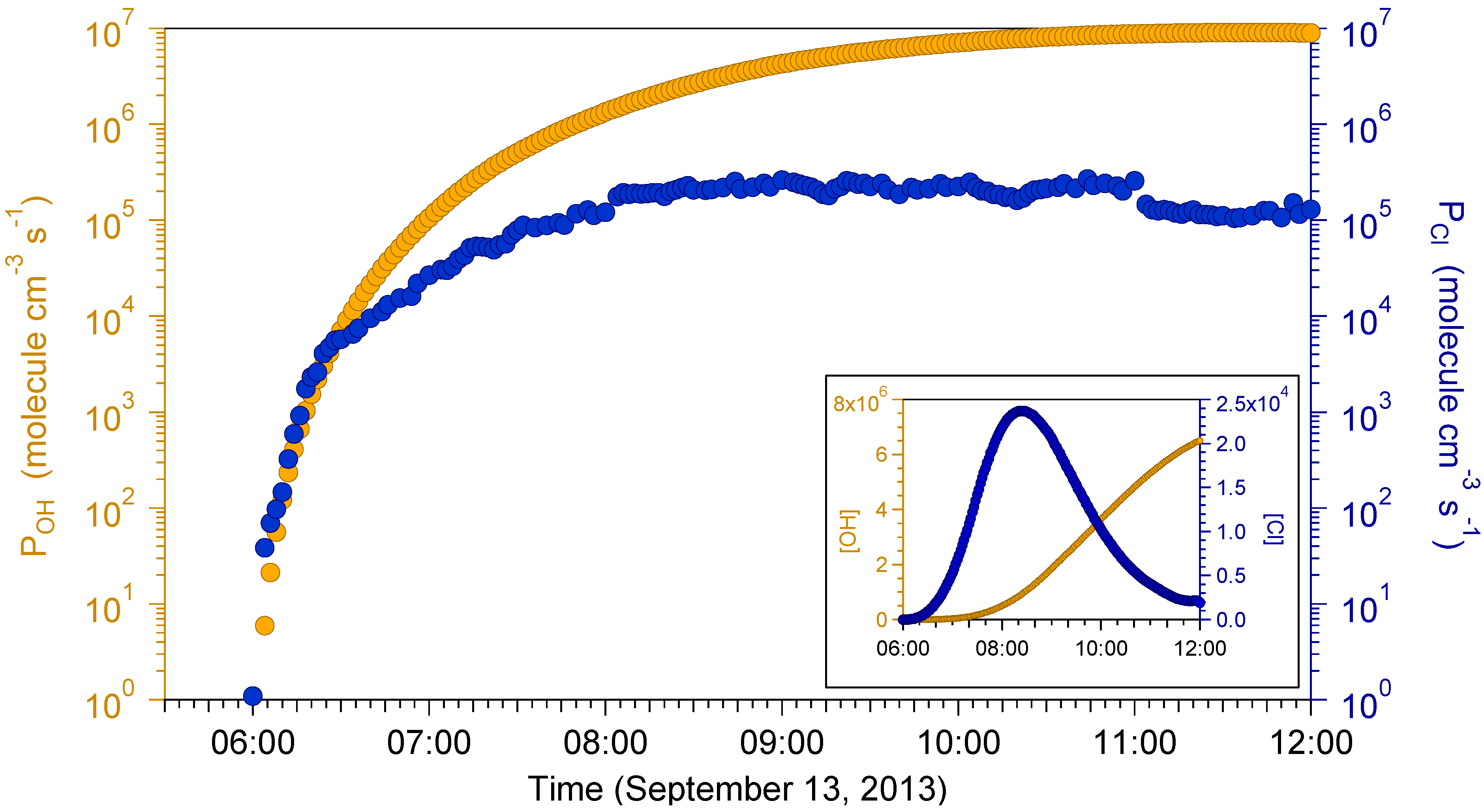
2.4. Box Modeling Results
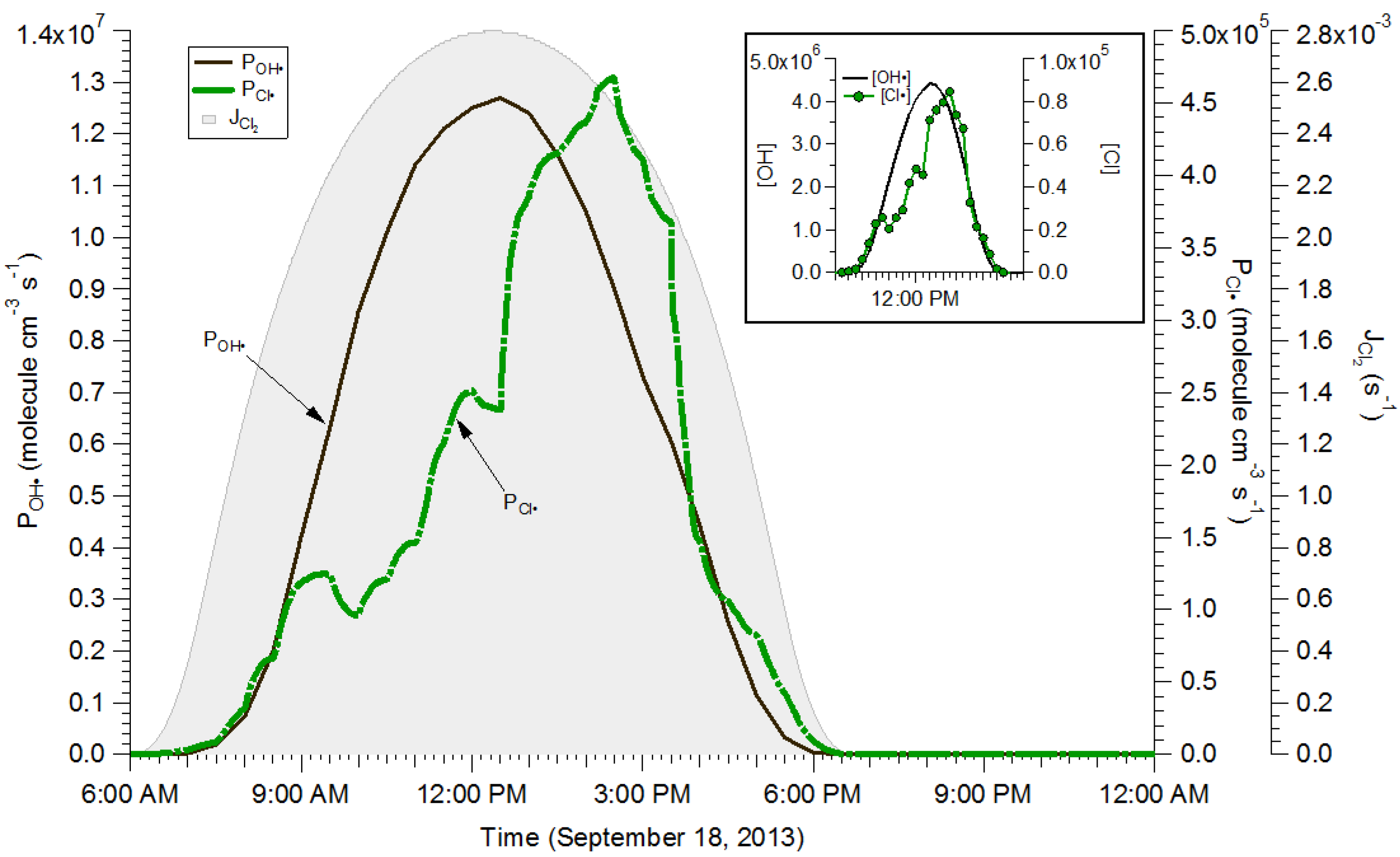
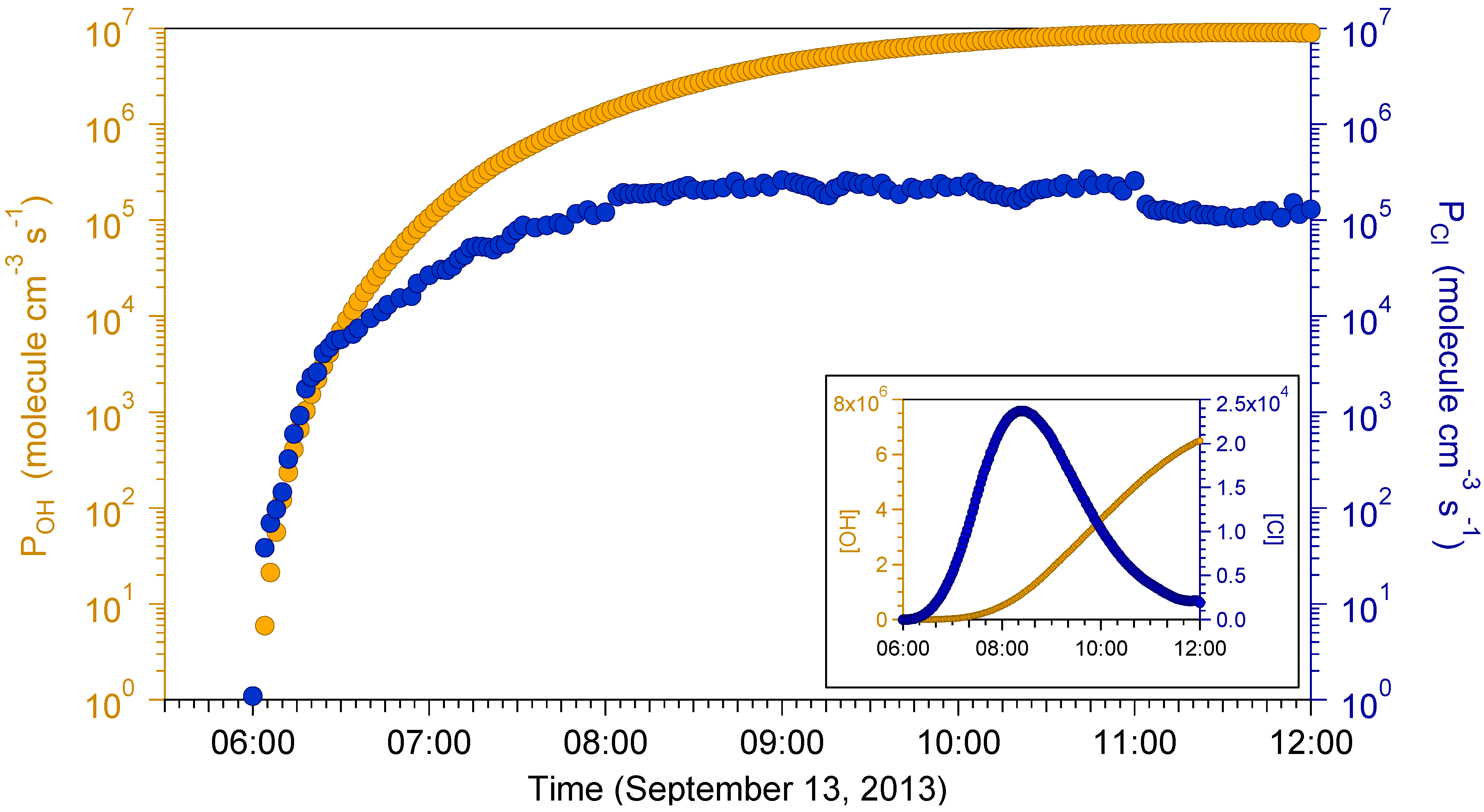

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Scenario | Chlorine Species | Comparison | [VOC] (ppbC) | ∆[O3]max (%) | ∆[RO2•]max (%) |
|---|---|---|---|---|---|
| 1 | -- | 1 | 0 | 10 | 50 |
| 2 | Cl2 | 0 | |||
| 3 | -- | 2 | 20 | 8 | 28 |
| 4 | Cl2 | 20 | |||
| 5 | -- | 3 | 20 | 6 | 0.4 |
| 6 | ClNO2 | 20 | |||
| 7 | -- | 4 | 20 | 12 | 1 |
| 8 | ClNO2 | 20 |
3. Experimental Section
4. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tanaka, P.L.; Riemer, D.D.; Chang, S.; Yarwood, G.; McDonald-Buller, E.C.; Apel, E.C.; Orlando, J.J.; Silva, P.J.; Jimenez, J.L.; Canagaratna, M.R.; et al. Direct evidence for chlorine-enhanced urban ozone formation in Houston, Texas. Atmos. Environ. 2003, 37, 1393–1400. [Google Scholar] [CrossRef]
- Wang, L.; Thompson, T.; McDonald-Buller, E.C.; Allen, D.T. Photochemical modeling of emissions trading of highly reactive volatile organic compounds in Houston, Texas. 2. Incorporation of chlorine emissions. Environ. Sci. Technol. 2007, 41, 2103–2107. [Google Scholar] [CrossRef] [PubMed]
- Faxon, C.B.; Allen, D.T. Chlorine chemistry in urban atmospheres: A review. Environ. Chem. 2013, 10, 221–233. [Google Scholar] [CrossRef]
- Wingenter, O.W.; Sive, B.C.; Blake, N.J.; Blake, D.R.; Rowland, F.S. Atomic chlorine concentrations derived from ethane and hydroxyl measurements over the equatorial Pacific Ocean: Implication for dimethyl sulfide and bromine monoxide. J. Geophys. Res. 2005, 110. [Google Scholar] [CrossRef]
- Graedel, T.E.; Keene, W.C. Tropospheric budget of reactive chlorine. Glob. Biogeochem. Cycles 1995, 9, 47–77. [Google Scholar] [CrossRef]
- Keene, C.; Aslam, M.; Khalil, K.; Erickson, J.; Archie, I.I.I.; Graedel, E.; Loberr, M.; Aucott, M.L.; Ling, S.; Harper, D.B.; et al. Composite global emissions of reactive chlorine from anthropogenic and natural sources : Reactive chlorine emissions inventory. J. Geophys. Res. 1999, 104, 8429–8440. [Google Scholar] [CrossRef]
- Sarwar, G.; Bhave, P.V. Modeling the effect of chlorine emissions on ozone levels over the eastern United States. J. Appl. Meteorol. Climatol. 2007, 46, 1009–1019. [Google Scholar] [CrossRef]
- Liu, C.L.; Smith, J.D.; Che, D.L.; Ahmed, M.; Leone, S.R.; Wilson, K.R. The direct observation of secondary radical chain chemistry in the heterogeneous reaction of chlorine atoms with submicron squalane droplets. Phys. Chem. Chem. Phys. 2011, 13, 8993–9007. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Arey, J.; Atkinson, R. Reactions of chlorine atoms with a series of aromatic hydrocarbons. Environ. Sci. Technol. 2005, 39, 5302–10. [Google Scholar] [CrossRef] [PubMed]
- Nelson, L.; Rattigan, O.; Neavyn, R.; Sidebottom, H. Absolute and relative rate constants for reactions of hydroxyl radicals and chlorine atoms with series of aliphatic alcohols and ethers at 298K. Int. J. Chem. Kinet. 1990, 22, 1111–1126. [Google Scholar] [CrossRef]
- Aschmann, S.M.; Atkinson, R. Rate Constants for the Gas-Phase Reactions of alkanes with Cl atoms at 296. Int. J. Chem. Kinet. 1995, 27, 613–622. [Google Scholar] [CrossRef]
- Canosa-Mas, C.E.; Hutton-Squire, H.R.; King, M.D.; Stewart, D.J.; Thompson, K.C.; Wayne, R.P. Laboratory Kinetic Studies of the Reactions of Cl Atoms with Species of Biogenic Origin: Δ3-Carene, Methacrolein and Methyl Vinyl Ketone. J. Atmos. Chem. 1999, 34, 163–170. [Google Scholar] [CrossRef]
- Ragains, M.L.; Finlayson-Pitts, B.J. Kinetics and mechanism of the reaction of Cl atoms with 2-Methyl-1,3-butadiene (Isoprene) at 298 K. J. Phys. Chem. A 1997, 5639, 1509–1517. [Google Scholar] [CrossRef]
- Chang, S.; McDonald-Buller, E.; Kimura, Y.; Yarwood, G.; Neece, J.; Russell, M.; Tanaka, P.; Allen, D. Sensitivity of urban ozone formation to chlorine emission estimates. Atmos. Environ. 2002, 36, 4991–5003. [Google Scholar] [CrossRef]
- Tanaka, P.L.; Allen, D.T.; Mullins, C.B. An environmental chamber investigation of chlorine-enhanced ozone formation in Houston, Texas. J. Geophys. Res. 2003, 108. [Google Scholar] [CrossRef]
- Knipping, E.M.; Dabdub, D. Impact of chlorine emissions from sea-salt aerosol on coastal urban ozone. Environ. Sci. Technol. 2003, 37, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Sander, S.P.; Friedl, R.R.; Barker, J.R.; Golden, D.M.; Kurylo, M.J.; Sciences, G.E.; Wine, P.H.; Abbatt, J.P.D.; Burkholder, J.B.; Kolb, C.E.; et al. Chemical Kinetics and Photochemical Data for Use in Atmospheric Studies: Evaluation Number 17. Available online: https://jpldataeval.jpl.nasa.gov/pdf/JPL%2010-6%20Final%2015Jun8011.pdf (accessed on 31 August 2015).
- Chang, S.; Allen, D.T. Chlorine chemistry in urban atmospheres: Aerosol formation associated with anthropogenic chlorine emissions in southeast Texas. Atmos. Environ. 2006, 40, 512–523. [Google Scholar] [CrossRef]
- Chang, S.; Tanaka, P.; Mcdonald-buller, E.; Allen, D.T. Emission inventory for atomic chlorine precursors in Southeast Texas. Available online: http://www.tceq.state.tx.us/assets/public/implementation/air/am/contracts/reports/oth/EmissioninventoryForAtomicChlorinePrecursors.pdf (accessed on 31 August 2015).
- Atkinson, R.; Baulch, D.L.; Cox, R.A.; Crowley, J.N.; Hampson, R.F.; Hynes, R.G.; Jenkin, M.E.; Rossi, M.J.; Troe, J. Evaluated kinetic and photochemical data for atmospheric chemistry: Volume III—Gas phase reactions of inorganic halogens. Atmos. Chem. Phys. 2007, 7, 981–1191. [Google Scholar] [CrossRef]
- Pszenny, A.A.P.; Keene, W.C.; Jacob, D.J.; Fan, S.; Maben, J.R.; Zetwo, M.P. Evidence of inorganic chlorine gases other than hydrogen chloride in marine surface air. Geopys. Res. Lett. 1993, 20, 699–702. [Google Scholar] [CrossRef]
- Spicer, C.; Chapman, E.; Finlayson-Pitts, B.; Plastrige, R.; Hubbe, J.; Fast, J.; Berkowitz, C. Unexpectedly high concentrations of molecular chlorine in coastal air. Nature 1998, 394, 353–356. [Google Scholar] [CrossRef]
- Finley, B.D.; Saltzman, E.S. Measurement of Cl2 in coastal urban air. Geophys. Res. Lett. 2006, 33. [Google Scholar] [CrossRef]
- Riedel, T.P.; Bertram, T.H.; Crisp, T.A.; Williams, E.J.; Lerner, B.M.; Vlasenko, A.; Li, S.M.; Gilman, J.; de Gouw, J.; Bon, D.M.; et al. A Nitryl chloride and molecular chlorine in the coastal marine boundary layer. Environ. Sci. Technol. 2012, 46, 10463–10470. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Huey, L.G.; Liu, Z.; Tanner, D.J.; Cantrell, C.A.; Orlando, J.J.; Flocke, F.M.; Shepson, P.B.; Weinheimer, A.J.; Hall, S.R.; et al. High levels of molecular chlorine in the Arctic atmosphere. Nat. Geosci. 2014, 7, 91–94. [Google Scholar] [CrossRef]
- Chang, S.; Allen, D.T. Atmospheric chlorine chemistry in southeast Texas: impacts on ozone formation and control. Environ. Sci. Technol. 2006, 40, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Oum, K.W.; Lakin, M.J.; DeHaan, D.O.; Brauers, T.; Finlayson-Pitts, B.J. Formation of molecular chlorine from the photolysis of ozone and aqueous sea-salt particles. Science 1998, 279, 74–76. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, H.; Majdik, Z.; Ervens, B.; Weise, D. Halogen production from aqueous tropospheric particles. Chemosphere 2003, 52, 485–502. [Google Scholar] [CrossRef]
- Sadanaga, Y.; Hirokawa, J.; Akimoto, H. Formation of molecular chlorine in dark condition: Heterogeneous reaction of ozone with sea salt in the presence of ferric ion. Geophys. Res. Lett. 2001, 28, 4433–4436. [Google Scholar] [CrossRef]
- Knipping, E.M. Experiments and simulations of ion-enhanced interfacial chemistry on aqueous NaCl aerosols. Science 2000, 288, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Behnke, W.; Zetzsch, C. Heterogeneous formation of chlorine atoms from various aerosols in the presence of O3 and HCl. J. Aerosol Sci. 1989, 20, 1167–1170. [Google Scholar] [CrossRef]
- Keene, W.C.; Pszenny, A.A.P.; Jacob, D.J.; Duce, R.A.; Galloway, J.N.; Schultz-Tokos, J.J.; Sievering, H.; Boatman, J.F. The geochemical cycling of reactive chlorine through the marine troposphere. Glob. Biogeochem. Cycles 1990, 4, 407–430. [Google Scholar] [CrossRef]
- Knipping, E.M. Modeling Cl2 formation from aqueous NaCl particles: Evidence for interfacial reactions and importance of Cl2 decomposition in alkaline solution. J. Geophys. Res. 2002, 107. [Google Scholar] [CrossRef]
- George, I.J.; Abbatt, J.P.D. Heterogeneous oxidation of atmospheric aerosol particles by gas-phase radicals. Nat. Chem. 2010, 2, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Kercher, J.P.; Riedel, T.P.; Thornton, J.A. Chlorine activation by N2O5: Simultaneous, in situ detection of ClNO2 and N2O5 by chemical ionization mass spectrometry. Atmos. Measu. Tech. 2009, 2, 193–204. [Google Scholar] [CrossRef]
- Thornton, J.A.; Abbatt, J.P.D. N2O5 reaction on submicron sea salt aerosol: Kinetics, products, and the effect of surface active organics. J. Phys. Chem. A 2005, 109, 10004–10012. [Google Scholar] [CrossRef] [PubMed]
- Osthoff, H.D.; Roberts, J.M.; Ravishankara, a. R.; Williams, E.J.; Lerner, B.M.; Sommariva, R.; Bates, T.S.; Coffman, D.; Quinn, P.K.; Dibb, J.E.; et al. High levels of nitryl chloride in the polluted subtropical marine boundary layer. Nat. Geosci. 2008, 1, 324–328. [Google Scholar] [CrossRef]
- Roberts, J.M.; Osthoff, H.D.; Brown, S.S.; Ravishankara, A.R.; Coffman, D.; Quinn, P.; Bates, T. Laboratory studies of products of N2O5 uptake on Cl-containing substrates. Geophys. Res. Lett. 2009, 36. [Google Scholar] [CrossRef]
- Sarwar, G.; Simon, H.; Bhave, P.; Yarwood, G. Examining the impact of heterogeneous nitryl chloride production on air quality across the United States. Atmos. Chem. Phys. 2012, 12, 6455–6473. [Google Scholar] [CrossRef]
- Simon, H.; Kimura, Y.; McGaughey, G.; Allen, D.T.; Brown, S.S.; Coffman, D.; Dibb, J.; Osthoff, H.D.; Quinn, P.; Roberts, J.M. Modeling heterogeneous ClNO2 formation, chloride availability, and chlorine cycling in Southeast Texas. Atmos. Environ. 2010, 44, 5476–5488. [Google Scholar] [CrossRef]
- Simon, H.; Kimura, Y.; McGaughey, G.; Allen, D.T.; Brown, S.S.; Osthoff, H.D.; Roberts, J.M.; Byun, D.; Lee, D. Modeling the impact of ClNO2 on ozone formation in the Houston area. J. Geophys. Res. 2009, 114. [Google Scholar] [CrossRef]
- Sarwar, G.; Simon, H.; Xing, J.; Mathur, R. Importance of tropospheric ClNO2 chemistry across the Northern Hemisphere. Geophys. Res. Lett. 2014, 41, 4050–4058. [Google Scholar] [CrossRef]
- Roberts, J.M.; Osthoff, H.D.; Brown, S.S.; Ravishankara, A.R. N2O5 oxidizes chloride to Cl2 in acidic atmospheric aerosol. Science 2008, 321, 1059–1059. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.L.; Jimenez-Aranda, A.; Finlayson-Pitts, B.J.; Dabdub, D. Gas-phase molecular halogen formation from NaCl and NaBr aerosols: when are interface reactions important? J. Phys. Chem. A 2006, 110, 1859–1867. [Google Scholar] [CrossRef] [PubMed]
- Phillips, G.J.; Tang, M.J.; Thieser, J.; Brickwedde, B.; Schuster, G.; Bohn, B.; Lelieveld, J.; Crowley, J.N. Significant concentrations of nitryl chloride observed in rural continental Europe associated with the influence of sea salt chloride and anthropogenic emissions. Geophys. Res. Lett. 2012, 39. [Google Scholar] [CrossRef]
- Thornton, J.A.; Kercher, J.P.; Riedel, T.P.; Wagner, N.L.; Cozic, J.; Holloway, J.S.; Dubé, W.P.; Wolfe, G.M.; Quinn, P.K.; Middlebrook, A.M.; et al. A large atomic chlorine source inferred from mid-continental reactive nitrogen chemistry. Nature 2010, 464, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Mielke, L.H.; Furgeson, A.; Osthoff, H.D. Observation of ClNO2 in a mid-continental urban environment. Environ. Sci. Technol. 2011, 45, 8889–8896. [Google Scholar] [CrossRef] [PubMed]
- Behnke, W.; George, C.; Scheer, V.; Zetzsch, C. Production and decay of ClNO2 from the reaction of gaseous N2O5 with NaCl solution: Bulk and aerosol experiments. J. Geophys. Res. 1997, 102, 3795–3804. [Google Scholar] [CrossRef]
- Behnke, W.; Krüger, H.-U.; Scheer, V.; Zetzsch, C. Formation of atomic Cl from sea spray via photolysis of nitryl chloride: Determination of the sticking coefficient of N2O5 on NaCl aerosol. J. Aerosol Sci. 1991, 22, S609–S612. [Google Scholar] [CrossRef]
- Frenzel, A.; Scheer, V.; Sikorski, R.; George, C.; Behnke, W.; Zetzsch, C. Heterogeneous interconversion reactions of BrNO2, ClNO2, Br2, and Cl2. J. Phys. Chem. A 1998, 102, 1329–1337. [Google Scholar] [CrossRef]
- Schweitzer, F.; Mirabel, P.; George, C. Multiphase chemistry of N2O5, ClNO2, and BrNO2. J. Phys. Chem. A 1998, 102, 3942–3952. [Google Scholar] [CrossRef]
- Bertram, T.H.; Thornton, J.A. Toward a general parameterization of N2O5 reactivity on aqueous particles: The competing effects of particle liquid water, nitrate and chloride. Atmos. Chem. Phys. Discuss. 2009, 9, 15181–15214. [Google Scholar] [CrossRef]
- Mielke, L.H.; Stutz, J.; Tsai, C.; Hurlock, S.C.; Roberts, J.M.; Veres, P.R.; Froyd, K.D.; Hayes, P.L.; Cubison, M.J.; Jimenez, J.L.; et al. Heterogeneous formation of nitryl chloride and its role as a nocturnal NOx reservoir species during CalNex-LA 2010. J. Geophys. Res. Atmos. 2013, 118, 10638–10652. [Google Scholar]
- Tham, Y.J.; Yan, C.; Xue, L.; Zha, Q.; Wang, X.; Wang, T. Presence of high nitryl chloride in Asian coastal environment and its impact on atmospheric photochemistry. Chin. Sci. Bull. 2013, 59, 356–359. [Google Scholar] [CrossRef]
- Riedel, T.P.; Wagner, N.L.; Dubé, W.P.; Middlebrook, A.M.; Young, C.J.; Öztürk, F.; Bahreini, R.; VandenBoer, T.C.; Wolfe, D.E.; Williams, E.J.; et al. Chlorine activation within urban or power plant plumes: Vertically resolved ClNO2 and Cl2 measurements from a tall tower in a polluted continental setting. J. Geophys. Res. Atmos. 2013, 118, 8702–8715. [Google Scholar] [CrossRef]
- National Aeronautics and Space Administration. Deriving Information on Surface Conditions from Column and Vertically Resolved Observations Relevant to Air Quality (DISCOVER-AQ). Available online: http://discover-aq.larc.nasa.gov/ (accessed on 31 August 2015).
- Ng, N.L.; Herndon, S.C.; Trimborn, a.; Canagaratna, M.R.; Croteau, P.L.; Onasch, T.B.; Sueper, D.; Worsnop, D.R.; Zhang, Q.; Sun, Y.L.; et al. An Aerosol Chemical Speciation Monitor (ACSM) for routine monitoring of the composition and mass concentrations of ambient aerosol. Aerosol Sci. Technol. 2011, 45, 780–794. [Google Scholar] [CrossRef]
- Finley, B.D.; Saltzman, E.S. Observations of Cl2, Br2, and I2 in coastal marine air. J. Geophys. Res. 2008, 113, 1–14. [Google Scholar]
- Jordan, C.E.; Pszenny, A.A.P.; Keene, W.C.; Cooper, O.R.; Deegan, B.; Maben, J.; Routhier, M.; Sander, R.; Young, A.H. Origins of aerosol chlorine during winter over north central Colorado, USA. J. Geophys. Res. Atmos. 2015, 120, 678–694. [Google Scholar] [CrossRef]
- Young, A.H.; Keene, W.C.; Pszenny, A.A.P.; Sander, R.; Thornton, J.A.; Riedel, T.P.; Maben, J.R. Phase partitioning of soluble trace gases with size-resolved aerosols in near-surface continental air over northern Colorado, USA, during winter. J. Geophys. Res. Atmos. 2013, 118, 9414–9427. [Google Scholar] [CrossRef]
- Pratt, K.A.; Prather, K.A. Aircraft measurements of vertical profiles of aerosol mixing states. J. Geophys. Res. Atmos. 2010, 115, 1–10. [Google Scholar] [CrossRef]
- Hildebrandt Ruiz, L.; Yarwood, G.; Koo, B.; Heo, G. Quality assurance project plan project 14-024—Sources of organic particulate matter in Houston: Evidence from DISCOVER-AQ data modeling and experiments. Available online: http://aqrp.ceer.utexas.edu/projectinfoFY14_15%5C14-024%5C14-024%20QAPP.pdf (accessed on 31 August 2015).
- Draxler, R.R.; Hess, G. Description of the HYSPLIT_4 Modeling System. Available online: https://ready.arl.noaa.gov/HYSPLIT.php; 2010 (accessed on 31 August 2015).
- Yarwood, G.; Jung, J.; Whitten, G.Z.; Heo, G.; Mellberg, J.; Estes, M. Updates to the Carbon Bond mechanism for version 6 (CB6). In Proceedings of the 9th Annual CMAS Conference, Chapel Hill, NC, USA, 11–13 October 2010; pp. 1–4.
- Yarwood, G.; Rao, S. Updates to the Carbon Bond Chemical Mechanism: CB05; RT-0400675; Final report to the US EPA; US EPA: Washington, DC, USA, 2005.
- Tanaka, P.L.; Allen, D.T.; McDonald-Buller, E.C.; Chang, S.; Kimura, Y.; Mullins, C.B.; Yarwood, G.; Neece, J.D. Development of a chlorine mechanism for use in the carbon bond IV chemistry model. J. Geophys. Res. 2003, 108. [Google Scholar] [CrossRef]
- Carter, W.P.L.; Luo, D.; Malkina, I.L.; Pierce, J.A. Environmental Chamber Studies of Atmospheric Reactivities of Volatile Organic Compounds: Effects of Varying Chamber and Light Source. 1995. Availavle online: https://www.cert.ucr.edu/~carter/pubs/explrept.pdf (accessed on 31 August 2015).
- Sokolov, O.; Hurley, M.D.; Wallington, T.J.; Kaiser, E.W.; Platz, J.; Nielsen, O.J.; Berho, F.; Rayez, M.T.; Lesclaux, R. Kinetics and mechanism of the gas-phase reaction of Cl atoms with benzene. J. Phys. Chem. A 1998, 102, 10671–10681. [Google Scholar] [CrossRef]
- Wingenter, O.W.; Blake, D.R.; Blake, N.J.; Sive, B.C.; Rowland, F.S.; Atlas, E.; Flocke, F. Tropospheric hydroxyl and atomic chlorine concentrations, and mixing timescales determined from hydrocarbon and halocarbon measurements made over the Southern Ocean. J. Geophys. Res. 1999, 104, 21,819–21,828. [Google Scholar] [CrossRef]
- Bertram, T.H.; Kimmel, J.R.; Crisp, T.A.; Ryder, O.S.; Yatavelli, R.L.N.; Thornton, J.A.; Cubison, M.J.; Gonin, M.; Worsnop, D.R. A field-deployable, chemical ionization time-of-flight mass spectrometer. Atmos. Meas. Tech. 2011, 4, 1471–1479. [Google Scholar] [CrossRef]
- Yatavelli, R.L.N.; Lopez-Hilfiker, F.; Wargo, J.D.; Kimmel, J.R.; Cubison, M.J.; Bertram, T.H.; Jimenez, J.L.; Gonin, M.; Worsnop, D.R.; Thornton, J.A. A chemical ionization high-resolution time-of-flight mass spectrometer coupled to a Micro Orifice Volatilization Impactor (MOVI-HRToF-CIMS) for analysis of gas and particle-phase organic species. Aerosol Sci. Technol. 2012, 46, 1313–1327. [Google Scholar] [CrossRef]
- Lee, B.H.; Lopez-Hilfiker, F.D.; Mohr, C.; Kurtén, T.; Worsnop, D.R.; Thornton, J.A. An iodide-adduct high-resolution time-of-flight chemical-ionization mass spectrometer: Application to atmospheric inorganic and organic compounds. Environ. Sci. Technol. 2014, 48, 6309–6317. [Google Scholar] [CrossRef] [PubMed]
- Aljawhary, D.; Lee, A.K.Y.; Abbatt, J.P.D. High-resolution chemical ionization mass spectrometry (ToF-CIMS): Application to study SOA composition and processing. Atmos. Meas. Tech. 2013, 6, 3211–3224. [Google Scholar] [CrossRef]
- Slusher, D.L.; Huey, L.G.; Tanner, D.J.; Flocke, F.M.; Roberts, J.M. A Thermal Dissociation-Chemical Ionization Mass Spectrometry (TD-CIMS) technique for the simultaneous measurement of peroxyacyl nitrates and dinitrogen pentoxide. J. Geophys. Res. 2004, 109. [Google Scholar] [CrossRef]
- Zheng, W.; Flocke, F.M.; Tyndall, G.S.; Swanson, A.; Orlando, J.J.; Roberts, J.M.; Huey, L.G.; Tanner, D.J. Characterization of a thermal decomposition chemical ionization mass spectrometer for the measurement of Peroxy Acyl Nitrates (PANs) in the atmosphere. Atmos. Chem. Phys. 2011, 11, 6529–6547. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, H.; Dube, W.P.; Ciciora, S.J.; Brown, S.S. Determination of inlet transmission and conversion efficiencies for in situ measurements of the nocturnal nitrogen oxides, NO3, N2O5 and NO2, via pulsed cavity ring-down spectroscopy. Anal. Chem. 2008, 80, 6010–6017. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faxon, C.B.; Bean, J.K.; Ruiz, L.H. Inland Concentrations of Cl2 and ClNO2 in Southeast Texas Suggest Chlorine Chemistry Significantly Contributes to Atmospheric Reactivity. Atmosphere 2015, 6, 1487-1506. https://doi.org/10.3390/atmos6101487
Faxon CB, Bean JK, Ruiz LH. Inland Concentrations of Cl2 and ClNO2 in Southeast Texas Suggest Chlorine Chemistry Significantly Contributes to Atmospheric Reactivity. Atmosphere. 2015; 6(10):1487-1506. https://doi.org/10.3390/atmos6101487
Chicago/Turabian StyleFaxon, Cameron B., Jeffrey K. Bean, and Lea Hildebrandt Ruiz. 2015. "Inland Concentrations of Cl2 and ClNO2 in Southeast Texas Suggest Chlorine Chemistry Significantly Contributes to Atmospheric Reactivity" Atmosphere 6, no. 10: 1487-1506. https://doi.org/10.3390/atmos6101487