3.1. Summary Statistics
Nucleation events were observed on approximately 69% (273 days) of days for days with complete measurements. The observed nucleation frequency at Duke Forest was significantly higher compared to that observed at forested areas elsewhere. From the total valid observations, 46% of the days at a mixed deciduous forest in southern Indiana [
22], 35% of the days at a deciduous forest in central Virginia [
19], 45% of the days [
26] at a boreal forest in Hyytyala in Finland, and 53% of the days [
11] at a forested site near Pittsburg, PA exhibited new particle formation characteristics. Month-to-month comparisons of event statistics are difficult as the total number of valid observation days in each month (labeled in
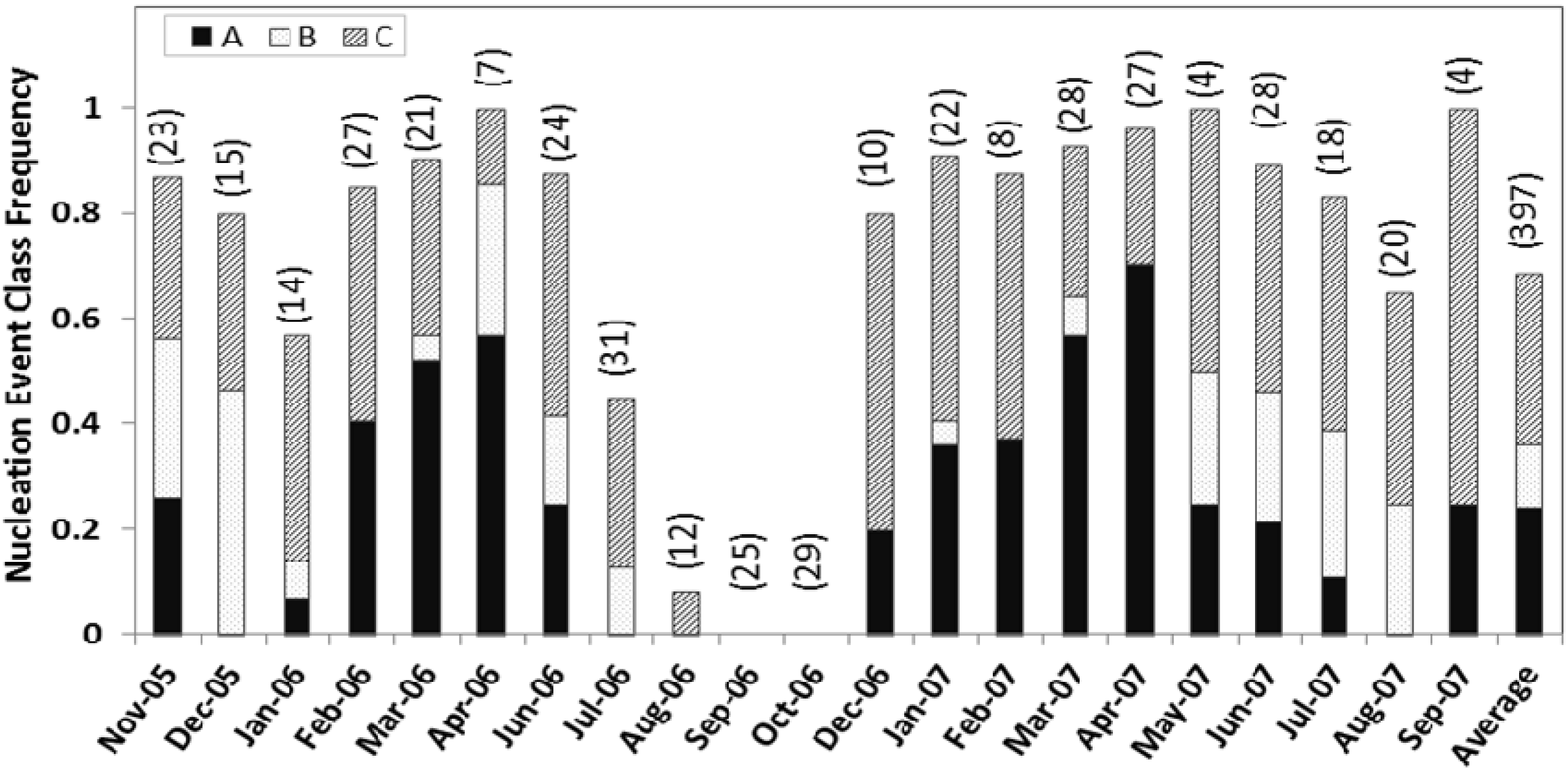
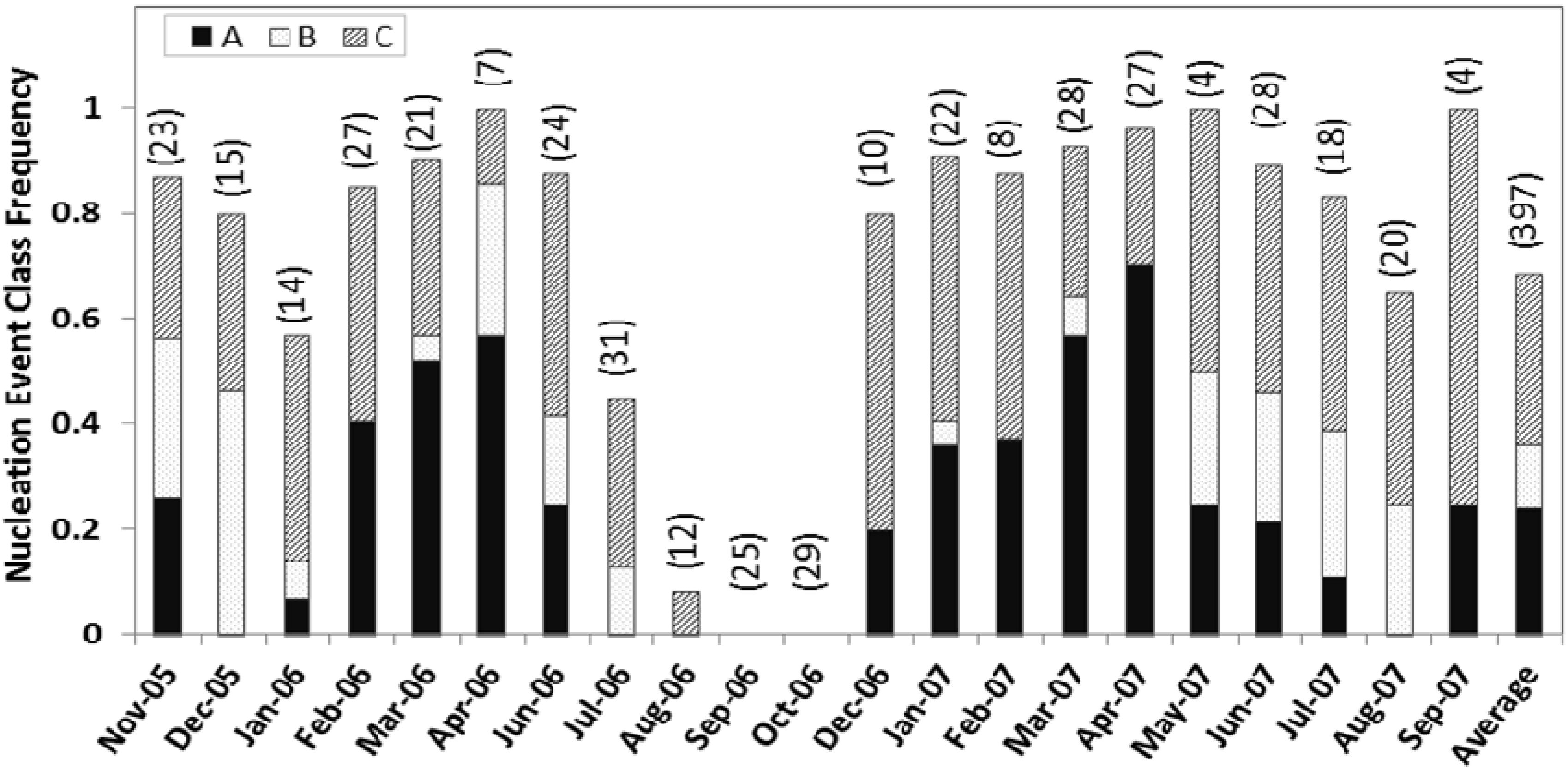
Figure 2) varied greatly, ranging from four days for May and September 2007 to 31 days for July 2006. No days in October had nucleation. However, the event classes show clear seasonal signatures.
Figure 2.
The frequency of nucleation event classes A, B, and C identified in each month is shown normalized with the total number of classifiable observation days for that particular month from November 2005 to September 2007. The total number of observation days for each month is labeled in the graph.
Figure 2.
The frequency of nucleation event classes A, B, and C identified in each month is shown normalized with the total number of classifiable observation days for that particular month from November 2005 to September 2007. The total number of observation days for each month is labeled in the graph.
The frequency of class A events is highest during spring (April–May) and lowest during mid-summer to mid-fall. This pattern is consistent with the observations of Pryor
et al. [
22] in Indiana. No class A events were observed in August. No, or a significantly lower number of, class B events were observed from mid-winter to mid-spring. Class C events were frequently observed from late fall through the early winter months. At Duke Forest, we observed the highest nucleation frequency for event class C (33%). Classes A and B event days total 144 (36%), of which 121 events are selected for further detailed analysis based on the growth pattern and growth rate. For quantitative analysis, classes A and B together account for about 36% of the valid observation days of which the contribution from class A events were about 26%. This is similar to the observations at Morgan-Monroe State Forest in southern Indiana [
22], where A and B events together accounted for about 32% of total classifiable observation days.
In May and August, the nucleation events were more frequent in the early mornings, whereas in winter months (e.g., December), the onset of nucleation was observed even in the afternoon, with later nucleation events being in December. The earliest nucleation onset was observed during August (~6:00 am) and, on average, in summer months, the onset of nucleation occurred in the early morning (~7:30 am); however, during winter months, the onset of nucleation occurred later in the day with the latest being observed at ~2:00 pm. The averaged total fine particle number concentration (
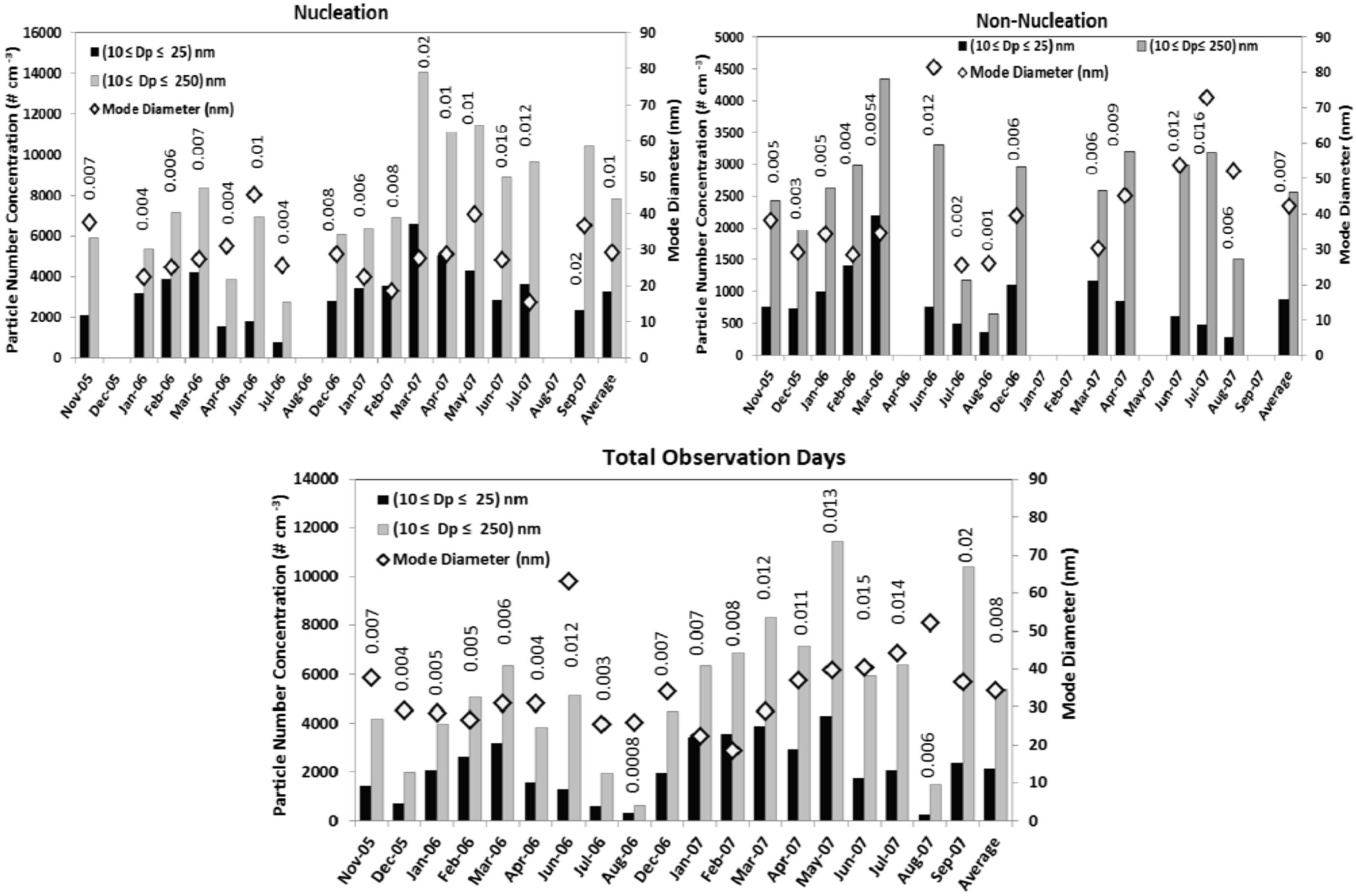
Figure 3) varied greatly between seasons.
Figure 3.
The monthly mean total number concentration of nucleation mode and fine mode particles are plotted along with the mode diameter for nucleation and non-nucleation events. The monthly mean condensation sink (cm−2) is also labeled on the top of each bar. The lower panel is the monthly means for all the observation days.
Figure 3.
The monthly mean total number concentration of nucleation mode and fine mode particles are plotted along with the mode diameter for nucleation and non-nucleation events. The monthly mean condensation sink (cm−2) is also labeled on the top of each bar. The lower panel is the monthly means for all the observation days.
The monthly averaged mode diameter shows a distinct seasonal pattern. The average mode diameter on nucleation days (29 nm) was significantly lower than that on non-nucleation days (42 nm). In general, we observed larger mode diameters in summer months. This may be due to increased availability of condensable vapors in the atmosphere which favor particle growth by condensation and coagulation. Monthly means of particle number concentration, mode diameters
etc. are shown in lower panel of
Figure 3.
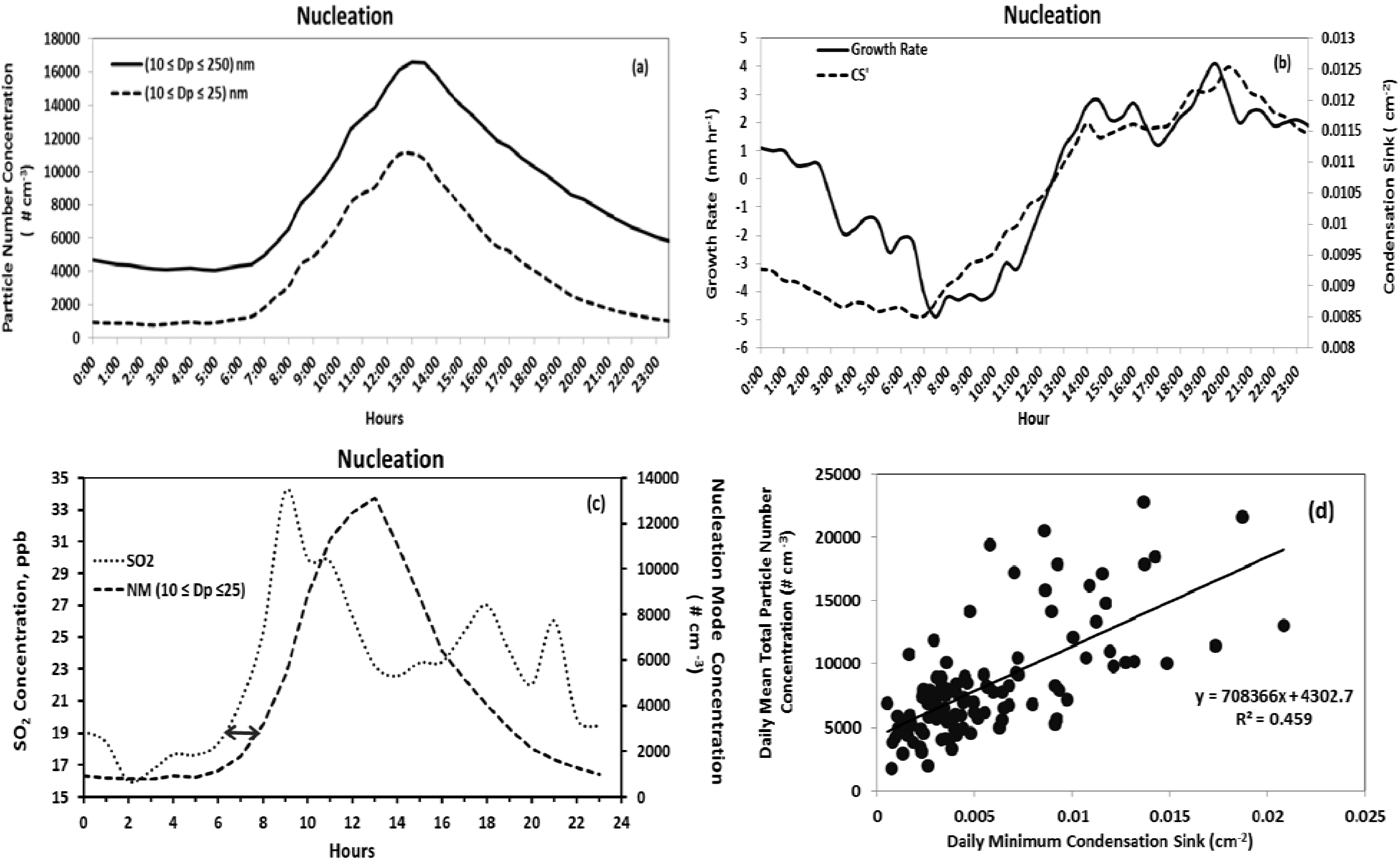
Figure 4(a) provides the grand average of the hourly mean total number concentration of particles with diameters 10 nm ≤
Dp ≤ 25 nm, and 10 nm ≤
Dp ≤ 250 nm.
Figure 4(b) shows the corresponding diurnal profile of condensation sink (cm
−2) and growth rate averaged for all the observed nucleation days. The particle number concentration peak is observed at approximately 1:30 pm. The grand average total particle number concentration on nucleation event days was 8,684 cm
−3 (10 nm ≤
Dp ≤ 250 nm) and 3,991 cm
−3 for the size range 10 nm ≤
Dp≤ 25 nm with a 45% contribution from NM particles. The geometric mean diameter and mode diameter were 35 nm and 28 nm, respectively. On non-nucleation days, the contribution from NM particles was 30% of the total number concentration and the values were 2,143 cm
−3 (10 nm ≤
Dp ≤ 250 nm) and 655 cm
−3 (10 nm ≤
Dp ≤ 25 nm). Both the geometric mean (49.6 nm) and mode (44.5 nm) diameters showed an increase of about 15 nm on non-nucleation days compared to nucleation days.
Figure 4.
Diurnal profiles of nucleation day (a) nucleation mode and fine mode particle number concentration, (b) particle growth rates and condensation sink, (c) NM particle number concentration and concentration of SO2, and (d) the relationship between condensation sink and total particle number concentration during nucleation. Diurnal profile data points represent hourly averages for all nucleation days.
Figure 4.
Diurnal profiles of nucleation day (a) nucleation mode and fine mode particle number concentration, (b) particle growth rates and condensation sink, (c) NM particle number concentration and concentration of SO2, and (d) the relationship between condensation sink and total particle number concentration during nucleation. Diurnal profile data points represent hourly averages for all nucleation days.
Condensation sink quantifies the strength of condensation processes occurring during the nucleation event and is a function of the particle size distribution. The hourly averaged condensation sink typically reaches a minimum at around 7:00 am and shows a continuous increase as the total particle concentration evolves. This is due to the contribution of newly formed particles to the condensation sink. It is evident from
Figure 4(a,b) that the daily minimum of CS is a good indicator of the time of onset of the new particle formation. This observation is consistent with the findings during BIOFOR campaign [
29]. No such behavior for the condensation sink was observed for non-nucleation days. Furthermore, while the daily minimum CS was a good indicator of the timing of nucleation onset, the strength of the event, indicated by the total particle concentration, was positively correlated with daily minimum CS (
Figure 4(d)). On the other hand, daily averaged CS values were not good predictors for nucleation events. In fact, during our analysis, the nucleation days were associated with higher condensation sink (0.009 cm
−2) compared to non-nucleation days (0.0064 cm
−2). This demonstrates a strong contribution of new particle formation events to the aerosol CS.
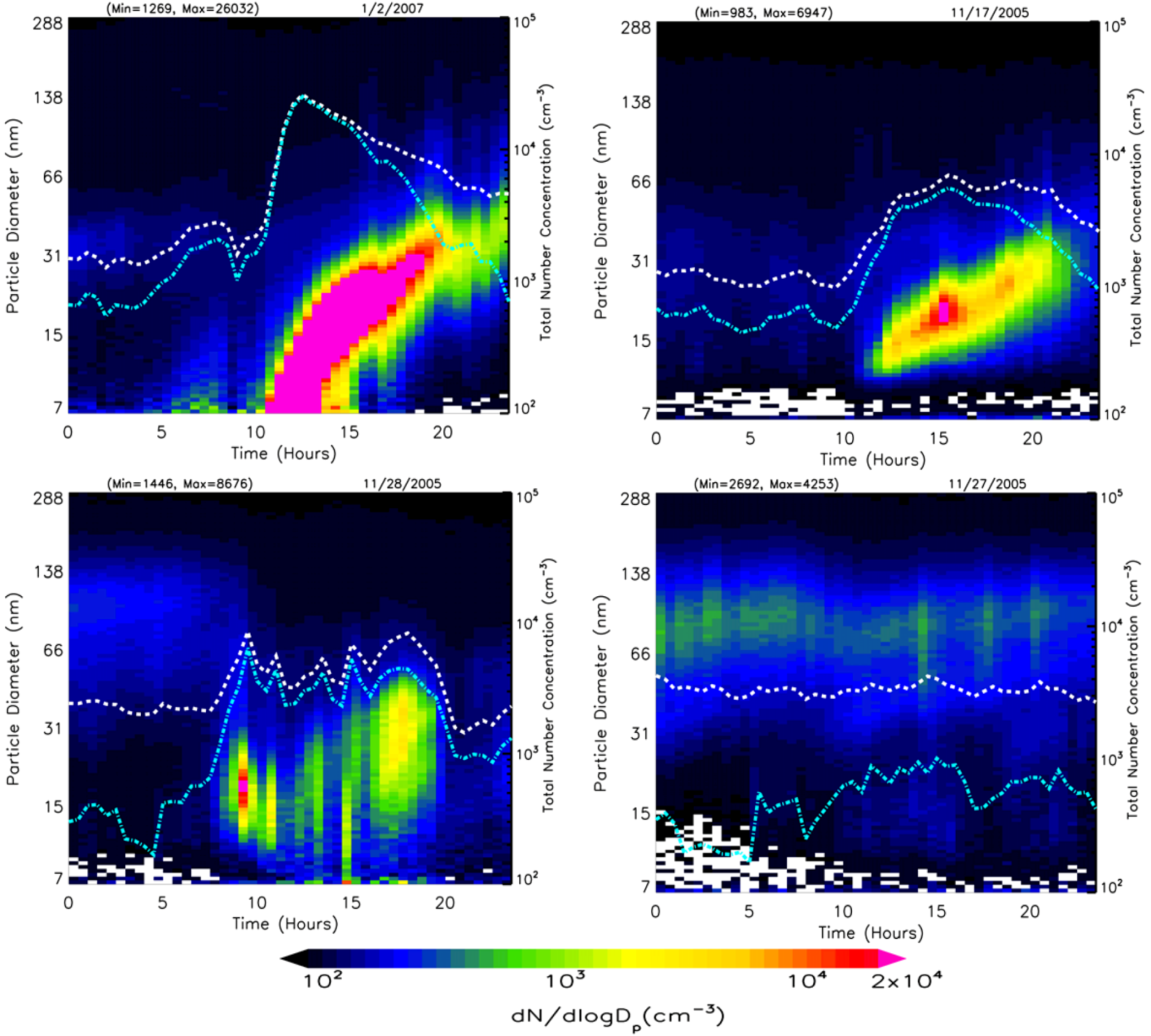
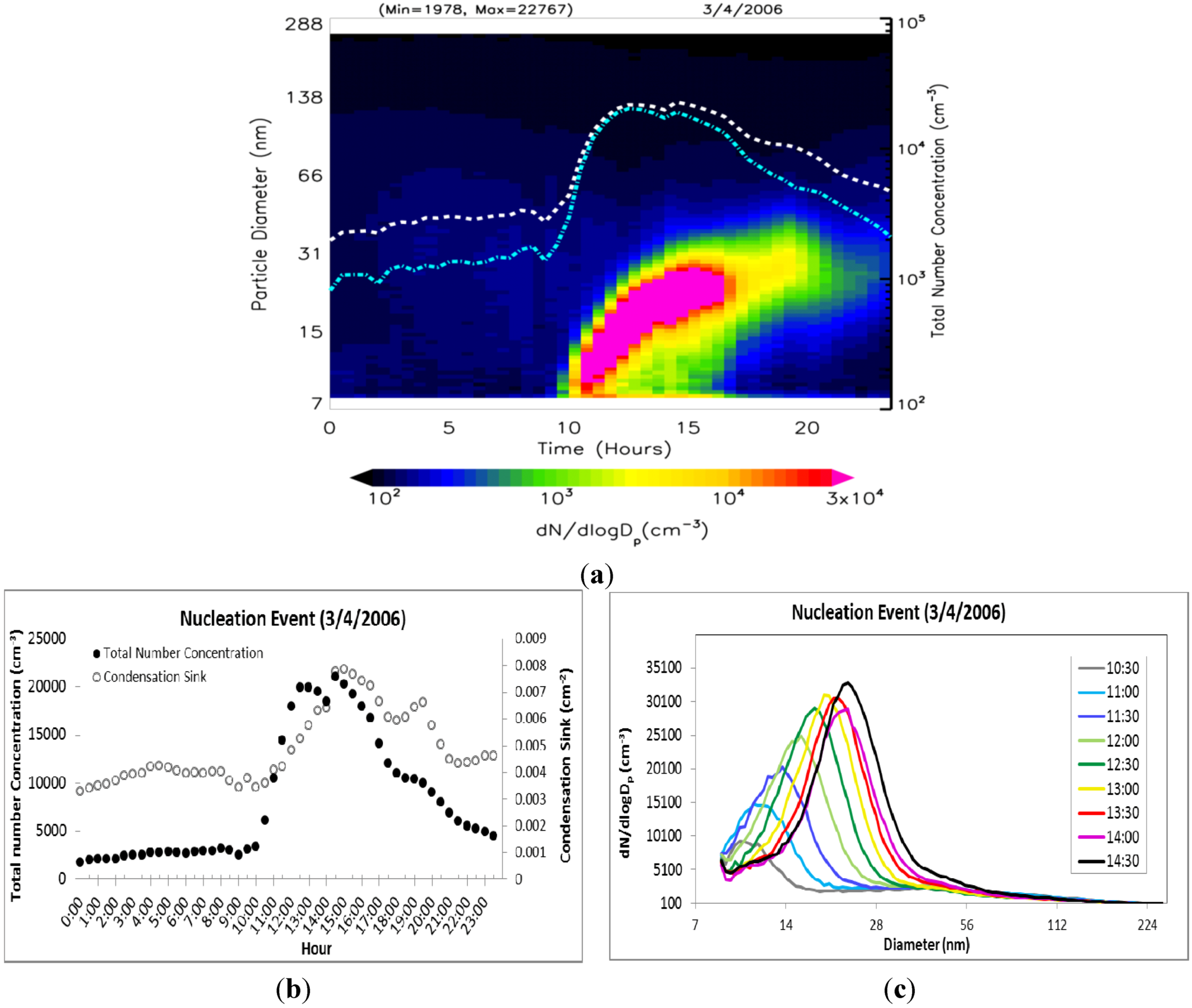
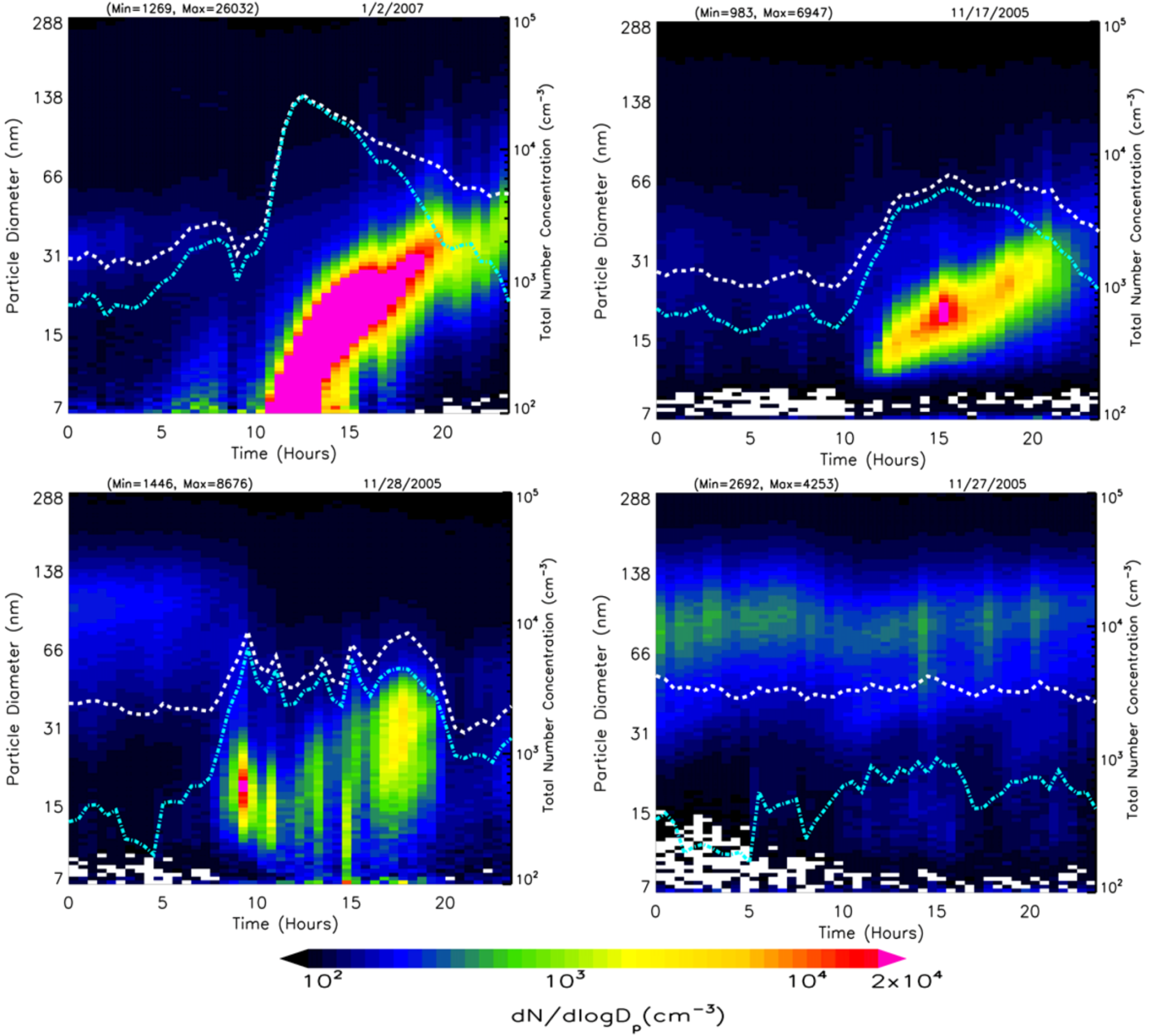
Evolution of the particle size distribution (dN/dlogD
p) during a typical nucleation event day on 4 March 2006 is shown in
Figure 5(a). The total number concentrations of nucleation mode (cyan) and fine (white) particles are shown on a log scale on the secondary vertical axis. There is no evidence of plume impact from local sources on the selected nucleation day. Nucleation started at ~10:00 am and continued until 15:30 pm. The evolution of the total number concentration and particle size distribution with time during the nucleation event is shown respectively in
Figure 5(b,c). In the first phase, the particle number concentration, as well as particle peak diameter, increases, thereby indicating nucleation and subsequent growth. After ~15:30 pm, the particle number concentration begins to decrease while peak diameter increases, thus indicating the growth of newly formed particles mainly through condensation. The average growth rate (
Table 1) during this nucleation event was calculated to be 2.7 ± 0.9 nm/h based on the peak diameter for each time interval.
Figure 5.
(a) Evolution of particle size distribution on a day with nucleation (4 March 2006) with dotted lines representing total particle number concentration (white) and nucleation mode particle number concentration (cyan), (b) corresponding hourly distribution of total number concentration and condensation sink, and (c) evolution of the corresponding size distribution dN/dlogDp (cm−3) as a function of diameter midpoint of each size channel.
Figure 5.
(a) Evolution of particle size distribution on a day with nucleation (4 March 2006) with dotted lines representing total particle number concentration (white) and nucleation mode particle number concentration (cyan), (b) corresponding hourly distribution of total number concentration and condensation sink, and (c) evolution of the corresponding size distribution dN/dlogDp (cm−3) as a function of diameter midpoint of each size channel.
Table 1.
Total number concentrations of fine mode (N) and nucleation mode (N25) particles, peak diameter (Dpeak), condensation sink (CS), and particle growth rate (GR) during the 4 March 2006 nucleation event.
Table 1.
Total number concentrations of fine mode (N) and nucleation mode (N25) particles, peak diameter (Dpeak), condensation sink (CS), and particle growth rate (GR) during the 4 March 2006 nucleation event.
| Time | N (cm−3) | N25 (cm−3) | CS | Dpeak | GR |
|---|
| (10nm ≤ Dp ≤ 250 nm) | (10 ≤ Dp ≤ 25 nm) | (cm−2) | (nm) | (nm·h−1) |
|---|
| 9:00 am | 2,530 | 1,559 | 0.0034 | 8.82 | - |
| 11:00 am | 10,522 | 9,462 | 0.0041 | 9.14 | 1.60 |
| 13:00 pm | 19,964 | 19,128 | 0.0058 | 15.70 | 3.28 |
| 15:00 pm | 20,307 | 18,975 | 0.0078 | 18.80 | 3.10 |
The hourly average growth rate (GR) (
Figure 4(b)) illustrates diurnal patterns in the rate of change of nucleation mode particle size and is dependent on the condensable vapor concentration and the number concentration of pre-existing particles that act as CS. Growth rate begins to increase with the onset of nucleation and generally reaches its peak 4 to 5 h later due to increasing photochemistry. However, there is about an hour delay (
Figure 4(a,b)) between the nucleation onset and increase in GR. The similar time lag (
Figure 4(c)) observed between NM particle concentration and concentration of SO
2 indicates condensational growth. This temporal lag may be due to the two stages in nucleation—the formation of new particles, and their growth through condensation to detectable larger sizes. Sihto
et al. [
33] observed a 1.4-h time lag between the number concentration of 3–6 nm particles and sulfuric acid concentration. Not all nucleation events lead to new particle formation. If the nucleated species can survive against condensation and coagulation loss to pre-existing particles, and if condensable vapors are available for condensation onto nucleated species, new particle formation may occur [
34].
The typical growth rates observed at continental sites are between 1 and 20 nm/h [
9]. The grand average growth rate during nucleation events was 2.7 ± 0.3 nm/h. The observed average growth rates are consistent with the range of the growth rates reported by Pryor
et al. [
22] (2.5 nm/h), and Place
et al. [
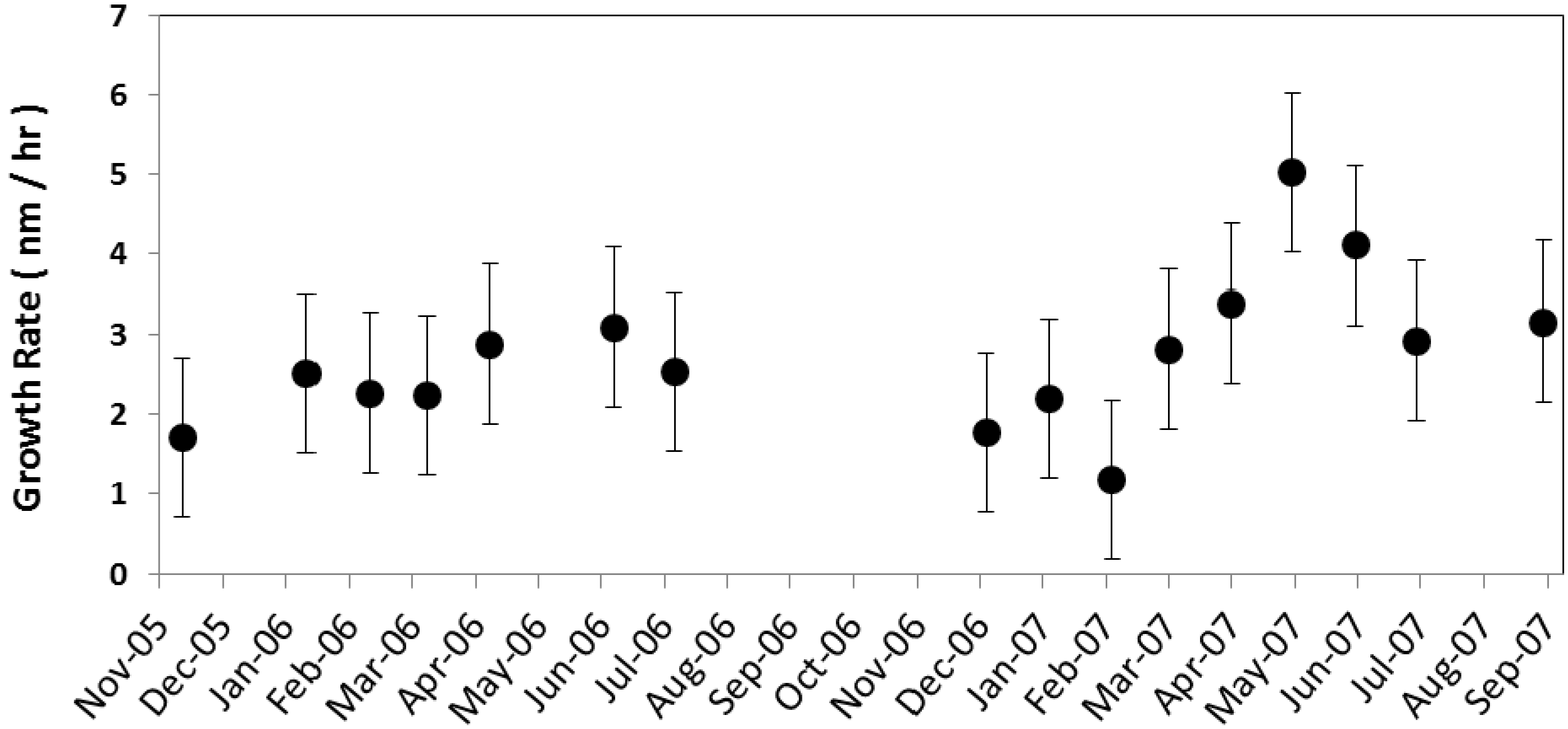
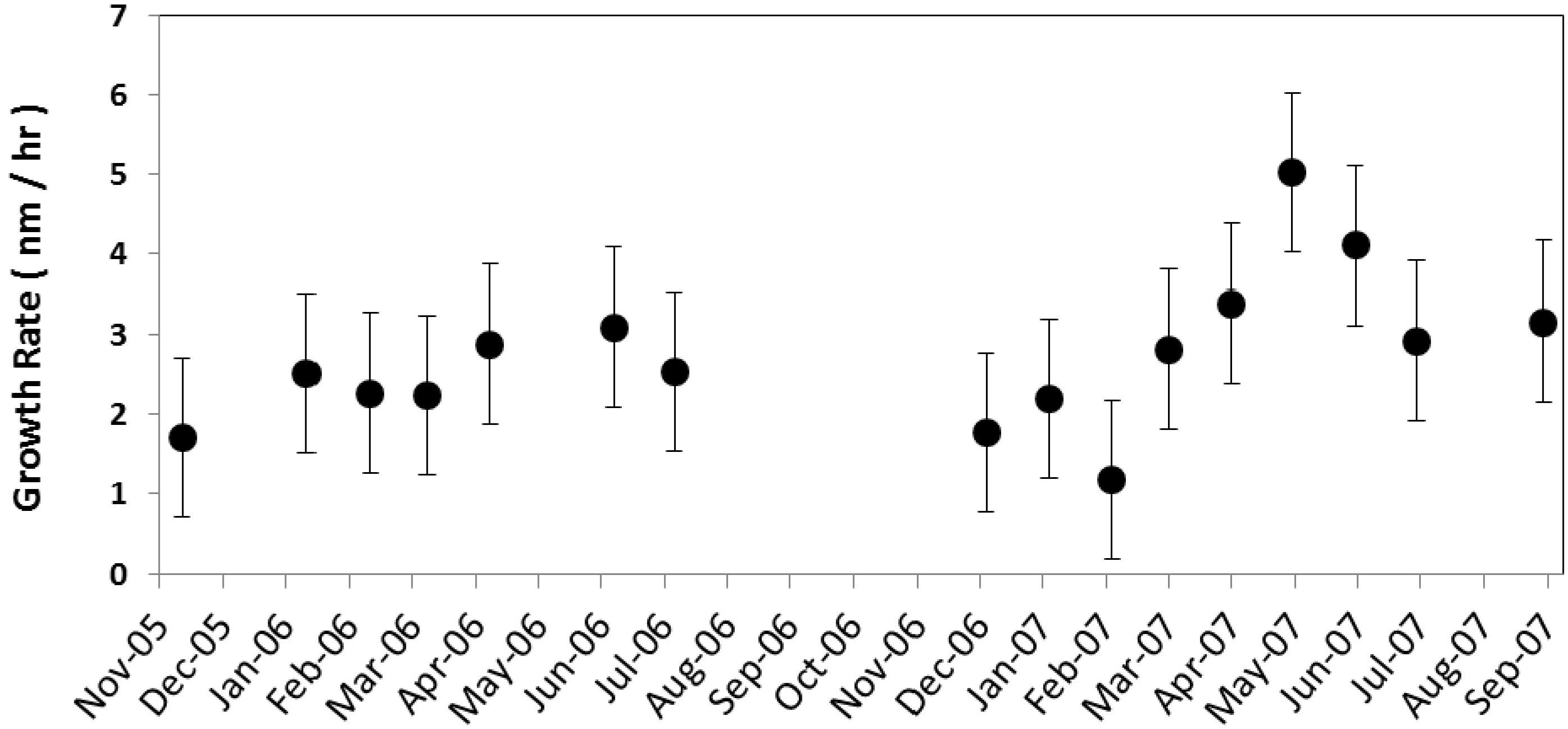
17] (2.7 nm/h).The particle growth rate at the Duke Forest site exhibits seasonal variability. The monthly average growth rate (
Figure 6) was at its maximum during late spring and the early summer months, with the highest growth rate being in May (5.0 ± 3.6 nm/h) followed by June (4.1 ± 3.9 nm/h), and the minimum growth rate occurring during the winter months, with the lowest being in February (1.2 ± 2.2 nm/h). This observed seasonality is consistent with Qian
et al. [
35] (6.7 ± 4.8 nm/h for summer and 1.8 ± 1.9 nm/h for winter). Higher growth rates in summer months may be due to the higher oxidizing capacity of the atmosphere and subsequent oxidation rates of precursors, such as SO
2, and other compounds, such as biogenic volatile organic compounds (VOC), that exhibit temperature or physiologically dependent (e.g., bud break or needle expansion) emission rates.
The particle growth rate depends on the availability of condensable vapor concentration and the new particle formation is observed when freshly nucleated particles can grow fast enough to be detected without being scavenged onto pre-existing aerosols. The condensable vapor species are produced from their precursor gases through oxidation by ozone, and hydroxyl (OH), and/or nitrate (NO
3) radicals [
9]. The concentration of these condensable vapor species depends on the concentration ratio between precursor gases and pre-existing aerosols [
9].
Figure 6.
Monthly mean particle growth rate (nm/h) averaged for all nucleation events for the corresponding month along with the standard error is shown.
Figure 6.
Monthly mean particle growth rate (nm/h) averaged for all nucleation events for the corresponding month along with the standard error is shown.
3.2. Relation to Meteorology
Previous studies have shown that nucleation is more likely to occur on days with low or no cloud cover [
17,
36] and lower humidity [
33,
37], relative to days on which nucleation is not observed. The link to solar radiation may reflect a dependence of nucleation on OH concentration, which drives the production of low vapor pressure gas phase [
38] and ionic [
6] precursors required for nucleation. The onset of nucleation is often observed in the mid to late morning, coincident with the break-up of the nocturnal boundary layer and downward mixing of nucleation precursors from aloft [
11,
22], which is more vigorous on days with high radiative flux (
i.e., cloudless days). The relationship between relative humidity and nucleation may reflect the role of water vapor in the characteristics of biogenic VOC oxidation and subsequent production of low vapor pressure nucleation precursors [
39,
40] or simply a pattern of lower relative humidity on cloudless days [
22]. Hamed
et al. [
41] recently showed that the inverse relationship between nucleation and relative humidity is primarily due to less intense solar radiation at high relative humidity, which limits the photochemical production of H
2SO
4 and organic nucleation precursors.
At Duke Forest, overall lower average relative humidity (43%) was observed on nucleation event days compared to (66%) non-event days, a pattern that was also observed in monthly averages (
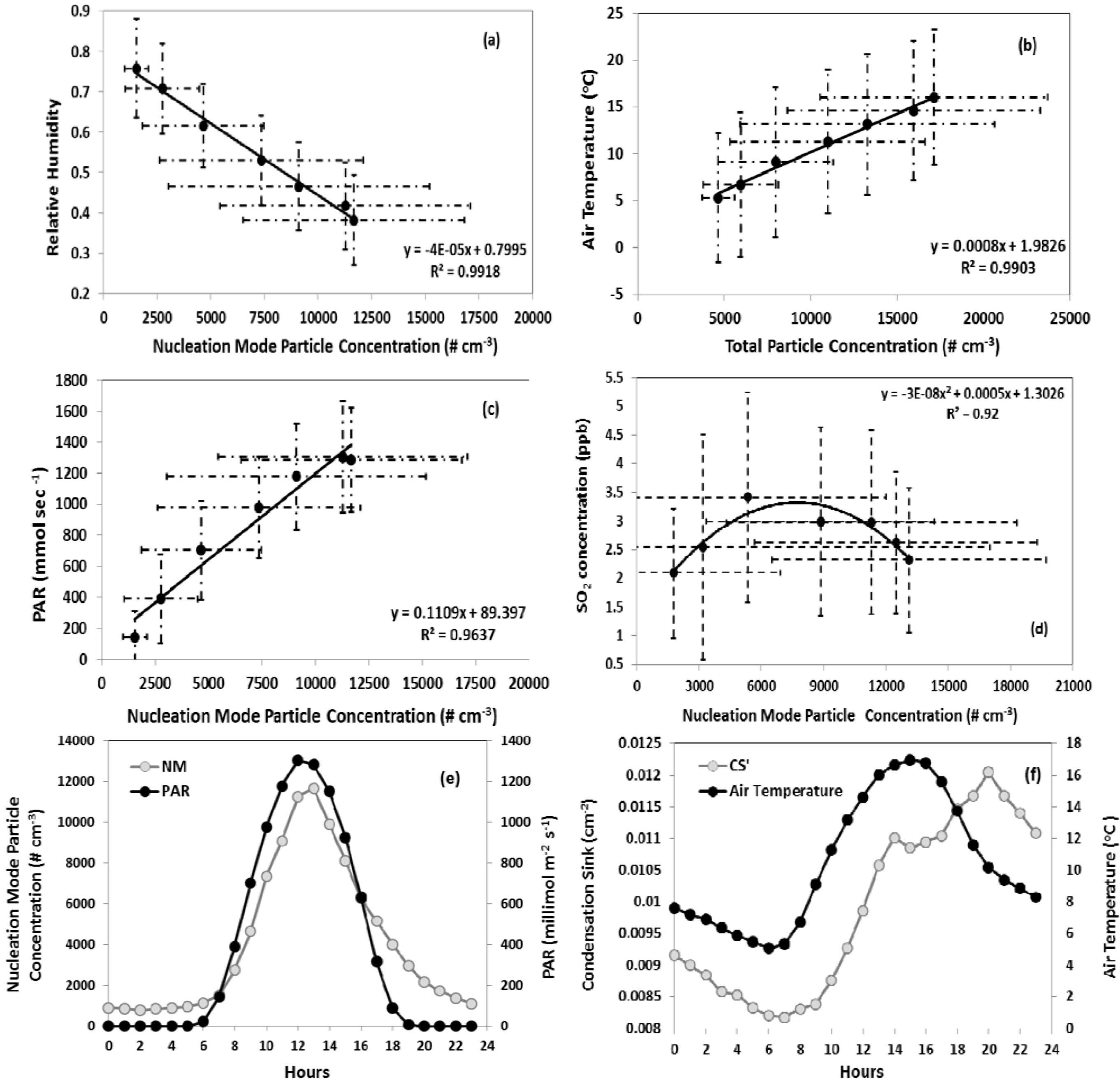
Figure 7). Consistent with other studies, nucleation events were more likely to occur on days with lower relative humidity. The role of humidity with respect to the intensity of nucleation, which may be quantified as the total particle number concentration during nucleation events, is also of interest. Total particle number concentration was found to be inversely correlated with relative humidity for all the temporally coincident data points, for the daily mean values, and for the daytime daily mean values (
Figure 8(a)), while no correlation was observed on non-event days. At high relative humidity, growth of pre-existing particles by condensation may further increase the CS and thus tends to prevent new particle formation.
Figure 7.
The grand average monthly mean daytime temperature and relative humidity for nucleation days and non-nucleation days.
Figure 7.
The grand average monthly mean daytime temperature and relative humidity for nucleation days and non-nucleation days.
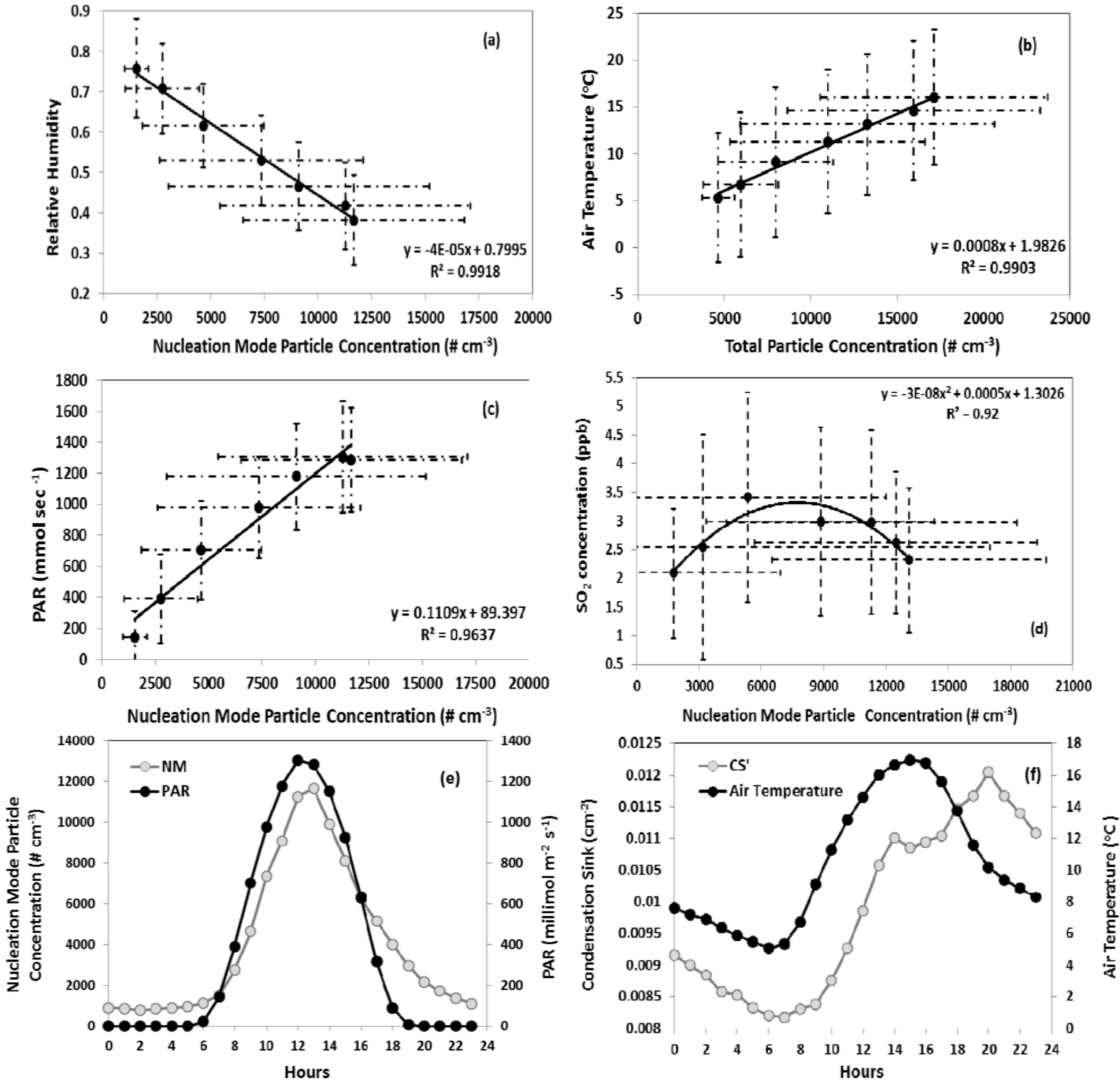
Figure 8.
Correlation of nucleation mode particles with (a) relative humidity, (b) air temperature, (c) PAR, (d) SO2. Data points represent averages between 7:00 am and 1:00 pm (approximately 1 hour prior to nucleation onset until the peak in number concentration), (e) diurnal profiles of PAR, nucleation mode particle number concentration, and (f) air temperature and condensation sink. Data represent hourly averages of all nucleation events. The error bars are standard deviation of that particular hour data during all nucleation events.
Figure 8.
Correlation of nucleation mode particles with (a) relative humidity, (b) air temperature, (c) PAR, (d) SO2. Data points represent averages between 7:00 am and 1:00 pm (approximately 1 hour prior to nucleation onset until the peak in number concentration), (e) diurnal profiles of PAR, nucleation mode particle number concentration, and (f) air temperature and condensation sink. Data represent hourly averages of all nucleation events. The error bars are standard deviation of that particular hour data during all nucleation events.
Total particle number concentration was positively correlated with air temperature on nucleation days (
Figure 8(b)). This pattern is expected, given the inverse relationship between relative humidity and temperature and therefore does not necessarily indicate a mechanistic linkage. However, emissions of biogenic VOC, which may play a role in nucleation, are also positively correlated with temperature. This potential linkage is discussed in more detail in the following section. Particle number concentration is positively correlated with PAR (
Figure 8(c)). As mentioned above, the correlation between nucleation intensity and radiative flux may indicate the importance of OH concentration in the formation of nucleation precursors or may simply be indicative of days characterized by more intense downward mixing during the morning boundary layer transition and lower relative humidity.
The nucleation day condensation sink typically reached its daily minimum at or before the onset of nucleation followed by an increase over the course of the event. In
Figure 8(e,f), the diurnal profile of PAR is plotted along with nucleation mode particle concentration and shows the time lagged relation between the diurnal temperature profile and condensation sink. The daily minimum in condensation sink and air temperature occur within 30 min of each other. However, there exists a time lag of more than a couple of hours between the continuous increase of these two parameters. This lag may be due to the time required for sufficient production of OH and subsequent nucleation of precursors such as H
2SO
4 after sunrise or downward mixing of existing precursors from above the nocturnal boundary layer. Condensation sink was positively correlated with PAR (R
2 = 0.14) and temperature (R
2 = 0.13), though the strength of the correlations was weak. No significant relationship between condensation sink and relative humidity was observed. Non-nucleation days are associated with an average precipitation of 0.197 mm, whereas the average precipitation on nucleation days was 0.0016 mm. When precipitation occurred on nucleation days, it was typically after the onset of nucleation and lasted only for a short period. Nucleation and non-nucleation days show no significant difference in wind speed and direction.
3.3. Relation to Chemistry
The relationships between nucleation events and concentrations of trace gases and PM were examined in an effort to characterize the chemical conditions that favor particle nucleation and growth.
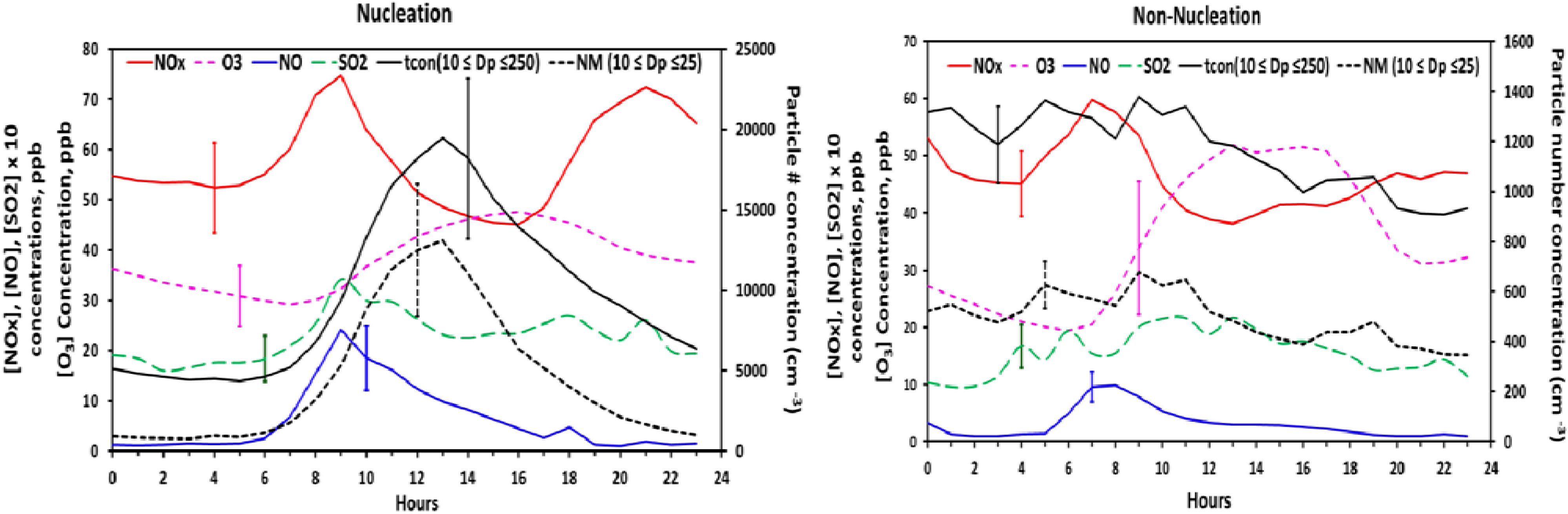
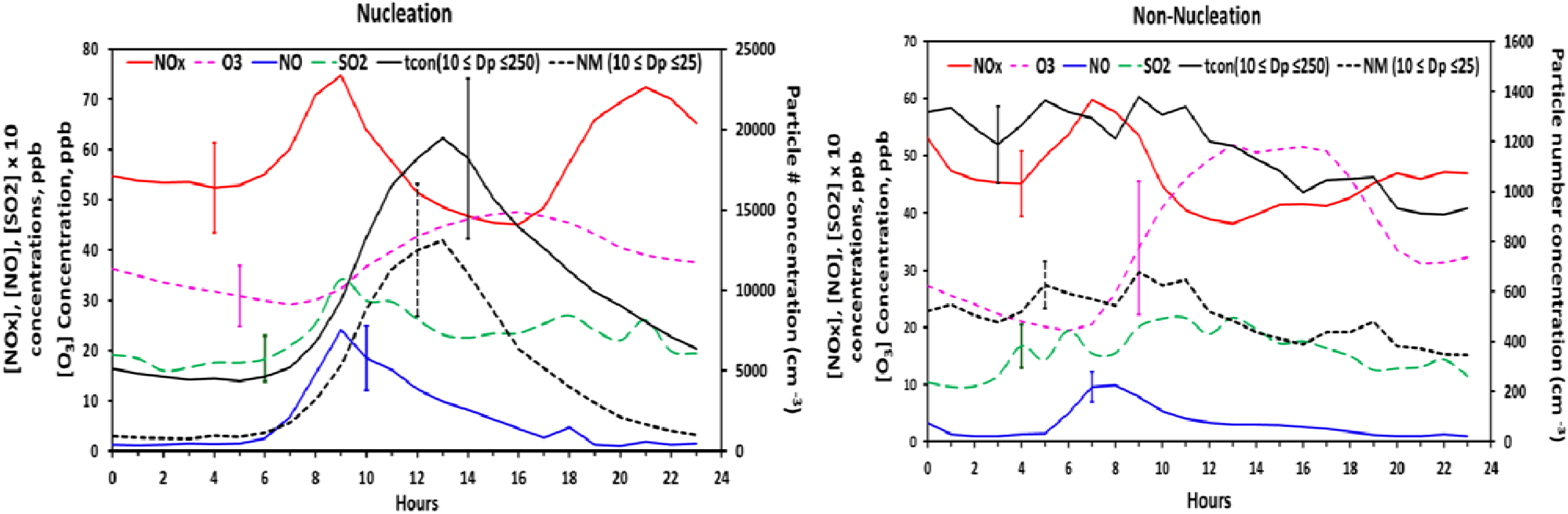
Figure 9 shows the diurnal variation of total nucleation mode and fine particle number concentration
versus O
3, SO
2, NO
x, and NO both for nucleation and non-nucleation days. The bi-modal profile of NO
x is indicative of mobile emissions during morning and evening rush hour. Meteorological differences between nucleation days (lower temperature, higher RH,
etc.) and non-nucleation days may be responsible for the differences in the diurnal structure of NO
x on these days. NO peaks during the morning rush hour while O
3 concentrations are still insufficient for significant NO
2 formation. A coincident bimodal pattern in particle number concentration, indicative of ultrafine particle emissions from mobile sources, is not evident. However, ultrafine particle emissions from mobile sources may not be expected to significantly affect particle number concentrations at the Duke Forest site. Emissions of ultrafine particles from light-duty vehicles are much lower than NO emissions and decrease exponentially with distance away from the point of emissions. Hagler
et al. [
42] conducted a study along a major roadway in nearby Raleigh, NC in which concentrations of NO, NO
2, and ultrafine particles (10–70 nm) were measured 20 m from the roadway and at increasing distances downwind. Very good correlation between NO and ultrafine particle number concentrations (UFP) was observed at a distance of 20 m, with regression slopes indicating a UFP (#/cm
−3)/NO(ppb) concentration ratio of ≈230. Using this ratio, the morning and evening peaks in NO and NOx shown in
Figure 9 would translate to small increases in particle number concentration and would be difficult to resolve within the temporal variability displayed by the particle number concentrations. A study by Geron 2009 [
23] observed that diesel traffic from major roads northeast of the site is partly responsible for a late morning peak in elemental carbon during spring and summer weekdays. A coincident peak in particle number concentration is not discernible in the average diurnal profiles of nucleation
versus non-nucleation days (
Figure 9). We conclude that it is not possible to clearly identify an effect of mobile emissions on particle number concentrations (<250 nm) at this site.
Figure 9.
The diurnal variation of number concentration and atmospheric gas species averaged for all the nucleation event days with coincident chemistry data.
Figure 9.
The diurnal variation of number concentration and atmospheric gas species averaged for all the nucleation event days with coincident chemistry data.
SO
2 concentrations display a distinct peak between 8:00 and 10:00 am on nucleation days and higher concentrations relative to non-nucleation days. This pattern of increasing SO
2 concentrations after sunrise is consistent with downward mixing of SO
2 from aloft as the nocturnal boundary layer breaks down. This period of increasing SO
2 concentrations often coincides with the daily minimum in condensation sink and onset of nucleation, particularly during warm months, which may be indicative of H
2SO
4-mediated nucleation. Hydroxyl radical (OH) concentrations were not measured during the study. However, OH concentrations would be expected to increase rapidly after sunrise on cloudless days when nucleation events were most frequently observed. The formation and growth of H
2SO
4 would be expected to lag slightly behind the increase in OH [
43] and SO
2. When hourly averaged for all the nucleation events (
Figure 8(d)), from approximately 1 h prior to the nucleation onset (7:00 am), until the peak in number concentration (1:00 pm), there exists a strong positive correlation between nucleation mode particles and SO
2 in the initial couple of hours at the start of nucleation; as the particle number concentration increases, the concentration of SO
2 decreases. The fact that on non-nucleation days no such relation exists between the two supports our suggestion of increased precursor concentrations on nucleation days.
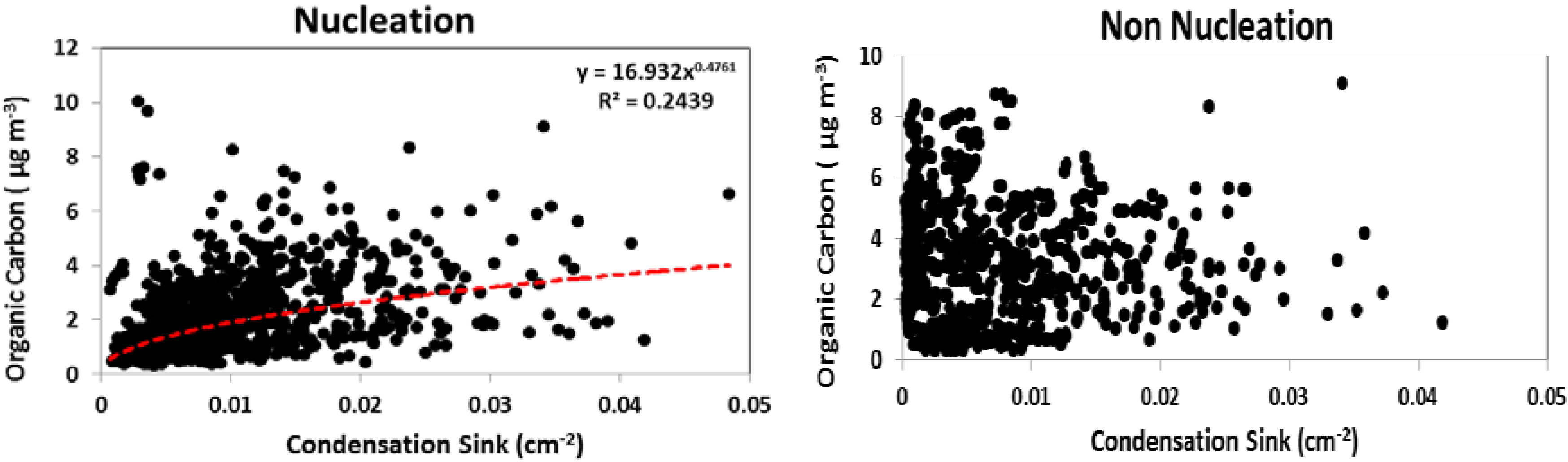
For particle growth to proceed following nucleation, sufficient concentrations of low volatility condensable gases are required. In less polluted areas, biogenic organic compounds may play a role in the formation [
44] and growth [
45] of new particles. To examine the potential role of organics at this site, we examined correlations between nucleation characteristics and organic carbon concentrations in PM
2.5, as well as O
3, a gas phase precursor to secondary organic aerosol. Condensation sink is positively correlated with organic carbon concentration (
Figure 10) on nucleation days (R
2 = 0.24) whereas no correlation is observed on non-nucleation days. In addition to the direct role of organic species in secondary aerosol formation, Zhang
et al. [
7] identified that the presence of organic acids also enhanced sulfuric acid nucleation. On days in which nucleation occurred, high values of CS, the rate at which vapor molecules condense onto existing particles, were associated with high organic carbon concentrations. Geron [
46] showed that oxidation products of biogenic VOC (e.g., isoprene, monoterpenes, sesquiterpenes) contribute significantly to organic carbon in PM at this forest site, which may partially explain the moderate correlation observed between CS and total organic carbon in PM
2.5 on nucleation days. While the predominance of nucleation events on days with high PAR likely reflects a photochemical control, such days would also exhibit higher emission rates of biogenic VOC relative to low PAR days at similar temperature [
47]. Furthermore, the highest particle growth rates were observed during warm months when VOC (mono- and sesquiterpene) emission rates from vegetation at this site are highest [
48].
Figure 10.
The correlation between organic carbon and condensation sink on nucleation days and non-nucleation days.
Figure 10.
The correlation between organic carbon and condensation sink on nucleation days and non-nucleation days.
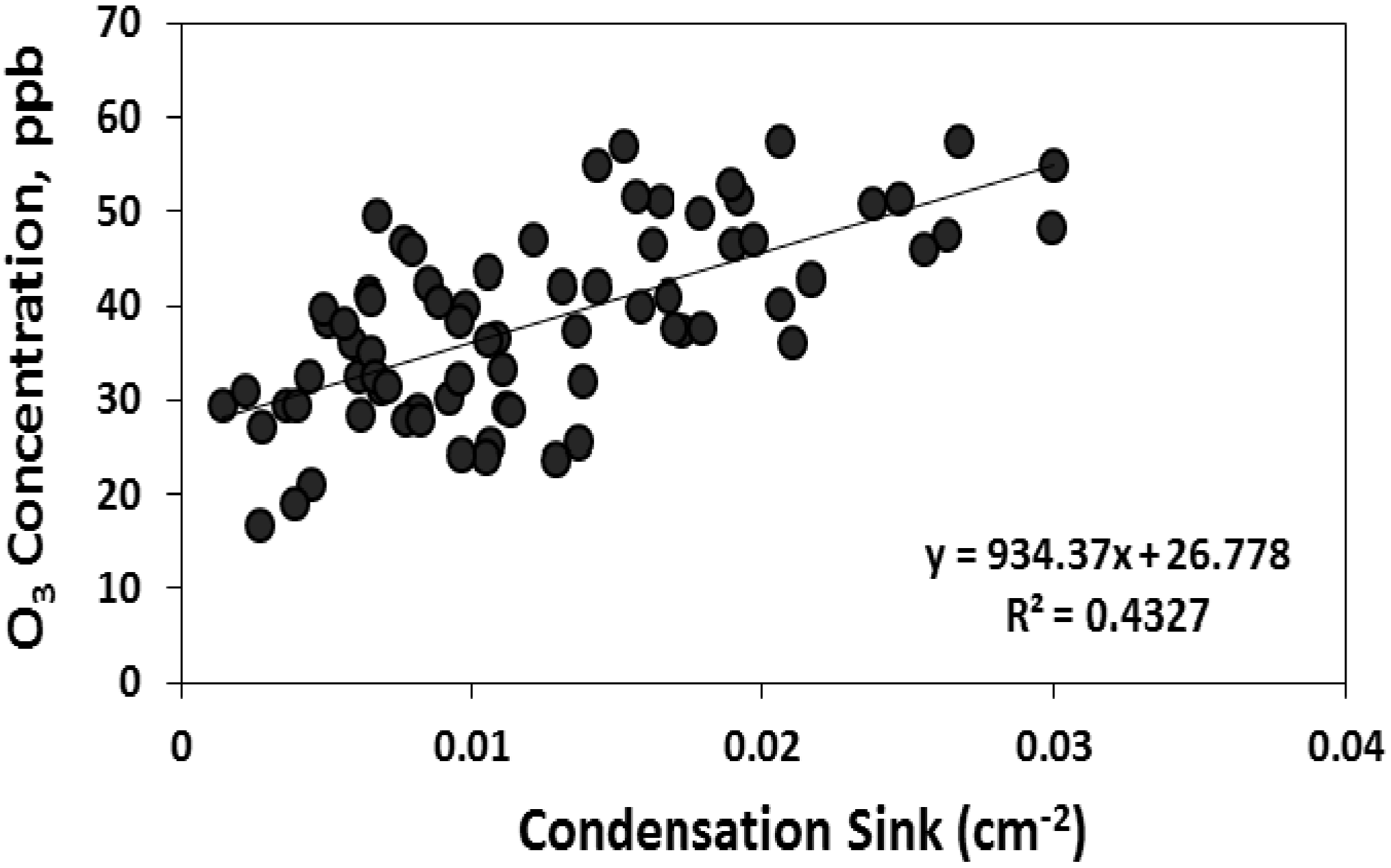
If biogenic VOC plays a role in particle formation and growth at this site, as suggested by the relationship between condensation sink and organic carbon in PM, a relationship between nucleation characteristics and atmospheric oxidative capacity may also be expected. Based on
in situ measurements collected during the Chemical Emission, Loss, Transformation, and Interactions with Canopies (CELTIC) campaign and subsequent longer term measurements of organic and elemental carbon, the modeling results from Geron [
46] show that isoprene, monoterpenes, and sesquiterpenes contribute 90%, 6%, and 4% of secondary organic aerosol at this site, which dominates total organic carbon in PM
2.5 during warm months [
23]. Also based on CELTIC measurements, Stroud
et al. [
49] concluded that local isoprene oxidation is dominated by OH, whereas O
3 and OH exert similar control over monoterpene oxidation, and sesquiterpene oxidation is dominated by O
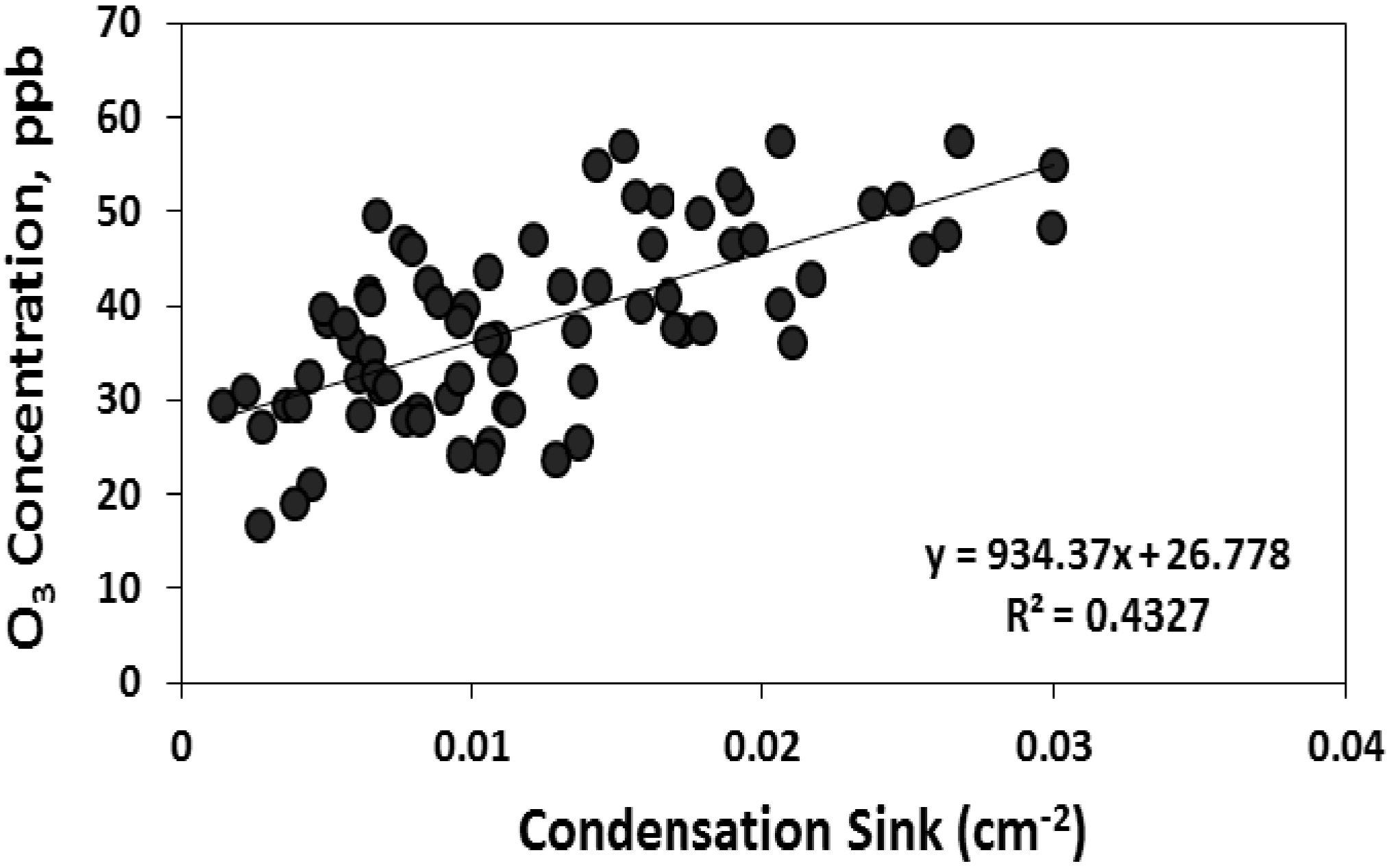
3. We found moderate positive correlation between the condensation sink and O
3 concentrations on nucleation days and no correlation on non-nucleation days (
Figure 11). This relationship suggests that on days when nucleation occurred, rapid and sustained particle growth was associated with conditions (
i.e., high O
3) that favor the oxidation of gas phase organic compounds, a pattern that is consistent with the relationship between CS and the concentration of organic carbon in PM.
Figure 11.
The correlation between condensation sink with total particle number concentration and O3 on nucleation days.
Figure 11.
The correlation between condensation sink with total particle number concentration and O3 on nucleation days.



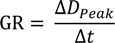

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}