Epigenetic Control of Pancreatic Regeneration in Diabetes
by
Shruti Balaji
, 
Tiziana Napolitano
,
Serena Silvano
,
Marika Elsa Friano
,
Anna Garrido-Utrilla
,
Josipa Atlija
and
Patrick Collombat
* Université Nice Sophia Antipolis, Inserm, CNRS, iBV, FR-06100 Nice, France
*
Author to whom correspondence should be addressed.
Genes 2018, 9(9), 448; https://doi.org/10.3390/genes9090448
Submission received: 31 July 2018
/
Revised: 31 August 2018
/
Accepted: 3 September 2018
/
Published: 7 September 2018
(This article belongs to the Special Issue Advances in Genetics of Regeneration in Metabesity)
{kind=link}
Abstract
:Both type 1 and type 2 diabetes are conditions that are associated with the loss of insulin-producing β-cells within the pancreas. An active research therefore aims at regenerating these β-cells with the hope that they could restore euglycemia. The approaches classically used consist in mimicking embryonic development, making use of diverse cell sources or converting pre-existing pancreatic cells. Despite impressive progresses and promising successes, it appears that we still need to gain further insight into the molecular mechanisms underlying β-cell development. This becomes even more obvious with the emergence of a relatively new field of research, epigenetics. The current review therefore focuses on the latest advances in this field in the context of β-cell (neo-)genesis research.
1. Introduction
The pancreas is an organ that contains both an exocrine and an endocrine compartment, all of its cells arising from a common epithelium [1]. The exocrine compartment secretes and transports digestive enzymes to the duodenum, whereas the endocrine tissue is organized into scattered clusters of cells, termed islets of Langerhans. The latter contain five different hormone-secreting cell subtypes: α-, β-, δ-, ε-, and pancreatic polypeptide (PP)-cells producing glucagon, insulin, somatostatin, ghrelin and PP, respectively. Among these different hormones, insulin acts to decrease blood sugar levels, whereas glucagon prevents hypoglycemia.
Diabetes is a condition that results from an alteration of insulin production or action. It is divided into two major types: Type 1 diabetes (T1D) corresponds to an autoimmune disorder wherein the β-cells are selectively destroyed whereas type 2 diabetes (T2D) is caused either by a loss of function of the β-cells or a resistance to insulin action in the peripheral target tissues (liver, muscle, adipose, etc.). Both forms of diabetes eventually result in β-cell loss and chronic hyperglycaemia [2]. Concerning T1D, while current therapeutic approaches (insulin supplementation, islet transplantation) save lives, the overall life expectancy of diabetic patients remains decreased, and their quality of life remains altered [3,4,5]. Numerous research laboratories therefore attempt to develop alternative therapies. Most of these aim at (re-)generating a functional insulin-producing β-cell mass by recapitulating endocrine pancreas development, as seen during embryogenesis [6,7,8,9,10]. Such an approach could also be useful for late-stage T2D patients, as these display β-cell loss as the disease progresses. However, it is clear that we need to gain further insight into the molecular mechanisms underlying β-cell development prior to an eventual application. This is mostly due to the fact that pancreas morphogenesis is a highly dynamic process that requires a precise pattern of activation/inactivation of several genes, quite often in a concomitant fashion.
In addition to this intricate genetic control, epigenetic regulation adds an additional layer of complexity. Epigenetics, as elegantly defined by others, corresponds to “the structural adaptation of chromosomal regions so as to register, signal or perpetuate altered activity states” [11]. Since chromatin consists of DNA wrapped around histone proteins, epigenetics also refers to DNA methylation and histone modifications that modulate gene expression without affecting the nucleotide sequence itself [12,13]. Several studies and reviews have been dedicated to the understanding of this relatively new field with a focus on the determination of whether particular regions of the genome are in an opened or closed state [11,14,15]. Indeed, chromatin that is not compacted (opened) allows for genes to be transcribed, while the opposite favors the silencing of genes. Opened chromatin marks include de-methylated DNA, histone methylation (H3K4me1/2/3), histone de-methylation (H3K27), and histone acetylation (H3K4/27ac). On the other hand, DNA methylation, histone methylation (H3K27me3), and histone deacetylation represent hallmarks of closed chromatin. The proteins that bring about these changes, as well as those that recognize the chromatin modifications at a specific genomic locus and regulate target gene expression by the recruitment of the required transcription factors (TFs), are known as epigenetic regulators. The histone modifying enzymes, long non-coding RNAs (lncRNAs), and chromatin status readers, such as bromodomain-containing proteins (BET) and lethal 3 MBT-like protein-1 (L3MBTL1), are examples of epigenetic regulators [16,17].
In the context of the pancreas, since all endocrine cells arise from a common progenitor pool where a balance of activation/inactivation of several concomitantly expressed TFs determines cell allocation, the role of epigenetics and epigenetic regulators is crucial. From a pharmacological and organ regeneration perspective, this may be even more important than attempting to control gene expression. The current review therefore aims at summarizing the advances that were made in recent years in the field of epigenetics with particular emphasis on the mechanisms underlying pancreatic β-cell (neo)genesis.
2. Advances in Our Understanding of the Epigenetics of Pancreas Development
The first morphological signs of pancreas development are detected at around embryonic day 9.5 (e9.5) in mice [1]. At this stage, the endoderm evaginates into the overlying mesenchyme, forming the dorsal bud. This is followed by the budding of the ventral pancreas under the control of a different molecular cascade. The two buds eventually fuse at around e12–e13 to form a single organ, the pancreas. Concomitantly, the process of endocrine specification is initiated, as outlined by the expression of several TFs, including Pancreas/duodenum homeobox protein 1 (Pdx1), Neurogenin 3 (Ngn3), Aristaless-related homeobox (Arx), and Paired box gene 4 (Pax4). While Pdx1 labels the early pancreatic progenitors, Ngn3 is a marker of pancreatic endocrine commitment [18,19]. Arx expression restricts the endocrine-committed cells to an α-cell fate, while Pax4 expression guides cells towards a β- or δ-cell lineage [8,20,21]. In mice, the proliferation of endocrine cells and their compaction into islets are known to continue for a few days postpartum.
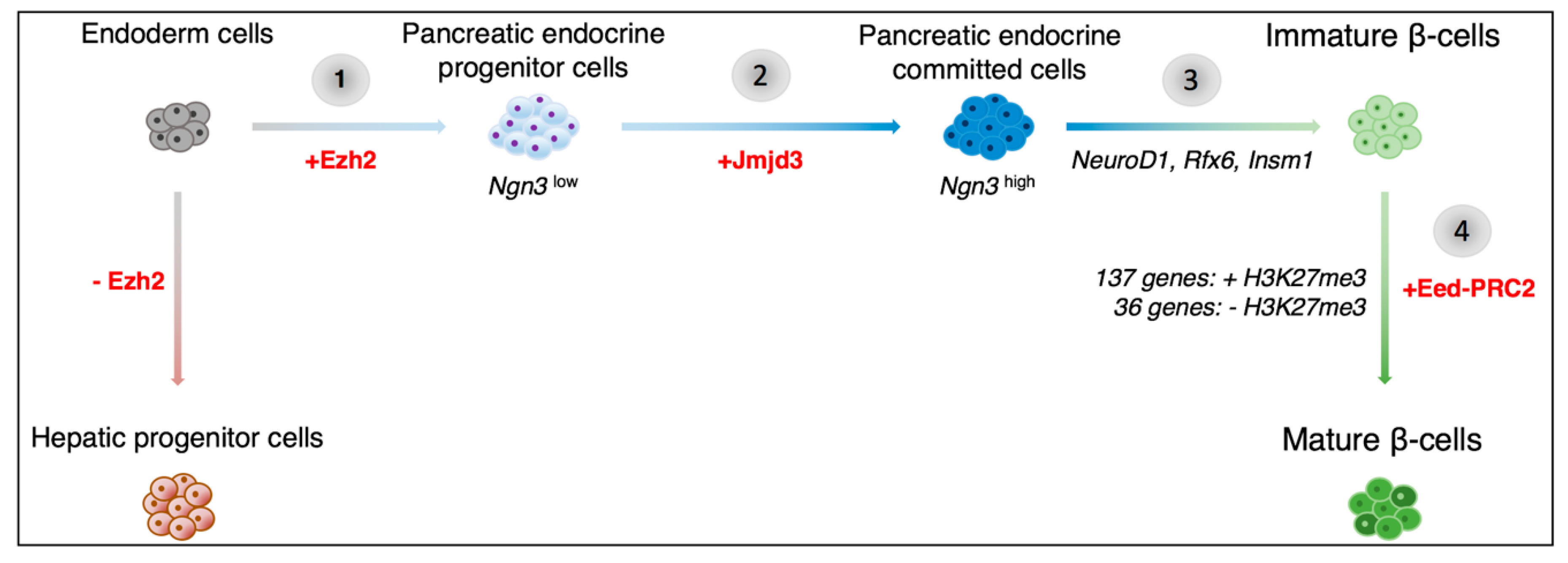
In terms of epigenetics, several studies have previously uncovered the bivalent status of pancreatic regulatory elements in the early developing pancreas, reviewed in [12,13]. Indeed, at early stages, the chromatin within these regulatory regions bears both active and repressive histone marks. The choice between the maintenance or loss of a given mark is made based on both inter- and intra-cellular developmental cues. Interestingly, Xu et al. have previously shown that the Polycomb repressor complex 2 (PRC2) protein, Enhancer of zeste homolog 2 (Ezh2), plays a key role in assigning a ventral pancreatic fate versus a hepatic fate in endodermal cells, Figure 1 [22,23]. By adding repressive H3K27me3 marks to Pdx1 regulatory elements, Ezh2 maintains the balance between hepatic and pancreatic progenitor cell numbers [22]. The addition of the inactive H3K27me3 marks specifically by the polycomb repressor proteins, has been shown to occur in a step-wise manner [24]. Pancreatic progenitor cells are thus progressively programmed towards a particular endocrine cell fate.
Recent work by Yu et al. allowed us to gain further insight into the Ngn3-induced transition of endocrine progenitor cells to endocrine-committed cells [25]. Neurogenin 3 is a basic helix-loop-helix-containing transcription factor whose targeted disruption leads to a lack of pancreatic endocrine cells in mice [18]. Yu et al. confirmed that, in the pancreatic context, the levels of Ngn3 directed cell fate [25]. A low level was associated with the maintenance of the pluripotent state, whereas a high level of Ngn3 expression was found to induce endocrine cell lineage allocation, Figure 1.
Interestingly, the transition from a Ngn3low to Ngn3high state was associated with the resolution of bivalency within the promoter region of 590 out of 2131 genes initially harboring bivalent marks. Interestingly, for 80% of these genes, bivalency was resolved by the sole loss of H3K27me3 repressive marks, while retaining the H3K4me3 active marks, in a Jumonji domain-containing protein 3 (Jmjd3)-dependant process, Figure 1. This is not surprising as Jmjd3 is a known H3K27me3-specific demethylase [26]. Ribonucleic acid-sequencing (RNA-seq) experiments subsequently demonstrated a concomitant increase in Ngn3 downstream target genes expression, such as Neurogenic differentiation factor 1 (NeuroD1), Regulatory factor X, 6 (Rfx6), and Insulinoma-associated 1 (Insm1), providing evidence of the key role exerted by chromatin regulation on the programming of pancreatic precursors towards an endocrine cell fate, Figure 1.
Several reports have suggested that mature β-cells can be subdivided into different populations that are based on the differential expression of marker genes or cell-surface antigens [27,28]. These distinct cell subtypes could represent differentially maturing and/or functioning β-cells. Interestingly, very little is known about the pancreatic α-cell maturation process. Qiu and colleagues therefore used single-cell RNA-seq to dissect the maturation mechanisms of α- and β-cells [29]. It was found that while α-cells mature primarily through the downregulation of genes expressed in immature α-cells, β-cell maturation involves the upregulation of additional genes, Figure 1. Interestingly, Assay for Transposase-Accessible Chromatin with high-throughput sequencing (ATAC-seq) data from mature α- and β-cells showed that 78% of open chromatin peaks found in β-cells could also be observed in α-cells, whereas only 46% of peaks detected in α-cells were also found in β-cells [30]. Additional work indicated that the difference between mature α- and β-cells may come down to less than 21% of active genes. Since Qiu et al. suggested that α-cell maturation involves the silencing of certain genes, while β-cell maturation involves the activation of new genes, one would assume that more open chromatin peaks would be found in mature β-cells as compared to α-cells [29]. However, Ackermann et al. found that nearly 27,000 ATAC-seq peak regions were classified as α-cell-specific, whereas only 1850 β-cell-specific regions were detected [30]. What is even more interesting is that combined with mRNA-seq data, it was suggested that open chromatin may be a better predictor of gene activation in mature α-cells than in β-cells, thus implying that differential regulation alone could explain the phenotypic and functional difference between the two cell subtypes.
Adding further to our understanding of the differential allocation to the various endocrine cell lineages, Neiman et al. studied the methylation pattern of CpG islands within the promoter region of various β-cell specific genes [31]. Surprisingly, while they found that the promoters of β-cell specific genes were highly methylated in non-endocrine tissue, such as in acinar cells, this difference was not visible when comparing the different endocrine populations themselves. Indeed, the INS and GCG gene promoters were found unmethylated in Fluorescence-activated cell sorting (FACS) sorted α-, β-, and δ-cells from human islets. However, the methylation of CpG sites downstream of the transcription start sites (TSS), i.e., at the enhancers, reflected the differential gene expression pattern amongst the endocrine subtypes. Methylation downstream of the INS TSS was thus found exclusively in α- and δ-cells, whereas methylation downstream of the GCG TSS was found in β- and δ-cells. Further, it appeared that such a methylation pattern was obtained progressively with age. Indeed, prior to birth, all Ngn3-positive progenitors displayed the same demethylation pattern. These observations are of interest as they further outline the cellular plasticity that is often observed in pathological conditions, as well as in attempts to transdifferentiate alternative endocrine cell subtypes into β-cells.
3. The Epigenetic State in Type 2 Diabetes and Obesity
Type 2 diabetes is caused either by a loss of insulin secretion or insulin resistance. Interestingly, recent in-depth analyses of the mouse and human genome by Lu et al. showed that in mice subjected to a high fat diet (HFD) and in humans with T2D, chromatin-state-defined transcriptome dysregulation led to a preferential loss of key β-cell TFs [32]. Equally important was the observation that such loss of identity was associated with a transition to a bivalent epigenetic state that is normally associated with stemness, suggestive of dedifferentiation [32]. This is in accordance with the finding that humans with a high BMI display a decline in β-cell function [33]. Interestingly, an embryonic ectoderm development protein (Eed)-containing PRC2 was shown to be required for the long-term maintenance of β-cell function in vivo. The Eed protein along with the Ezh2 histone methyltransferase and other proteins, form the PRC2. The PRC2 catalyzes the trimethylation of H3K27, thus leading to target gene repression. The loss of the Eed-PRC2 complex was found to result in the expression of islet dedifferentiation factors, like GLI-Kruppel family member 2 (Gli2), and in the loss of the terminal β-cell identity [32]. Importantly, the simultaneous inhibition of a polycomb opposer, such as MII (a H3K4 methyltransferase), could rescue this loss of a β-cell phenotype, thereby raising hope in the context of T2D research. Equally noteworthy was the finding that multiple T2D risk loci are, in fact, α-cell-specific, as demonstrated while using ATAC-seq, further suggesting that T2D may also be associated with α-cell dysfunction and not just β-cell failure [30].
A few years ago, mutations within the high mobility group (HMG) box-containing protein 20A (HMG20A) gene were reported in T2D populations [34,35]. This protein is a known chromatin modifier in neurons, but it was not until recently that its role in the maintenance of pancreatic β-cell function was reported [36]. It has an opposing role to another chromatin modifier, RE-1 silencing transcription factor (Rest) [37]. Whereas Rest is completely absent in mature islets, HMG20A is expressed in islets from normal individuals. There, HMG20A normally represses β-cell development-associated genes, like Pax4 and NeuroD1, and instead activates β-cell maturation-associated genes, such as v-maf musculoaponeurotic fibrosarcoma oncogene family, protein A (MafA) [36]. Mutations in HMG20A that are associated with T2D could thus alter its function in repressing the ‘disallowed’ genes and result in impaired glucose-stimulated insulin secretion (GSIS) and other functional markers of mature β-cell function. This could occur by the deposition of active H3K4me2 marks at the promoters of genes, such as Pax4 [36].
Upon a glucose bolus, β-cells sense glucose levels in the systemic circulation via their Glucose transporter 2 (Glut2 receptors) [38], which eventually triggers the exocytosis of insulin. Insulin then mobilizes peripheral tissues, viz. liver, fat and muscle, to take up glucose thereby leading to a reduction of glucose in the peripheral circulation [39]. Insulin resistance results from the unresponsiveness and/or loss of insulin receptors in peripheral tissues [40,41]. Recently, Kang et al. sought to study the epigenetic basis of insulin resistance, making use of two agents with seemingly opposite actions: Dexamethasone (Dex) and the tumor necrosis factor α (TNF-α), the former being an anti-inflammatory agent and the latter displaying pro-inflammatory properties [42]. Surprisingly, the authors found that treating adipocytes in vitro with either agent resulted in some common epigenetic changes. For instance, an increase in opened-chromatin H3K27ac marks in the distal enhancers of common target genes, viz. Transmembrane protein 176a/b (Tmem176a/b) and Family with sequence similarity 46 member b (Fam46b), was noted and associated with a decrease in insulin-stimulated glucose uptake. This suggests that although the two agents could act via different signaling pathways, they eventually converge towards a common downstream target. Even more surprisingly, a number of common enhancer sites were found to be bound by the glucocorticoid receptor (GR), previously thought only to be activated by Dex [42]. Accordingly, the expression of some of the common insulin resistance-associated genes, including Vitamin D receptor (Vdr), was found to be reduced by treating cells with Rosiglitazone, which is normally used as an insulin-sensitising agent in T2D. Thus, in addition to providing an epigenetic basis for insulin resistance, these studies also provided several therapeutic research paths, such as the use of anti-inflammatory agents, insulin-sensitizing agents, GR inhibitors, alone or in combination.
As previously mentioned, gaining further insight into the role of epigenetic regulators is also an important aspect of studying the epigenetic causes underlying diseases. lncRNAs being one such class of epigenetic regulators, Akerman and colleagues studied gene expression datasets that included human islet samples from donors that were diagnosed with T2D or impaired glucose tolerance (IGT) [43]. They found that the expression of 15 lncRNAs was strongly reduced in T2D islets, whereas 100 lncRNAs were insufficiently expressed in IGT islets when compared to islets from control individuals. In particular, the lncRNA PDX1 locus upstream transcript (PLUTO) was found to be among the most markedly downregulated lncRNAs in islets from T2D or IGT donors. By combining several experimental approaches in the human EndoC-βH1 cell line and in dispersed human islets, they established that inhibiting PLUTO expression also severely inhibited PDX1 expression. This was attributed to a loss of contact between the PDX1 promoter and its enhancer cluster, i.e., a loss of three-dimensional (3D) chromatin conformation. The loss of lncRNA expression resulting in the loss of expression of key β-cell-associated TFs is thus worth looking into in order to uncover the pathogenic basis of non-genetic diabetes.
While more information is available about the genetic and epigenetic modifications that accompany T2D, much is yet to be understood of the epigenetic basis of T1D. In a recent review, Wu et al. dedicated a short paragraph to this topic [44]. They reviewed the available literature and found that in T1D twins, DNA methylation of B lymphocytes could contribute to T1D pathogenesis. They also postulated that the currently available chromatin modifying drugs, such as 5-Aza and Histone deacetylase inhibitors (HDACs), could potentially be of interest for T1D therapy. Other studies in T1D individuals also supported the notion that epigenetic changes in the immune effector cells are responsible for the immune destruction of the pancreatic β-cells [45,46,47,48]. No studies so far have, however, examined the accompanying epigenetic changes, if any, in the β-cells themselves.
4. Epigenetics and Pancreatic Regeneration
In the context of T1D research, numerous laboratories aim at regenerating lost insulin-producing β-cells by either mimicking embryonic development or converting pre-existing cells. Several reports suggest that ectopically expressing key pancreatic differentiation factors, such as Ngn3, in immature cells could force these to adopt an endocrine fate. Recently, Fontcuberta-PiSunyer et al. described the use of both inhibitors of histone demethylases, as well as ectopic expression of NGN3 to improve pancreatic differentiation protocols [49]. Interestingly, both the timing and the cellular context seem to influence the robustness of the protocol. Indeed, only the simultaneous inhibition of Ezh2 and the concomitant activation of NGN3 led to an activation of Ngn3 downstream target genes in mouse pancreatic (mPac) cells and mouse embryonic fibroblasts (MEFs). The inhibition of Ezh2 prior to NGN3 activation was however found to be required in the induced pluripotent stem cell (iPSC) context. Further, although the authors described the increased expression of islet hormone genes, it remains to be determined whether the differentiated cells are functional. Nevertheless, this approach appears as a promising strategy, as confirmed by studies demonstrating that Ezh2 is required for the proliferation of immature β-cells [29].
While it is desirable to find highly proliferative and ethically-sound alternative tissue sources for the mass production of β-like cells, it is also worthwhile to pursue the re- or trans-differentiation of existing pools of pancreatic cells within the patient body. Not only could this avoid the need for surgery, it could also avoid several differentiation steps given the important differences in methylation patterns between non-endocrine and endocrine cell subtypes [31]. In light of this, a study of Qiu et al. highlighted the proliferative capacity of mature β-cells, thus providing ideas for the use of existing/leftover β-cells in diabetes therapy [29]. They found that although both α- and β-cells are most proliferative a few days after birth, the expression of the histone methyltransferase gene Ezh2 was maintained at a low level even in adult, non-proliferative cells. An activation of the Ezh2 gene could therefore potentially silence those genes that are associated with α- and β-cell maturation and provide a way for these cells to revert back to the immature, proliferative state [50].
As previously mentioned, Arx is a marker required for the α-cell allocation and maintenance of the α-cell phenotype, while DNA methyltransferase 1 (Dnmt1) is a methyltransferase that recognizes hemimethylated CpGs and re-methylates them [20,51,52]. Building up on previous research, Chakravarthy et al. showed that the simultaneous loss of Dnmt1 and Arx could induce a reprogramming of α-cells into β-cells in vivo [53]. The heterogenous population of β-like cells so obtained expressed many of the mature, functional markers of β-cells within just 8 weeks. Further, they were able to show that the reprogrammed α-cells were able to respond to glucose like native β-cells. Their observations were confirmed in T1D patients with five years of diabetes or more. In these individuals, they found a subset of glucagon+ cells lacking both DNMT1 and ARX expression and ectopically expressing β-cell markers, including insulin. While these results suggest a putative attempt at regenerating lost β-cells in type 1 diabetic patients, whether this conversion is eventually completed or whether these converted cells are also attacked by the immune system remains to be determined. As the authors point out though, it is likely that the conditional loss of Arx and Dnmt1 alone would be insufficient to reprogram α-cells into β-cells. However, guided by this information, there is a potential to find out additional (epi)genetic factors that may be key for α-cell-mediated β-like cell neogenesis.
5. The Influence of Culture Substrate on Epigenetic Regulation of Differentiation
While it is well known that the culture substrate influences the differentiation process in vitro ,reviewed in [54], the epigenetic basis of this has only recently been investigated. Thus, Pennarossa et al. checked the global DNA methylation status upon culturing mouse dermal fibroblasts that had been treated with an epigenetic eraser 5-azacytidine on a plastic (stiff) and gel (soft) matrix [55]. Interestingly, they found that, while global methylation decreased as expected upon 5-aza treatment, there was a difference in methylation status when comparing cells cultured on the different substrates, with cells grown on the soft matrix exhibiting lower DNA methylation overall. This was accompanied by a decrease in Histone deacetylase 1 (Hdac1) and an increase in Histone acetyltransferase 1 (Hat1) and Tet methylcytosine dioxygenase 2 (Tet2) gene expression. The cells also acquired a dedifferentiated-like state with an increased expression of pluripotency markers, such as Octamer-binding transcription factor 4 (Oct4), Nanog, and Sex determining region Y-box 2 (Sox2). Interestingly, the induction of pancreatic differentiation while using a three-step protocol showed that the cells grown on the soft matrix were more efficiently differentiated down the pancreatic lineage. Indeed, these cells eventually formed islet-like structures that are composed of a heterogenous population of mostly mono-hormonal cells. These islet-like clusters were further able to respond well to a high glucose challenge, thereby opening new research avenues for β-cell differentiation protocols.
6. Conclusions
Although a number of pharmaceutical agents are currently available to treat the consequences of β-cell loss in diabetic patients, improved alternative treatments remain required. In order to achieve this goal, an active research is ongoing, spanning from designing insulin/glucagon pumps to improving islet transplantation [4,56]. However, numerous reports have highlighted the need to go beyond the currently available solutions due to their many disadvantages [57]. One such avenue of disease therapy is tissue regeneration. The field of regenerative medicine is abuzz with efforts to differentiate or trans-differentiate cells from the pancreas and unrelated tissues [6,8,9,10,58,59,60]. However, despite impressive progresses, regenerating fully functional β-cells remains challenging [61]. Clearly, a better understanding of the molecular mechanisms underlying pancreas development, including the accompanying epigenetic alterations, is required.
In this short review, we described some of the latest discoveries that were made in the field of epigenetics, these clearly demonstrating that epigenetic manipulations could represent a new approach in order to induce β-cell neogenesis. While it is clear that much work remains to be done due to the further increased complexity, new research avenues with great potential have certainly been opened. It would also be worthwhile to further focus on the altered epigenetic status of T2D patients to uncover potential therapeutic avenues. Similarly, a thorough analysis of the epigenetic status of β-cells in T1D individuals is also severely lacking and such studies could also provide new research strategies.
Funding
This research was funded by the Juvenile Diabetes Research foundation (#C1-1-SRA-2018-536-M-R, #3-SRA-2014-282-Q-R, #3-SRA-2017-415-S-B, #2-SRA-2017-416-S-B, #17-2013-426), the INSERM AVENIR program, the INSERM, the European Research Council (#StG-2011-281265), the FRM (#DRC20091217179), the ANR (#2009 GENO 105 01/01KU0906, #ANR-17-ERC2-0004-01, #ANR-16-CE18-0005-02, #Betaplasticity), the “Investments for the Future” LABEX SIGNALIFE (#ANR-11-LABX-0028-01), Club Isatis, and Dorato, and de Marffy-Mantuano family, the Dujean family, the Fondation Générale de Santé, and the Foundation Schlumberger pour l'Education et la Recherche.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gittes, G.K. Developmental biology of the pancreas: A comprehensive review. Dev. Biol. 2009, 326, 4–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weir, G.C.; Bonner-Weir, S. Islet β cell mass in diabetes and how it relates to function, birth, and death. Ann. N. Y. Acad. Sci. 2013, 1281, 92–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawlyk, A.C.; Giacomini, K.N.; McKeon, C.; Shuldiner, A.R.; Florez, J.C. Metformin pharmacogenomics: Current status and future directions. Diabetes 2014, 63, 2590–2599. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, L.; Fleming, G.A.; Petrie, J.R.; Holl, R.W.; Bergenstal, R.M.; Peters, A.L. Insulin pump risks and benefits: A clinical appraisal of pump safety standards, adverse event reporting, and research needs. Diabetes Care 2015, 38, 716–722. [Google Scholar] [CrossRef] [PubMed]
- Pratley, R.E.; Heller, S.R.; Miller, M.A. Treatment of type 2 diabetes mellitus in the older adult: A review. Endocr. Pract. 2014, 20, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Balaji, S.; Zhou, Y.; Opara, E.C.; Soker, S. Combinations of activin A or nicotinamide with the pancreatic transcription factor PDX1 support differentiation of human amnion epithelial cells toward a pancreatic lineage. Cell. Reprogram. 2017, 19, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Sambathkumar, R.; Akkerman, R.; Dastidar, S.; Roelandt, P.; Kumar, M.; Bajaj, M.; Rosa, A.R.M.; Helsen, N.; Vanslembrouck, V.; Kalo, E.; et al. Generation of hepatocyte- and endocrine pancreatic-like cells from human induced endodermal progenitor cells. PLoS ONE 2018, 13. [Google Scholar] [CrossRef] [PubMed]
- Collombat, P.; Xu, X.; Ravassard, P.; Sosa-Pineda, B.; Dussaud, S.; Billestrup, N.; Madsen, O.D.; Serup, P.; Heimberg, H.; Mansouri, A. The ectopic expression of Pax4 in the mouse pancreas converts progenitor cells into α and Subsequently β Cells. Cell 2009, 138, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Al-Hasani, K.; Pfeifer, A.; Courtney, M.; Ben-Othman, N.; Gjernes, E.; Vieira, A.; Druelle, N.; Avolio, F.; Ravassard, P.; Leuckx, G.; et al. Adult duct-lining cells can reprogram into β-like cells able to counter repeated cycles of toxin-induced diabetes. Dev. Cell 2013, 26, 86–100. [Google Scholar] [CrossRef] [PubMed]
- Balaji, S.; Zhou, Y.; Ganguly, A.; Opara, E.C.; Soker, S. The combined effect of PDX1, epidermal growth factor and poly-l-ornithine on human amnion epithelial cells’ differentiation. BMC Dev. Biol. 2016, 16. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. Perceptions of epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Xie, R.; Carrano, A.C.; Sander, M. A systems view of epigenetic networks regulating pancreas development and β-cell function. Wiley Interdiscip. Rev. Syst. Biol. Med. 2015, 7, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quilichini, E.; Haumaitre, C. Implication of epigenetics in pancreas development and disease. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 883–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jerram, S.T.; Dang, M.N.; Leslie, R.D. The role of epigenetics in type 1 diabetes. Curr. Diab. Rep. 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Kwak, S.H.; Park, K.S. Recent progress in genetic and epigenetic research on type 2 diabetes. Exp. Mol. Med. 2016, 48. [Google Scholar] [CrossRef] [PubMed]
- Hiragami-Hamada, K.; Fischle, W. RNAs—Physical and functional modulators of chromatin reader proteins. Biochim. Biophys. Acta 2014, 1839, 737–742. [Google Scholar] [CrossRef] [PubMed]
- James, L.I.; Frye, S.V. Targeting chromatin readers. Clin. Pharmacol. Ther. 2013, 93, 312–314. [Google Scholar] [CrossRef] [PubMed]
- Gradwohl, G.; Dierich, A.; LeMeur, M.; Guillemot, F. Neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc. Natl. Acad. Sci. USA 2000, 97, 1607–1611. [Google Scholar] [CrossRef] [PubMed]
- Ahlgren, U.; Jonsson, J.; Edlund, H. The morphogenesis of the pancreatic mesenchyme is uncoupled from that of the pancreatic epithelium in IPF1/PDX1-deficient mice. Development 1996, 122, 1409–1416. [Google Scholar] [PubMed]
- Collombat, P.; Mansouri, A.; Hecksher-Sørensen, J.; Serup, P.; Krull, P.; Gradwohl, G.; Gruss, P. Opposing actions of Arx and Pax4 in endocrine pancreas development. Genes Dev. 2003, 17, 2591–2603. [Google Scholar] [CrossRef] [PubMed]
- Sosa-Pineda, B.; Chowdhury, K.; Torres, M.; Oliver, G.; Gruss, P. The Pax4 gene is essential for differentiation of insulin-producing β cells in the mammalian pancreas. Nature 1997, 386, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.-R.; Cole, P.A.; Meyers, D.J.; Kormish, J.; Dent, S.; Zaret, K.S. Chromatin “pre-pattern” and histone modifiers in a fate choice for liver and pancreas. Science 2011, 332, 963–966. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.R.; Li, L.C.; Donahue, G.; Ying, L.; Zhang, Y.W.; Gadue, P.; Zaret, K.S. Dynamics of genomic H3K27me3 domains and role of EZH2 during pancreatic endocrine specification. EMBO J. 2014, 33, 2157–2170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Arensbergen, J.; García-Hurtado, J.; Moran, I.; Maestro, M.A.; Xu, X.; Van de Casteele, M.; Skoudy, A.L.; Palassini, M.; Heimberg, H.; Ferrer, J. Derepression of polycomb targets during pancreatic organogenesis allows insulin-producing beta-cells to adopt a neural gene activity program. Genome Res. 2010, 20, 722–732. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.X.; Qiu, W.L.; Yang, L.; Li, L.C.; Zhang, Y.W.; Xu, C.R. Dynamics of chromatin marks and the role of JMJD3 during pancreatic endocrine cell fate commitment. Development 2018, 145. [Google Scholar] [CrossRef] [PubMed]
- Agger, K.; Cloos, P.A.; Christensen, J.; Pasini, D.; Rose, S.; Rappsilber, J.; Issaeva, I.; Canaani, E.; Salcini, A.E.; Helin, K. UTX and JMJD3 are histone H3K27 demethylases involved in HOX gene regulation and development. Nature 2007, 449, 731–734. [Google Scholar] [CrossRef] [PubMed]
- Bader, E.; Migliorini, A.; Gegg, M.; Moruzzi, N.; Gerdes, J.; Roscioni, S.S.; Bakhti, M.; Brandl, E.; Irmler, M.; Beckers, J.; et al. Identification of proliferative and mature β-cells in the islets of langerhans. Nature 2016, 535, 430–434. [Google Scholar] [CrossRef] [PubMed]
- Dorrell, C.; Schug, J.; Canaday, P.S.; Russ, H.A.; Tarlow, B.D.; Grompe, M.T.; Horton, T.; Hebrok, M.; Streeter, P.R.; Kaestner, K.H.; et al. Human islets contain four distinct subtypes of β cells. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.-L.; Zhang, Y.-W.; Feng, Y.; Li, L.-C.; Yang, L.; Xu, C.-R. Deciphering Pancreatic Islet β Cell and α Cell Maturation Pathways and Characteristic Features at the Single-Cell Level. Cell Metab. 2017, 25, 1194–1205.e4. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, A.M.; Wang, Z.; Schug, J.; Naji, A.; Kaestner, K.H. Integration of ATAC-seq and RNA-seq identifies human α cell and β cell signature genes. Mol. Metab. 2016, 5, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Neiman, D.; Moss, J.; Hecht, M.; Maggenheim, J.; Piyanzin, S.; Shapiro, A.M.J.; de Koning, E.J.P.; Razin, A.; Cedar, H.; Shemer, R.; et al. Islet cells share promoter hypomethylation independently of expression, but exhibit cell-type-specific methylation in enhancers. Proc. Natl. Acad. Sci. USA 2017, 114, 13525–13530. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.T.; Heyne, S.; Dror, E.; Casas, E.; Leonhardt, L.; Boenke, T.; Yang, C.H.; Sagar; Arrigoni, L.; Dalgaard, K.; et al. The polycomb-dependent epigenome controls β cell dysfunction, dedifferentiation, and diabetes. Cell Metab. 2018, 27, 1294–1308. [Google Scholar] [CrossRef] [PubMed]
- Avrahami, D.; Li, C.; Zhang, J.; Schug, J.; Avrahami, R.; Rao, S.; Sadler, M.B.; Burger, L.; Schübeler, D.; Glaser, B.; et al. Aging-dependent demethylation of regulatory elements correlates with chromatin state and improved β cell function. Cell Metab. 2015, 22, 619–632. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.P.; Voight, B.F.; Teslocvich, T.M.; Ferreira, T.; Segré, A.V.; Steinthorsdottir, V.; Strawbridge, R.J.; Khan, H.; Grallert, H.; Mahajan, A.; et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat. Genet. 2012, 44, 981–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kooner, J.S.; Saleheen, D.; Sim, X.; Sehmi, J.; Zhang, W.; Frossard, P.; Been, L.F.; Chia, K.S.; Dimas, A.S.; Hassanali, N.; et al. Genome-wide association study in individuals of South Asian ancestry identifies six new type 2 diabetes susceptibility loci. Nat. Genet. 2011, 43, 984–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mellado-Gil, J.M.; Fuente-Martín, E.; Lorenzo, P.I.; Cobo-Vuilleumier, N.; López-Noriega, L.; Martín-Montalvo, A.; Gómez, I.G.H.; Ceballos-Chávez, M.; Gómez-Jaramillo, L.; Campos-Caro, A.; et al. The type 2 diabetes-associated HMG20A gene is mandatory for islet β cell functional maturity. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.; Grapin-Botton, A. The importance of REST for development and function of β cells. Front. Cell Dev. Biol. 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Gilbert, E.R.; Liu, D. Regulation of insulin synthesis and secretion and pancreatic β-cell dysfunction in diabetes. Curr. Diabetes Rev. 2013, 9, 25–53. [Google Scholar] [CrossRef] [PubMed]
- Kahn, C.R. The Molecular Mechanism of Insulin Action. Annu. Rev. Med. 1985, 36, 429–451. [Google Scholar] [CrossRef] [PubMed]
- Guilherme, A.; Virbasius, J.V.; Puri, V.; Czech, M.P. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat. Rev. Mol. Cell Biol. 2008, 9, 367–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bisbis, S.; Bailbe, D.; Tormo, M.A.; Picarel-Blanchot, F.; Derouet, M.; Simon, J.; Portha, B. Insulin resistance in the GK rat: Decreased receptor number but normal kinase activity in liver. Am. J. Physiol. Endocrinol. Metab. 1993, 265, E807–E813. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Tsai, L.T.; Zhou, Y.; Evertts, A.; Xu, S.; Griffin, M.J.; Issner, R.; Whitton, H.J.; Garcia, B.A.; Epstein, C.B.; et al. Identification of nuclear hormone receptor pathways causing insulin resistance by transcriptional and epigenomic analysis. Nat. Cell Biol. 2015, 17, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Akerman, I.; Tu, Z.; Beucher, A.; Rolando, D.M.Y.; Sauty-Colace, C.; Benazra, M.; Nakic, N.; Yang, J.; Wang, H.; Pasquali, L.; et al. Human pancreatic β cell lncRNAs control cell-specific regulatory networks. Cell Metab. 2017, 25, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Deng, Y.; Feng, Y.; Long, D.; Ma, K.; Wang, X.; Zhao, M.; Lu, L.; Lu, Q. Epigenetic regulation in B-cell maturation and its dysregulation in autoimmunity. Cell. Mol. Immunol. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, D.S.; Teschendorff, A.E.; Dang, M.A.N.; Lowe, R.; Hawa, M.I.; Ecker, S.; Beyan, H.; Cunningham, S.; Fouts, A.R.; Ramelius, A.; et al. Increased DNA methylation variability in type 1 diabetes across three immune effector cell types. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Huang, G.; Wang, Z.; Luo, S.; Zheng, P.; Zhou, Z. Epigenetic regulation of Toll-like receptors and its roles in type 1 diabetes. J. Mol. Med. 2018, 96, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Zurawek, M.; Dzikiewicz-Krawczyk, A.; Izykowska, K.; Ziolkowska-Suchanek, I.; Skowronska, B.; Czainska, M.; Podralska, M.; Fichna, P.; Przybylski, G.; Fichna, M.; et al. miR-487a-3p upregulated in type 1 diabetes targets CTLA4 and FOXO3. Diabetes Res. Clin. Pract. 2018, 142, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Lu, Q. Genetic and epigenetic influences on the loss of tolerance in autoimmunity. Cell. Mol. Immunol. 2018, 15, 575–585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontcuberta-PiSunyer, M.; Cervantes, S.; Miguel, E.; Mora-Castilla, S.; Laurent, L.C.; Raya, A.; Gomis, R.; Gasa, R. Modulation of the endocrine transcriptional program by targeting histone modifiers of the H3K27me3 mark. Biochim. Biophys. Acta 2018, 1861, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Gu, X.; Su, I.H.; Bottino, R.; Contreras, J.L.; Tarakhovsky, A.; Kim, S.K. Polycomb protein Ezh2 regulates pancreatic β-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes Dev. 2009, 23, 975–985. [Google Scholar] [CrossRef] [PubMed]
- Courtney, M.; Gjernes, E.; Druelle, N.; Ravaud, C.; Vieria, A.; Ben-Othman, N.; Pfeifer, A.; Avolio, F.; Leuckx, G.; Lacas-Gervais, S.; et al. The inactivation of Arx in pancreatic α-Cells triggers their neogenesis and conversion into functional β-like cells. PLoS Genet. 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, S.; Georgia, S.; Tschen, S.I.; Fan, G.; Bushan, A. Pancreatic β cell identity is maintained by DNA methylation-mediated repression of Arx. Dev. Cell 2011, 20, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Chakravarthy, H.; Gu, X.; Enge, M.; Dai, X.; Wang, Y.; Damond, N.; Downie, C.; Liu, K.; Wang, J.; Xing, Y.; et al. Converting adult pancreatic islet α cells into β cells by targeting both Dnmt1 and Arx. Cell Metab. 2017, 25, 622–634. [Google Scholar] [CrossRef] [PubMed]
- Chai, C.; Leong, K.W. Biomaterials approach to expand and direct differentiation of stem cells. Mol. Ther. 2007, 15, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Pennarossa, G.; Santoro, R.; Manzoni, E.F.M.; Pesce, M.; Gandolfi, F.; Brevini, T.A.L. Epigenetic erasing and pancreatic differentiation of dermal fibroblasts into insulin-producing cells are boosted by the use of low-stiffness substrate. Stem Cell Rev. 2018, 14, 398–411. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, A.M.; Lakey, J.R.; Ryan, E.A.; Kortbutt, G.S.; Toth, E.; Warnock, G.L.; Kneteman, N.M.; Rajotte, R.V. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N. Engl. J. Med. 2000, 343, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Ryan, E.A.; Oaty, B.W.; Senior, P.A.; Bigam, D.; Alfadhli, E.; Kneteman, N.M.; Lakey, J.R.; Shapiro, A.M. Five-year follow-up after clinical islet transplantation. Diabetes 2005, 54, 2060–2069. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Brown, J.; Kanarek, A.; Rajagopal, J.; Melton, D.A. In vivo reprogramming of adult pancreatic exocrine cells to β-cells. Nature 2008, 455, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Mack, D.L.; Williams, J.K.; Mirmalek-Sani, S.H.; Moorefield, E.; Chun, S.Y.; Wang, J.; Lorenzetti, D.; Furth, M.; Atala, A.; et al. Genetic modification of primate amniotic fluid-derived stem cells produces pancreatic progenitor cells in vitro. Cells Tissues Organs 2013, 197, 269–282. [Google Scholar] [CrossRef] [PubMed]
- Pagliuca, F.W.; Millman, J.R.; Gürtler, M.; Segel, M.; Van Dervort, A.; Ryu, J.H.; Peterson, Q.P.; Greiner, D.; Melton, D.A. Generation of functional human pancreatic β cells in vitro. Cell 2014, 159, 428–439. [Google Scholar] [CrossRef] [PubMed]
- Cito, M.; Pellegrini, S.; Piemonti, L.; Sordi, V. The potential and challenges of alternative sources of β cells for the cure of type 1 diabetes. Endocr. Connect. 2018, 7, R114–R125. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematics of the potential epigenetic mechanisms involved in pancreatic β-cell development. Previous research has shown that the transition from endoderm cells to pancreatic progenitor cells involves the activation of the Enhancer of zeste homolog 2 (Ezh2) histone methyltransferase (1). Subsequently, Neurogenin 3 (Ngn3)low pancreatic endocrine progenitor cells transition to a Ngn3high cell state and are thereby committed to the endocrine cell lineage (2). The histone demethylase, Jumonji domain-containing protein 3 (Jmjd3), was found to be involved in this process. A high level of Ngn3 expression leads to the activation Ngn3 downstream targets, such as Neurogenic differentiation factor 1 (NeuroD1), Regulatory factor X, 6 (Rfx6), and Insulinoma-associated 1 (Insm1) (3). The resulting immature β-cells further develop through the silencing of the immaturity-associated genes (via the addition of H3K27me3 marks) and the de novo activation of maturity-associated genes (with the loss of H3K27me3 marks) (4). The epigenetic changes associated with this stage in development are regulated by the polycomb repressor complex (EeD-PRC2).
Figure 1.
Schematics of the potential epigenetic mechanisms involved in pancreatic β-cell development. Previous research has shown that the transition from endoderm cells to pancreatic progenitor cells involves the activation of the Enhancer of zeste homolog 2 (Ezh2) histone methyltransferase (1). Subsequently, Neurogenin 3 (Ngn3)low pancreatic endocrine progenitor cells transition to a Ngn3high cell state and are thereby committed to the endocrine cell lineage (2). The histone demethylase, Jumonji domain-containing protein 3 (Jmjd3), was found to be involved in this process. A high level of Ngn3 expression leads to the activation Ngn3 downstream targets, such as Neurogenic differentiation factor 1 (NeuroD1), Regulatory factor X, 6 (Rfx6), and Insulinoma-associated 1 (Insm1) (3). The resulting immature β-cells further develop through the silencing of the immaturity-associated genes (via the addition of H3K27me3 marks) and the de novo activation of maturity-associated genes (with the loss of H3K27me3 marks) (4). The epigenetic changes associated with this stage in development are regulated by the polycomb repressor complex (EeD-PRC2).
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Balaji, S.; Napolitano, T.; Silvano, S.; Friano, M.E.; Garrido-Utrilla, A.; Atlija, J.; Collombat, P. Epigenetic Control of Pancreatic Regeneration in Diabetes. Genes 2018, 9, 448. https://doi.org/10.3390/genes9090448
AMA Style
Balaji S, Napolitano T, Silvano S, Friano ME, Garrido-Utrilla A, Atlija J, Collombat P. Epigenetic Control of Pancreatic Regeneration in Diabetes. Genes. 2018; 9(9):448. https://doi.org/10.3390/genes9090448
Chicago/Turabian StyleBalaji, Shruti, Tiziana Napolitano, Serena Silvano, Marika Elsa Friano, Anna Garrido-Utrilla, Josipa Atlija, and Patrick Collombat. 2018. "Epigenetic Control of Pancreatic Regeneration in Diabetes" Genes 9, no. 9: 448. https://doi.org/10.3390/genes9090448
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.