Gene Therapy for Chronic HBV—Can We Eliminate cccDNA?


Wits/SAMRC Antiviral Gene Therapy Research Unit, School of Pathology, Faculty of Health Sciences, University of the Witwatersrand, Private Bag 3, Johannesburg, WITS 2050, South Africa
*
Author to whom correspondence should be addressed.
Genes 2018, 9(4), 207; https://doi.org/10.3390/genes9040207
Submission received: 15 March 2018
/
Revised: 5 April 2018
/
Accepted: 9 April 2018
/
Published: 12 April 2018
(This article belongs to the Special Issue Hepatitis B Virus Infection: An Update on Epidemiology, Diagnosis, Treatment and Prevention)
Abstract
:Chronic infection with the hepatitis B virus (HBV) is a global health concern and accounts for approximately 1 million deaths annually. Amongst other limitations of current anti-HBV treatment, failure to eliminate the viral covalently closed circular DNA (cccDNA) and emergence of resistance remain the most worrisome. Viral rebound from latent episomal cccDNA reservoirs occurs following cessation of therapy, patient non-compliance, or the development of escape mutants. Simultaneous viral co-infections, such as by HIV-1, further complicate therapeutic interventions. These challenges have prompted development of novel targeted hepatitis B therapies. Given the ease with which highly specific and potent nucleic acid therapeutics can be rationally designed, gene therapy has generated interest for antiviral application. Gene therapy strategies developed for HBV include gene silencing by harnessing RNA interference, transcriptional inhibition through epigenetic modification of target DNA, genome editing by designer nucleases, and immune modulation with cytokines. DNA-binding domains and effectors based on the zinc finger (ZF), transcription activator-like effector (TALE), and clustered regularly interspaced short palindromic repeat (CRISPR) systems are remarkably well suited to targeting episomal cccDNA. This review discusses recent developments and challenges facing the field of anti-HBV gene therapy, its potential curative significance and the progress towards clinical application.
1. Introduction
Viral hepatitis accounts for up to 1.34 million deaths per year and remains the major cause of morbidity and mortality from cirrhosis and hepatocellular carcinoma [1]. Hepatitis B virus (HBV) infections contribute significantly to the global health problem and, with an estimated 257 million people chronically infected, it is a major health priority. More than 50 years have passed since the discovery of the Australia antigen [2], and while valuable progress has been made in vaccine and antiviral development, there is still no reliable cure for HBV infection. Worldwide prophylactic vaccination programs have reduced the prevalence of HBV in children under the age of five, but inadequate coverage in hyper-endemic African countries means that prevalence remains high in some countries [1]. Management of chronic HBV infection involves use of immune modulators or direct-acting antivirals, in the form of interferons or nucleoside/nucleotide analogs (NAs). The rationale for combining these therapies is to manage both immune dysregulation as well as viral pathogenesis. Currently, approved therapeutics include interferon α, pegylated interferon α, lamivudine, telbivudine, adenovir dipivoxil, entecavir, tenofovir disoproxil fumarate, and tenofovir alafenamide. Recommendations for first-line monotherapies are pegylated interferon α and the NAs with high barriers to resistance, which are entecavir, tenofovir disoproxil fumarate, and tenofovir alafenamide [3,4,5]. Guidelines for administration of combination therapies sometimes vary as a result of conflicting opinions about long-term efficacy, influence of patient selection, and whether simultaneous or sequential administration is favored (reviewed by [6,7]).
The critical limitation of licensed therapeutics is their inability to reliably achieve a virological cure [3]. While NAs inhibit posttranscriptional stages of viral replication, they do not target the stable episomal covalently closed circular DNA (cccDNA). This key HBV replication intermediate forms a minichromosome in the nucleus of hepatocytes [8,9,10], and may undergo epigenetic modifications [11,12,13]. The hepatitis B X protein (HBx) plays a role in stabilizing cccDNA by inactivating components of the structural maintenance of chromosomes (SMC) complex [14]. The cccDNA associates with host transcription factors and viral proteins to enable viral gene expression and replication. Associations with factors regulating methylation or heterochromatin formation may also render cccDNA inactive, and lead to persistent latent or occult HBV infections. Cessation or interruption of antiviral therapy, development of viral escape mutants, or immunodeficiency could all lead to reactivation of HBV replication, highlighting the need for cccDNA-specific therapies. This review focuses on recent advances aimed at generating HBV-targeting designer nucleases, epigenetic modifications to the viral DNA and nucleic acid-based immune modulation to treat chronic HBV infection.
2. Hepatitis B Virus Therapies Under Development
Several novel anti-HBV therapeutics are in preclinical development or early clinical trial (reviewed by [15,16]). Most candidate drugs are small molecule drugs designed to impede various stages of HBV replication. Affordable next-generation NAs, which inhibit the viral polymerase with reduced toxicity and higher barriers to HBV resistance are currently the preferred first-line of therapy. Other direct-acting antivirals include HBV core protein allosteric modulators [17,18], HBV surface antigen (HBsAg) release inhibitors [19,20] and nucleic acid polymers which also inhibit viral entry [21]. With the discovery that the sodium taurocholate co-transporting polypeptide (NTCP) facilitates HBV entry into hepatocytes [22], peptide inhibitors such as Myrcludex-B (NCT02881008 and NCT02888106) are also being developed for therapeutic application [23]. Another popular host-related strategy has been to recondition the immune system using interferons, cytokines, and peptides as immune modulators which are discussed in more detail below (Section 3.3).
Use of gene therapy to disable HBV replication has shown promise and generated considerable interest. Different strategies that have been employed include HBV-specific gene silencing, gene editing, epigenome modification, and nucleic acid-based vaccination. Harnessing the RNA interference (RNAi) pathway is a well-established strategy that has been used extensively to silence genes of HBV. RNAi is an endogenous gene regulatory pathway that can be reprogrammed by exogenous RNA molecules. Feasibility of using RNAi to treat HBV has been established and extensively reviewed elsewhere [24,25] and will not be covered in detail here. Expressed or synthetic antiviral sequences may mimic primary microRNAs, precursor microRNAs, or mature microRNAs (miRs) [26,27,28,29]. Because of easier large-scale production, dose control and delivery to the cytoplasmic site of action, development of synthetic short interfering RNAs (siRNAs) has advanced rapidly. These simulate mature miRs, and are now in Phase 2 clinical trials. Anti-HBV siRNA formulations with impressive antiviral activity include ARC-520 (NCT02065336), ALN-HBV (NCT02826018), and ARB-001467 (NCT02631096). However, since their effect is transient, repeated administration of siRNAs will be required for long term anti-HBV efficacy. To increase durability of an anti-HBV effect expressed HBV-targeting sequences have been developed. For example, adeno-associated viral vectors (AAVs) have been used to deliver cassettes that express HBV-targeting primary microRNA mimics [30]. Safe and sustained inhibition of HBV replication in HBV transgenic mice indicates that this approach has potential for clinical translation. Strategies targeting host factors have also shown significant promise in reducing cccDNA levels. RNAi-mediated gene silencing of tyrosyl-DNA-phosphodiesterase 2, a DNA repair enzyme thought to mediate polymerase release from the relaxed circular DNA, delays its conversion to cccDNA in HepG2 cells [31]. A recent study has also demonstrated that silencing the expression of pre-mRNA processing factor 31, an HBx-interacting partner, reduces cccDNA levels in HepAD38 cells [32].
3. Gene-Based Therapies to Target Covalently Closed Circular DNA
While the HBV therapeutic landscape is vast, few approaches are being developed to disable cccDNA directly. Targeted mutagenesis by sequence-specific RNA-guided nucleases (RGNs) and proteins has thus generated considerable interest, as the technology potentially provides the means to cure HBV infection by permanently disabling cccDNA [33,34].
3.1. Designer Nucleases
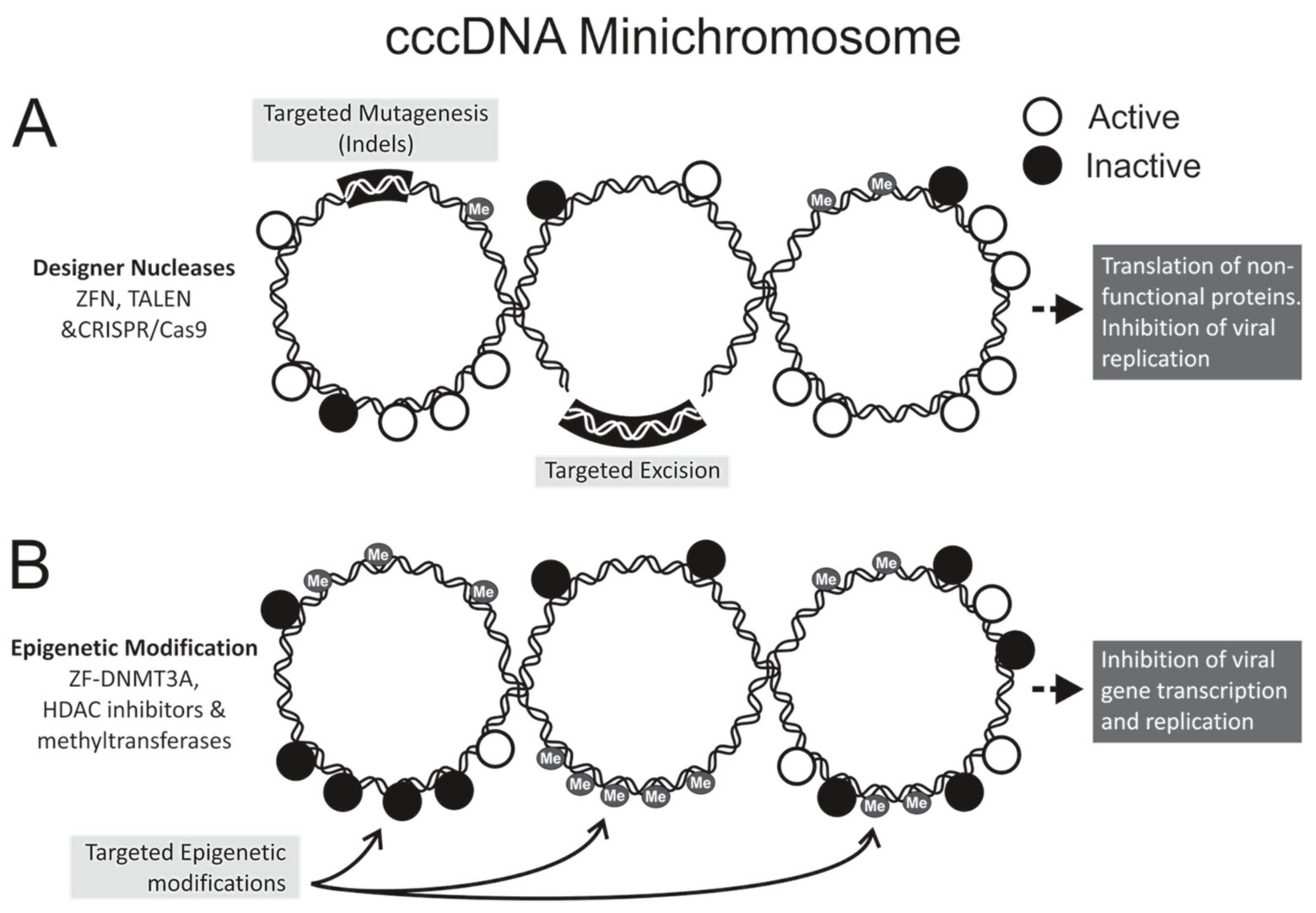
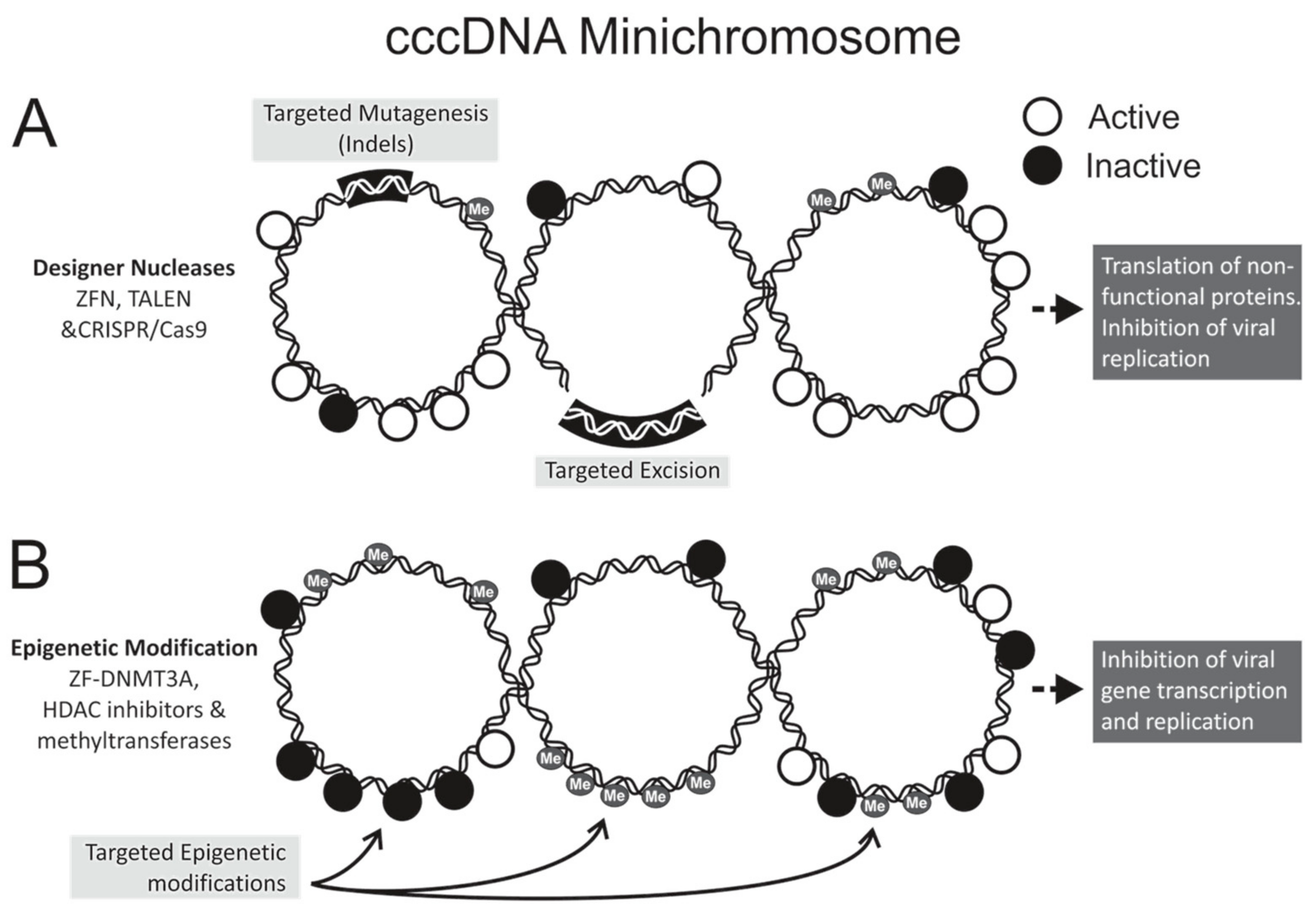
Designer nucleases have dominated the anti-HBV gene editing field, with zinc finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and clustered regularly interspaced short palindromic repeats (CRISPR) with CRISPR-associated (Cas) systems all showing antiviral efficacy (Table 1). Nucleases act by inducing double stranded breaks at a pre-defined target site within the HBV genome (Figure 1A). By exploiting the host cells’ error-prone non-homologous end joining repair machinery, targeted mutagenesis is realized. HBV cccDNA is a primary candidate for nuclease gene editing, owing to its episomal minichromosome configuration and limited sequence plasticity. The compact viral genome and overlapping reading frames restrict development of escape mutants, despite the low fidelity of the viral reverse transcriptase [35]. Insertions and deletions (Indels) within the viral genome may give rise to aberrant or truncated proteins, which in turn interfere with viral replication.
Zinc fingers (ZFs) are abundant multifunctional mammalian proteins that occur naturally as transcription factors. By exploiting the specific targeting of nucleotide triplets of single ZFs, these proteins may be engineered to form arrays with defined DNA binding properties [36]. Addition of a FokI effector domain at the C-terminus of a ZF yields an engineered nuclease that cuts one strand of a DNA duplex. ZFN dimers, with cognates on complementary strands of the DNA duplex, may thus be used to create double stranded breaks [37]. Anti-HBV ZFNs were first described in a proof-of-concept experiment where 36% of plasmid-derived viral sequences were disrupted in a cell culture model of viral replication [38]. More recently Weber et al. reported on delivery of self-complementary adeno-associated viral vectors (scAAVs) encoding ZFNs targeting the polymerase/X (1), polymerase/core (2) and polymerase (3) viral open reading frames (ORFs) [39]. Using inducible liver-derived HepAD38 cells to mimic natural HBV infection [40], ZFN pairs 1, 2 and 3 cleaved 9.8, 34, and 28% of the viral targets respectively. These results were confirmed using single molecule real time sequencing which additionally identified potential off targets, albeit at low frequencies. Interestingly ZFN pair 2 resulted in the highest detectable targeted disruption, yet it was also found to be cytotoxic. Only ZFN pair 3 showed antiviral efficacy but cleavage of cccDNA could not be verified.
As with ZFNs, TALENs are dimeric engineered nucleases that comprise a DNA-binding protein fused to an endonuclease domain. The transcriptional activator-like effector (TALE) is derived from the Xanthomonas bacteria where individual repeat domains comprising 33–35 amino acids recognize a single base pair [41,42]. The nucleotide binding specificity of these repeats is predetermined by repeat-variable di-residues (RVDs) located at amino acid positions 12 and 13 [43,44]. Linking multiple repeats in a defined order generates engineered TALEs with highly specific DNA binding properties. Unlike with ZFNs, nucleotide binding affinity of each monomer comprising the DNA binding domain is not influenced by a neighboring unit. Our group first described antiviral efficacy of engineered TALENs on HBV cccDNA in cultured cells and inhibition of viral replication in a murine model [45]. TALEN dimers designed to bind within the surface (S) and core (C) ORFs showed optimal cleavage activity in the HepG2.2.15 cell line without measurable cytotoxicity. Importantly, cccDNA targeted disruption frequencies of 35% and 12% were achieved with the S and C TALEN pairs respectively. Using the murine hydrodynamic injection (HDI) model, co-administration of HBV replication-competent plasmids with TALEN-encoding sequences demonstrated in vivo antiviral efficacy of the nucleases and there was no evidence of liver toxicity. A significant and substantial reduction in serum concentrations of HBsAg and circulating viral particle equivalents was observed in TALEN-treated mice, and targeted mutagenesis of up to 87% was achieved. Deep sequencing verified large deletions in viral DNA, which were likely to inactivate HBV replication. A subsequent study by Chen et al. confirmed the cccDNA-specific antiviral potential of TALENs designed to target conserved regions within the polymerase (RNaseH sequence) and C ORFs [46]. This was achieved in liver-derived Huh7 cells transfected with linear viral sequences that generate cccDNA and recapitulate HBV replication [11,12]. A reduction in viral protein expression was observed across genotypes A, B, C, and D, emphasizing the applicability of anti-HBV TALENs to a variety of viral isolates. Moreover, a synergistic effect was shown when combining IFN-α with core TALENs, to result in an approximately 60% reduction in copies of cccDNA. In another study, co-transfection of linear donor sequences encoding trimeric gene silencers significantly augmented anti-HBV efficacy of S or C TALEN pairs in HepG2.2.15 cells [47]. This approach exploited the homology directed repair pathway to introduce the artificial primary microRNA-encoding sequences directly into viral DNAs. In doing so, the viral genome may be permanently disrupted and after homologous recombination, HBV DNA transcribes an antiviral sequence from its own rearranged genome.
Use of RGNs is now the most popular method of inactivating HBV gene expression. This bacterial CRISPR/Cas9 system relies on an RNA guided DNA binding domain with associated Cas9 endonuclease [48]. These RGNs typically comprise a CRISPR RNA (crRNA) with sequences that are complementary to a pre-defined DNA target sequence and a trans-activating crRNA (tracrRNA). Annealing of the RNA to its cognate enables Cas9-mediated double-stranded target DNA cleavage. Fusing crRNA and tracrRNA to form a single guide RNA (gRNA) has further simplified the system, which has made RGNs the easiest nucleases to engineer. Since first reported by Lin et al. in 2014 [46], more than 16 publications have described anti-HBV efficacy of RGNs [49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65] (Table 1). Targeted mutagenesis of cccDNA in mammalian cell cultures was first demonstrated by Seeger and Sohn [49], who designed CRISPR/Cas9 constructs to bind the conserved enhancer II/core promoter and precore ORFs. Subsequent studies confirmed that single or multiple gRNAs spanning the entire HBV genome cleaved cccDNA to cause indel formation and resultant reduced viral protein expression [50,51,52,54,55,56,59,62,63,64,65]. However, methods used to detect mutagenic events vary greatly between individual studies, which makes it difficult to compare efficiency of different gRNA constructs directly. Nevertheless, as with all designer nucleases, targeting conserved regions is advantageous as antiviral efficacy is achieved across different genotypes [53,55]. A second study by Seeger and Sohn used next generation sequencing to map CRISPR/Cas9-induced cccDNA mutations [58]. The majority of indels were identified as small deletions when extremely high targeted cleavage (up to 90%) was achieved. Although off-target cleavage may be frequently associated with CRISPR/Cas9 action [66,67,68], data on the potential of HBV RGNs to create nonspecific mutations is scarce. Using multiple gRNAs to improve antiviral efficacy [46] or to excise integrated viral DNA fragments (Figure 1A) [62] may exacerbate this issue. As expected off-target cleavage events were detected by deep sequencing when multiple gRNAs were used [57,62]. However, other studies report no detectable genotoxic activity [54,61,63], which may be a result of differences between gRNA design and properties of the viral target sequences. Replacing the Cas9 endonuclease with engineered nickases may improve specificity of HBV RGNs. With this configuration, gRNA and Cas9 nickases comprising heterodimers are required to effect double stranded DNA cleavage [51,57]. Another challenge with the CRISPR/Cas9 system relates to delivery of the combined gRNA and the large Cas9 endonuclease. Sequences encoding these two components exceed the transgene capacity of the popular AAVs. A recent publication by Scott et al. demonstrated that the smaller Cas9 derived from Staphylococcus aureus (Sa), together with expression cassettes encoding short gRNAs targeting the HBV surface ORF, could be packaged into single stranded AAVs (ssAAV) [63]. Efficient delivery and expression of the RGN-encoding ssAAVs in HepG2.2.15 and hNTCP-HepG2 cells resulted in decreased viral replication, target specific cccDNA cleavage, and reduced cccDNA copy numbers. Analysis of sequences at predicted off target sites did not reveal non-specific mutagenesis.
3.2. Epigenetic Gene Silencing
Natural epigenetic modification of DNA may silence gene expression and is a host defence mechanism against expression of viral genes. Epigenetic modification is a stable and heritable gene regulatory mechanism found in many different organisms. It involves chemical alteration of DNA or associated proteins without changing the encoded genetic information. It is involved in typical cell development and multiple normal or abnormal cellular responses (reviewed by [69]). There is accumulating evidence to indicate that epigenetic machinery controls transcription of HBV cccDNA (Figure 1B), and contributes to the outcome of chronic HBV infection [70,71,72]. Use of exogenous epigenetic modifiers has thus attracted interest as a mode of disabling cccDNA. Epigenetic modifications include histone acetylation or deacetylation, histone methylation or demethylation, cccDNA methylation, and cccDNA minichromosome acetylation (reviewed by [73,74]). In cooperation with viral factors such as HBx and the core antigen (HBc), the major epigenetic modifiers of HBV DNA include histone acetyltransferases/deacetylases (HATs/HDACs) [12], lysine methyltransferases [75], protein arginine methyltransferases [70,76], and DNA methyltransferases (DNMTs) [77].
Hypoacetylation of cccDNA-bound histone 3/4 results in low HBV viremia in hepatitis B patients, whereas hyperacetylation increases HBV replication [11]. HDAC inhibitors have been shown to suppress cccDNA transcription in duck hepatitis B virus (DHBV) [82,83]. Methylation of arginine 3 on cccDNA-bound histone 4 prevents RNA polymerase binding and transcription in an arginine methyltransferase 5 and HBc-dependent manner [70,76]. Transcriptional inhibition by methylation can be direct, by blocking binding of transcriptional factors or RNA-polymerase loading, as well as indirect, by recruiting histone-modifying and chromatin remodelling complexes to the methylated DNA (reviewed by [69]). Epigenetic gene silencing may facilitate reduction of viral reservoirs through normal hepatocyte turnover and prevention of replenishment of the cccDNA pool. Furthermore, epigenetic modifications may accelerate cccDNA decay, as was shown following IFN-α treatment of DHBV infections [82]. HBV cccDNA contains three CpG islands: island I, island II and island III [84,85]. Island I overlaps the start site of the S gene, island II encompasses enhancer I, the HBx gene promoter and the core promoter, whereas island III harbors the Sp1 promoter and start codon of the polymerase gene. Methylation of island I is rare and variable across genotypes, whereas methylation of island II and III is more conserved. Island III methylation is associated with reduced serum HBsAg concentrations in chronically infected patients and correlates with hepatocarcinogenesis. Island II methylation is associated with reduced pregenomic RNA (pgRNA) transcription, viral replication and viremia [72,86]. In vitro studies showed that several DNMTs are upregulated in response to HBV infection, leading to viral DNA methylation, decreased HBV gene expression, and diminished viral replication [77,87]. Importantly, similar reduction in viral gene expression and viremia was observed in human tissue samples with methylated HBV DNA [85,88,89].
Despite the importance of HBV DNA epigenetic modification for disease progression, evidence supporting the feasibility of using epigenetic modifiers against HBV is currently limited. Few studies have taken advantage of the sequence-specific binding domains of designer nucleases for their repurposing as epigenetic silencers. By replacing a nuclease domain of a designer nuclease with an epigenetic modulator, ZFs, TALEs, and CRISPR/Cas have usually been modified for epigenetic editing of endogenous genes (reviewed by [90]). However, this strategy is theoretically applicable to targeted epigenetic modification of HBV DNA. One study reported successful epigenetic editing of HBV DNA after fusing the catalytic domain of DNMT3a to a ZF that targeted the HBx promoter sequence [79]. The engineered sequence caused methylation of targeted CpG sites, downregulation of viral mRNAs and proteins, and a decrease in viral replication in cell culture and in HBV transgenic mice. Repressors based on the Krüppel associated box (KRAB) domain have also been investigated as ZF [80] and TALE [81] fusions. Although these KRAB-repressors inhibit viral replication, verification of heterochromatin formation, which is important to achieve permanent gene silencing, remains to be confirmed. Studies demonstrating antiviral efficacy and sustainability of epigenetic editors on HBV replication are preliminary. However, investigations aimed at developing the approach to treat other chronic viral infections, such as HIV [91], are more advanced. Moreover, Food and Drug Administration (FDA) approval of HDAC inhibitors for cancer treatment [92] supports the notion that epigenetic editing has clinical potential for treatment of HBV infection.
3.3. Immune Modulation for Covalently Closed Circular DNA Attenuation
Administration of recombinant IFN-α or its pegylated derivatives remains the only immunomodulatory drugs licensed for management of chronic HBV infection. IFN-α is therefore the only licensed anti-HBV therapy capable of eliminating cccDNA. Immunomodulation has been shown to augment innate and adaptive immune responses against the virus. Stimulating T-cell-mediated elimination of infected hepatocytes, and indirectly cccDNA, is thus a promising immune-based strategy to achieve functional cure from HBV infection. However, the success rate of IFN therapy remains low and side effects represent a major shortcoming of this therapy. Gene therapy to enable durable expression of immune modulators may be useful to attenuate cccDNA.
Expression of IFN-α-encoding sequences in the liver has been explored as a means of improving anti-HBV efficacy and reducing side effects of conventional IFN-α treatment. One of the first studies explored expression of murine IFN-α2 under transcriptional control of the liver-specific transthyretin promoter [93]. The IFN-α expression construct was delivered to the livers of mice using a helper-dependent adenovirus (HDAd), and the interferon response genes 2′,5′-oligoadenylate synthetase and tumor necrosis factor α (TNF-α) were effectively induced. As a surrogate for assessing anti-HBV potential of this system the authors challenged mice with a murine coronavirus, MHV-2. Mice pre-treated with the HDAd carrying IFN-α were protected from infection and did not exhibit any toxic side effects. Fiedler et al. assessed usefulness of a gene therapy-based approach to express IFN-α or IFN-γ in a woodchuck hepatitis virus (WHV) model of HBV infection [94]. Sequences encoding woodchuck IFN-α or IFN-γ (wIFN-α or wIFN-γ) were delivered to woodchucks with an HDAd. In animals chronically infected with WHV, viral replicative intermediates in the liver and serum were considerably diminished. This was a significant finding as WHV maintains very high viral loads in its host, much higher than in chronic HBV carriers. Furthermore, no obvious side effects were observed and wIFN-α expression lasted for at least a year. In contrast to the promising results achieved with wIFN-α, expression of wIFN-γ did not have a significant effect on WHV replication. This differs from the results of Dumortier et al. who demonstrated that IFN-γ expression was able to limit HBV replication in mice [95].
Subsequent work assessed utility of AAV vector for delivery of interferon expression cassettes. Berraondo et al. tested intrahepatic and intramuscular expression of AAV8-delivered wIFN-α5 following administration to woodchucks chronically infected with WHV [96]. Interferon was readily expressed at both sites of delivery, but expression in the liver was superior. Furthermore, only expression of interferon from the liver was associated with an antiviral response. Although the higher vector dose had a more pronounced antiviral effect, it was also associated with greater toxicity. The woodchuck with the greatest decrease in viremia exhibited such severe side effects that it required euthanasia prior to completion of the study. Another limitation of the approach was the transient nature of the antiviral effect, which was attributed to immune-mediated clearance of AAV-infected hepatocytes. To improve their delivery system Berraondo et al. developed a modified IFN-α which was fused to sequences encoding apolipoprotein A (InterApo) [97]. Initial assessment demonstrated that InterApo exhibited an improved safety profile while maintaining antiviral efficacy in HBV transgenic mice. In the woodchucks chronically infected with WHV, AAV-delivered InterApo was well-tolerated but showed little efficacy. This was attributed to the high WHV load in woodchucks with chronic hepatitis. Pre-treatment of woodchucks with the NA entecavir with subsequent administration of AAV-InterApo facilitated immune-mediated clearance. However, the antiviral effect was transient and viral rebound to pre-treatment levels eventually occurred.
Further evaluation of gene therapy-based immune modulation explored antiviral effects of sequences encoding interleukins. Crettaz et al. assessed efficacy of HDAd-delivered interleukin-12 (IL-12) in the WHV model [98]. The authors used a murine IL-12 sequence, under transcriptional control of an inducible promoter, and delivered the cassette directly to the liver with the viral vector. Interestingly, woodchucks with a viral load lower than 1010 viral genome equivalents per ml of serum responded well to treatment, whereas those with higher viral loads did not. Extensive characterization revealed decreased intrahepatic viral DNA and RNA levels. Liver inflammation was reduced, and woodchuck hepatitis e and surface antigens were cleared from the serum. Significantly, disappearance of intrahepatic core antigen was attributed to a T-cell response against the virus, which was induced by IFN-γ. Importantly, the treatment was shown to be well-tolerated. The same group subsequently evaluated efficacy of IL-12 expressed from a Semliki Forest Virus vector [99]. While the aim was to assess the anti-tumor effects of the constructs in WHV-induced HCC, the authors also showed strong antiviral responses resulting from intrahepatic IL-12 expression. As before, the anti-tumor and antiviral effects were attributed to the induction of T-cell responses. The potential of using cytokines to treat HBV infection was further highlighted by a recent study that evaluated efficacy of dual expression of IFN-α and IL-15 to counter HBV replication in transgenic mice [100]. Co-administration of an AAV encoding the IFN-α gene and an AAV carrying the IL-15 sequence resulted in near complete clearance of intrahepatic HBc and viral DNA replication intermediates. More importantly, IFN-α and IL-15 expression were shown to induce an antibody response and a functional antiviral CD8+ T-cell response. The authors further assessed their combination strategy using recombinant IFN-α and IL-15 on samples from patients with chronic HBV infection. After stimulation of peripheral blood mononuclear cells from these patients with an HBc peptide in the presence of IFN-α and IL-15, effector function was restored to HBV specific CD8+ T-cells.
Reconstituting the cytotoxic T-cell response against HBV holds great promise as a strategy for the immune-mediated clearance of the virus in chronic carriers. While engineered T-cells with chimeric T-cell receptors against HBV antigens have been developed, investigations are yet to progress beyond preclinical development of the technology [101,102,103,104,105]. DNA vaccines have the potential to induce strong antibody and T-cell-mediated immune responses and thereby clear chronic HBV infections (reviewed by [106]). While studies exploring this strategy are few, inducing strong antiviral immunity with therapeutic vaccines in conjunction with cytokine-mediated stimulation of an immune response undoubtedly has potential for management of chronic hepatitis B.
4. Discussion/Perspective
Realization of a functional or complete cure for chronic HBV infections requires innovative therapeutic approaches aimed at disabling and eliminating the persistent episomal cccDNA. Drugs that act directly on the viral genome, such as designer nucleases and epigenetic modifiers, have the potential to disable viral replication permanently. Restoration of the anti-HBV immune response may also facilitate clearance of infected hepatocytes and thus diminish or eradicate the cccDNA pool. A combination of gene-based immune and cccDNA-targeting gene therapy may provide the means to achieve this goal.
Other challenges, which are broadly associated with implementing gene-based therapies [107], also need to be met for the approach to be successful against HBV. Efficient liver-specific delivery using viral or non-viral vectors still remains a challenge [108,109]. This will be particularly important if multiple doses of gene therapy are required. Improving DNA-binding specificity, particularly for designer nucleases, and defining off target effects are vital to limit unintended side effects [110]. The lack of suitable chronic HBV infection models also complicates the development of gene therapies for the treatment of the disease. The CRISPR/Cas9 system has been shown to target cccDNA efficiently in primary duck hepatocytes infected with DHBV [65]. Although DHBV infection of ducks provides a useful model of chronic HBV infection, it does not recapitulate all aspects of the condition in humans. Despite the challenges facing clinical translation of gene-based curative therapy for chronic HBV infection, the field is gaining momentum and significant progress seems imminent.
Acknowledgments
Financial assistance of the authors’ laboratory, which was received from the South African Medical Research Council, National Research Foundation, Johnson & Johnson Innovation and the Poliomyelitis Research Foundation is gratefully acknowledged. Costs to cover open access publication were covered by the South African Medical Research Council.
Author Contributions
K.B. was responsible for compiling the manuscript after receiving written input and editing comments from M.B.M., A.E., and P.A.
Conflicts of Interest
The authors declare no conflict of interest.
References
- WHO. Global Hepatitis Report. 2017, pp. 1–83. Available online: http://www.who.int/hepatitis/ publications/global-hepatitis-report2017/en/ (accessed on 6 January 2018).
- Blumberg, B.S.; Alter, H.J.; Visnich, S. A “new” antigen in leukemia sera. JAMA 1965, 191, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Terrault, N.A.; Bzowej, N.H.; Chang, K.M.; Hwang, J.P.; Jonas, M.M.; Murad, M.H. Aasld guidelines for treatment of chronic hepatitis B. Hepatology 2016, 63, 261–283. [Google Scholar] [CrossRef] [PubMed]
- EASL. 2017 clinical practice guidelines on the management of hepatitis B virus infection. J. Hepatol. 2017, 67, 370–398. [Google Scholar]
- Sarin, S.K.; Kumar, M.; Lau, G.K.; Abbas, Z.; Chan, H.L.; Chen, C.J.; Chen, D.S.; Chen, H.L.; Chen, P.J.; Chien, R.N.; et al. Asian-pacific clinical practice guidelines on the management of hepatitis B: A 2015 update. Hepatol. Int. 2016, 10, 1–98. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Ning, Q. Toward a cure for hepatitis B virus infection: Combination therapy involving viral suppression and immune modulation and long-term outcome. J. Infect. Dis. 2017, 216, S771–S777. [Google Scholar] [CrossRef] [PubMed]
- Hagiwara, S.; Nishida, N.; Kudo, M. Antiviral therapy for chronic hepatitis B: Combination of nucleoside analogs and interferon. World J. Hepatol. 2015, 7, 2427–2431. [Google Scholar] [CrossRef] [PubMed]
- Tuttleman, J.S.; Pourcel, C.; Summers, J. Formation of the pool of covalently closed circular viral DNA in hepadnavirus-infected cells. Cell 1986, 47, 451–460. [Google Scholar] [CrossRef]
- Newbold, J.E.; Xin, H.; Tencza, M.; Sherman, G.; Dean, J.; Bowden, S.; Locarnini, S. The covalently closed duplex form of the hepadnavirus genome exists in situ as a heterogeneous population of viral minichromosomes. J. Virol. 1995, 69, 3350–3357. [Google Scholar] [PubMed]
- Bock, C.T.; Schranz, P.; Schroder, C.H.; Zentgraf, H. Hepatitis B virus genome is organized into nucleosomes in the nucleus of the infected cell. Virus Genes 1994, 8, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Pollicino, T.; Belloni, L.; Raffa, G.; Pediconi, N.; Squadrito, G.; Raimondo, G.; Levrero, M. Hepatitis B virus replication is regulated by the acetylation status of hepatitis B virus cccDNA-bound H3 and H4 histones. Gastroenterology 2006, 130, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Belloni, L.; Pollicino, T.; De Nicola, F.; Guerrieri, F.; Raffa, G.; Fanciulli, M.; Raimondo, G.; Levrero, M. Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc. Natl. Acad. Sci. USA 2009, 106, 19975–19979. [Google Scholar] [CrossRef] [PubMed]
- Tropberger, P.; Mercier, A.; Robinson, M.; Zhong, W.; Ganem, D.E.; Holdorf, M. Mapping of histone modifications in episomal HBV cccDNA uncovers an unusual chromatin organization amenable to epigenetic manipulation. Proc. Natl. Acad. Sci. USA 2015, 112, E5715–5724. [Google Scholar] [CrossRef] [PubMed]
- Decorsiere, A.; Mueller, H.; van Breugel, P.C.; Abdul, F.; Gerossier, L.; Beran, R.K.; Livingston, C.M.; Niu, C.; Fletcher, S.P.; Hantz, O.; et al. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature 2016, 531, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Durantel, D.; Zoulim, F. New antiviral targets for innovative treatment concepts for hepatitis B virus and hepatitis delta virus. J. Hepatol. 2016, 64, S117–S131. [Google Scholar] [CrossRef] [PubMed]
- Brahmania, M.; Feld, J.; Arif, A.; Janssen, H.L. New therapeutic agents for chronic hepatitis B. Lancet. Infect. Dis. 2016, 16, e10–e21. [Google Scholar] [CrossRef]
- Cole, A.G. Modulators of HBV capsid assembly as an approach to treating hepatitis B virus infection. Curr. Opin. Pharmacol. 2016, 30, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Zlotnick, A.; Venkatakrishnan, B.; Tan, Z.; Lewellyn, E.; Turner, W.; Francis, S. Core protein: A pleiotropic keystone in the HBV lifecycle. Antiviral Res. 2015, 121, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, J.F.; Keeffe, E.B. Thiazolides: A new class of drugs for the treatment of chronic hepatitis B and C. Future Microbiol. 2008, 3, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Goddard, C.; Clearfield, E.; Mills, C.; Xiao, T.; Guo, H.; Morrey, J.D.; Motter, N.E.; Zhao, K.; Block, T.M.; et al. Design, synthesis, and biological evaluation of triazolo-pyrimidine derivatives as novel inhibitors of hepatitis B virus surface antigen (HBsAg) secretion. J. Med. Chem. 2011, 54, 5660–5670. [Google Scholar] [CrossRef] [PubMed]
- Vaillant, A. Nucleic acid polymers: Broad spectrum antiviral activity, antiviral mechanisms and optimization for the treatment of hepatitis B and hepatitis D infection. Antiviral Res. 2016, 133, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 2012, 3, e05570. [Google Scholar] [CrossRef] [PubMed]
- Volz, T.; Allweiss, L.; Ben, M.M.; Warlich, M.; Lohse, A.W.; Pollok, J.M.; Alexandrov, A.; Urban, S.; Petersen, J.; Lutgehetmann, M.; et al. The entry inhibitor myrcludex-B efficiently blocks intrahepatic virus spreading in humanized mice previously infected with hepatitis B virus. J. Hepatol. 2013, 58, 861–867. [Google Scholar] [CrossRef] [PubMed]
- Maepa, M.B.; Roelofse, I.; Ely, A.; Arbuthnot, P. Progress and prospects of Anti-HBV gene therapy development. Int. J. Mol. Sci 2015, 16, 17589–17610. [Google Scholar] [CrossRef] [PubMed]
- Ivacik, D.; Ely, A.; Arbuthnot, P. Countering hepatitis B virus infection using RNAi: How far are we from the clinic? Rev. Med. Virol. 2011, 21, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Carmona, S.; Ely, A.; Crowther, C.; Moolla, N.; Salazar, F.H.; Marion, P.L.; Ferry, N.; Weinberg, M.S.; Arbuthnot, P. Effective inhibition of HBV replication in vivo by anti-HBx short hairpin RNAs. Mol. Ther. 2006, 13, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Ely, A.; Naidoo, T.; Arbuthnot, P. Efficient silencing of gene expression with modular trimeric Pol II expression cassettes comprising microRNA shuttles. Nucleic Acids Res. 2009, 37, e91. [Google Scholar] [CrossRef] [PubMed]
- Marimani, M.D.; Ely, A.; Buff, M.C.; Bernhardt, S.; Engels, J.W.; Arbuthnot, P. Inhibition of hepatitis B virus replication in cultured cells and in vivo using 2’-O-guanidinopropyl modified siRNAs. Bioorg. Med. Chem. 2013, 21, 6145–6155. [Google Scholar] [CrossRef] [PubMed]
- Grimm, D.; Streetz, K.L.; Jopling, C.L.; Storm, T.A.; Pandey, K.; Davis, C.R.; Marion, P.; Salazar, F.; Kay, M.A. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature 2006, 441, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Maepa, M.B.; Ely, A.; Grayson, W.; Arbuthnot, P. Sustained inhibition of HBV replication in vivo after systemic injection of AAVs encoding artificial antiviral primary microRNAs. Mol. Ther. Nucleic Acids 2017, 7, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Koniger, C.; Wingert, I.; Marsmann, M.; Rosler, C.; Beck, J.; Nassal, M. Involvement of the host DNA-repair enzyme TDP2 in formation of the covalently closed circular DNA persistence reservoir of hepatitis B viruses. Proc. Natl. Acad. Sci. USA 2014, 111, E4244–4253. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, W.; Ogura, N.; Watashi, K.; Wakita, T. Host factor PRPF31 is involved in cccDNA production in HBV-replicating cells. Biochem. Biophys. Res. Commun. 2017, 482, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Ruiz de Galarreta, M.; Lujambio, A. Therapeutic editing of hepatocyte genome in vivo. J. Hepatol. 2017, 67, 818–828. [Google Scholar] [CrossRef] [PubMed]
- Moyo, B.; Bloom, K.; Scott, T.; Ely, A.; Arbuthnot, P. Advances with using CRISPR/Cas-mediated gene editing to treat infections with hepatitis B virus and hepatitis C virus. Virus Res. 2018, 244, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Locarnini, S.; Zoulim, F. Molecular genetics of HBV infection. Antivir. Ther. 2010, 15 (Suppl. S3), 3–14. [Google Scholar] [CrossRef] [PubMed]
- Beerli, R.R.; Barbas, C.F., 3rd. Engineering polydactyl zinc-finger transcription factors. Nat. Biotechnol. 2002, 20, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Bitinaite, J.; Wah, D.A.; Aggarwal, A.K.; Schildkraut, I. Foki dimerization is required for DNA cleavage. Proc. Natl. Acad. Sci. USA 1998, 95, 10570–10575. [Google Scholar] [CrossRef] [PubMed]
- Cradick, T.J.; Keck, K.; Bradshaw, S.; Jamieson, A.C.; McCaffrey, A.P. Zinc-finger nucleases as a novel therapeutic strategy for targeting hepatitis B virus DNAs. Mol. Ther. 2010, 18, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Weber, N.D.; Stone, D.; Sedlak, R.H.; De Silva Feelixge, H.S.; Roychoudhury, P.; Schiffer, J.T.; Aubert, M.; Jerome, K.R. AAV-mediated delivery of zinc finger nucleases targeting hepatitis B virus inhibits active replication. PLoS ONE 2014, 9, e97579. [Google Scholar] [CrossRef] [PubMed]
- Ladner, S.K.; Otto, M.J.; Barker, C.S.; Zaifert, K.; Wang, G.H.; Guo, J.T.; Seeger, C.; King, R.W. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: A novel system for screening potential inhibitors of HBV replication. Antimicrob. Agents Chemother. 1997, 41, 1715–1720. [Google Scholar] [PubMed]
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 2009, 326, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Moscou, M.J.; Bogdanove, A.J. A simple cipher governs DNA recognition by TAL effectors. Science 2009, 326, 1501. [Google Scholar] [CrossRef] [PubMed]
- Deng, D.; Yan, C.; Pan, X.; Mahfouz, M.; Wang, J.; Zhu, J.K.; Shi, Y.; Yan, N. Structural basis for sequence-specific recognition of DNA by TAL effectors. Science 2012, 335, 720–723. [Google Scholar] [CrossRef] [PubMed]
- Mak, A.N.; Bradley, P.; Cernadas, R.A.; Bogdanove, A.J.; Stoddard, B.L. The crystal structure of TAL effector PthXo1 bound to its DNA target. Science 2012, 335, 716–719. [Google Scholar] [CrossRef] [PubMed]
- Bloom, K.; Ely, A.; Mussolino, C.; Cathomen, T.; Arbuthnot, P. Inactivation of hepatitis B virus replication in cultured cells and in vivo with engineered transcription activator-like effector nucleases. Mol. Ther. 2013, 21, 1889–1897. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, W.; Lin, J.; Wang, F.; Wu, M.; Chen, C.; Zheng, Y.; Peng, X.; Li, J.; Yuan, Z. An efficient antiviral strategy for targeting hepatitis B virus genome using transcription activator-like effector nucleases. Mol. Ther. 2014, 22, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Dreyer, T.; Nicholson, S.; Ely, A.; Arbuthnot, P.; Bloom, K. Improved antiviral efficacy using TALEN-mediated homology directed recombination to introduce artificial primary mirnas into DNA of hepatitis B virus. Biochem. Biophys. Res. Commun. 2016, 478, 1563–1568. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Seeger, C.; Sohn, J.A. Targeting hepatitis B virus with CRISPR/Cas9. Mol. Ther. Nucleic Acids 2014, 3, e216. [Google Scholar] [CrossRef] [PubMed]
- Dong, C.; Qu, L.; Wang, H.; Wei, L.; Dong, Y.; Xiong, S. Targeting hepatitis B virus cccDNA by CRISPR/Cas9 nuclease efficiently inhibits viral replication. Antiviral Res. 2015, 118, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Karimova, M.; Beschorner, N.; Dammermann, W.; Chemnitz, J.; Indenbirken, D.; Bockmann, J.H.; Grundhoff, A.; Luth, S.; Buchholz, F.; Schulze zur Wiesch, J.; et al. Crispr/cas9 nickase-mediated disruption of hepatitis B virus open reading frame S and X. Sci. Rep. 2015, 5, 13734. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, E.M.; Bassit, L.C.; Mueller, H.; Kornepati, A.V.; Bogerd, H.P.; Nie, T.; Chatterjee, P.; Javanbakht, H.; Schinazi, R.F.; Cullen, B.R. Suppression of hepatitis B virus DNA accumulation in chronically infected cells using a bacterial CRISP/Cas RNA-guided DNA endonuclease. Virology 2015, 476, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Hao, R.; Chen, S.; Guo, D.; Chen, Y. Inhibition of hepatitis B virus by the CRISPR/Cas9 system via targeting the conserved regions of the viral genome. J. Gen. Virol. 2015, 96, 2252–2261. [Google Scholar] [CrossRef] [PubMed]
- Ramanan, V.; Shlomai, A.; Cox, D.B.; Schwartz, R.E.; Michailidis, E.; Bhatta, A.; Scott, D.A.; Zhang, F.; Rice, C.M.; Bhatia, S.N. CRISPR/Cas9 cleavage of viral DNA efficiently suppresses hepatitis B virus. Sci. Rep. 2015, 5, 10833. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xu, Z.W.; Liu, S.; Zhang, R.Y.; Ding, S.L.; Xie, X.M.; Long, L.; Chen, X.M.; Zhuang, H.; Lu, F.M. Dual gRNAs guided CRISPR/Cas9 system inhibits hepatitis B virus replication. World J. Gastroenterol. 2015, 21, 9554–9565. [Google Scholar] [CrossRef] [PubMed]
- Zhen, S.; Hua, L.; Liu, Y.H.; Gao, L.C.; Fu, J.; Wan, D.Y.; Dong, L.H.; Song, H.F.; Gao, X. Harnessing the clustered regularly interspaced short palindromic repeat (CRISPR)/CRISPR-associated Cas9 system to disrupt the hepatitis B virus. Gene Ther. 2015, 22, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Sakuma, T.; Masaki, K.; Abe-Chayama, H.; Mochida, K.; Yamamoto, T.; Chayama, K. Highly multiplexed CRISPR-Cas9-nuclease and Cas9-nickase vectors for inactivation of hepatitis B virus. Genes Cells 2016, 21, 1253–1262. [Google Scholar] [CrossRef] [PubMed]
- Seeger, C.; Sohn, J.A. Complete spectrum of CRISPR/Cas9-induced mutations on HBV cccDNA. Mol. Ther. 2016, 24, 1258–1266. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Xie, K.; Xu, Y.; Wang, L.; Chen, K.; Zhang, L.; Fang, J. CRISPR/Cas9 produces anti-hepatitis B virus effect in hepatoma cells and transgenic mouse. Virus Res. 2016, 217, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Sheng, C.; Liu, H.; Liu, G.; Du, X.; Du, J.; Zhan, L.; Li, P.; Yang, C.; Qi, L.; et al. An effective molecular target site in hepatitis B virus S gene for Cas9 cleavage and mutational inactivation. Int. J. Biol. Sci. 2016, 12, 1104–1113. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Mei, M.; Li, B.; Zhu, X.; Zu, W.; Tian, Y.; Wang, Q.; Guo, Y.; Dong, Y.; Tan, X. A non-viral CRISPR/Cas9 delivery system for therapeutically targeting HBV DNA and pcsk9 in vivo. Cell Res. 2017, 27, 440–443. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Sheng, C.; Wang, S.; Yang, L.; Liang, Y.; Huang, Y.; Liu, H.; Li, P.; Yang, C.; Yang, X.; et al. Removal of integrated hepatitis B virus DNA using CRISPR-Cas9. Front. Cell. Infect. Microbiol. 2017, 7, 91. [Google Scholar] [CrossRef] [PubMed]
- Scott, T.; Moyo, B.; Nicholson, S.; Maepa, M.B.; Watashi, K.; Ely, A.; Weinberg, M.S.; Arbuthnot, P. ssAAVs containing cassettes encoding SaCas9 and guides targeting hepatitis B virus inactivate replication of the virus in cultured cells. Sci. Rep. 2017, 7, 7401. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, R.; Zhang, R.; Ding, S.; Zhang, T.; Yuan, Q.; Guan, G.; Chen, X.; Zhang, T.; Zhuang, H.; et al. The gRNA-miRNA-gRNA ternary cassette combining CRISPR/Cas9 with RNAi approach strongly inhibits hepatitis B virus replication. Theranostics 2017, 7, 3090–3105. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Bai, L.; Zheng, S.; Liu, M.; Zhang, J.; Wang, T.; Xu, Z.; Chen, Y.; Li, J.; Duan, Z. Efficient inhibition of duck hepatitis B virus DNA by the CRISPR/Cas9 system. Mol Med Rep 2017, 16, 7199–7204. [Google Scholar] [CrossRef] [PubMed]
- Kuscu, C.; Arslan, S.; Singh, R.; Thorpe, J.; Adli, M. Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat. Biotechnol. 2014, 32, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Cradick, T.J.; Brown, M.T.; Deshmukh, H.; Ranjan, P.; Sarode, N.; Wile, B.M.; Vertino, P.M.; Stewart, F.J.; Bao, G. CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res. 2014, 42, 7473–7485. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaissiere, T.; Sawan, C.; Herceg, Z. Epigenetic interplay between histone modifications and DNA methylation in gene silencing. Mutat. Res. 2008, 659, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Chen, J.; Wu, M.; Zhang, X.; Zhang, M.; Yue, L.; Li, Y.; Liu, J.; Li, B.; Shen, F.; et al. PRMT5 restricts hepatitis B virus replication through epigenetic repression of covalently closed circular DNA transcription and interference with pregenomic RNA encapsidation. Hepatology 2017, 66, 398–415. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.H.; Li, Y.N.; Zhao, J.R.; Zhang, J.; Yan, Z. HBc binds to the CpG islands of HBV cccDNA and promotes an epigenetic permissive state. Epigenetics 2011, 6, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Mao, R.; Yan, R.; Cai, D.; Zhang, Y.; Zhu, H.; Kang, Y.; Liu, H.; Wang, J.; Qin, Y.; et al. Transcription of hepatitis B virus covalently closed circular DNA is regulated by CpG methylation during chronic infection. PLoS ONE 2014, 9, e110442. [Google Scholar] [CrossRef] [PubMed]
- Koumbi, L.; Karayiannis, P. The epigenetic control of hepatitis B virus modulates the outcome of infection. Front. Microbiol. 2015, 6, 1491. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Kim, E.S.; Guo, H. Epigenetic regulation of hepatitis B virus covalently closed circular DNA: Implications for epigenetic therapy against chronic hepatitis B. Hepatology 2017, 66, 2066–2077. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Deng, L.; Chen, M.; Gan, X.; Shinozaki, K.; Shoji, I.; Hotta, H. Interaction of the hepatitis B virus X protein with the lysine methyltransferase SET and MYND domain-containing 3 induces activator protein 1 activation. Microbiol. Immunol. 2016, 60, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Lubyova, B.; Hodek, J.; Zabransky, A.; Prouzova, H.; Hubalek, M.; Hirsch, I.; Weber, J. PRMT5: A novel regulator of hepatitis B virus replication and an arginine methylase of HBV core. PLoS ONE 2017, 12, e0186982. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Hu, Y.; Shen, X.; Lao, Y.; Zhang, L.; Qiu, X.; Hu, J.; Gong, P.; Cui, H.; Lu, S.; et al. HBx represses RIZ1 expression by DNA methyltransferase 1 involvement in decreased miR-152 in hepatocellular carcinoma. Oncol. Rep. 2017, 37, 2811–2818. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.R.; Yang, H.C.; Kuo, Y.T.; Liu, C.J.; Yang, T.Y.; Sung, K.C.; Lin, Y.Y.; Wang, H.Y.; Wang, C.C.; Shen, Y.C.; et al. The CRISPR/Cas9 system facilitates clearance of the intrahepatic HBV templates in vivo. Mol. Ther. Nucleic Acids 2014, 3, e186. [Google Scholar] [CrossRef] [PubMed]
- Xirong, L.; Rui, L.; Xiaoli, Y.; Qiuyan, H.; Bikui, T.; Sibo, Z.; Naishuo, Z. Hepatitis B virus can be inhibited by DNA methyltransferase 3a via specific zinc-finger-induced methylation of the X promoter. Biochemistry 2014, 79, 111–123. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhao, Z.; Guo, J.; Huang, P.; Zhu, X.; Zhou, X.; Yang, Z.; Zhao, L.; Xu, L.; Xu, J.; et al. Creation of a six-fingered artificial transcription factor that represses the hepatitis B virus HBx gene integrated into a human hepatocellular carcinoma cell line. J. Biomol. Screen. 2013, 18, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Moyo, B.; Nicholson, S.; Roelofse, I.; Crowther, C.; Bloom, K.; Mussolino, C.; Cathomen, T.; Watashi, K.; Ely, A.; Arbuthnot, P. 483. Epigenetic silencing of hepatitis B cccDNA in vitro and in vivo using AAV-delivered engineered repressor transcription activator-like effector. Mol. Ther. 2015, 23, S192. [Google Scholar] [CrossRef]
- Liu, F.; Campagna, M.; Qi, Y.; Zhao, X.; Guo, F.; Xu, C.; Li, S.; Li, W.; Block, T.M.; Chang, J.; et al. α-interferon suppresses hepadnavirus transcription by altering epigenetic modification of cccDNA minichromosomes. PLoS Pathog. 2013, 9, e1003613. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Zhao, X.; Zang, L.; Fang, X.; Zhao, J.; Yang, X.; Wang, Q.; Zheng, L.; Chang, J. Anti-hepatitis B virus activities of α-DDB-FNC, a novel nucleoside-biphenyldicarboxylate compound in cells and ducks, and its anti-immunological liver injury effect in mice. Antiviral Res. 2012, 96, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, C.; Zhang, Y.; Zhu, H.; Kang, Y.; Liu, H.; Wang, J.; Qin, Y.; Mao, R.; Xie, Y.; et al. Comparative analysis of CpG islands among HBV genotypes. PLoS ONE 2013, 8, e56711. [Google Scholar] [CrossRef] [PubMed]
- Vivekanandan, P.; Thomas, D.; Torbenson, M. Hepatitis B viral DNA is methylated in liver tissues. J. Viral Hepat. 2008, 15, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Jain, S.; Chang, T.T.; Chen, S.; Boldbaatar, B.; Clemens, A.; Lin, S.Y.; Yan, R.; Hu, C.T.; Guo, H.; Block, T.M.; et al. Comprehensive DNA methylation analysis of hepatitis B virus genome in infected liver tissues. Sci. Rep. 2015, 5, 10478. [Google Scholar] [CrossRef] [PubMed]
- Vivekanandan, P.; Kannangai, R.; Ray, S.C.; Thomas, D.L.; Torbenson, M. Comprehensive genetic and epigenetic analysis of occult hepatitis B from liver tissue samples. Clin. Infect. Dis. 2008, 46, 1227–1236. [Google Scholar] [CrossRef] [PubMed]
- Kaur, P.; Paliwal, A.; Durantel, D.; Hainaut, P.; Scoazec, J.Y.; Zoulim, F.; Chemin, I.; Herceg, Z. DNA methylation of hepatitis B virus (HBV) genome associated with the development of hepatocellular carcinoma and occult HBV infection. J. Infect. Dis. 2010, 202, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Li, Y.; Mu, S.; Zhang, J.; Yan, Z. Evidence that methylation of hepatitis B virus covalently closed circular DNA in liver tissues of patients with chronic hepatitis B modulates HBV replication. J. Med. Virol. 2009, 81, 1177–1183. [Google Scholar] [CrossRef] [PubMed]
- Thakore, P.I.; Black, J.B.; Hilton, I.B.; Gersbach, C.A. Editing the epigenome: Technologies for programmable transcriptional modulation and epigenetic regulation. Nature methods 2016, 13, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Archin, N.M.; Keedy, K.S.; Espeseth, A.; Dang, H.; Hazuda, D.J.; Margolis, D.M. Expression of latent human immunodeficiency type 1 is induced by novel and selective histone deacetylase inhibitors. AIDS 2009, 23, 1799–1806. [Google Scholar] [CrossRef] [PubMed]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Aurisicchio, L.; Delmastro, P.; Salucci, V.; Paz, O.G.; Rovere, P.; Ciliberto, G.; La Monica, N.; Palombo, F. Liver-specific alpha 2 interferon gene expression results in protection from induced hepatitis. J. Virol. 2000, 74, 4816–4823. [Google Scholar] [CrossRef] [PubMed]
- Fiedler, M.; Rodicker, F.; Salucci, V.; Lu, M.; Aurisicchio, L.; Dahmen, U.; Jun, L.; Dirsch, O.; Putzer, B.M.; Palombo, F.; et al. Helper-dependent adenoviral vector-mediated delivery of woodchuck-specific genes for alpha interferon (IFN-α) and IFN-γ: IFN-α but not IFN-γ reduces woodchuck hepatitis virus replication in chronic infection in vivo. J. Virol. 2004, 78, 10111–10121. [Google Scholar] [CrossRef] [PubMed]
- Dumortier, J.; Schonig, K.; Oberwinkler, H.; Low, R.; Giese, T.; Bujard, H.; Schirmacher, P.; Protzer, U. Liver-specific expression of interferon γ following adenoviral gene transfer controls hepatitis b virus replication in mice. Gene Ther. 2005, 12, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Berraondo, P.; Ochoa, L.; Crettaz, J.; Rotellar, F.; Vales, A.; Martinez-Anso, E.; Zaratiegui, M.; Ruiz, J.; Gonzalez-Aseguinolaza, G.; Prieto, J. IFN-α gene therapy for woodchuck hepatitis with adeno-associated virus: Differences in duration of gene expression and antiviral activity using intraportal or intramuscular routes. Mol. Ther. 2005, 12, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Berraondo, P.; Di Scala, M.; Korolowicz, K.; Thampi, L.M.; Otano, I.; Suarez, L.; Fioravanti, J.; Aranda, F.; Ardaiz, N.; Yang, J.; et al. Liver-directed gene therapy of chronic hepadnavirus infection using interferon alpha tethered to apolipoprotein A-I. J. Hepatol. 2015, 63, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Crettaz, J.; Otano, I.; Ochoa-Callejero, L.; Benito, A.; Paneda, A.; Aurrekoetxea, I.; Berraondo, P.; Rodriguez-Madoz, J.R.; Astudillo, A.; Kreppel, F.; et al. Treatment of chronic viral hepatitis in woodchucks by prolonged intrahepatic expression of interleukin-12. J. Virol. 2009, 83, 2663–2674. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Madoz, J.R.; Liu, K.H.; Quetglas, J.I.; Ruiz-Guillen, M.; Otano, I.; Crettaz, J.; Butler, S.D.; Bellezza, C.A.; Dykes, N.L.; Tennant, B.C.; et al. Semliki forest virus expressing interleukin-12 induces antiviral and antitumoral responses in woodchucks with chronic viral hepatitis and hepatocellular carcinoma. J. Virol. 2009, 83, 12266–12278. [Google Scholar] [CrossRef] [PubMed]
- Di Scala, M.; Otano, I.; Gil-Farina, I.; Vanrell, L.; Hommel, M.; Olague, C.; Vales, A.; Galarraga, M.; Guembe, L.; Ortiz de Solorzano, C.; et al. Complementary effects of interleukin-15 and alpha interferon induce immunity in hepatitis b virus transgenic mice. J. Virol. 2016, 90, 8563–8574. [Google Scholar] [CrossRef] [PubMed]
- Bohne, F.; Chmielewski, M.; Ebert, G.; Wiegmann, K.; Kurschner, T.; Schulze, A.; Urban, S.; Kronke, M.; Abken, H.; Protzer, U. T cells redirected against hepatitis B virus surface proteins eliminate infected hepatocytes. Gastroenterology 2008, 134, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Gehring, A.J.; Xue, S.A.; Ho, Z.Z.; Teoh, D.; Ruedl, C.; Chia, A.; Koh, S.; Lim, S.G.; Maini, M.K.; Stauss, H.; et al. Engineering virus-specific T cells that target HBV infected hepatocytes and hepatocellular carcinoma cell lines. J. Hepatol. 2011, 55, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Kah, J.; Koh, S.; Volz, T.; Ceccarello, E.; Allweiss, L.; Lutgehetmann, M.; Bertoletti, A.; Dandri, M. Lymphocytes transiently expressing virus-specific T cell receptors reduce hepatitis B virus infection. J. Clin. Investig. 2017, 127, 3177–3188. [Google Scholar] [CrossRef] [PubMed]
- Krebs, K.; Bottinger, N.; Huang, L.R.; Chmielewski, M.; Arzberger, S.; Gasteiger, G.; Jager, C.; Schmitt, E.; Bohne, F.; Aichler, M.; et al. T cells expressing a chimeric antigen receptor that binds hepatitis B virus envelope proteins control virus replication in mice. Gastroenterology 2013, 145, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Wisskirchen, K.; Metzger, K.; Schreiber, S.; Asen, T.; Weigand, L.; Dargel, C.; Witter, K.; Kieback, E.; Sprinzl, M.F.; Uckert, W.; et al. Isolation and functional characterization of hepatitis B virus-specific T-cell receptors as new tools for experimental and clinical use. PLoS ONE 2017, 12, e0182936. [Google Scholar] [CrossRef] [PubMed]
- Kosinska, A.D.; Liu, J.; Lu, M.; Roggendorf, M. Therapeutic vaccination and immunomodulation in the treatment of chronic hepatitis B: Preclinical studies in the woodchuck. Med. Microbiol. Immunol. 2015, 204, 103–114. [Google Scholar] [CrossRef] [PubMed]
- Cornu, T.I.; Mussolino, C.; Cathomen, T. Refining strategies to translate genome editing to the clinic. Nat. Med. 2017, 23, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Hardee, L.C.; Arévalo-Soliz, M.L.; Hornstein, D.B.; Zechiedrich, L. Advances in non-viral DNA vectors for gene therapy. Genes 2017, 8, 65. [Google Scholar] [CrossRef] [PubMed]
- Baruteau, J.; Waddington, S.N.; Alexander, I.E.; Gissen, P. Delivering efficient liver-directed AAV-mediated gene therapy. Gene Ther. 2017, 24, 263. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.M.; Cradick, T.J.; Fine, E.J.; Bao, G. Nuclease target site selection for maximizing on-target activity and minimizing off-target effects in genome editing. Mol. Ther. 2016, 24, 475–487. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Strategies for hepatitis B virus (HBV) gene editing and epigenome modifications. The covalently closed circular DNA (cccDNA), which may be methylated, forms a minichromosome with transcriptionally active (open circles) and inactive (closed circles) chromatin. (A) Designer nucleases cleave at pre-defined sequences within the HBV genome to effect targeted mutagenesis. Employing multiple nucleases to digest different sequences may lead to targeted excision. Mutated cccDNA may be transcribed, but mutant viral proteins cannot carry out viral replication; (B) Epigenetic modification involves conversion of actively transcribed DNA to a transcriptionally inactive state without altering the nucleotide sequence the viral DNA. Targeted modifications occur when DNA-binding domains guide epigenetic effectors to pre-defined sequences of cccDNA. Histone modification and cccDNA methylation may affect epigenetic modifications by acting directly on the cccDNA or on associated histone proteins. Indels: insertions and deletions; ZFN: zinc finger nuclease; TALEN: transcription activator-like effector nuclease; CRISPR/Cas: clustered regularly interspaced palindromic repeats with CRISPR-associated protein; HDAC: histone deacetylase; DNMT: DNA methyltransferase; Me: methyl.
Figure 1.
Strategies for hepatitis B virus (HBV) gene editing and epigenome modifications. The covalently closed circular DNA (cccDNA), which may be methylated, forms a minichromosome with transcriptionally active (open circles) and inactive (closed circles) chromatin. (A) Designer nucleases cleave at pre-defined sequences within the HBV genome to effect targeted mutagenesis. Employing multiple nucleases to digest different sequences may lead to targeted excision. Mutated cccDNA may be transcribed, but mutant viral proteins cannot carry out viral replication; (B) Epigenetic modification involves conversion of actively transcribed DNA to a transcriptionally inactive state without altering the nucleotide sequence the viral DNA. Targeted modifications occur when DNA-binding domains guide epigenetic effectors to pre-defined sequences of cccDNA. Histone modification and cccDNA methylation may affect epigenetic modifications by acting directly on the cccDNA or on associated histone proteins. Indels: insertions and deletions; ZFN: zinc finger nuclease; TALEN: transcription activator-like effector nuclease; CRISPR/Cas: clustered regularly interspaced palindromic repeats with CRISPR-associated protein; HDAC: histone deacetylase; DNMT: DNA methyltransferase; Me: methyl.

{kind=link}
Table 1.
Overview of designer nucleases for HBV gene therapy. RVDs: repeat-variable di-residues; tracrRNA: trans-activating crRNA; HDI: hydrodynamic injection; RGN: RNA-guided nucleases
Table 1.
Overview of designer nucleases for HBV gene therapy. RVDs: repeat-variable di-residues; tracrRNA: trans-activating crRNA; HDI: hydrodynamic injection; RGN: RNA-guided nucleases
| ZFN | TALEN | CRISPR/Cas | ||
|---|---|---|---|---|
| DNA binding domain |
|
|
| |
| Nuclease domain |
|
|
| |
| Advantages |
|
|
| |
| Disadvantages |
|
|
| |
| HBV model systems |
| |||
| cccDNA | Cleavage (%) |
| ||
| Reduction (%) |
|
| ||
| Alternative effector domain |
| |||
* Varying methods of introducing the NTCP receptor into HepG2 cells. ** Results from single and/or multiple gRNAs. *** Incorporates co-administration of NAs.
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bloom, K.; Maepa, M.B.; Ely, A.; Arbuthnot, P. Gene Therapy for Chronic HBV—Can We Eliminate cccDNA? Genes 2018, 9, 207. https://doi.org/10.3390/genes9040207
AMA Style
Bloom K, Maepa MB, Ely A, Arbuthnot P. Gene Therapy for Chronic HBV—Can We Eliminate cccDNA? Genes. 2018; 9(4):207. https://doi.org/10.3390/genes9040207
Chicago/Turabian StyleBloom, Kristie, Mohube Betty Maepa, Abdullah Ely, and Patrick Arbuthnot. 2018. "Gene Therapy for Chronic HBV—Can We Eliminate cccDNA?" Genes 9, no. 4: 207. https://doi.org/10.3390/genes9040207
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.