
Controlling the Master: Chromatin Dynamics at the MYC Promoter Integrate Developmental Signaling


1
ACRF Department of Cancer Biology and Therapeutics, The John Curtin School of Medical Research, The Australian National University, Canberra, ACT 2600, Australia
2
School of Biomedical Sciences, University of Melbourne, Parkville 3010, Australia
*
Author to whom correspondence should be addressed.
Genes 2017, 8(4), 118; https://doi.org/10.3390/genes8040118
Submission received: 27 January 2017
/
Revised: 15 March 2017
/
Accepted: 7 April 2017
/
Published: 11 April 2017
(This article belongs to the Special Issue MYC Networks)
{kind=link}
Abstract
:The transcription factor and cell growth regulator MYC is potently oncogenic and estimated to contribute to most cancers. Decades of attempts to therapeutically target MYC directly have not resulted in feasible clinical applications, and efforts have moved toward indirectly targeting MYC expression, function and/or activity to treat MYC-driven cancer. A multitude of developmental and growth signaling pathways converge on the MYC promoter to modulate transcription through their downstream effectors. Critically, even small increases in MYC abundance (<2 fold) are sufficient to drive overproliferation; however, the details of how oncogenic/growth signaling networks regulate MYC at the level of transcription remain nebulous even during normal development. It is therefore essential to first decipher mechanisms of growth signal-stimulated MYC transcription using in vivo models, with intact signaling environments, to determine exactly how these networks are dysregulated in human cancer. This in turn will provide new modalities and approaches to treat MYC-driven malignancy. Drosophila genetic studies have shed much light on how complex networks signal to transcription factors and enhancers to orchestrate Drosophila MYC (dMYC) transcription, and thus growth and patterning of complex multicellular tissue and organs. This review will discuss the many pathways implicated in patterning MYC transcription during development and the molecular events at the MYC promoter that link signaling to expression. Attention will also be drawn to parallels between mammalian and fly regulation of MYC at the level of transcription.
Keywords:
MYC; Drosophila dMYC; FUBP1/Psi; FIR/Hfp; TFIIH; Mediator; transcription; signaling; development; DNA topology1. MYC: A Potent Driver of Cell and Tissue Growth
MYC upregulation is a key driver of tumorigenesis [1,2,3]. Fundamental to MYC’s impact on both normal development and cancer cell physiology is a potent capacity to promote cell and tissue growth through activation of the genes necessary for protein synthesis for accumulation of cellular biomass [4,5,6,7] and cell cycle machinery to drive proliferation [8]. MYC abundance therefore correlates with proliferation [9], consistent with the observation that MYC’s transcriptional activity decreases in differentiated cells [10,11]. MYC’s capacity to promote growth and cell division is essential for tissue patterning during organ growth and, given that elevated MYC is inexorably linked to cancer initiation and progression, developmental signals must normally ensure tight control of MYC expression [12,13].
MYC heterodimerizes with its partner bHLH protein MAX in order to bind DNA and activate transcription [14,15]. MYC-driven transcriptional activation is, in part, regulated by a related bHLH protein MAD which forms heterodimers with MAX, therefore limiting the pool of available MAX protein [15]. Other binding partners possess capacity to modulate MYC activity; for example, binding to MIZ-1 (ZBTB17) leads to transcriptional downregulation of MYC targets, providing a mechanism for MYC’s capacity as a transcriptional repressor of certain targets [16,17]. Early studies conducted in Drosophila, prior to the advent of genome-wide deep sequencing, implicated dMYC in controlling expression of 10%–15% of all genes [18,19]. The prominent and conserved transcriptional signature (i.e., cell growth and proliferation [4,5,6,7]) was attributed to MYC’s capacity to selectively upregulate a common set of cellular targets [13,20], enriched for the presence of the consensus “E-box” enhancer sequence [21,22]. Nevertheless, genome-wide chromatin immunoprecipitation (ChIP)-sequencing studies, detecting MYC on all promoters, enhancers and intergenic regions with an open chromatin structure [23,24,25,26], appear to challenge MYC’s role as gene-specific transcriptional regulator. Naïve mouse lymphocytes carrying endogenous MYC tagged with green fluorescent protein (GFP) were stimulated to induce Myc expression, revealing a global positive correlation between level of gene expression, active chromatin marks and MYC binding across the genome [23]. This observation is supported by studies in human Burkitt’s lymphoma tumour cell models [24], and together these studies suggest that MYC behaves as a broad amplifier of transcription.
So how does the transcriptional signature associated with increased MYC arise in both normal and tumour cells? At the broadest level, global amplification as a result of elevated MYC levels will increase the output of previously active transcriptional programs, potentiating the growth and proliferation programs already operational in developing organs, proliferating cell culture systems or tumours. The observation that MYC binding may not always result in productive transcriptional outcomes [27] could also enable fine tuning of the MYC signature in the event of global binding to active genes. However, a recent study revealed that MYC’s differing affinity for certain promoters likely potentiates MYC-specific expression profiles [28]. Specifically, genes containing high-affinity promoters are predicted to be fully occupied by physiological levels of MYC; thus, introducing additional MYC protein can only increase binding at low affinity targets, generating a signature by enhancing expression of these weakly-bound MYC-targets. Together these studies explain why MYC has such potent oncogenic capacity. Although MYC overexpression alone does not induce aggressive tumours, in the context of dysregulated growth signaling, the ability of MYC to amplify all active transcription could lead to tumourigenesis. For instance, elevation of MYC will not only enhance cellular overgrowth and tissue hyperplasia (by binding promoters that are active in proliferating tumour cells), but will also upregulate expression of weakly expressed cancer-promoting targets to accelerate tumour progression. The expanse of MYC’s transcriptional capacity is reviewed comprehensively elsewhere [29], but the message is clear regardless of nuances; moderate increases in MYC can broadly influence the cell’s transcriptome. Thus, to maintain normal cell growth patterning during animal development, expression of MYC must be tightly controlled.
2. MYC Functional Redundancy Depends on Transcriptional Patterning
In mammals, the MYC family benefits somewhat from the redundancy of three family members, MYC, MYCN and MYCL (reviewed elsewhere [12,30]). Myc haploinsufficiency results in viable mice, which are small compared with wildtype due to impaired proliferation [31]. Some MYC is essential for viability, as null mice display severe developmental defects and die before midgestation [31]. Interestingly, the lethality associated with Myc nulls is actually a result of placental insufficiency, since embryo-specific knockout (in the epiblast) in the context of wild type extraembryonic structures (trophectoderm and primitive endoderm), generates embryonic organs without gross developmental abnormalities [32]. The exception is the hematopoietic lineage, which is particularly sensitive to Myc depletion; thus, placental rescue pups are severely anaemic due to loss of hematopoietic stem cells (HSCs) [32]. Strikingly, gene-replacement of Myc with Mycn results in viability, demonstrating that MYCN can provide functional compensation when MYC is absent [33].
The sensitivity of certain lineages to Myc depletion, and inability of Mycn expressed from its endogenous locus to compensate in the placenta and hematopoietic lineage, likely reflects large differences in expression levels of the paralogs between tissues. Myc is expressed broadly in developing mice, with particularly high levels of mRNA in thymus, spleen, and liver, while Mycn is highly expressed in the brain and kidney [34]. The cerebellum is more sensitive to conditional deletion of Mycn, which impairs neural stem and progenitor cell proliferation, while deletion of Myc does not alter these lineages [35]. In the tumour context, MYC and MYCN-driven medulloblastomas exhibit distinct phenotypes due to the preferential ability of MYC to inhibit expression of certain genes via binding to the MIZ-1 transcriptional repressor [36].
In the hematopoietic lineage, Mycn and Myc are only coexpressed in immature HSCs, and Mycn transcripts decrease following HSC differentiation [37]. In accordance with functional redundancy in early development, the Mycn and Myc double knockout hematopoietic phenotype is significantly more severe than individual knockout of either gene [37]. Although deletion of Myc prevents HSCs from differentiating into progenitors due to inability to exit the niche [38], the remaining endogenous MYCN enables maintenance of proliferation and self-renewal capacity, indicating that MYCN can provide all MYC-family functions during HSC maintenance. However, in contrast to Myc, deletion of Mycn alone does not impair HSC number or steady-state hematopoiesis [37]. Therefore, MYCN is only required in very primitive HSCs, while other functions are provided by MYC throughout the hematopoietic lineage.
Thus, despite the ability of MYCN to functionally replace MYC during development [33], and the observation that Myc depletion can lead to increased Mycn abundance [39], MYC and MYCN do not play overlapping roles in some mammalian organs. How, then, does the insertion of Mycn into the endogenous Myc locus rescue the Myc null? Expression of the Mycn gene from the Myc locus reflects the endogenous Myc patterning, suggesting that transcriptional control of the Myc locus is critical for normal development.
3. Patterning MYC Transcription during Animal Development
In Drosophila, the single orthologue (dMYC) has enabled MYC function to be discerned without concern for redundancy. The functional conservation of MYC between invertebrates and vertebrates is stunning. Not only can expression of human MYC rescue lethal dMYC mutations in flies [40], but expression of dMYC can provide competitive advantage to Myc null mouse fibroblasts [39] and cooperate with activated RasV12 alleles to rescue proliferation defects and induce oncogenic transformation in Myc null rat fibroblasts [41]. Thus, like mammalian MYC, Drosophila dMYC regulates cell growth and progression through the cell cycle [42,43,44,45]. Indeed, Drosophila was the first organism where cell growth impairment phenotypes (i.e., a reduced cell size) were associated with dMYC reduction and diminished organ and tissue growth [42]. Specifically, hypomorphic dMYC mutant flies are smaller than wildtype, with normal organ proportions, albeit composed of cells reduced in size [42]. In line with the impaired growth, dMYC targets the machinery required for increasing cellular biomass, particularly for building ribosomes [6,46,47,48]. Conversely, global dMYC overexpression increases ribosome biogenesis and tissue growth to result in overgrown flies [6,49].
The potent oncogenic capacity of MYC is the downside of its efficiency to normally drive growth and proliferation in developing organisms, and once again highlights the need for precise regulation of MYC levels. Even small changes in MYC levels are sufficient for cell and tissue overgrowth [50,51], in contrast to other proto-oncogenes such as Ras or EGFR where mutations constitutively activate the protein [52]. To enable normal development, MYC expression must not only be tightly restricted, but also maintain capacity to rapidly respond to developmental growth signals according to the cell’s requirements. Since transcriptional upregulation of MYC expression can trigger and propagate tumour pathogenesis, we focused the next part of this review on the mechanisms orchestrating expression of MYC under normal circumstances and reflect on their disarray in the context of cancer.
4. Developmental Signals Patterning MYC Expression
For MYC to drive the major cellular growth and proliferation events orchestrating development, abundance is predominantly regulated at the levels of transcription and mRNA/protein stability. To attain the level of MYC appropriate to cell fate, signaling inputs to MYC transcription must be integrated with those altering mRNA and protein abundance. mRNA stability and translation of both mammalian MYC and Drosophila dMYC is modulated by numerous microRNAs (miRNAs) [53,54,55,56,57,58,59,60]. Further to this, the generally short half-life of the MYC protein [61] can be increased as a consequence of stabilising phosphorylation directly by kinases in the Ras/ERK pathway [62,63,64,65] and/or PI3K/AKT pathway via inhibition of GSK3-β, which, when active, targets MYC and dMYC for ubiquitin-mediated proteasome degradation [66,67,68,69]. Indeed, point mutations in the MYC coding sequence in the context of cancer [70,71] tend to cluster at the T58 phosphorylation site within the MYC transactivation domain. This can stabilise MYC protein as a result of reduced degradation [72,73], although certain point mutations at T58 do not extend the half-life of the protein [74], but might impair the ability of MYC to interact with regulatory binding factors to alter transcriptional activity.
Ultimately, increased MYC mRNA/protein relies on production of mRNA via transcriptional changes in response to extracellular stimuli and developmental growth signals. The orchestration of cell growth and cell cycle for tissue and organ development of multicellular organisms requires MYC patterning; MYC must be appropriately expressed in a subset of tissues and repressed in others. Early human cell culture studies demonstrated that MYC expression is rapidly activated by growth factors in serum, and maintained in proliferating stem cells/progenitor cells, but becomes downregulated in response to differentiation signals [75,76,77]. An overview of the multitude of developmental signals converging on the MYC promoter to pattern cell and tissue growth is outlined briefly below, but is more comprehensively reviewed elsewhere [44].
Links between upstream pathways controlling dMYC expression have been particularly well studied in the Drosophila wing imaginal disc, where dMYC drives cell growth and G1 to S phase progression [42,78,79]. The Wingless (Wg, Wnt pathway in mammals) morphogen drives cell cycle exit and differentiation in the wing by repressing dMYC expression [80]. On the other hand, activating Wg in the adjacent hinge region of the wing can drive tissue overgrowth [81], highlighting the complex outcomes elicited by the same developmental signal. Similar nuances in the orthologous Wnt pathway are observed in mammals, where the Wnt proteins can behave as oncogenes or tumour suppressors depending on cellular context. For instance, while most studies from the mammalian intestine highlight the capacity of Wnt to activate β-catenin/TCF4 binding to the MYC promoter in association with tissue overgrowth and tumour predisposition [82,83,84,85], Wnt signals drive terminal differentiation in Paneth cells in the intestinal crypt [86], inducing Myc in the crypt and CyclinD1 in the villi distinct cell populations [87].
Drosophila studies have enabled the dissection of interplay between Wg/Wnt and the Notch pathway, another major architect of development frequently linked with tumour progression [88,89]. Notch can inactivate dMYC indirectly via the Wg/Wnt pathway to control dMYC expression, and thus promote cell cycle arrest and differentiation in the fly wing [90,91,92,93,94]. On the other hand, stimulation of the Notch pathway via overexpression of cleaved Notch intracellular domain promotes binding of the Su(H) transcription factor to the dMYC gene, increasing dMYC expression and inducing wing overgrowth independently of Wg/Wnt [95]. In Drosophila muscle cell precursors, Notch activation also increases dMYC expression [96]. Notch further refines dMYC expression via induction of the E(spl)m8 transcriptional repressor, which negatively regulates dMYC in the wing [95]. In the context of gastric cancer, NOTCH4 induces Wnt signaling to result in activation of Myc by β-catenin [97]. Mammalian NOTCH1 can also directly activate Myc transcription by interacting with the Su(H) orthologue CBF1 in murine mammary cells [98]. Moreover, MYC is an essential NOTCH1 target for tumour progression in mammalian T cell lymphoblastic leukaemia (T-ALL) cells [99] and mouse models [100,101], where the Myc promoter is directly activated as a result of binding by intracellular domain of NOTCH and CBF1. In mouse models for pancreatic neoplasia, specific NOTCH2 activation of Myc is also required for tumour development [102].
Further to the responsiveness of the MYC promoter to the master developmental regulators WNT and NOTCH, a myriad of cellular signalling cascades possess capacity to influence MYC transcription. In the wing disc, the Decapentaplegic (Dpp, TGFβ in mammals) morphogen controls dMYC expression, albeit indirectly by downregulating the transcriptional repressor Brinker (Brk), which normally acts to suppress dMYC [103]. Testament to the context dependency of such morphogens, TGFβ can also repress mammalian MYC via direct activity of the downstream target SMAD3/SMAD4 on the MYC promoter [104]. In Drosophila midgut, JAK/Stat and EGFR activity increase dMYC promoter activity [105], while the mammalian MYC promoter is activated in response to JAK signaling, but independently of STAT3 [106]. The insulin/TOR pathway induces MYC expression via direct binding of the downstream transcription factor FOXO [107]. Drosophila dMYC transcription is also activated directly by the downstream effector of Hippo pathway, Yorkie (Yki) [108], and overexpression of the mammalian Yki orthologue, YAP, increases MYC abundance in mouse models [109]. The functional conservation and utility of Drosophila genetics has enabled dissection of complex networks controlling MYC abundance in multicellular organisms under conditions of normal development, thus enhancing our understanding of the mechanisms of MYC dysregulation in disease.
5. MYC Promoter Architecture Reflects Signaling Inputs
Early interest in MYC promoter architecture came from the observation that MYC-driven malignancy was often associated with rearrangements of the MYC locus and, thus, dysregulation of transcription. In particular, the B-cell malignancy Burkitt’s Lymphoma is associated with reciprocal chromosomal translocations between MYC and one of 3 immunoglobulin loci [110,111]. The breakpoints relative to both MYC and the immunoglobulin loci vary considerably, being either internal to the MYC transcriptional unit or up to several hundred kilobases away. Interestingly, the translocated MYC alleles contain either truncated or mutated exon 1, which alleviates the tightly regulated attenuation of RNA Polymerase II (Pol II) transcriptional elongation [112], enabling constitutive Pol II transcriptional read-through and increased MYC expression [77,113,114]. In accordance, a block to Pol II elongation in the endogenous MYC promoter induces transcriptional down-regulation of MYC and differentiation in human leukaemia cell lines [115]. This block to progression of engaged Pol II, or promoter proximal paused Pol II, was first interrogated using models of the human MYC promoter [77,116]. In vitro studies using an intact MYC locus identified Pol II accumulation downstream of the MYC transcription start site, engaged but unable to proceed to productive elongation [117]. As MYC receives multiple activation signals, post initiation control via Pol II pausing provides opportunity for rapid transcriptional activation in response to developmental signaling, which could not be achieved by Pol II recruitment alone.
6. FUBP1—A Hyperactivator of MYC Transcription
At promoters, melting of duplex DNA is obligatory for Pol II entry and transcriptional initiation. Indeed, DNase hypersensitivity of the human MYC promoter correlates with activity, being more sensitive in cells actively transcribing MYC compared with MYC-silent cells [77,115,118]. Thus, major changes in DNA architecture, particularly generation of nuclease-sensitive, non-B DNA elements, will arise as the multitude of growth and developmental signaling networks converge on the MYC promoter. The disruption to DNase hypersensitive elements in Burkitt’s lymphoma cells [115] led to the characterization of chromatin modifications and mapping of the DNA elements regulating the endogenous MYC promoter as a means of understanding mechanisms of this MYC-driven cancer. Of the many sequence-specific binding sites predicted for the MYC promoter based on the nuclease sensitivity assays, induction of differentiation and downregulation of MYC expression is only associated with lost binding activity of the far upstream sequence element (FUSE) 1.5kb upstream of the P1 promoter [119].
Oligonucleotide affinity chromatography using the FUSE retrieve the FUSE binding protein (initially FBP and recently renamed FUBP1) from proliferating cell extracts, with binding dramatically decreasing following induction of differentiation and down regulation of MYC [120]. Preferential FUBP1 binding to the noncoding, single-stranded FUSE provoked a closer examination of FUSE structure and function. Early studies revealed broad regions of specific S1 nuclease (single-strand nucleic acid) sensitivity in chromatin upstream of the human MYC gene [76]; however, mapping using potassium permanganate, which reacts preferentially with thymine in single-stranded DNA, enables single-base resolution [121]. Indeed, such permanganate assays were used in the first studies confirming melting of DNA strands in transcription bubbles associated with paused Pol II downstream of P2 promoter in human MYC [122]. Permanganate mapping of the MYC coding strand reveals hyperreactive thymidine residues in FUSE, consistent with an open single strand extending from FUSE toward P1 [121]. In contrast, the noncoding strand is predominantly hyporeactive; particularly protected are the nucleotides preferentially bound by FUBP1 in vitro, i.e., consistent with FUBP1 binding the single-stranded noncoding strand of FUSE in vivo [120]. Interestingly, FUBP1 is able to alter DNA conformation, possessing ability to force separation of the FUSE contained in supercoiled plasmid DNA and drive further opening of dsDNA at distances over 2.8 kilobases away [121]. Indeed, subsequent studies (described below) demonstrated that FUBP1-dependent remodeling of the MYC promoter structure is essential for tight regulation of MYC transcription.
The observation that FUBP1 binds the single-stranded FUSE, in preference to the double-stranded sequence, suggests that formation of the FUBP1-DNA complex requires prior unwinding of the DNA helix [123]. In accordance, FUSE is contained within a region of helical instability predicted to partially unwind in negatively supercoiled DNA, which would provide a platform for more extensive double-strand separation and stabilisation driven by FUBP1. Strand separation associated with targeted melting of the A-T rich FUSE, and subsequent binding activity of FUBP1, would further drive supercoiling and generate torsional energy within adjacent double-stranded DNA (dsDNA) strands. Indeed, double-strand opening is only observed in vitro when FUSE is surrounded by supercoiled—not unwound—double-stranded plasmid DNA [123]. As conversion of negatively supercoiled plasmid to the relaxed form using Topoisomerase I (Topo I) abolishes all spontaneous and FUBP1-induced strand melting, the helical stabilisation effect on FUSE in supercoiled DNA must be a consequence of non-B-DNA induced by FUBP1 at the more distant sites. Analysis of the interaction between FUBP1 and torsionally stressed supercoiled DNA, but not linear duplexes, suggests FUBP1 might directly link alterations in DNA conformation and topology with changes in MYC expression [121]. Factors that recognize topological strain, such as FUBP1, can thus act as sensors of MYC promoter activity. Under the circumstances where MYC transcription initiates (i.e., Pol II engages with the open P1 and P2 duplex but does not immediately escape the promoter) FUBP1 binding to this pre-activated MYC promoter would enable release of the paused Pol II to drive promoter escape and enhance transcriptional elongation.
Hence, in addition to conventional dsDNA-binding transcriptional regulators, activity of single-strand nucleic acid binding proteins can drive MYC transcription. Moreover, energy generated from promoter unwinding can be harnessed as a productive force in MYC transcription [124]. Specifically, increased MYC expression will be associated with region-specific destabilisation of B-DNA in torsionally strained regions of the active MYC promoter, and forward movement of Pol II will generate and transmit negative supercoils to the FUSE to enable FUBP1 binding. Thus, MYC promoter activity associated with transcription initiation enables FUBP1 binding to FUSE to maximize Pol II release and activation of MYC transcription [125].
7. Developmental Function of FUBP1
Interaction between FUBP1 and the single-stranded FUSE is essential for maximal activation of the MYC promoter; however, the physiological outcomes of single-strand-specific FUBP1 binding during development is still relatively obscure. Early studies demonstrated that reduced FUBP1 levels and/or activity diminishes MYC expression and cell proliferation in ex vivo cell culture systems [126]. FUBP1 knockdown in human hepatocellular carcinoma (HCC) cell lines also decreases proliferation, and impairs tumour formation in mouse xenograft models [127]. Essential functions in HSC self-renewal were revealed using Fubp1 gene trap mice, with embryonic lethality (around E15.5) associated with anaemia [128]. Secondary transplantation assays for long-term repopulating hematopoietic stem cells (LT-HSCs) revealed reduced blood cell reconstitution for Fubp1 knockdown. Fubp1-deficient adult HSCs exhibited increased expression of key cell cycle (cyclin-dependent kinase inhibitor, p21) and pro-apoptotic (Noxa) genes based on mRNA expression profile data [128]. Given that the haematopoietic lineage displays specific sensitivity to MYC levels [32], and the ability of MYC to repress p21 expression [129], the Fubp1 knockout phenotype suggests that MYC could be a key downstream target that mediates these effects. Further studies are therefore required to determine if these transcriptional changes are direct, or occur indirectly, via altered MYC expression.
Fubp1 knockouts display variable penetrance; although mice start dying at embryonic day 10.5 they can survive until birth [130]. Broader phenotypic analysis revealed a diverse range of defects including small body size, pulmonary hypoplasia, hypoplastic spleen/thymus/bone marrow, cerebral hyperplasia, pale livers, cardiac hypertrophy and placental distress, all characteristic of severe anaemia. The Fubp1 knockout had normal numbers of HSCs, but transplantation of Fubp1 loss-of-function HSCs into irradiated mice failed to reconstitute haematopoiesis [130]. The discrepancy with the Fubp1 gene trap, which reduces but does not eliminate reconstitution capacity [128], suggests the gene trap might be hypomorphic.
Interestingly, Myc mRNA levels varied dramatically between individual ex vivo cultures of Fubp1 knockout mouse embryo fibroblasts (MEFs) harvested from different Fubp1 embryos [130]. The reason for this variability is currently unknown, but it is possible that reduced FUBP1 binding on Myc impairs recruitment of FIR (FUBP-interacting repressor, discussed below) and results in a failure to shut Myc down, thus generating transcriptional noise. Another possibility is redundancy and/or compensatory activity of the other FUBP family members, FUBP2 and FUBP3. Like FUBP1, FUBP2 and FUBP3 can bind sequence specifically to the non-coding strand of FUSE, and possess potent transcriptional activation domains in vitro [131]. FUBP2 would be predicted to be most likely to compensate for loss of FUBP1 function as these two family members share the most functional similarity; in contrast, FUBP3 is only weakly localised to the nucleus and does not bind FIR [132]. Given FUBP1 and 2 regulate common target genes in vivo, including MYC, it will be of great interest to determine whether FUBP1 loss-of-function leads to heightened FUBP2 activity and the phenotypic outcome of double knockout. In short, these mouse studies leave the major function of FUBP1 during development requiring clarification.
8. FUBP-Interacting Repressor (FIR)—the FUBP1 Antagonist Keeps MYC Quiet
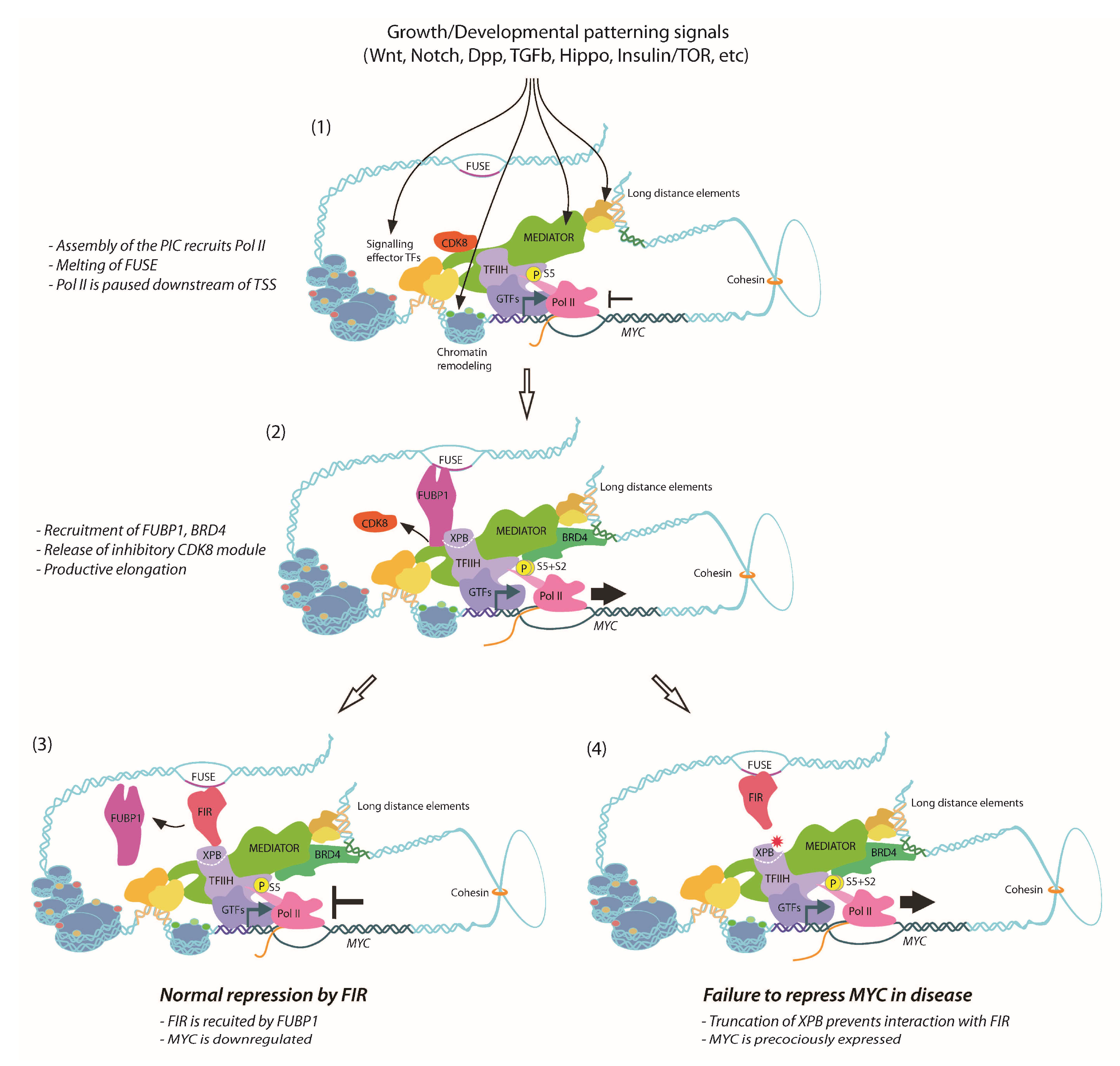
Yeast two-hybrid screens, using FUBP1 as bait, identified the FUBP1 interacting repressor (FIR) as a potent binding partner [133]. FIR behaves as an FUBP1 antagonist, blocking activator-dependent, but not basal MYC transcription. A co-immunoprecipitation (co-IP) screen in HeLa cells using purified components of the basal transcription apparatus available at the time (i.e., the general transcription factors or GTFs) detected association of FIR with TFIIH, but not with GTF complexes necessary at earlier stages of promoter recognition and preinitiation complex (PIC) formation [133] (e.g., TBP, TAFs, TFIIB, TFIIF [134,135,136]). However, FIR did not alter TFIIH-mediated phosphorylation of the carboxyl-terminal domain (CTD) of the largest Pol II subunit at S5 [133] required for initiation and promoter escape [137,138,139], so was unlikely to cause repression of the cyclin-dependent kinase 7 (CDK7) kinase module of TFIIH. Rather, FIR inhibited the helicase activity of TFIIH’s xeroderma pigmentosum type B (XPB)/P89 module, which is essential for both transcription initiation and promoter escape [140,141,142].
FIR therefore blocks FUBP1-dependent MYC activation by decreasing XPB helicase activity, to permit only basal transcription [133]. The MYC promoter integrates multiple signaling inputs, but regulation by TFIIH at the level of promoter clearance would provide an opportunity to control transcription post-initiation. Repression of TFIIH by FIR, late in the transcription cycle, would provide an additional regulatory mechanism to safeguard against inappropriate or excessive signal-induced MYC activation. In context with the post-initiation block controlling MYC transcription [77,112,114], FIR could encumber TFIIH release, enable Pol II pausing and delay the subsequent progression to elongation. Indeed, FIR is an essential MYC repressor, as FIR knockdown is associated with MYC dysregulation ex vivo [143] and loss-of-function FIR mutations are associated with colorectal cancer displaying increased MYC abundance [144].
9. Dynamics of FUBP1, FIR and TFIIH Activity on the MYC Promoter
Serum-stimulation of mammalian tissue culture cells results in rapid activation of the endogenous MYC promoter and a pulse (i.e., up- followed by down-regulation) of MYC transcription [75]. Elegant ex vivo ChIP time-course experiments revealed the sequence of events on the MYC promoter following signal-stimulated MYC transcription, achieved by serum stimulation of previously starved human fibroblasts [143]. MYC enhancers and activators are first to load, enrichment for FUBP1 is subsequently detected, prior to decreased Pol II loading (i.e., consistent with release of paused Pol II) and increased abundance of MYC mRNA [143]. Following the peak in MYC mRNA levels, FUBP1 and FIR initially co-localise; however, once MYC returns to basal levels FUBP1 exits and only FIR is detected on FUSE. Thus, maximal activation of MYC transcription correlates with dissociation of RNA Pol II and recruitment of FUBP1 [143]. Pol II depletion from the transcription start site (TSS) and maximal enrichment for FUBP1, prior to the peak in MYC mRNA levels, is consistent with FUBP1 promoting RNA Pol II release to hyperactivate MYC transcription.
The time course ChIP data supported findings from electrophoretic mobility shift assays (EMSA), demonstrating that FUBP1 and FIR binding occurs simultaneously; FIR addition enhances FUBP1 binding with FUSE; i.e., FUBP1’s affinity within the ternary FUBP1-FIR-FUSE complex is stronger than the FUBP1-FUSE interaction alone [133]. Moreover, the observation that FIR and FUBP1 enrichment is detected both on FUSE and proximal to the TSS suggests the FUBP1-FIR-XPB/TFIIH complex could generate a protein bridge between FUSE and TFIIH/Pol II bound to the TSS. Such a bridge would result in a closed topological DNA domain, to generate stress on the DNA duplex in the event of Pol II movement, which when transmitted as writhe would further melt FUSE to optimise the signle-stranded DNA (ssDNA) structure recognised by FIR. As FUBP1 leaves the complex, torsional strain would be lost and, as FIR engages, transcription would be returned to basal levels. The interaction between the FUBP1 activator and the repressor protein FIR can thus modify DNA topology and fine-tune MYC expression in response to signaling inputs.
10. Defective FIR-Dependent Repression of MYC in XPB-Related Disease
We predict that FUBP1 normally increases MYC transcription by stimulating XPB helicase activity and decreasing the frequency of Pol II pausing normally associated with the MYC promoter. FIR interacts with XPB/TFIIH to delay escape of Pol II from the promoter post-initiation. Such fine-tuning of transcription is essential for genes receiving multiple activation signals and requiring exquisite regulation like MYC, where recruitment alone would not provide sufficiently stringent control. Indeed, the early analysis of MYC dysregulation in Burkitt’s lymphoma revealed the importance of transcriptional regulation post-initiation, with precocious Pol II escape associated with MYC-driven pathology [77,112,114].
In addition to the essential function of TFIIH in basal transcription, the complex is essential for DNA excision repair [145]. Mutations in TFIIH subunits, including the XPB helicase, are associated with the inherited genetic disease Xeroderma Pigmentosum (XP), where failure of DNA repair and accelerated DNA damage drive carcinogenesis, especially in the skin to cause melanomas, squamous cell carcinomas, and basal cell carcinomas [145,146]. Although XP patient cells are responsive to other MYC transcriptional activators, they are defective in both FUBP1 activation and FIR repression [147]. In addition to chromosomal instability induced by defective DNA repair, the mutation at the 3′ end of XPB coding sequence in XP patients negates the XPB-FIR interaction and repression by FIR [147]. The FUBP1 and FIR enrichment normally detected on FUSE are no longer observed at the MYC TSS in cell lines derived from XP patients with C-terminally truncated XPB, suggesting a failure to form the FUBP1-FIR-XPB/TFIIH bridge as a consequence of the defective interaction between XPB and FIR. Interestingly, three different XP patients with mutations causing loss of the wild type C-terminus of XPB all display UV sensitivity, developmental and aging disorders, but have very different cancer predispositions (one severe, one moderate, one with no cancer) [148]. Together with the knowledge that all patients in these families have reduced NER, these observations suggest that cancer phenotypes are not due to NER defects per se, but likely due to a second mutation. Specifically, the cancer phenotypes could arise when XPB-dependent DNA repair defects alter hyperproliferative input(s); e.g., defective function of the FUBP1/FIR/TFIIH nexus would be predicted to disrupt repression of the MYC oncogene and contribute to XP-related neoplasia.
11. Defective MYC Repression and Tissue Overgrowth in Drosophila Models of XPB-Related Disease
The Drosophila ortholog of FIR, Half pint (Hfp), behaves as a tumour suppressor, with reduced abundance of Hfp resulting in tissue overproliferation and larval overgrowth [149,150]. Haywire (Hay) is the Drosophila XPB homolog [151,152], and loss-of-function Hay mutants display phenotypes consistent with DNA repair and transcription defects [152,153]. Furthermore, Hay forms a complex with Hfp in vivo and co-ablation of Hay in Hfp loss-of-function cells reduces dMYC expression and cell growth [150], suggesting conservation between mammalian FIR-XPB and Drosophila Hfp-Hay in terms of transcriptional regulation of MYC. The Drosophila models therefore provide the means to dissect molecular mechanism(s) of defective repression of dMYC and tissue overgrowth associated with Hfp loss-of-function. Not only did Hay/XPB mutants—C-terminally truncated like the human disease alleles—strongly enhance cellular overproliferation and tissue overgrowth normally associated with the Hfp depletion, but also further impaired Pol II pausing and exacerbated dMYC derepression [154]. Thus, consistent with Pol II pausing attenuating MYC transcription, Hfp interacts with XPB/TFIIH to maintain a pool of engaged Pol II on the dMYC promoter in vivo [154]. An overview of the current understanding of MYC control by FUBP1/FIR is shown in Figure 1. Given the conserved nature of the FIR-XPB interaction, we would predict impaired Pol II pausing and defective transcriptional repression of MYC might contribute to hyperproliferation and cancer associated with XPB-related human diseases.
12. Drosophila FUBP1/Psi Interacts with Mediator to Control MYC Transcription
The capacity of the MYC promoter to integrate extracellular and developmental signals is fundamental to patterning of growth in multicellular animals. Although connections between signaling and patterning of dMYC transcription have been well delineated in Drosophila [44,79,155], until recently it was unknown whether signaling was integrated with dMYC transcription via P-element somatic inhibitor (Psi), the sole ortholog of the three mammalian FUBP proteins. Psi was originally ascribed the function of modulating splicing of transposable P-elements in Drosophila [156]. However, given that P-elements are a recent addition to Drosophila melanogaster, only entering the genome within the past 50 years [157], this function cannot reflect evolutionary pressures. Indeed, subsequent studies revealed broader roles for Psi in pre-mRNA splicing for many genes, including those required for courtship behaviour [158]. In accordance with Psi behaving in a functionally analogous manner to FUBP1, Psi is also essential for dMYC-dependent control of cell and tissue growth during Drosophila development [159]. The Psi interactome (determined by co-IP-mass spectrometry [160]) was predominantly comprised of Pol II transcriptional machinery [159]. Psi also has potent transcriptional activator capacity in vitro, mediated by conserved tyrosine-rich domains (YM1 and YM2 repeat motifs) in the C terminus; i.e., as observed for FUBP1 [161].
Of the Psi-interactors with designated functions in Pol II transcription (63% of the top 65 Psi-interactors), 32% were part of the chromatin-remodeling machinery, 12% gene specific transcriptional regulators, but by far most proteins (56%) comprised subunits of the transcriptional Mediator (MED) complex [159]. The MED complex interacts with the Pol II machinery to modulate transcription in all eukaryotes [162,163]. Although the MED complex is required for most (if not all) Pol II dependent transcription, the MED/CDK8 module can act as a sensor of developmental and environmental cues to activate specific transcriptional programs [164,165,166]. The MED complex bridges transcriptional enhancers and the Pol II machinery, to integrate developmental and environmental cues into specific transcriptional outcomes [167,168,169,170]. In line with this, MED responds to specific signaling networks to control developmental patterning in Drosophila [169,171,172,173,174]. In flies, kohtalo and skuld, which encode Drosophila homologs of the MED12 and MED13 subunits of the kinase module, are essential for the transcription of Wg/Wnt and Notch pathway targets and, thus, required to establish compartment boundaries of the wing imaginal disc [172,175,176]. More recently, a specific reduction in the expression of genes involved in wing margin formation was observed for MED26 null mutant wing disc clones [173]. At the level of promoters, direct interplay between gene/tissue-specific Hox transcription factors and MED19 is essential for regulating expression of both embryonic and larval imaginal disc patterning genes [174]. A genetic screen for factors affecting wing growth, in the background sensitized with a copy of a Minute locus mutation, revealed a requirement for MED15 in expression of selected Dpp target genes, but not EGFR or Wg target genes [175].
In general, the large (~30 subunit) MED complex can behave either as an activator or inhibitor of Pol II-dependent transcription. The “small” or core MED complex is required for activation of Pol II transcription. The “large” complex has been predominantly characterised as a transcriptional repressor and it contains an additional 4 proteins; the kinase module comprising the Cyclin dependent kinase complex (CDK8/CycC) and 2 additional MED subunits (MED12, and MED13) [166]. The impaired growth phenotype associated with Psi depletion is suppressed following either co-depletion of subunits from the transcriptionally repressive CDK8/CycC kinase module or overexpression of core MED subunits, suggesting that Psi/dMYC-dependent tissue growth depends on MED abundance and activity [159]. Moreover, the decreased cell and tissue growth associated with Psi depletion is suppressed by dMYC overexpression and enhanced by co-knockdown of dMYC. In line with an activating role in dMYC transcription, Psi is required for maintaining dMYC mRNA at endogenous levels, with the latter being significantly decreased following Psi knockdown. In accordance with dMYC depletion being due to diminished transcriptional activity, ChIP revealed that depletion of Psi decreases enrichment of initiating Pol II (S5 phosphorylated) and elongating Pol II (S2 phosphorylated) across the dMYC gene. The observation that Psi activates from a single DNA binding site within a minimal promoter [159] (as observed for FUBP1 [161]), compared with most G4-activators that require tandem sites, suggests rather than acting at the level of Pol II recruitment, Psi drives dMYC transcription downstream of pre-initiation complex assembly.
Psi interacts with core Pol II machinery to maintain cell and tissue growth, which is of great interest given the proficiency of the MYC to drive cell growth [6,42]. We predict that dMYC dysregulation is capable of such potent modification of the Psi knockdown phenotype due to MYC’s capacity to act as a transcriptional amplifier [23,24]. In the context of rapidly proliferating wing disc cells, the major program of dMYC-modulated transcription will include genes required for cell and tissue growth. Thus, in the wing Psi and MED integrate growth signals to maintain developmentally regulated dMYC transcription, cell and tissue growth. Further studies are required to determine whether human FUBP1 also interacts with MED to modulate expression of the mammalian MYC oncogene. As even subtle increases in MYC expression (>2 fold) can promote the cell and tissue overgrowth fundamental to cancer initiation and progression, these observations will have implications for human disease [1,12].
13. MYC Control at A Distance—3D Genomic Architecture in MYC Regulation
Our understanding of transcriptional regulation has been greatly extended by elucidation of the 3-dimensional structure of the mammalian genome [177,178,179]. In particular, the capacity of promoter-enhancer loops to shape local DNA structure, revealed by techniques such as 3C and Hi-C, has enabled mapping of physically adjacent chromosome segments and determination of how their arrangement in topologically associated domains (TADs) affects gene expression [180]. Boundaries of TADs are generally delineated by cohesin-CCCTC-binding factor (cohesin-CTCF) loops [181]. Genes located within a common TAD naturally make contact more frequently and are more often co-expressed than genes located in different TADs, which suggests chromosomes arrange to establish environments enriched for transcriptional regulators to enable enhancer sharing within local insulated neighbourhoods [182,183].
The Drosophila genome is similarly arranged into physical domains that form visible structures in polytene chromosomes of the salivary glands [177,179]. Drosophila cohesin maintains genome structure by interacting with genes bound by paused Pol II to regulate gene expression within TADs [184]. Drosophila CTCF also binds distinct sites to enable mitotic bookmarking (i.e., maintenance of active and inactive chromatin marks [185]), in proliferating cells [186]. Highlighting the versatility of enhancer-promoter interactions, activation from an enhancer can occur bi-directionally in the Drosophila genome [187]. In the context of enhancer sharing, complexes such as MED, which can bridge enhancers to the general transcriptional machinery, will be key to integrating 3D interactions to modulate Pol II activity.
In regard to the MYC locus, early studies identified CTCF as a repressor of the chicken MYC gene [188]. Regulation of MYC transcription by cohesin is also conserved in zebrafish [189]. Studies of long distance regulation in the mouse implicated the 8q24 region of the genome in regulation of the mammalian Myc gene [190,191]. The recent efforts, focused on the role TADs play in chromosome organization, revealed that mutations in CTCF binding sites in nonmalignant cells can initiate expression of oncogenes, including MYC, located within the insulated neighbourhood associated with T-cell acute lymphoblastic leukemia (T-ALL) pathogenesis [192]. Moreover, looping of the MYC locus is rearranged as a consequence of infection with the lymphoma-associated Epstein-Barr virus (EBV) [193]. Transactivators encoded by the virus hijack endogenous transcription machinery by recruiting SWI/SNF remodelers, to enable MYC promoter interactions with several upstream long range enhancers (ranging from -556kb to -168kb upstream of TSS) to drive MYC activation [193]. Thus, by allowing distant enhancers to come in proximity with promoters from ubiquitously expressed genes, disruption of TADs associated with MYC transcriptional control could drive cancer initiation. There are emerging links between the 3D genomic architecture and developmental signaling inputs, with NOTCH-mediated MYC activation occurring through direct binding at long-distance elements capable of forming loops with the MYC promoter [94,194]. Future studies investigating the response of the genome in 3D to signaling inputs implicated in MYC patterning, both specifically on the MYC locus and more globally, are awaited with great interest.
14. Targeting MYC for Cancer Treatment
Links to upstream pathways that input MYC transcriptional regulators would be informative for many current cancer therapies, where inhibition of signaling pathway components (particularly kinases) results in rapid development of drug resistance [195,196]. Commonly, cancer cells become resistant by accumulating mutations that activate a parallel pathway, ultimately providing compensatory signals that converge on common downstream effectors [197,198]. Therefore, co-targeting MYC in addition to upstream oncogenic signaling pathways would provide an avenue for treatment.
As an excellent proof of principle, MYC can be targeted in transgenic mouse models of disease by expression of the dominant negative Omomyc [199,200]. This basic helix-loop-helix (bHLH) protein, which is able to form heterodimers with endogenous MYC, lacks capacity to bind DNA and thus interferes with MYC-MAX association and transcriptional activation, but enables MYC to still interact with the repressor network via MIZ-1 [200]. Omomyc was efficacious at eliminating Ras-driven adenocarcinoma in mice [201,202], and more recently global expression of Omomyc has been used to successfully treat Ras-induced astrocytoma in mouse models [203]. Although expression of Omomyc did reduce proliferation in normal tissues, it was generally well tolerated, and thus provides evidence that widespread dampening of MYC activity could be an effective cancer treatment. However, other attempts to target MYC protein itself have proven to be challenging [204], and individually inhibiting targets of MYC is difficult due to their number and variety [205]. Thus, targeting regulatory networks that control MYC transcription could be a promising treatment avenue.
The potential success of targeting MYC at the transcriptional level has been demonstrated using the bromodomain and extraterminal (BET) protein inhibitor JQ1. Although initially explored as a means of modulating MYC activity by interfering with MYC-associated acetylated histones, JQ1 regulates BRD4 to limit MYC expression in cancer cells [206]. BRD4 modifies MYC transcription by binding a super-enhancer 1.7Mb downstream of the MYC promoter [207,208] (Figure 1), providing another mechanism for MYC-specific activation by a factor originally characterized as playing general transcriptional co-activator roles [209]. Moreover, treatment with JQ1 kills T-ALL leukaemia cells with acquired resistance to γ-secretase, an inhibitor of NOTCH-mediated MYC activation [210] which is known to operate through control of promoter-enhancer looping [94,194].
Through 4C mapping, the long-distance BRD4-bound enhancer was found to form a loop preferentially interacting with the MYC promoter [207], and BRD4 drives MYC transcription by associating with the MED complex and the transcriptional elongation factor p-TEFb [211,212,213]. However, inhibition of BRD4 by JQ1 abolishes binding of the entire MED complex at multiple sites across the genome of acute myeloid leukemia (AML) cells, including the MYC promoter [214]. Consequently, BRD4 inhibition or downregulation results in cell cycle exit and differentiation, which can be replicated by knockdown of certain MED subunits e.g., MED23 and MED13 [214]. Tissue-specific requirements for MED23 have also been reported in Ras-driven lung tumorigenesis [215]. Future studies investigating whether Ras-driven tumours in the lung are sensitive to MYC modulation via BRD4 treatment would therefore be of great interest.
15. Concluding Remarks and Future Considerations
Here we have brought together a snapshot of the vast web of signals integrated by the MYC promoter, with a focus on mechanisms required to fine-tune promoter output. In particular, the interplay between the two non-canonical MYC transcriptional regulators FUBP1 and FIR provides a mechanism for integration of cellular signals by enabling both rapid Pol II release (via FUBP1) and reestablishment of pausing (via FIR). The importance of these single-stranded nucleic acid binding proteins to MYC control in vivo is highlighted by their conservation between mammals and invertebrates, with Drosophila FIR/Hfp being essential for repression of dMYC [149,150,154], and Psi/FUBP1 required to maintain dMYC mRNA levels under signaling conditions conducive to growth in the fly wing [159]. The capacity of Psi/FUBP1 to modulate cell and tissue growth through interaction with the MED complex would provide a hub for integration of the developmental signaling inputs essential for the patterning of MYC transcription.
Intriguingly, the bromodomain complex proteins were also detected in complex with Psi/FUBP1 in Drosophila mass spectrometry screens [160], including the first identified bromodomain protein Brm and the core members of the Brm complex, Moira and Ebi [159]. Investigation of the physiological importance of this interaction is compelling, particularly whether Psi/FUBP1 plays a role in recruiting BRD4/Brm to the MYC promoter to elicit MYC-specific regulatory effects and/or modifies BRD4/Brm activity in order to promote release of Pol II. In addition, 4C analysis of the Drosophila dMYC promoter will undoubtedly identify long-distance elements interacting with the Brm complex, MED and Psi/FUBP1.
Recently the RNA-recognition motif (RRM)-containing protein B52, orthologous to mammalian Serine/arginine-rich splicing factors (SRSF1-6), was implicated in regulating dMYC expression and promoting cell growth [216]. B52 is essential for viability in Drosophila development [217], B52 overexpression increased dMYC promoter activity measured by dMYC-lacZ reporter activity and enrichment of phosphorylated Pol II across the dMYC promoter [216]. Determining whether B52, like the KH domain protein FUBP1 and RRM protein FIR, also possesses capacity to sequence-specifically bind ssDNA at the MYC promoter to modulate DNA topology and Pol II activity will be extremely important. Of great interest will be investigation of potential interactions with MED and the other single-stranded DNA-binding proteins implicated in MYC transcription, i.e., FUBP1/Psi and Hfp/FIR. We therefore invite future studies interrogating mechanisms by which FUBP1/Psi, FIR/Hfp and B52 sense the cellular signaling environment and how these factors modulate MYC promoter architecture to control cell and tissue growth during development, as these will shed light on MYC-dysregulation in cancer.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Levens, D. You Don’t Muck with MYC. Genes Cancer 2010, 1, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Gabay, M.; Li, Y.; Felsher, D.W. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harbor Perspect. Med. 2014, 4, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Dang, C.V. MYC on the path to cancer. Cell 2012, 149, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Grandori, C.; Gomez-Roman, N.; Felton-Edkins, Z.A.; Ngouenet, C.; Galloway, D.A.; Eisenman, R.N.; White, R.J. c-Myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nat. Cell Biol. 2005, 7, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Poortinga, G.; Wall, M.; Sanij, E.; Siwicki, K.; Ellul, J.; Brown, D.; Holloway, T.P.; Hannan, R.D.; McArthur, G.A. c-MYC coordinately regulates ribosomal gene chromatin remodeling and Pol I availability during granulocyte differentiation. Nucleic Acids Res. 2011, 39, 3267–3281. [Google Scholar] [CrossRef] [PubMed]
- Grewal, S.S.; Li, L.; Orian, A.; Eisenman, R.N.; Edgar, B.A. Myc-dependent regulation of ribosomal RNA synthesis during Drosophila development. Nat. Cell Biol. 2005, 7, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Poortinga, G.; Hannan, K.M.; Snelling, H.; Walkley, C.R.; Jenkins, A.; Sharkey, K.; Wall, M.; Brandenburger, Y.; Palatsides, M.; Pearson, R.B.; et al. MAD1 and c-MYC regulate UBF and rDNA transcription during granulocyte differentiation. EMBO J. 2004, 23, 3325–3335. [Google Scholar] [CrossRef] [PubMed]
- Bretones, G.; Delgado, M.D.; León, J. Myc and cell cycle control. Biochim. Biophys. Acta 2015, 1849, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Shichiri, M.; Hanson, K.D.; Sedivy, J.M. Effects of c-myc expression on proliferation, quiescence, and the G0 to G1 transition in nontransformed cells. Cell Growth Differ. 1993, 4, 93–104. [Google Scholar] [PubMed]
- Gowda, S.D.; Koler, R.D.; Bagby, G.C. Regulation of C-myc expression during growth and differentiation of normal and leukemic human myeloid progenitor cells. J. Clin. Investig. 1986, 77, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.I.; Lin, Y.; Calame, K. Repression of c-myc is necessary but not sufficient for terminal differentiation of B lymphocytes in vitro. Mol. Cell. Biol. 2000, 20, 8684–8695. [Google Scholar] [CrossRef] [PubMed]
- Eilers, M.; Eisenman, R.N. Myc’s broad reach. Genes Dev. 2008, 22, 2755–2766. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Woo, A.J.; Chu, J.; Snow, J.W.; Fujiwara, Y.; Kim, C.G.; Cantor, A.B.; Orkin, S.H. A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell 2010, 143, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Blackwood, E.M.; Eisenman, R.N. Max: A helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science 1991, 251, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Grandori, C.; Cowley, S.M.; James, L.P.; Eisenman, R.N. The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu. Rev. Cell Dev. Biol. 2000, 16, 653–699. [Google Scholar] [CrossRef] [PubMed]
- Si, J.; Yu, X.; Zhang, Y.; DeWille, J.W. Myc interacts with Max and Miz1 to repress C/EBPdelta promoter activity and gene expression. Mol. Cancer 2010, 9, 92. [Google Scholar] [CrossRef] [PubMed]
- Herkert, B.; Eilers, M. Transcriptional repression: The dark side of myc. Genes Cancer 2010, 1, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Orian, A. Genomic binding by the Drosophila Myc, Max, Mad/Mnt transcription factor network. Genes Dev. 2003, 17, 1101–1114. [Google Scholar] [CrossRef] [PubMed]
- Orian, A.; Grewal, S.S.; Knoepfler, P.S.; Edgar, B.A.; Parkhurst, S.M.; Eisenman, R.N. Genomic binding and transcriptional regulation by the Drosophila Myc and Mnt transcription factors. Cold Spring Harbor Symp. Quant. Biol. 2005, 70, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Wu, G.; Zhan, X.; Nolan, A.; Koh, C.; De Marzo, A.; Doan, H.M.; Fan, J.; Cheadle, C.; Fallahi, M.; et al. Cell-type independent MYC target genes reveal a primordial signature involved in biomass accumulation. PLoS ONE 2011, 6, e26057. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, G.C.; Ziff, E.B. Methylation-sensitive sequence-specific DNA binding by the c-Myc basic region. Science 1991, 251, 186–189. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, P.C.; Frank, S.R.; Wang, L.; Schroeder, M.; Liu, S.; Greene, J.; Cocito, A.; Amati, B. Genomic targets of the human c-Myc protein. Genes Dev. 2003, 17, 1115–1129. [Google Scholar] [CrossRef] [PubMed]
- Nie, Z.; Hu, G.; Wei, G.; Cui, K.; Yamane, A.; Resch, W.; Wang, R.; Green, D.R.; Tessarollo, L.; Casellas, R.; et al. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell 2012, 151, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Lovén, J.; Rahl, P.B.; Paranal, R.M.; Burge, C.B.; Bradner, J.E.; Lee, T.I.; Young, R.A. Transcriptional amplification in tumor cells with elevated c-Myc. Cell 2012, 151, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Sabo, A.; Amati, B. Genome Recognition by MYC. Cold Spring Harbor Perspect. Med. 2014, 4, a014191. [Google Scholar] [CrossRef] [PubMed]
- Walz, S.; Lorenzin, F.; Morton, J.; Wiese, K.E.; von Eyss, B.; Herold, S.; Rycak, L.; Dumay-Odelot, H.; Karim, S.; Bartkuhn, M.; et al. Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature 2014, 511, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Kress, T.R.; Sabò, A.; Amati, B. MYC: Connecting selective transcriptional control to global RNA production. Nat. Rev. Cancer 2015, 15, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Lorenzin, F.; Benary, U.; Baluapuri, A.; Walz, S.; Jung, L.A.; von Eyss, B.; Kisker, C.; Wolf, J.; Eilers, M.; Wolf, E. Different promoter affinities account for specificity in MYC-dependent gene regulation. Elife 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Wolf, E.; Lin, C.Y.; Eilers, M.; Levens, D.L. Taming of the beast: Shaping Myc-dependent amplification. Trends Cell Biol. 2015, 25, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Mathsyaraja, H.; Eisenman, R.N. Parsing Myc Paralogs in Oncogenesis. Cancer Cell 2016, 29, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.C.; Wims, M.; Spotts, G.D.; Hann, S.R.; Bradley, A. A null c-myc mutation causes lethality before 10.5 days of gestation in homozygotes and reduced fertility in heterozygous female mice. Genes Dev. 1993, 7, 671–682. [Google Scholar] [CrossRef] [PubMed]
- Dubois, N.C.; Adolphe, C.; Ehninger, A.; Wang, R.A.; Robertson, E.J.; Trumpp, A. Placental rescue reveals a sole requirement for c-Myc in embryonic erythroblast survival and hematopoietic stem cell function. Development 2008, 135, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Malynn, B.A.; de Alboran, I.M.; O'Hagan, R.C.; Bronson, R.; Davidson, L.; DePinho, R.A.; Alt, F.W. N-myc can functionally replace c-myc in murine development, cellular growth, and differentiation. Genes Dev. 2000, 14, 1390–1399. [Google Scholar] [PubMed]
- Zimmerman, K.A.; Yancopoulos, G.D.; Collum, R.G.; Smith, R.K.; Kohl, N.E.; Denis, K.A.; Nau, M.M.; Witte, O.N.; Toran-Allerand, D.; Gee, C.E. Differential expression of myc family genes during murine development. Nature 1986, 319, 780–783. [Google Scholar] [CrossRef] [PubMed]
- Hatton, B.A.; Knoepfler, P.S.; Kenney, A.M.; Rowitch, D.H.; de Alborán, I.M.; Olson, J.M.; Eisenman, R.N. N-myc is an essential downstream effector of Shh signaling during both normal and neoplastic cerebellar growth. Cancer Res. 2006, 66, 8655–8661. [Google Scholar] [CrossRef] [PubMed]
- Vo, B.T.; Wolf, E.; Kawauchi, D.; Gebhardt, A.; Rehg, J.E.; Finkelstein, D.; Walz, S.; Murphy, B.L.; Youn, Y.H.; Han, Y.-G.; et al. The Interaction of Myc with Miz1 Defines Medulloblastoma Subgroup Identity. Cancer Cell 2016, 29, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Laurenti, E.; Varnum-Finney, B.; Wilson, A.; Ferrero, I.; Blanco-Bose, W.E.; Ehninger, A.; Knoepfler, P.S.; Cheng, P.F.; MacDonald, H.R.; Eisenman, R.N.; et al. Hematopoietic stem cell function and survival depend on c-Myc and N-Myc activity. Cell Stem Cell 2008, 3, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.; Laurenti, E.; Oser, G.; van der Wath, R.C.; Blanco-Bose, W.; Jaworski, M.; Offner, S.; Dunant, C.F.; Eshkind, L.; Bockamp, E.; et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell 2008, 135, 1118–1129. [Google Scholar] [CrossRef] [PubMed]
- Trumpp, A.; Refaeli, Y.; Oskarsson, T.; Gasser, S.; Murphy, M.; Martin, G.R.; Bishop, J.M. c-Myc regulates mammalian body size by controlling cell number but not cell size. Nature 2001, 414, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Benassayag, C.; Montero, L.; Colombie, N.; Gallant, P.; Cribbs, D.; Morello, D. Human c-Myc Isoforms Differentially Regulate Cell Growth and Apoptosis in Drosophila melanogaster. Mol. Cell. Biol. 2005, 25, 9897–9909. [Google Scholar] [CrossRef] [PubMed]
- Schreiber-Agus, N.; Stein, D.; Chen, K.; Goltz, J.S.; Stevens, L.; DePinho, R.A. Drosophila Myc is oncogenic in mammalian cells and plays a role in the diminutive phenotype. Proc. Natl. Acad. Sci. USA 1997, 94, 1235–1240. [Google Scholar] [CrossRef] [PubMed]
- Johnston, L.A.; Prober, D.A.; Edgar, B.A.; Eisenman, R.N.; Gallant, P. Drosophila myc regulates cellular growth during development. Cell 1999, 98, 779–790. [Google Scholar] [CrossRef]
- Prober, D.A.; Edgar, B.A. Interactions between Ras1, dMyc, and dPI3K signaling in the developing Drosophila wing. Genes Dev. 2002, 16, 2286–2299. [Google Scholar] [CrossRef] [PubMed]
- Er Amanda Lee, J.; May Parsons, L.; M Quinn, L. MYC function and regulation in flies: How Drosophila has enlightened MYC cancer biology. AIMS Genet. 2014, 1, 81–98. [Google Scholar] [CrossRef]
- Grifoni, D.; Bellosta, P. Drosophila Myc: A master regulator of cellular performance. Biochim. Biophys. Acta 2015, 1849, 570–581. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Sung, E.; Donlin-Asp, P.G.; Corces, V.G. A subset of Drosophila Myc sites remain associated with mitotic chromosomes colocalized with insulator proteins. Nat. Commun. 2013, 4, 1464. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Kim, H.D.; Jung, Y.; Kim, J.; Chung, J. Drosophila Low Temperature Viability Protein 1 (LTV1) Is Required for Ribosome Biogenesis and Cell Growth Downstream of Drosophila Myc (dMyc). J. Biol. Chem. 2015, 290, 13591–13604. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, N.C.; Tchoubrieva, E.B.; Chahal, A.; Woods, S.; Lee, A.; Lin, J.I.; Parsons, L.; Jastrzebski, K.; Poortinga, G.; Hannan, K.M.; et al. S6 Kinase is essential for MYC-dependent rDNA transcription in Drosophila. Cell. Signal. 2015, 27, 2045–2053. [Google Scholar] [CrossRef] [PubMed]
- de la Cova, C.; Abril, M.; Bellosta, P.; Gallant, P.; Johnston, L.A. Drosophila myc regulates organ size by inducing cell competition. Cell 2004, 117, 107–116. [Google Scholar] [CrossRef]
- Levens, D. Cellular MYCro economics: Balancing MYC function with MYC expression. Cold Spring Harbor Perspect. Med. 2013, 3, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Posternak, V.; Ung, M.H.; Cheng, C.; Cole, M.D. MYC Mediates mRNA Cap Methylation of Canonical Wnt/beta-catenin Signaling Transcripts by Recruiting CDK7 and RNA Methyltransferase. Mol. Cancer Res. 2016, 15, 213. [Google Scholar] [CrossRef] [PubMed]
- Croce, C.M. Oncogenes and cancer. N. Engl. J. Med. 2008, 358, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Sampson, V.B.; Rong, N.H.; Han, J.; Yang, Q.; Aris, V.; Soteropoulos, P.; Petrelli, N.J.; Dunn, S.P.; Krueger, L.J. MicroRNA let-7a down-regulates MYC and reverts MYC-induced growth in Burkitt lymphoma cells. Cancer Res. 2007, 67, 9762–9770. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, S.; Saini, S.; Majid, S.; Hirata, H.; Ueno, K.; Deng, G.; Dahiya, R. MicroRNA-34a modulates c-Myc transcriptional complexes to suppress malignancy in human prostate cancer cells. PLoS ONE 2012, 7, e29722. [Google Scholar] [CrossRef] [PubMed]
- Daneshvar, K.; Nath, S.; Khan, A.; Shover, W.; Richardson, C.; Goodliffe, J.M. MicroRNA miR-308 regulates dMyc through a negative feedback loop in Drosophila. Biol. Open 2013, 2, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Zhao, X.; Tao, J. c-MYC-miRNA circuitry: A central regulator of aggressive B-cell malignancies. Cell Cycle 2014, 13, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Jackstadt, R.; Hermeking, H. MicroRNAs as regulators and mediators of c-MYC function. Biochim. Biophys. Acta 2015, 1849, 544–553. [Google Scholar] [CrossRef] [PubMed]
- Herranz, H.; Hong, X.; Pérez, L.; Ferreira, A.; Olivieri, D.; Cohen, S.M.; Milán, M. The miRNA machinery targets Mei-P26 and regulates Myc protein levels in the Drosophila wing. EMBO J. 2010, 29, 1688–1698. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.; Boulan, L.; Pérez, L.; Milán, M. Mei-P26 mediates tissue-specific responses to the Brat tumor suppressor and the dMyc proto-oncogene in Drosophila. Genetics 2014, 198, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Schwamborn, J.C.; Berezikov, E.; Knoblich, J.A. The TRIM-NHL protein TRIM32 activates microRNAs and prevents self-renewal in mouse neural progenitors. Cell 2009, 136, 913–925. [Google Scholar] [CrossRef] [PubMed]
- Gregory, M.A.; Hann, S.R. c-Myc proteolysis by the ubiquitin-proteasome pathway: Stabilization of c-Myc in Burkitt’s lymphoma cells. Mol. Cell. Biol. 2000, 20, 2423–2435. [Google Scholar] [CrossRef] [PubMed]
- Yeh, E.; Cunningham, M.; Arnold, H.; Chasse, D.; Monteith, T.; Ivaldi, G.; Hahn, W.C.; Stukenberg, P.T.; Shenolikar, S.; Uchida, T.; et al. A signalling pathway controlling c-Myc degradation that impacts oncogenic transformation of human cells. Nat. Cell Biol. 2004, 6, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Lutterbach, B.; Hann, S.R. Hierarchical phosphorylation at N-terminal transformation-sensitive sites in c-Myc protein is regulated by mitogens and in mitosis. Mol. Cell. Biol. 1994, 14, 5510–5522. [Google Scholar] [CrossRef] [PubMed]
- Sears, R.; Leone, G.; DeGregori, J.; Nevins, J.R. Ras enhances Myc protein stability. Mol. Cell 1999, 3, 169–179. [Google Scholar] [CrossRef]
- Sears, R.; Nuckolls, F.; Haura, E.; Taya, Y.; Tamai, K.; Nevins, J.R. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000, 14, 2501–2514. [Google Scholar] [CrossRef] [PubMed]
- Welcker, M.; Orian, A.; Jin, J.; Grim, J.E.; Grim, J.A.; Harper, J.W.; Eisenman, R.N.; Clurman, B.E. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc. Natl. Acad. Sci. USA 2004, 101, 9085–9090. [Google Scholar] [CrossRef] [PubMed]
- Yada, M.; Hatakeyama, S.; Kamura, T.; Nishiyama, M.; Tsunematsu, R.; Imaki, H.; Ishida, N.; Okumura, F.; Nakayama, K.; Nakayama, K.I. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J. 2004, 23, 2116–2125. [Google Scholar] [CrossRef] [PubMed]
- Gregory, M.A.; Qi, Y.; Hann, S.R. Phosphorylation by glycogen synthase kinase-3 controls c-myc proteolysis and subnuclear localization. J. Biol. Chem. 2003, 278, 51606–51612. [Google Scholar] [CrossRef] [PubMed]
- Parisi, F.; Riccardo, S.; Daniel, M.; Saqcena, M.; Kundu, N.; Pession, A.; Grifoni, D.; Stocker, H.; Tabak, E.; Bellosta, P. Drosophila insulin and target of rapamycin (TOR) pathways regulate GSK3 beta activity to control Myc stability and determine Myc expression in vivo. BMC Biol. 2011, 9, 65. [Google Scholar] [CrossRef] [PubMed]
- Pulverer, B.J.; Fisher, C.; Vousden, K.; Littlewood, T.; Evan, G.; Woodgett, J.R. Site-specific modulation of c-Myc cotransformation by residues phosphorylated in vivo. Oncogene 1994, 9, 59–70. [Google Scholar] [PubMed]
- Yano, T.; Sander, C.A.; Clark, H.M.; Dolezal, M.V.; Jaffe, E.S.; Raffeld, M. Clustered mutations in the second exon of the MYC gene in sporadic Burkitt’s lymphoma. Oncogene 1993, 8, 2741–2748. [Google Scholar] [PubMed]
- Flinn, E.M.; Busch, C.M.; Wright, A.P. myc boxes, which are conserved in myc family proteins, are signals for protein degradation via the proteasome. Mol. Cell. Biol. 1998, 18, 5961–5969. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Jiang, C.; Pan, J.; Wang, X.; Jin, J.; Zhao, L.; Pan, W.; Liao, G.; Cai, X.; Li, X.; et al. Regulation of c-Myc protein stability by proteasome activator REGγ. Cell Death Differ. 2015, 22, 1000–1011. [Google Scholar] [CrossRef] [PubMed]
- Cowling, V.H.; Turner, S.A.; Cole, M.D. Burkitt's lymphoma-associated c-Myc mutations converge on a dramatically altered target gene response and implicate Nol5a/Nop56 in oncogenesis. Oncogene 2014, 33, 3519–3527. [Google Scholar] [CrossRef] [PubMed]
- Kelly, K.; Cochran, B.H.; Stiles, C.D.; Leder, P. Cell-specific regulation of the c-myc gene by lymphocyte mitogens and platelet-derived growth factor. Cell 1983, 35, 603–610. [Google Scholar] [CrossRef]
- Grosso, L.E.; Pitot, H.C. Chromatin structure of the c-myc gene in HL-60 cells during alterations of transcriptional activity. Cancer Res. 1985, 45, 5035–5041. [Google Scholar] [PubMed]
- Bentley, D.L.; Groudine, M. A block to elongation is largely responsible for decreased transcription of c-myc in differentiated HL60 cells. Nature 1986, 321, 702–706. [Google Scholar] [CrossRef] [PubMed]
- Duman-Scheel, M.; Johnston, L.A.; Du, W. Repression of dMyc expression by Wingless promotes Rbf-induced G1 arrest in the presumptive Drosophila wing margin. Proc. Natl. Acad. Sci. USA 2004, 101, 3857–3862. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.C.; Johnston, L.A. Control of wing size and proportions by Drosophila myc. Genetics 2010, 184, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Herranz, H.; Pérez, L.; Martín, F.A.; Milán, M. A Wingless and Notch double-repression mechanism regulates G1-S transition in the Drosophila wing. EMBO J. 2008, 27, 1633–1645. [Google Scholar] [CrossRef] [PubMed]
- Johnston, L.A.; Sanders, A.L. Wingless promotes cell survival but constrains growth during Drosophila wing development. Nat. Cell Biol. 2003, 5, 827–833. [Google Scholar] [CrossRef] [PubMed]
- Sansom, O.J.; Meniel, V.S.; Muncan, V.; Phesse, T.J.; Wilkins, J.A.; Reed, K.R.; Vass, J.K.; Athineos, D.; Clevers, H.; Clarke, A.R. Myc deletion rescues APC deficiency in the small intestine. Nature 2007, 446, 676–679. [Google Scholar] [CrossRef] [PubMed]
- He, T.C.; Sparks, A.B.; Rago, C.; Hermeking, H.; Zawel, L.; da Costa, L.T.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Identification of c-MYC as a target of the APC pathway. Science 1998, 281, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Korinek, V.; Barker, N.; Morin, P.J.; van Wichen, D.; de Weger, R.; Kinzler, K.W.; Vogelstein, B.; Clevers, H. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/- colon carcinoma. Science 1997, 275, 1784–1787. [Google Scholar] [CrossRef] [PubMed]
- Sansom, O.J.; Reed, K.R.; Hayes, A.J.; Ireland, H.; Brinkmann, H.; Newton, I.P.; Batlle, E.; Simon-Assmann, P.; Clevers, H.; Nathke, I.S.; et al. Loss of APC in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes Dev. 2004, 18, 1385–1390. [Google Scholar] [CrossRef] [PubMed]
- Van Es, J.H.; Jay, P.; Gregorieff, A.; van Gijn, M.E.; Jonkheer, S.; Hatzis, P.; Thiele, A.; van den Born, M.; Begthel, H.; Brabletz, T.; et al. Wnt signalling induces maturation of Paneth cells in intestinal crypts. Nat. Cell Biol. 2005, 7, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Andreu, P.; Colnot, S.; Godard, C.; Gad, S.; Chafey, P.; Niwa-Kawakita, M.; Laurent-Puig, P.; Kahn, A.; Robine, S.; Perret, C.; et al. Crypt-restricted proliferation and commitment to the Paneth cell lineage following APC loss in the mouse intestine. Development 2005, 132, 1443–1451. [Google Scholar] [CrossRef] [PubMed]
- Roy, M.; Pear, W.S.; Aster, J.C. The multifaceted role of Notch in cancer. Curr. Opin. Genet. Dev. 2007, 17, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, Y.; Banerjee, S.; Sarkar, F.H. Exploitation of the Notch signaling pathway as a novel target for cancer therapy. Anticancer Res. 2008, 28, 3621–3630. [Google Scholar] [PubMed]
- De Celis, J.F.; Garcia-Bellido, A.; Bray, S.J. Activation and function of Notch at the dorsal-ventral boundary of the wing imaginal disc. Development 1996, 122, 359–369. [Google Scholar] [PubMed]
- Herranz, H.; Stamataki, E.; Feiguin, F.; Milán, M. Self-refinement of Notch activity through the transmembrane protein Crumbs: Modulation of gamma-secretase activity. EMBO Rep. 2006, 7, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Baonza, A.; Garcia-Bellido, A. Notch signaling directly controls cell proliferation in the Drosophila wing disc. Proc. Natl. Acad. Sci. USA 2000, 97, 2609–2614. [Google Scholar] [CrossRef] [PubMed]
- Neumann, C.J.; Cohen, S.M. A hierarchy of cross-regulation involving Notch, wingless, vestigial and cut organizes the dorsal/ventral axis of the Drosophila wing. Development 1996, 122, 3477–3485. [Google Scholar] [PubMed]
- Herranz, D.; Ambesi-Impiombato, A.; Palomero, T.; Schnell, S.A.; Belver, L.; Wendorff, A.A.; Xu, L.; Castillo-Martin, M.; Llobet-Navás, D.; Cordon-Cardo, C.; et al. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat. Med. 2014, 20, 1130–1137. [Google Scholar] [CrossRef] [PubMed]
- Djiane, A.; Krejci, A.; Bernard, F.; Fexova, S.; Millen, K.; Bray, S.J. Dissecting the mechanisms of Notch induced hyperplasia. EMBO J. 2013, 32, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Aradhya, R.; Zmojdzian, M.; Da Ponte, J.P.; Jagla, K. Muscle niche-driven Insulin-Notch-Myc cascade reactivates dormant Adult Muscle Precursors in Drosophila. Elife 2015, 4, e08497. [Google Scholar] [CrossRef] [PubMed]
- Qian, C.; Liu, F.; Ye, B.; Zhang, X.; Liang, Y.; Yao, J. Notch4 promotes gastric cancer growth through activation of Wnt1/β-catenin signaling. Mol. Cell. Biochem. 2015, 401, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Klinakis, A.; Szabolcs, M.; Politi, K.; Kiaris, H.; Artavanis-Tsakonas, S.; Efstratiadis, A. Myc is a Notch1 transcriptional target and a requisite for Notch1-induced mammary tumorigenesis in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 9262–9267. [Google Scholar] [CrossRef] [PubMed]
- Palomero, T.; Lim, W.K.; Odom, D.T.; Sulis, M.L.; Real, P.J.; Margolin, A.; Barnes, K.C.; O’Neil, J.; Neuberg, D.; Weng, A.P.; et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc. Natl. Acad. Sci. USA 2006, 103, 18261–18266. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.M.; Calvo, J.A.; Draheim, K.M.; Cunningham, L.A.; Hermance, N.; Beverly, L.; Krishnamoorthy, V.; Bhasin, M.; Capobianco, A.J.; Kelliher, M.A. Notch1 contributes to mouse T-cell leukemia by directly inducing the expression of c-myc. Mol. Cell. Biol. 2006, 26, 8022–8031. [Google Scholar] [CrossRef] [PubMed]
- Weng, A.P.; Millholland, J.M.; Yashiro-Ohtani, Y.; Arcangeli, M.L.; Lau, A.; Wai, C.; Del Bianco, C.; Rodriguez, C.G.; Sai, H.; Tobias, J.; et al. c-Myc is an important direct target of Notch1 in T-cell acute lymphoblastic leukemia/lymphoma. Genes Dev. 2006, 20, 2096–2109. [Google Scholar] [CrossRef] [PubMed]
- Mazur, P.K.; Einwächter, H.; Lee, M.; Sipos, B.; Nakhai, H.; Rad, R.; Zimber-Strobl, U.; Strobl, L.J.; Radtke, F.; Klöppel, G.; et al. Notch2 is required for progression of pancreatic intraepithelial neoplasia and development of pancreatic ductal adenocarcinoma. Proc. Natl. Acad. Sci. USA 2010, 107, 13438–13443. [Google Scholar] [CrossRef] [PubMed]
- Doumpas, N.; Ruiz-Romero, M.; Blanco, E.; Edgar, B.; Corominas, M.; Teleman, A.A. Brk regulates wing disc growth in part via repression of Myc expression. EMBO Rep. 2013, 14, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-R.; Kang, Y.; Siegel, P.M.; Massagué, J. E2F4/5 and p107 as Smad cofactors linking the TGFbeta receptor to c-myc repression. Cell 2002, 110, 19–32. [Google Scholar] [CrossRef]
- Ren, F.; Shi, Q.; Chen, Y.; Jiang, A.; Ip, Y.T.; Jiang, H.; Jiang, J. Drosophila Myc integrates multiple signaling pathways to regulate intestinal stem cell proliferation during midgut regeneration. Cell Res. 2013, 23, 1133–1146. [Google Scholar] [CrossRef] [PubMed]
- Rui, L.; Drennan, A.C.; Ceribelli, M.; Zhu, F.; Wright, G.W.; Huang, D.W.; Xiao, W.; Li, Y.; Grindle, K.M.; Lu, L.; et al. Epigenetic gene regulation by Janus kinase 1 in diffuse large B-cell lymphoma. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, E7260–E7267. [Google Scholar] [CrossRef] [PubMed]
- Teleman, A.A.; Hietakangas, V.; Sayadian, A.C.; Cohen, S.M. Nutritional control of protein biosynthetic capacity by insulin via Myc in Drosophila. Cell Metable 2008, 7, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Neto-Silva, R.M.; de Beco, S.; Johnston, L.A. Evidence for a growth-stabilizing regulatory feedback mechanism between Myc and Yorkie, the Drosophila homolog of Yap. Dev. Cell 2010, 19, 507–520. [Google Scholar] [CrossRef] [PubMed]
- Xiao, W.; Wang, J.; Ou, C.; Zhang, Y.; Ma, L.; Weng, W.; Pan, Q.; Sun, F. Mutual interaction between YAP and c-Myc is critical for carcinogenesis in liver cancer. Biochem. Biophys. Res. Commun. 2013, 439, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Dalla-Favera, R.; Bregni, M.; Erikson, J.; Patterson, D.; Gallo, R.C.; Croce, C.M. Human c-myc onc gene is located on the region of chromosome 8 that is translocated in Burkitt lymphoma cells. Proc. Natl. Acad. Sci. USA 1982, 79, 7824–7827. [Google Scholar] [CrossRef] [PubMed]
- Pelicci, P.G.; Knowles, D.M.; Magrath, I.; Dalla-Favera, R. Chromosomal breakpoints and structural alterations of the c-myc locus differ in endemic and sporadic forms of Burkitt lymphoma. Proc. Natl. Acad. Sci. USA 1986, 83, 2984–2988. [Google Scholar] [CrossRef] [PubMed]
- Spencer, C.A.; LeStrange, R.C.; Novak, U.; Hayward, W.S.; Groudine, M. The block to transcription elongation is promoter dependent in normal and Burkitt's lymphoma c-myc alleles. Genes Dev. 1990, 4, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Spencer, C.A.; Groudine, M. Molecular analysis of the c-myc transcription elongation block. Implications for the generation of Burkitt’s lymphoma. Ann. N. Y. Acad. Sci. 1990, 599, 12–28. [Google Scholar] [CrossRef] [PubMed]
- Spencer, C.A.; Groudine, M. Control of c-myc regulation in normal and neoplastic cells. Adv. Cancer Res. 1991, 56, 1–48. [Google Scholar] [PubMed]
- Siebenlist, U.; Hennighausen, L.; Battey, J.; Leder, P. Chromatin structure and protein binding in the putative regulatory region of the c-myc gene in Burkitt lymphoma. Cell 1984, 37, 381–391. [Google Scholar] [CrossRef]
- Schneider, E.E.; Albert, T.; Wolf, D.A.; Eick, D. Regulation of c-myc and immunoglobulin kappa gene transcription by promoter-proximal pausing of RNA polymerase II. Curr. Top. Microbiol. Immunol. 1999, 246, 225–231. [Google Scholar] [PubMed]
- Krumm, A.; Hickey, L.B.; Groudine, M. Promoter-proximal pausing of RNA polymerase II defines a general rate-limiting step after transcription initiation. Genes Dev. 1995, 9, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Grosso, L.E.; Pitot, H.C. Transcriptional regulation of c-myc during chemically induced differentiation of HL-60 cultures. Cancer Res. 1985, 45, 847–850. [Google Scholar] [PubMed]
- Avigan, M.I.; Strober, B.; Levens, D. A far upstream element stimulates c-myc expression in undifferentiated leukemia cells. J. Biol. Chem. 1990, 265, 18538–18545. [Google Scholar] [PubMed]
- Duncan, R.; Bazar, L.; Michelotti, G.; Tomonaga, T.; Krutzsch, H.; Avigan, M.; Levens, D. A sequence-specific, single-strand binding protein activates the far upstream element of c-myc and defines a new DNA-binding motif. Genes Dev. 1994, 8, 465–480. [Google Scholar] [CrossRef] [PubMed]
- Michelotti, G.A.; Michelotti, E.F.; Pullner, A.; Duncan, R.C.; Eick, D.; Levens, D. Multiple single-stranded cis elements are associated with activated chromatin of the human c-myc gene in vivo. Mol. Cell. Biol. 1996, 16, 2656–2669. [Google Scholar] [CrossRef] [PubMed]
- Krumm, A.; Meulia, T.; Brunvand, M.; Groudine, M. The block to transcriptional elongation within the human c-myc gene is determined in the promoter-proximal region. Genes Dev. 1992, 6, 2201–2213. [Google Scholar] [CrossRef] [PubMed]
- Bazar, L.; Meighen, D.; Harris, V.; Duncan, R.; Levens, D.; Avigan, M. Targeted melting and binding of a DNA regulatory element by a transactivator of c-myc. J. Biol. Chem. 1995, 270, 8241–8248. [Google Scholar] [CrossRef] [PubMed]
- Kouzine, F.; Sanford, S.; Elisha-Feil, Z.; Levens, D. The functional response of upstream DNA to dynamic supercoiling in vivo. Nat. Struct. Mol. Biol. 2008, 15, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Levens, D. How the c-myc promoter works and why it sometimes does not. J. Natl. Cancer Inst. Monogr. 2008, 2008, 41–43. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Liu, J.; Collins, I.; Sanford, S.; O'Connell, B.; Benham, C.J.; Levens, D. Loss of FBP function arrests cellular proliferation and extinguishes c-myc expression. EMBO J. 2000, 19, 1034–1044. [Google Scholar] [CrossRef] [PubMed]
- Rabenhorst, U.; Beinoraviciute-Kellner, R.; Brezniceanu, M.-L.; Joos, S.; Devens, F.; Lichter, P.; Rieker, R.J.; Trojan, J.; Chung, H.-J.; Levens, D.L.; et al. Overexpression of the far upstream element binding protein 1 in hepatocellular carcinoma is required for tumor growth. Hepatology 2009, 50, 1121–1129. [Google Scholar] [CrossRef] [PubMed]
- Rabenhorst, U.; Thalheimer, F.B.; Gerlach, K.; Kijonka, M.; Böhm, S.; Krause, D.S.; Vauti, F.; Arnold, H.-H.; Schroeder, T.; Schnütgen, F.; et al. Single-Stranded DNA-Binding Transcriptional Regulator FUBP1 Is Essential for Fetal and Adult Hematopoietic Stem Cell Self-Renewal. Cell Rep. 2015, 11, 1847–1855. [Google Scholar] [CrossRef] [PubMed]
- Gartel, A.L.; Ye, X.; Goufman, E.; Shianov, P.; Hay, N.; Najmabadi, F.; Tyner, A.L. Myc represses the p21(WAF1/CIP1) promoter and interacts with Sp1/Sp3. Proc. Natl. Acad. Sci. USA 2001, 98, 4510–4515. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Chung, Y.-J.; Castellar, E.R.P.; Zheng, Y.; Chung, H.-J.; Bandle, R.; Liu, J.; Tessarollo, L.; Batchelor, E.; Aplan, P.D.; et al. Far Upstream Element Binding Protein Plays aCrucial Role in Embryonic Development, Hematopoiesis, and Stabilizing MYC Expression Levels. Am. J. Pathol. 2016, 186, 701–715. [Google Scholar] [CrossRef] [PubMed]
- Davis-Smyth, T.; Duncan, R.C.; Zheng, T.; Michelotti, G.; Levens, D. The far upstream element-binding proteins comprise an ancient family of single-strand DNA-binding transactivators. J. Biol. Chem. 1996, 271, 31679–31687. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.-J.; Liu, J.; Dundr, M.; Nie, Z.; Sanford, S.; Levens, D. FBPs are calibrated molecular tools to adjust gene expression. Mol. Cell. Biol. 2006, 26, 6584–6597. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; He, L.; Collins, I.; Ge, H.; Libutti, D.; Li, J.; Egly, J.M.; Levens, D. The FBP interacting repressor targets TFIIH to inhibit activated transcription. Mol. Cell 2000, 5, 331–341. [Google Scholar] [CrossRef]
- Hahn, S. Structure and mechanism of the RNA polymerase II transcription machinery. Nat. Struct. Mol. Biol. 2004, 11, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Fazal, F.M.; Meng, C.A.; Murakami, K.; Kornberg, R.D.; Block, S.M. Real-time observation of the initiation of RNA polymerase II transcription. Nature 2015, 525, 274–277. [Google Scholar] [CrossRef] [PubMed]
- Hahn, S.; Buratowski, S. Structural biology: Snapshots of transcription initiation. Nature 2016, 533, 331. [Google Scholar] [CrossRef] [PubMed]
- Hirose, Y.; Ohkuma, Y. Phosphorylation of the C-terminal domain of RNA polymerase II plays central roles in the integrated events of eucaryotic gene expression. J. Biochem. 2007, 141, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Larivière, L.; Seizl, M.; Cramer, P. A structural perspective on Mediator function. Curr. Opin. Cell Biol. 2012, 24, 305–313. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.H.; Jin, Y.; Struhl, K. TFIIH Phosphorylation of the Pol II CTD Stimulates Mediator Dissociation from the Preinitiation Complex and Promoter Escape. Mol. Cell 2014, 54, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Drapkin, R.; Reinberg, D. The multifunctional TFIIH complex and transcriptional control. Trends Biochem. Sci. 1994, 19, 504–508. [Google Scholar] [CrossRef]
- Tirode, F.; Busso, D.; Coin, F.; Egly, J.M. Reconstitution of the transcription factor TFIIH: Assignment of functions for the three enzymatic subunits, XPB, XPD, and cdk7. Mol. Cell 1999, 3, 87–95. [Google Scholar] [CrossRef]
- Bradsher, J.; Coin, F.; Egly, J.M. Distinct roles for the helicases of TFIIH in transcript initiation and promoter escape. J. Biol. Chem. 2000, 275, 2532–2538. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kouzine, F.; Nie, Z.; Chung, H.-J.; Elisha-Feil, Z.; Weber, A.; Zhao, K.; Levens, D. The FUSE/FBP/FIR/TFIIH system is a molecular machine programming a pulse of c-myc expression. EMBO J. 2006, 25, 2119–2130. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, K.; Tomonaga, T.; Shimada, H.; Shioya, A.; Higashi, M.; Matsubara, H.; Harigaya, K.; Nomura, F.; Libutti, D.; Levens, D.; et al. An essential role of alternative splicing of c-myc suppressor FUSE-binding protein-interacting repressor in carcinogenesis. Cancer Res. 2006, 66, 1409–1417. [Google Scholar] [CrossRef] [PubMed]
- Compe, E.; Egly, J.-M. TFIIH: When transcription met DNA repair. Nat. Rev. Mol. Cell Biol. 2012, 13, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Coin, F.; Bergmann, E.; Tremeau-Bravard, A.; Egly, J.M. Mutations in XPB and XPD helicases found in xeroderma pigmentosum patients impair the transcription function of TFIIH. EMBO J. 1999, 18, 1357–1366. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Akoulitchev, S.; Weber, A.; Ge, H.; Chuikov, S.; Libutti, D.; Wang, X.W.; Conaway, J.W.; Harris, C.C.; Conaway, R.C.; et al. Defective interplay of activators and repressors with TFIH in xeroderma pigmentosum. Cell 2001, 104, 353–363. [Google Scholar] [CrossRef]
- Oh, K.-S.; Khan, S.G.; Jaspers, N.G.J.; Raams, A.; Ueda, T.; Lehmann, A.; Friedmann, P.S.; Emmert, S.; Gratchev, A.; Lachlan, K.; et al. Phenotypic heterogeneity in the XPB DNA helicase gene (ERCC3): xeroderma pigmentosum without and with Cockayne syndrome. Hum. Mutat. 2006, 27, 1092–1103. [Google Scholar] [CrossRef] [PubMed]
- Quinn, L.M.; Dickins, R.A.; Coombe, M.; Hime, G.R.; Bowtell, D.D.L.; Richardson, H. Drosophila Hfp negatively regulates dmyc and stg to inhibit cell proliferation. Development 2004, 131, 1411–1423. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, N.C.; Johanson, T.M.; Cranna, N.J.; Er, A.L.J.; Richardson, H.E.; Hannan, R.D.; Quinn, L.M. Hfp inhibits Drosophila myc transcription and cell growth in a TFIIH/Hay-dependent manner. Development 2010, 137, 2875–2884. [Google Scholar] [CrossRef] [PubMed]
- Mounkes, L.C.; Jones, R.S.; Liang, B.C.; Gelbart, W.; Fuller, M.T. A Drosophila model for xeroderma pigmentosum and Cockayne’s syndrome: Haywire encodes the fly homolog of ERCC3, a human excision repair gene. Cell 1992, 71, 925–937. [Google Scholar] [CrossRef]
- Mounkes, L.C.; Fuller, M.T. Molecular characterization of mutant alleles of the DNA repair/basal transcription factor haywire/ERCC3 in Drosophila. Genetics 1999, 152, 291–297. [Google Scholar] [PubMed]
- Merino, C.; Reynaud, E.; Vázquez, M.; Zurita, M. DNA repair and transcriptional effects of mutations in TFIIH in Drosophila development. Mol. Biol. Cell 2002, 13, 3246–3256. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.A.; Mitchell, N.C.; Zaytseva, O.; Chahal, A.; Mendis, P.; Cartier-Michaud, A.; Parsons, L.M.; Poortinga, G.; Levens, D.L.; Hannan, R.D.; et al. Defective Hfp-dependent transcriptional repression of dMYC is fundamental to tissue overgrowth in Drosophila XPB models. Nat. Commun. 2015, 6, 7404. [Google Scholar] [CrossRef] [PubMed]
- Gallant, P. Drosophila Myc. Adv. Cancer Res. 2009, 103, 111–144. [Google Scholar] [PubMed]
- Siebel, C.W.; Kanaar, R.; Rio, D.C. Regulation of tissue-specific P-element pre-mRNA splicing requires the RNA-binding protein PSI. Genes Dev. 1994, 8, 1713–1725. [Google Scholar] [CrossRef] [PubMed]
- Brookfield, J.F.; Montgomery, E.; Langley, C.H. Apparent absence of transposable elements related to the P elements of D. melanogaster in other species of Drosophila. Nature 1984, 310, 330–332. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Taliaferro, J.M.; Klibaite, U.; Hilgers, V.; Shaevitz, J.W.; Rio, D.C. The PSI-U1 snRNP interaction regulates male mating behavior in Drosophila. Proc. Natl. Acad. Sci. USA 2016, 113, 5269–5274. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Zaysteva, O.; Nie, Z.; Mitchell, N.C.; Amanda Lee, J.E.; Ware, T.; Parsons, L.; Luwor, R.; Poortinga, G.; Hannan, R.D.; et al. Defining the essential function of FBP/KSRP proteins: Drosophila Psi interacts with the mediator complex to modulate MYC transcription and tissue growth. Nucleic Acids Res. 2016, 44, 7646–7658. [Google Scholar] [CrossRef] [PubMed]
- Guruharsha, K.G.; Rual, J.-F.; Zhai, B.; Mintseris, J.; Vaidya, P.; Vaidya, N.; Beekman, C.; Wong, C.; Rhee, D.Y.; Cenaj, O.; et al. A protein complex network of Drosophila melanogaster. Cell 2011, 147, 690–703. [Google Scholar] [CrossRef] [PubMed]
- Duncan, R.; Collins, I.; Tomonaga, T.; Zhang, T.; Levens, D. A unique transactivation sequence motif is found in the carboxyl-terminal domain of the single-strand-binding protein FBP. Mol. Cell. Biol. 1996, 16, 2274–2282. [Google Scholar] [CrossRef] [PubMed]
- Boube, M.; Joulia, L.; Cribbs, D.L.; Bourbon, H.-M. Evidence for a mediator of RNA polymerase II transcriptional regulation conserved from yeast to man. Cell 2002, 110, 143–151. [Google Scholar] [CrossRef]
- Bourbon, H.-M. Comparative genomics supports a deep evolutionary origin for the large, four-module transcriptional mediator complex. Nucleic Acids Res. 2008, 36, 3993–4008. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.W.; Veschambre, P.; Erdjument-Bromage, H.; Tempst, P.; Conaway, J.W.; Conaway, R.C.; Kornberg, R.D. Mammalian mediator of transcriptional regulation and its possible role as an end-point of signal transduction pathways. Proc. Natl. Acad. Sci. USA 1998, 95, 8538–8543. [Google Scholar] [CrossRef] [PubMed]
- Taatjes, D.J. The human Mediator complex: A versatile, genome-wide regulator of transcription. Trends Biochem. Sci. 2010, 35, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Allen, B.L.; Taatjes, D.J. The Mediator complex: A centralintegrator of transcription. Nat. Rev. Mol. Cell Biol. 2015, 16, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Van de Peppel, J.; Kettelarij, N.; van Bakel, H.; Kockelkorn, T.T.J.P.; van Leenen, D.; Holstege, F.C.P. Mediator expression profiling epistasis reveals a signal transduction pathway with antagonistic submodules and highly specific downstream targets. Mol. Cell 2005, 19, 511–522. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Xu, X.; Hecht, A.; Boyer, T.G. Mediator is a transducer of Wnt/beta-catenin signaling. J. Biol. Chem. 2006, 281, 14066–14075. [Google Scholar] [CrossRef] [PubMed]
- Carrera, I.; Janody, F.; Leeds, N.; Duveau, F.; Treisman, J.E. Pygopus activates Wingless target gene transcription through the mediator complex subunits Med12 and Med13. Proc. Natl. Acad. Sci. USA 2008, 105, 6644–6649. [Google Scholar] [CrossRef] [PubMed]
- Firestein, R.; Bass, A.J.; Kim, S.Y.; Dunn, I.F.; Silver, S.J.; Guney, I.; Freed, E.; Ligon, A.H.; Vena, N.; Ogino, S.; et al. CDK8 is a colorectal cancer oncogene that regulates beta-catenin activity. Nature 2008, 455, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Janody, F.; Martirosyan, Z.; Benlali, A.; Treisman, J.E. Two subunits of the Drosophila mediator complex act together to control cell affinity. Development 2003, 130, 3691–3701. [Google Scholar] [CrossRef] [PubMed]
- Janody, F.; Treisman, J.E. Requirements for mediator complex subunits distinguish three classes of notch target genes at the Drosophila wing margin. Dev. Dyn. 2011, 240, 2051–2059. [Google Scholar] [CrossRef] [PubMed]
- Marr, S.K.; Lis, J.T.; Treisman, J.E.; Marr, M.T. The metazoan-specific mediator subunit 26 (Med26) is essential for viability and is found at both active genes and pericentric heterochromatin in Drosophila melanogaster. Mol. Cell. Biol. 2014, 34, 2710–2720. [Google Scholar] [CrossRef] [PubMed]
- Boube, M.; Hudry, B.; Immarigeon, C.; Carrier, Y.; Bernat-Fabre, S.; Merabet, S.; Graba, Y.; Bourbon, H.-M.; Cribbs, D.L. Drosophila melanogaster Hox transcription factors access the RNA polymerase II machinery through direct homeodomain binding to a conserved motif of mediator subunit Med19. PLoS Genet. 2014, 10, e1004303. [Google Scholar] [CrossRef] [PubMed]
- Terriente-Félix, A.; López-Varea, A.; de Celis, J.F. Identification of genes affecting wing patterning through a loss-of-function mutagenesis screen and characterization of med15 function during wing development. Genetics 2010, 185, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Mao, F.; Yang, X.; Fu, L.; Lv, X.; Zhang, Z.; Wu, W.; Yang, S.; Zhou, Z.; Zhang, L.; Zhao, Y. The Kto-Skd complex can regulate ptc expression by interacting with Cubitus interruptus (Ci) in the Hedgehog signaling pathway. J. Biol. Chem. 2014, 289, 22333–22341. [Google Scholar] [CrossRef] [PubMed]
- Hou, C.; Li, L.; Qin, Z.S.; Corces, V.G. Gene density, transcription, and insulators contribute to the partition of the Drosophila genome into physical domains. Mol. Cell 2012, 48, 471–484. [Google Scholar] [CrossRef] [PubMed]
- De Laat, W.; Dekker, J. 3C-based technologies to study the shape of the genome. Methods 2012, 58, 189–191. [Google Scholar] [CrossRef] [PubMed]
- Sexton, T.; Yaffe, E.; Kenigsberg, E.; Bantignies, F.; Leblanc, B.; Hoichman, M.; Parrinello, H.; Tanay, A.; Cavalli, G. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell 2012, 148, 458–472. [Google Scholar] [CrossRef] [PubMed]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Ong, C.-T.; Corces, V.G. CTCF: An architectural protein bridging genome topology and function. Nat. Rev. Genet. 2014, 15, 234–246. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Day, D.S.; Young, R.A. Insulated Neighborhoods: Structuraland Functional Units of Mammalian Gene Control. Cell 2016, 167, 1188–1200. [Google Scholar] [CrossRef] [PubMed]
- Burgess, D.J. Chromosomes: Dynamically in the loop. Nat. Rev. Genet. 2014, 15, 38–40. [Google Scholar] [CrossRef] [PubMed]
- Fay, A.; Misulovin, Z.; Li, J.; Schaaf, C.A.; Gause, M.; Gilmour, D.S.; Dorsett, D. Cohesin selectively binds and regulates genes with paused RNA polymerase. Curr. Biol. 2011, 21, 1624–1634. [Google Scholar] [CrossRef] [PubMed]
- Michelotti, E.F.; Sanford, S.; Levens, D. Marking of active genes on mitotic chromosomes. Nature 1997, 388, 895–899. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Wang, D.; Ye, B.; Shi, M.; Zhang, Y.; Zhao, Z. A possible role of Drosophila CTCF in mitotic bookmarking and maintaining chromatin domains during the cell cycle. Biol. Res. 2015, 48, 27. [Google Scholar] [CrossRef] [PubMed]
- Fukaya, T.; Lim, B.; Levine, M. Enhancer Control of Transcriptional Bursting. Cell 2016, 166, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Lobanenkov, V.V.; Nicolas, R.H.; Adler, V.V.; Paterson, H.; Klenova, E.M.; Polotskaja, A.V.; Goodwin, G.H. A novel sequence-specific DNA binding protein which interacts with three regularly spaced direct repeats of the CCCTC-motif in the 5′-flanking sequence of the chicken c-myc gene. Oncogene 1990, 5, 1743–1753. [Google Scholar] [PubMed]
- Rhodes, J.M.; Bentley, F.K.; Print, C.G.; Dorsett, D.; Misulovin, Z.; Dickinson, E.J.; Crosier, K.E.; Crosier, P.S.; Horsfield, J.A. Positive regulation of c-Myc by cohesin is direct, and evolutionarily conserved. Dev. Biol. 2010, 344, 637–649. [Google Scholar] [CrossRef] [PubMed]
- Sotelo, J.; Esposito, D.; Duhagon, M.A.; Banfield, K.; Mehalko, J.; Liao, H.; Stephens, R.M.; Harris, T.J.R.; Munroe, D.J.; Wu, X. Long-range enhancers on 8q24 regulate c-Myc. Proc. Natl. Acad. Sci. USA 2010, 107, 3001–3005. [Google Scholar] [CrossRef] [PubMed]
- Uslu, V.V.; Petretich, M.; Ruf, S.; Langenfeld, K.; Fonseca, N.A.; Marioni, J.C.; Spitz, F. Long-range enhancers regulating Myc expression are required for normal facial morphogenesis. Nat. Genet. 2014, 46, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Weintraub, A.S.; Day, D.S.; Valton, A.-L.; Bak, R.O.; Li, C.H.; Goldmann, J.; Lajoie, B.R.; Fan, Z.P.; Sigova, A.A.; et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science 2016, 351, 1454–1458. [Google Scholar] [CrossRef] [PubMed]
- Wood, C.D.; Veenstra, H.; Khasnis, S.; Gunnell, A.; Webb, H.M.; Shannon-Lowe, C.; Andrews, S.; Osborne, C.S.; West, M.J. MYC activation and BCL2L11 silencing by a tumour virus through the large-scale reconfiguration of enhancer-promoter hubs. Elife 2016, 5, e18270. [Google Scholar] [CrossRef] [PubMed]
- Yashiro-Ohtani, Y.; Wang, H.; Zang, C.; Arnett, K.L.; Bailis, W.; Ho, Y.; Knoechel, B.; Lanauze, C.; Louis, L.; Forsyth, K.S.; et al. Long-range enhancer activity determines Myc sensitivity to Notch inhibitors in T cell leukemia. Proc. Natl. Acad. Sci. USA 2014, 111, E4946–E4953. [Google Scholar] [CrossRef] [PubMed]
- Gainor, J.F.; Shaw, A.T. Emerging paradigms in the development of resistance to tyrosine kinase inhibitors in lung cancer. J. Clin. Oncol. 2013, 31, 3987–3996. [Google Scholar] [CrossRef] [PubMed]
- Foo, J.; Michor, F. Evolution of acquired resistance to anti-cancer therapy. J. Theor. Biol. 2014, 355, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Kolch, W.; Halasz, M.; Granovskaya, M.; Kholodenko, B.N. The dynamic control of signal transduction networks in cancer cells. Nat. Rev. Cancer 2015, 15, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Zahreddine, H.; Borden, K.L.B. Mechanisms and insights into drug resistance in cancer. Front. Pharmacol. 2013, 4, 28. [Google Scholar] [CrossRef] [PubMed]
- Soucek, L.; Helmer-Citterich, M.; Sacco, A.; Jucker, R.; Cesareni, G.; Nasi, S. Design and properties of a Myc derivative that efficiently homodimerizes. Oncogene 1998, 17, 2463–2472. [Google Scholar] [CrossRef] [PubMed]
- Savino, M.; Annibali, D.; Carucci, N.; Favuzzi, E.; Cole, M.D.; Evan, G.I.; Soucek, L.; Nasi, S. The action mechanism of the Myc inhibitor termed Omomyc may give clues on how to target Myc for cancer therapy. PLoS ONE 2011, 6, e22284. [Google Scholar] [CrossRef] [PubMed]
- Soucek, L.; Jucker, R.; Panacchia, L.; Ricordy, R.; Tatò, F.; Nasi, S. Omomyc, a potential Myc dominant negative, enhances Myc-induced apoptosis. Cancer Res. 2002, 62, 3507–3510. [Google Scholar] [PubMed]
- Soucek, L.; Whitfield, J.; Martins, C.P.; Finch, A.J.; Murphy, D.J.; Sodir, N.M.; Karnezis, A.N.; Swigart, L.B.; Nasi, S.; Evan, G.I. Modelling Myc inhibition as a cancer therapy. Nature 2008, 455, 679–683. [Google Scholar] [CrossRef] [PubMed]
- Annibali, D.; Whitfield, J.R.; Favuzzi, E.; Jauset, T.; Serrano, E.; Cuartas, I.; Redondo-Campos, S.; Folch, G.; Gonzàlez-Juncà, A.; Sodir, N.M.; et al. Myc inhibition is effective against glioma and reveals a role for Myc in proficient mitosis. Nat. Commun. 2014, 5, 4632. [Google Scholar] [CrossRef] [PubMed]
- Posternak, V.; Cole, M.D. Strategically targeting MYC in cancer. F1000Res 2016, 5, 408. [Google Scholar] [CrossRef] [PubMed]
- Hartl, M. The Quest for Targets Executing MYC-Dependent Cell Transformation. Front Oncol 2016, 6, 132. [Google Scholar] [CrossRef] [PubMed]
- Delmore, J.E.; Issa, G.C.; Lemieux, M.E.; Rahl, P.B.; Shi, J.; Jacobs, H.M.; Kastritis, E.; Gilpatrick, T.; Paranal, R.M.; Qi, J.; et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 2011, 146, 904–917. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Whyte, W.A.; Zepeda-Mendoza, C.J.; Milazzo, J.P.; Shen, C.; Roe, J.-S.; Minder, J.L.; Mercan, F.; Wang, E.; Eckersley-Maslin, M.A.; et al. Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev. 2013, 27, 2648–2662. [Google Scholar] [CrossRef] [PubMed]
- Zuber, J.; Shi, J.; Wang, E.; Rappaport, A.R.; Herrmann, H.; Sison, E.A.; Magoon, D.; Qi, J.; Blatt, K.; Wunderlich, M.; et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011, 478, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Lovén, J.; Hoke, H.A.; Lin, C.Y.; Lau, A.; Orlando, D.A.; Vakoc, C.R.; Bradner, J.E.; Lee, T.I.; Young, R.A. Selective Inhibition ofTumor Oncogenes by Disruption of Super-Enhancers. Cell 2013, 153, 320–334. [Google Scholar] [CrossRef] [PubMed]
- Knoechel, B.; Roderick, J.E.; Williamson, K.E.; Zhu, J.; Lohr, J.G.; Cotton, M.J.; Gillespie, S.M.; Fernandez, D.; Ku, M.; Wang, H.; et al. An epigenetic mechanism of resistance to targeted therapy in T cell acute lymphoblastic leukemia. Nat. Genet. 2014, 46, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Devaiah, B.N.; Lewis, B.A.; Cherman, N.; Hewitt, M.C.; Albrecht, B.K.; Robey, P.G.; Ozato, K.; Sims, R.J.; Singer, D.S. BRD4 is an atypical kinase that phosphorylates serine2 of the RNA polymerase II carboxy-terminal domain. Proc. Natl. Acad. Sci. USA 2012, 109, 6927–6932. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.K.; Mochizuki, K.; Zhou, M.; Jeong, H.-S.; Brady, J.N.; Ozato, K. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol. Cell 2005, 19, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.C.; Debrosse, M.; Smith, M.; Dey, A.; Huynh, W.; Sarai, N.; Heightman, T.D.; Tamura, T.; Ozato, K. BRD4 coordinates recruitment of pause release factor P-TEFb and the pausing complex NELF/DSIF to regulate transcription elongation of interferon-stimulated genes. Mol. Cell. Biol. 2013, 33, 2497–2507. [Google Scholar] [CrossRef] [PubMed]
- Bhagwat, A.S.; Roe, J.-S.; Mok, B.Y.L.; Hohmann, A.F.; Shi, J.; Vakoc, C.R. BET Bromodomain Inhibition Releases the Mediator Complex from Select cis-Regulatory Elements. Cell Rep. 2016, 15, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Lin, C.; Wang, L.; Guo, H.; Wang, X. Hypoxia and hypoxia-inducible factors in glioblastoma multiforme progression and therapeutic implications. Exp. Cell Res. 2012, 318, 2417–2426. [Google Scholar] [CrossRef] [PubMed]
- Fernando, C.; Audibert, A.; Simon, F.; Tazi, J.; Juge, F. A role for the serine/arginine-rich (SR) protein B52/SRSF6 in cell growth and myc expression in Drosophila. Genetics 2015, 199, 1201–1211. [Google Scholar] [CrossRef] [PubMed]
- Ring, H.Z.; Lis, J.T. The SR protein B52/SRp55 is essential for Drosophila development. Mol. Cell. Biol. 1994, 14, 7499–7506. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
(1) For expression of MYC at basal levels, the pre-initiation complex (PIC) is assembled, consisting of the Mediator complex and general transcription factors (GTFs) which together recruit hypophosphorylated Pol II holoenzyme to be paused at the promoter. Recruitment of TFIIH promotes phosphorylation of Pol II at S5 residues. Upon activation by growth signals, chromatin is further remodeled, and downstream effector transcription factors (TFs) bind to enhancer elements, interacting with the MYC promoter via the Mediator complex, resulting in transcription and torsional strain on promoter, which promotes melting of the far upstream sequence element (FUSE) element. (2) In response to the growth signals, FUSE binding protein (FUBP1) recognizes and binds single-stranded FUSE, interacting with xeroderma pigmentosum type B (XBP) helicase subunit of TFIIH complex at the promoter and modulating nucleic acid architecture to facilitate exit of inhibitory cyclin-dependent kinase 8 (CDK8) module. Concurrently, bromodomain containing 4 (BRD4) interacts with the MYC promoter via Mediator complex, together with TFIIH promoting phosphorylation of Pol II at S2 residues. Thus, Pol II proceeds into productive elongation, and maximal expression of MYC is achieved. (3) FUBP1 recruits the FUBP-interacting repressor (FIR), which also binds to regulatory FUSE. FIR represses expression of MYC by negatively regulating TFIIH activity, reducing the rate of promoter escape by Pol II. (4) When the C-terminus of XPB is truncated due to mutations, interaction with FIR no longer occurs. Therefore, MYC promoter remains in a perpetual hyperactive state, increasing risk of tumourigenesis. TSS, transcription start site.
Figure 1.
(1) For expression of MYC at basal levels, the pre-initiation complex (PIC) is assembled, consisting of the Mediator complex and general transcription factors (GTFs) which together recruit hypophosphorylated Pol II holoenzyme to be paused at the promoter. Recruitment of TFIIH promotes phosphorylation of Pol II at S5 residues. Upon activation by growth signals, chromatin is further remodeled, and downstream effector transcription factors (TFs) bind to enhancer elements, interacting with the MYC promoter via the Mediator complex, resulting in transcription and torsional strain on promoter, which promotes melting of the far upstream sequence element (FUSE) element. (2) In response to the growth signals, FUSE binding protein (FUBP1) recognizes and binds single-stranded FUSE, interacting with xeroderma pigmentosum type B (XBP) helicase subunit of TFIIH complex at the promoter and modulating nucleic acid architecture to facilitate exit of inhibitory cyclin-dependent kinase 8 (CDK8) module. Concurrently, bromodomain containing 4 (BRD4) interacts with the MYC promoter via Mediator complex, together with TFIIH promoting phosphorylation of Pol II at S2 residues. Thus, Pol II proceeds into productive elongation, and maximal expression of MYC is achieved. (3) FUBP1 recruits the FUBP-interacting repressor (FIR), which also binds to regulatory FUSE. FIR represses expression of MYC by negatively regulating TFIIH activity, reducing the rate of promoter escape by Pol II. (4) When the C-terminus of XPB is truncated due to mutations, interaction with FIR no longer occurs. Therefore, MYC promoter remains in a perpetual hyperactive state, increasing risk of tumourigenesis. TSS, transcription start site.
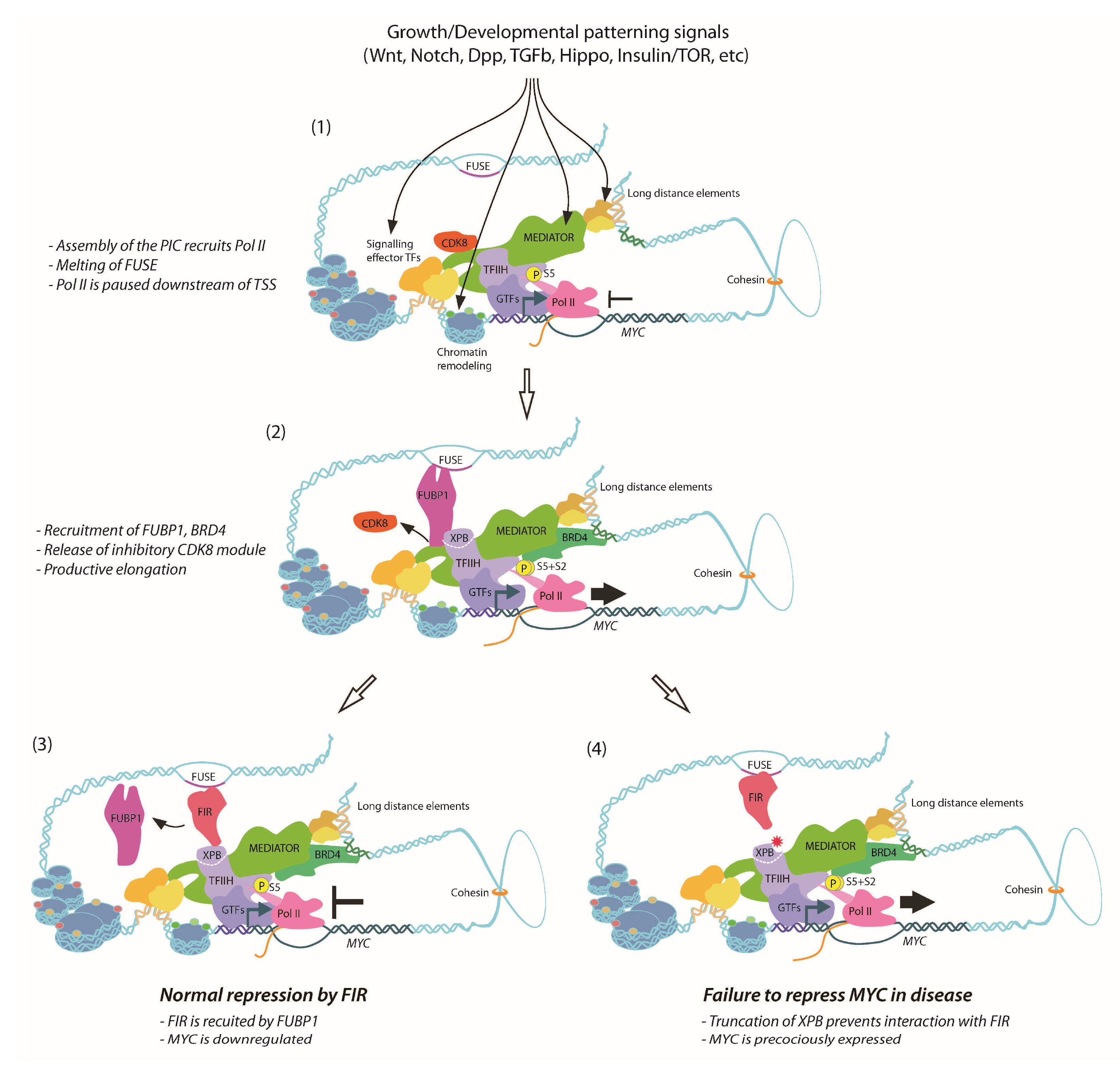
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zaytseva, O.; Quinn, L.M. Controlling the Master: Chromatin Dynamics at the MYC Promoter Integrate Developmental Signaling. Genes 2017, 8, 118. https://doi.org/10.3390/genes8040118
AMA Style
Zaytseva O, Quinn LM. Controlling the Master: Chromatin Dynamics at the MYC Promoter Integrate Developmental Signaling. Genes. 2017; 8(4):118. https://doi.org/10.3390/genes8040118
Chicago/Turabian StyleZaytseva, Olga, and Leonie M. Quinn. 2017. "Controlling the Master: Chromatin Dynamics at the MYC Promoter Integrate Developmental Signaling" Genes 8, no. 4: 118. https://doi.org/10.3390/genes8040118
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.