
Exonization of an Intronic LINE-1 Element Causing Becker Muscular Dystrophy as a Novel Mutational Mechanism in Dystrophin Gene

 , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Patient Samples
2.2. RNA Studies
2.3. Bioinformatics
2.4. LINE-1 Characterization
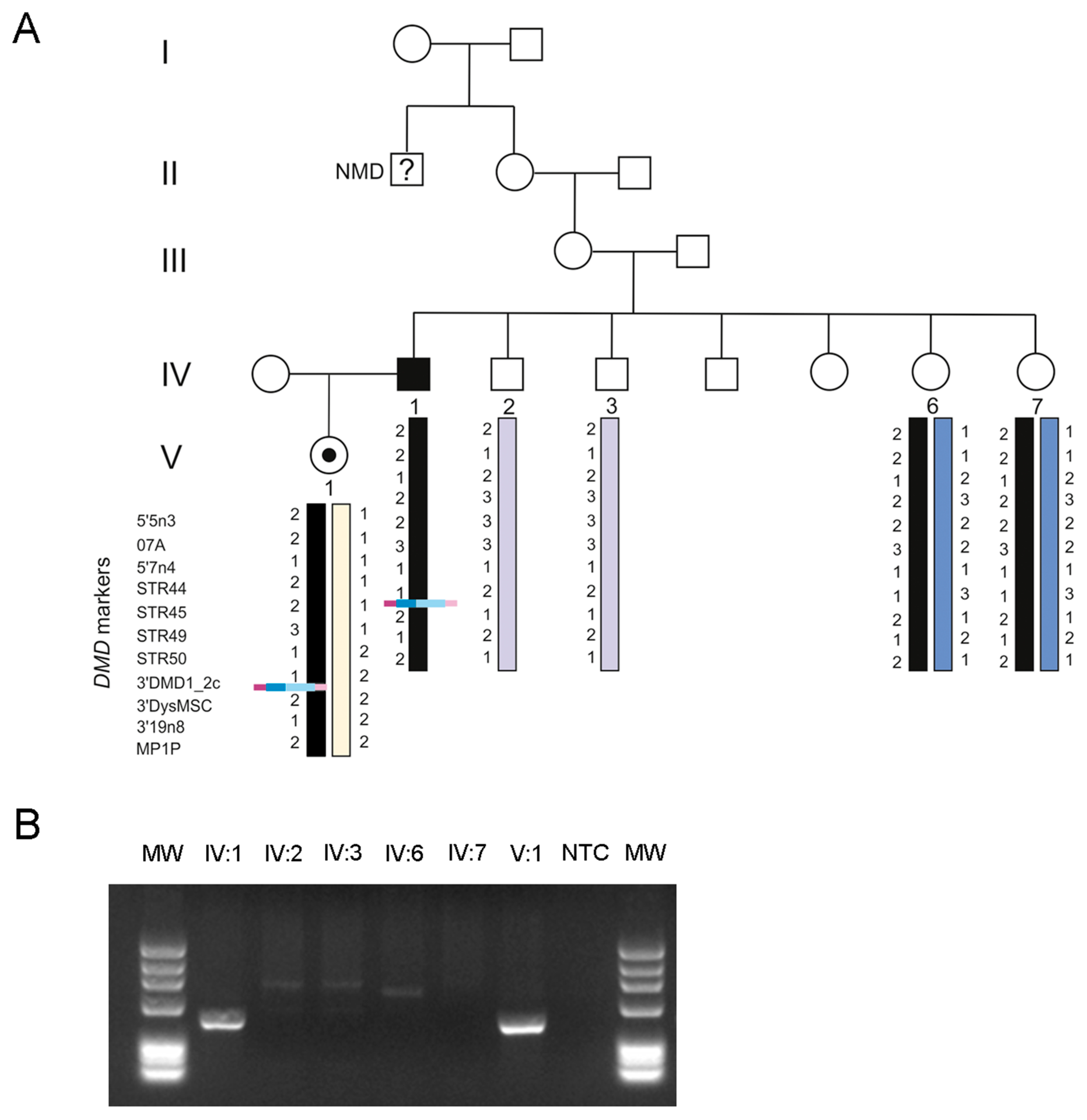
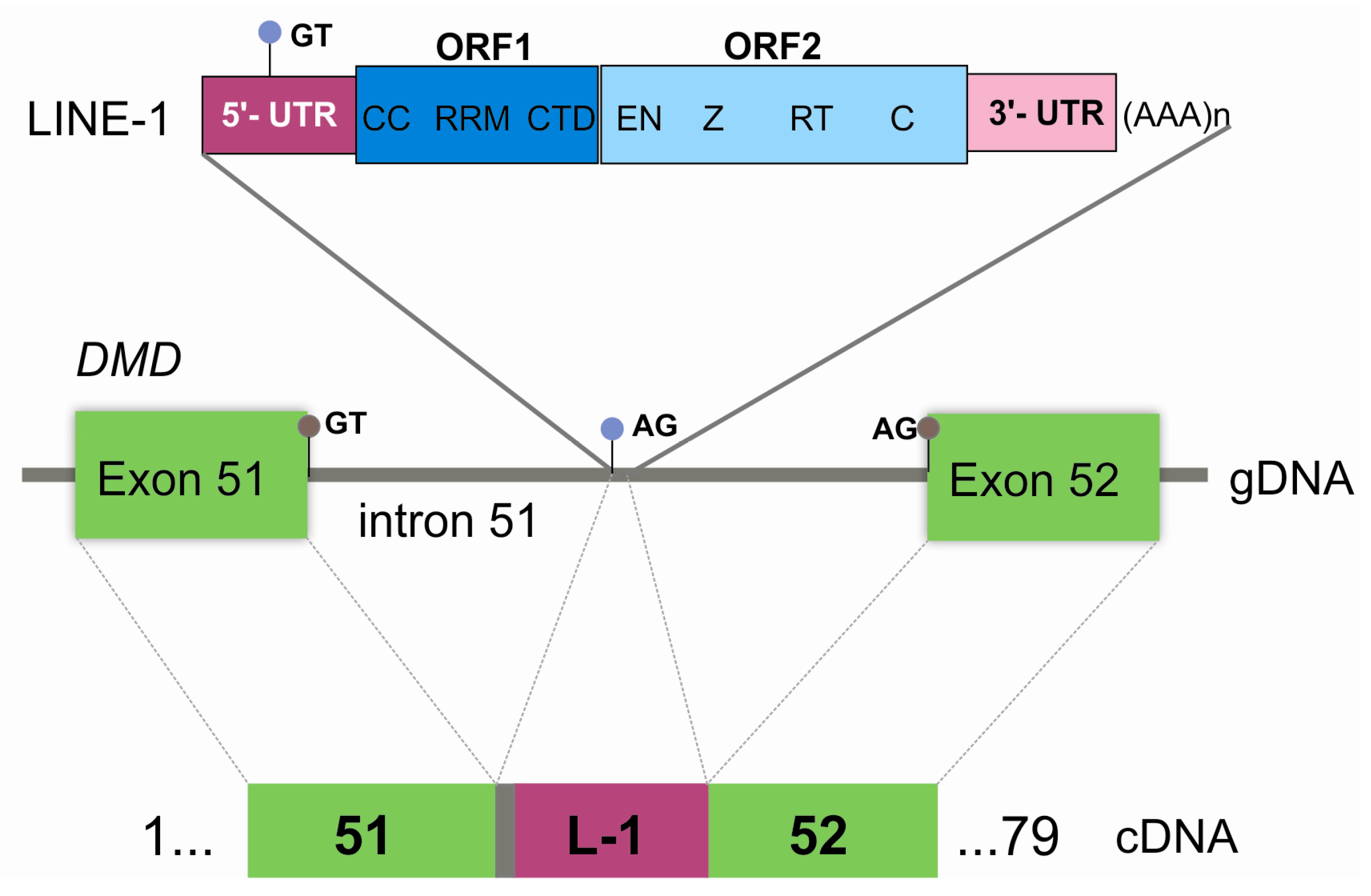
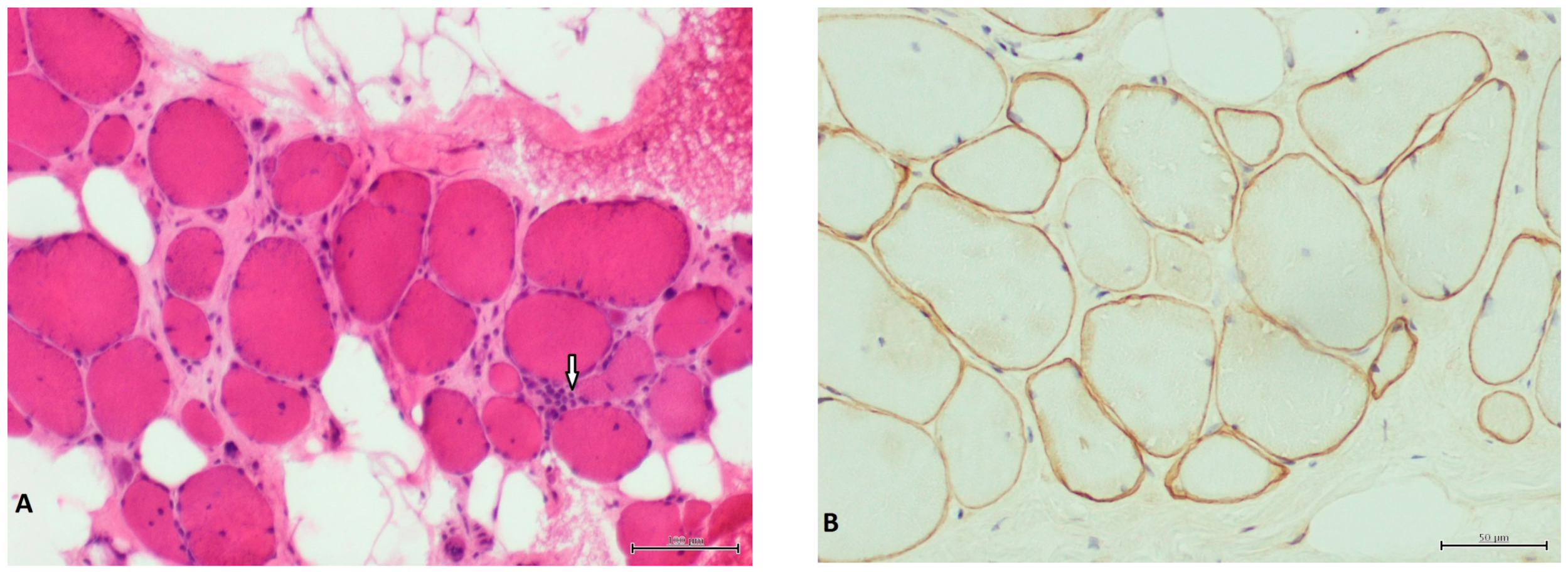
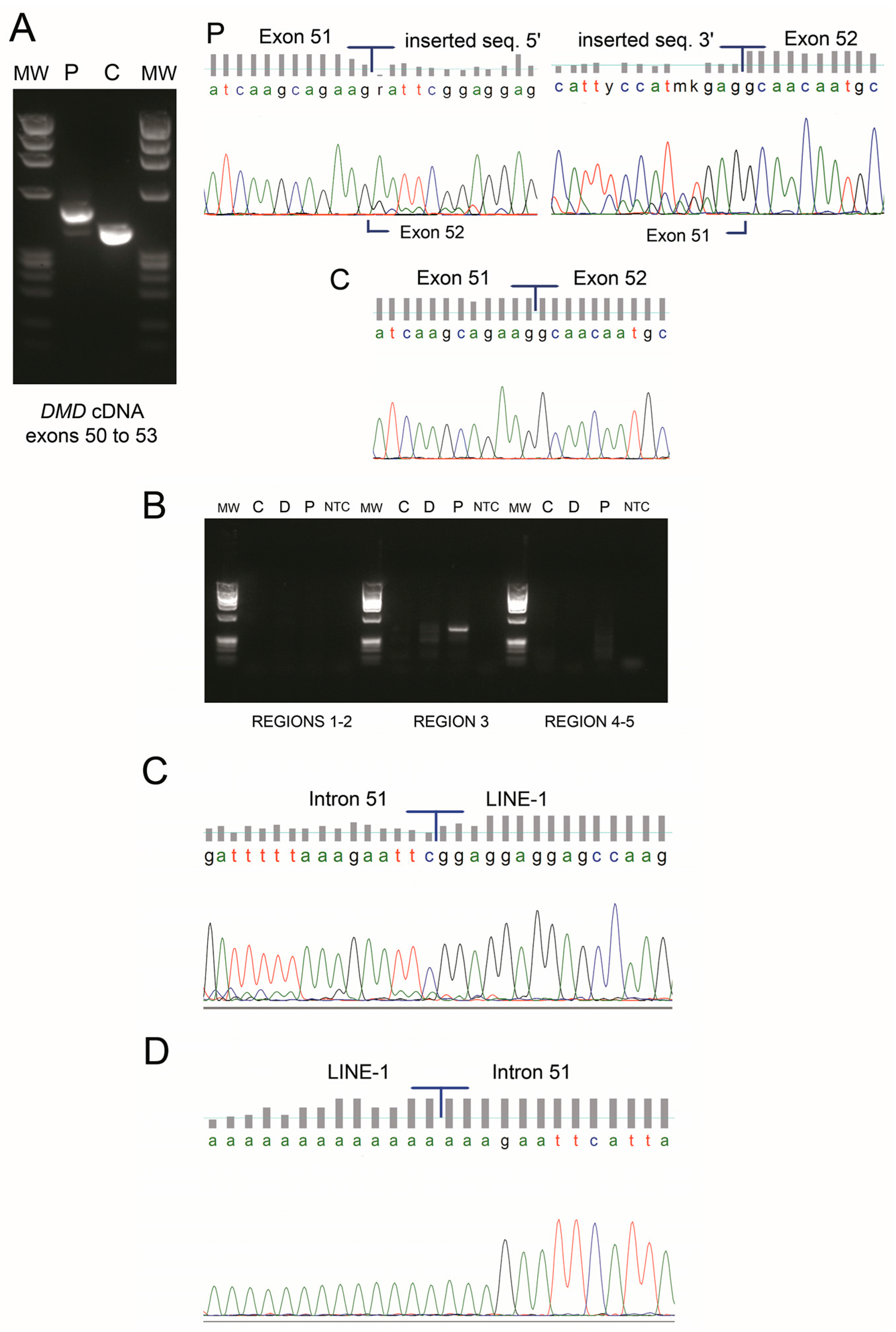
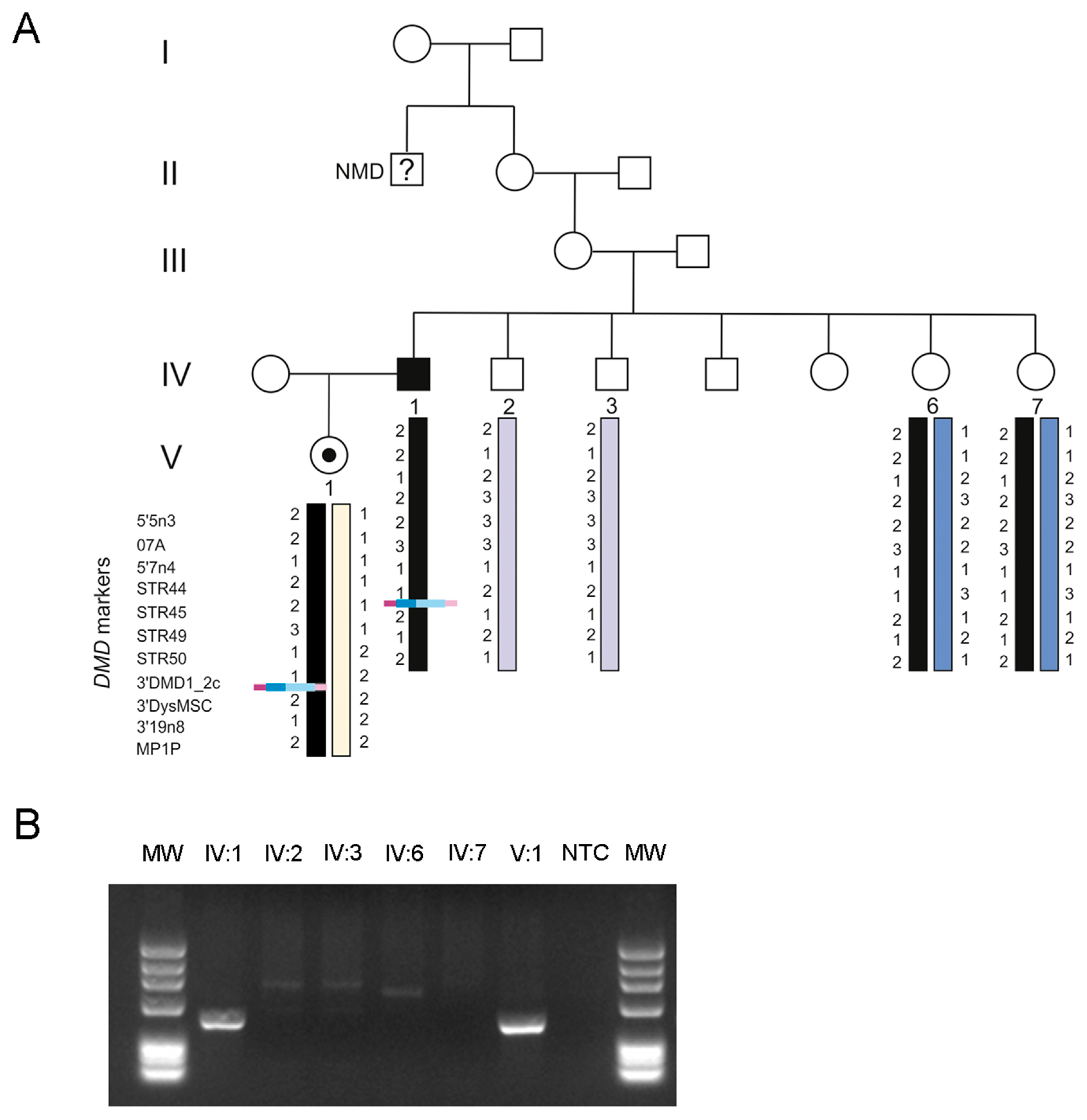
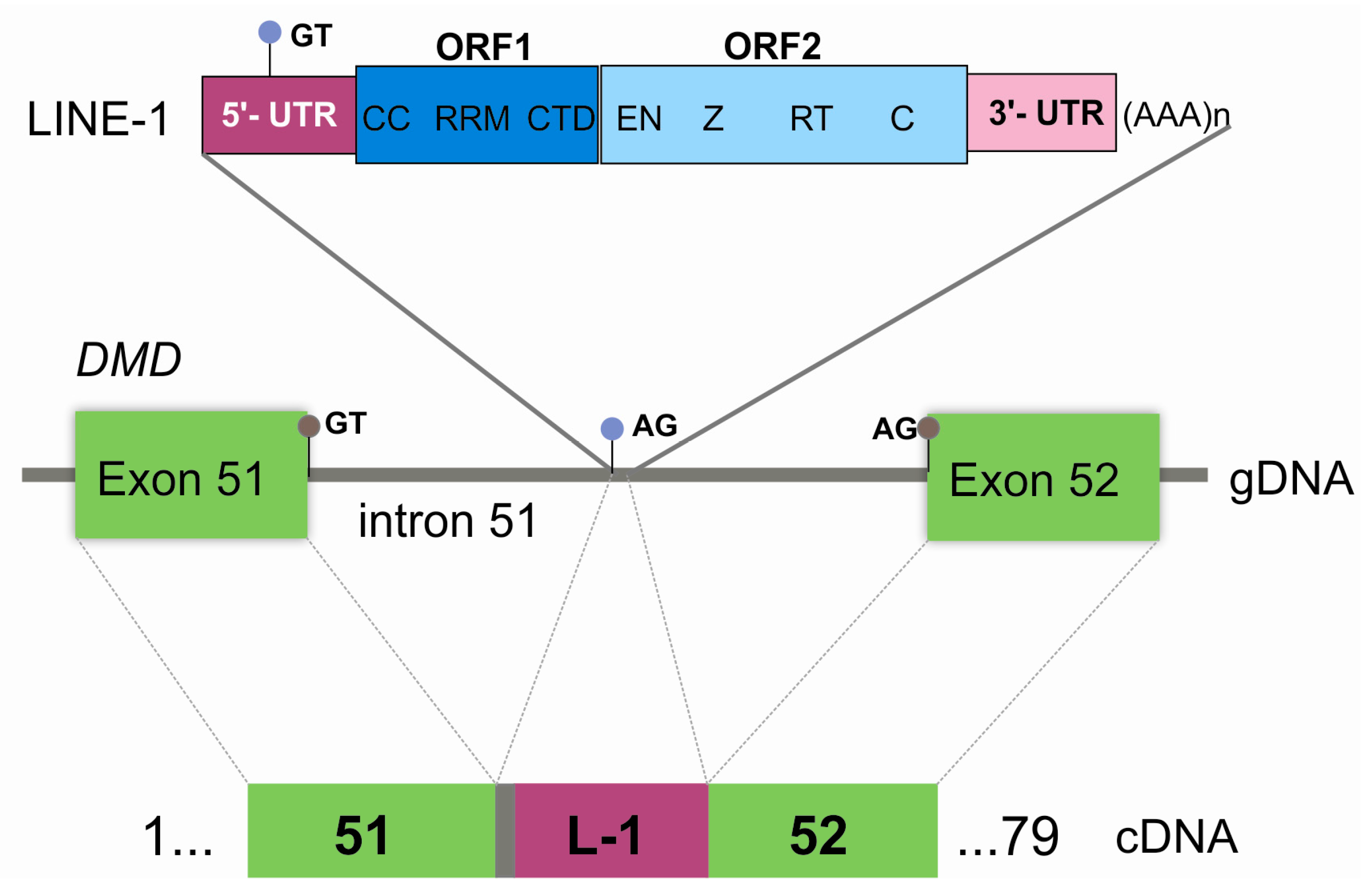
3. Results
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mendell, J.R.; Shilling, C.; Leslie, N.D.; Flanigan, K.M.; al-Dahhak, R.; Gastier-Foster, J.; Kneile, K.; Dunn, D.M.; Duval, B.; Aoyagi, A.; et al. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann. Neurol. 2012, 71, 304–313. [Google Scholar] [CrossRef] [PubMed]
- Flanigan, K.M. Duchenne and Becker muscular dystrophies. Neurol. Clin. 2014, 32, 671–688. [Google Scholar] [CrossRef] [PubMed]
- Solyom, S.; Ewing, A.D.; Hancks, D.C.; Takeshima, Y.; Awano, H.; Matsuo, M.; Kazazian, H.H., Jr. Pathogenic orphan transduction created by a nonreference LINE-1 retrotransposon. Hum. Mutat. 2012, 33, 369–371. [Google Scholar] [CrossRef] [PubMed]
- Ishmukhametova, A.; Chen, J.M.; Bernard, R.; de Massy, B.; Baudat, F.; Boyer, A.; Méchin, D.; Thorel, D.; Chabrol, B.; Vincent, M.C.; et al. Dissecting the structure and mechanism of a complex duplication-triplication rearrangement in the DMD gene. Hum. Mutat. 2013, 34, 1080–1084. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.; Gonçalves, A.; Oliveira, J.; Vieira, E.; Vieira, J.P.; Evangelista, T.; Moreno, T.; Santos, M.; Fineza, I.; Bronze-da-Rocha, E. New variants, challenges and pitfalls in DMD genotyping: Implications in diagnosis, prognosis and therapy. J. Hum. Genet. 2014, 59, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36. [Google Scholar] [CrossRef] [PubMed]
- Kohany, O.; Gentles, A.J.; Hankus, L.; Jurka, J. Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinform. 2006, 7, 474. [Google Scholar] [CrossRef] [PubMed]
- RepeatMasker. Available online: http://www.repeatmasker.org (accessed on 3 March 2017).
- Penzkofer, T.; Dandekar, T.; Zemojtel, T. L1Base: From functional annotation to prediction of active LINE-1 elements. Nucleic Acids Res. 2005, 33, 498–500. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Lee, J.; Han, K. Transposable Elements: No More ‘Junk DNA’. Genomics Inform. 2012, 10, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Hancks, D.C.; Kazazian, H.H., Jr. Roles for retrotransposon insertions in human disease. Mob. DNA 2016, 7, 9. [Google Scholar] [CrossRef] [PubMed]
- Narita, N.; Nishio, H.; Kitoh, Y.; Ishikawa, Y.; Ishikawa, Y.; Minami, R.; Nakamura, H.; Matsuo, M. Insertion of a 5′ truncated L1 element into the 3′ end of exon 44 of the dystrophin gene resulted in skipping of the exon during splicing in a case of Duchenne muscular dystrophy. J. Clin. Investig. 1993, 91, 1862–1867. [Google Scholar] [CrossRef] [PubMed]
- Musova, Z.; Hedvicakova, P.; Mohrmann, M.; Tesarova, M.; Krepelova, A.; Zeman, J.; Sedlacek, Z. A novel insertion of a rearranged L1 element in exon 44 of the dystrophin gene: Further evidence for possible bias in retroposon integration. Biochem. Biophys. Res. Commun. 2006, 347, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Awano, H.; Malueka, R.G.; Yagi, M.; Okizuka, Y.; Takeshima, Y.; Matsuo, M. Contemporary retrotransposition of a novel non-coding gene induces exon-skipping in dystrophin mRNA. J. Hum. Genet. 2010, 55, 785–790. [Google Scholar] [CrossRef] [PubMed]
- Holmes, S.E.; Dombroski, B.A.; Krebs, C.M.; Boehm, C.D.; Kazazian, H.H., Jr. A new retrotransposable human L1 element from the LRE2 locus on chromosome 1q produces a chimaeric insertion. Nat. Genet. 1994, 7, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Nakamura, A.; Yazaki, M.; Ikeda, S.; Takeda, S. Insertional mutation by transposable element, L1, in the DMD gene results in X-linked dilated cardiomyopathy. Hum. Mol. Genet. 1998, 7, 1129–1132. [Google Scholar] [CrossRef] [PubMed]
- Ferlini, A.; Muntoni, F. The 5′ Region of intron 11 of the dystrophin gene contains target sequences for mobile elements and three overlapping ORFs. Biochem, Biophys. Res. Commun. 1998, 242, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Kimberland, M.L.; Divoky, V.; Prchal, J.; Schwahn, U.; Berger, W.; Kazazian, H.H., Jr. Full-length human L1 insertions retain the capacity for high frequency retrotransposition in cultured cells. Hum. Mol. Genet. 1999, 8, 1557–1560. [Google Scholar] [CrossRef] [PubMed]
- Meischl, C.; Boer, M.; Ahlin, A.; Roos, D. A new exon created by intronic insertion of a rearranged LINE-1 element as the cause of chronic granulomatous disease. Eur. J. Hum. Genet. 2000, 8, 697–703. [Google Scholar] [CrossRef] [PubMed]
- Samuelov, L.; Fuchs-Telem, D.; Sarig, O.; Sprecher, E. An exceptional mutational event leading to Chanarin-Dorfman syndrome in a large consanguineous family. Br. J. Dermatol. 2011, 164, 1390–1392. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Martín, C.; Cidre, F.; Fernández-Teijeiro, A.; Gómez-Mariano, G.; de la Vega, L.; Ramos, P.; Zaballos, Á.; Monzón, S.; Alonso, J. Familial retinoblastoma due to intronic LINE-1 insertion causes aberrant and noncanonical mRNA splicing of the RB1 gene. J. Hum. Genet. 2016, 61, 463–466. [Google Scholar] [CrossRef] [PubMed]
- Tica, J.; Lee, E.; Untergasser, A.; Meiers, S.; Garfield, D.A.; Gokcumen, O.; Furlong, E.E.; Park, P.J.; Stütz, A.M.; Korbel, J.O. Next-generation sequencing-based detection of germline L1-mediated transductions. BMC Genomics 2016, 17, 342. [Google Scholar] [CrossRef] [PubMed]
- Tuffery-Giraud, S.; Miro, J.; Koenig, M.; Claustres, M. Normal and altered pre-mRNA processing in the DMD gene. Hum. Genet. 2017. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonçalves, A.; Oliveira, J.; Coelho, T.; Taipa, R.; Melo-Pires, M.; Sousa, M.; Santos, R. Exonization of an Intronic LINE-1 Element Causing Becker Muscular Dystrophy as a Novel Mutational Mechanism in Dystrophin Gene. Genes 2017, 8, 253. https://doi.org/10.3390/genes8100253
Gonçalves A, Oliveira J, Coelho T, Taipa R, Melo-Pires M, Sousa M, Santos R. Exonization of an Intronic LINE-1 Element Causing Becker Muscular Dystrophy as a Novel Mutational Mechanism in Dystrophin Gene. Genes. 2017; 8(10):253. https://doi.org/10.3390/genes8100253
Chicago/Turabian StyleGonçalves, Ana, Jorge Oliveira, Teresa Coelho, Ricardo Taipa, Manuel Melo-Pires, Mário Sousa, and Rosário Santos. 2017. "Exonization of an Intronic LINE-1 Element Causing Becker Muscular Dystrophy as a Novel Mutational Mechanism in Dystrophin Gene" Genes 8, no. 10: 253. https://doi.org/10.3390/genes8100253