
The Complex Relationship between Virulence and Antibiotic Resistance


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Antibiotic Resistance
2.1. Types of Resistance
2.1.1. Innate Resistance
2.1.2. Acquired Resistance
2.2. Mechanisms
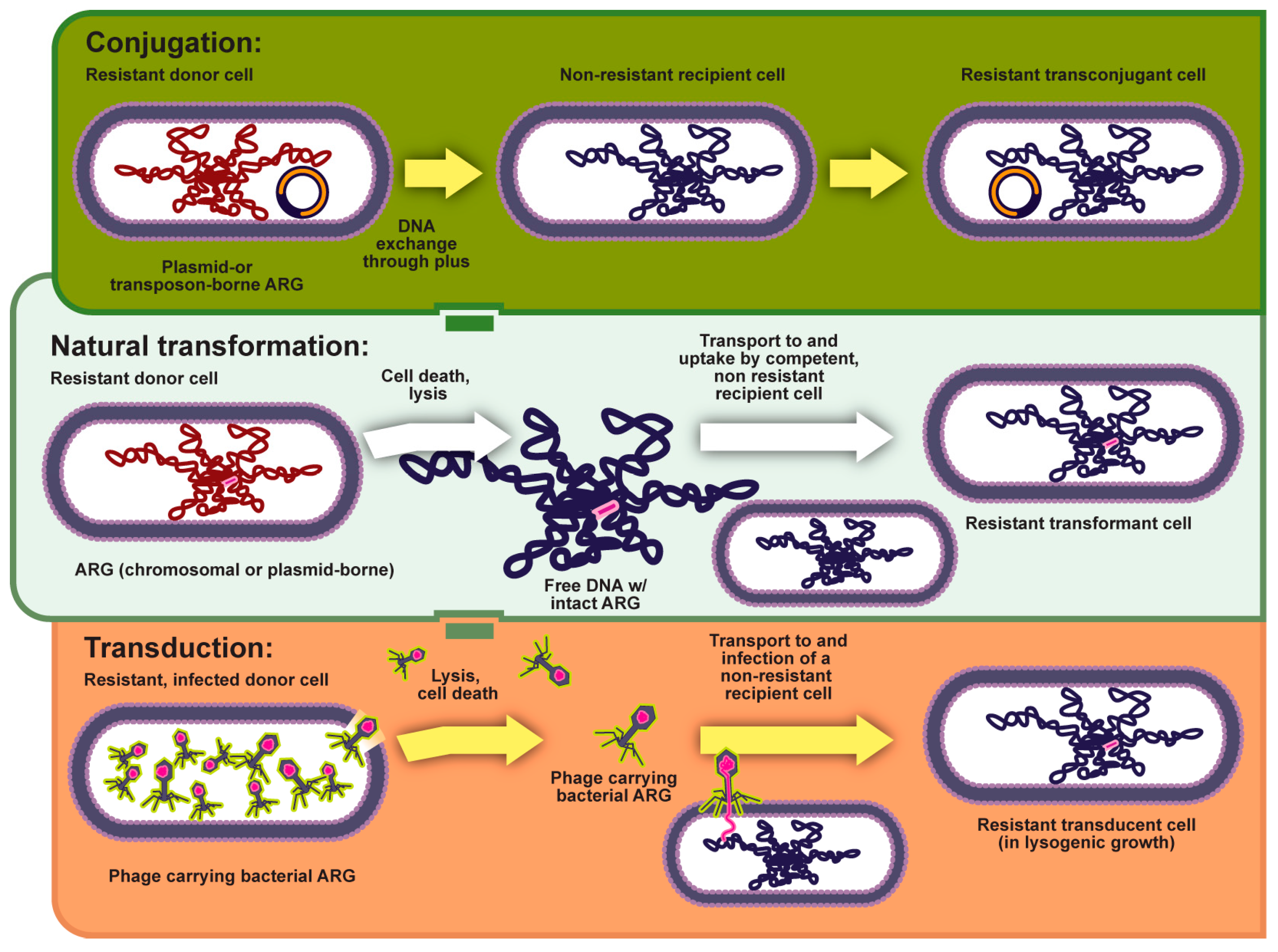
2.2.1. Horizontal Gene Transfer (HGT)
2.2.2. Elevated Mutation Rates
2.3. Adaptive Resistance
Drug Resistance in Biofilms
2.4. Antibiotic Inactivation
2.4.1. Alteration of the Antibiotic Target
2.4.2. Changes in Cell Permeability and Efflux
3. Virulence Mechanisms
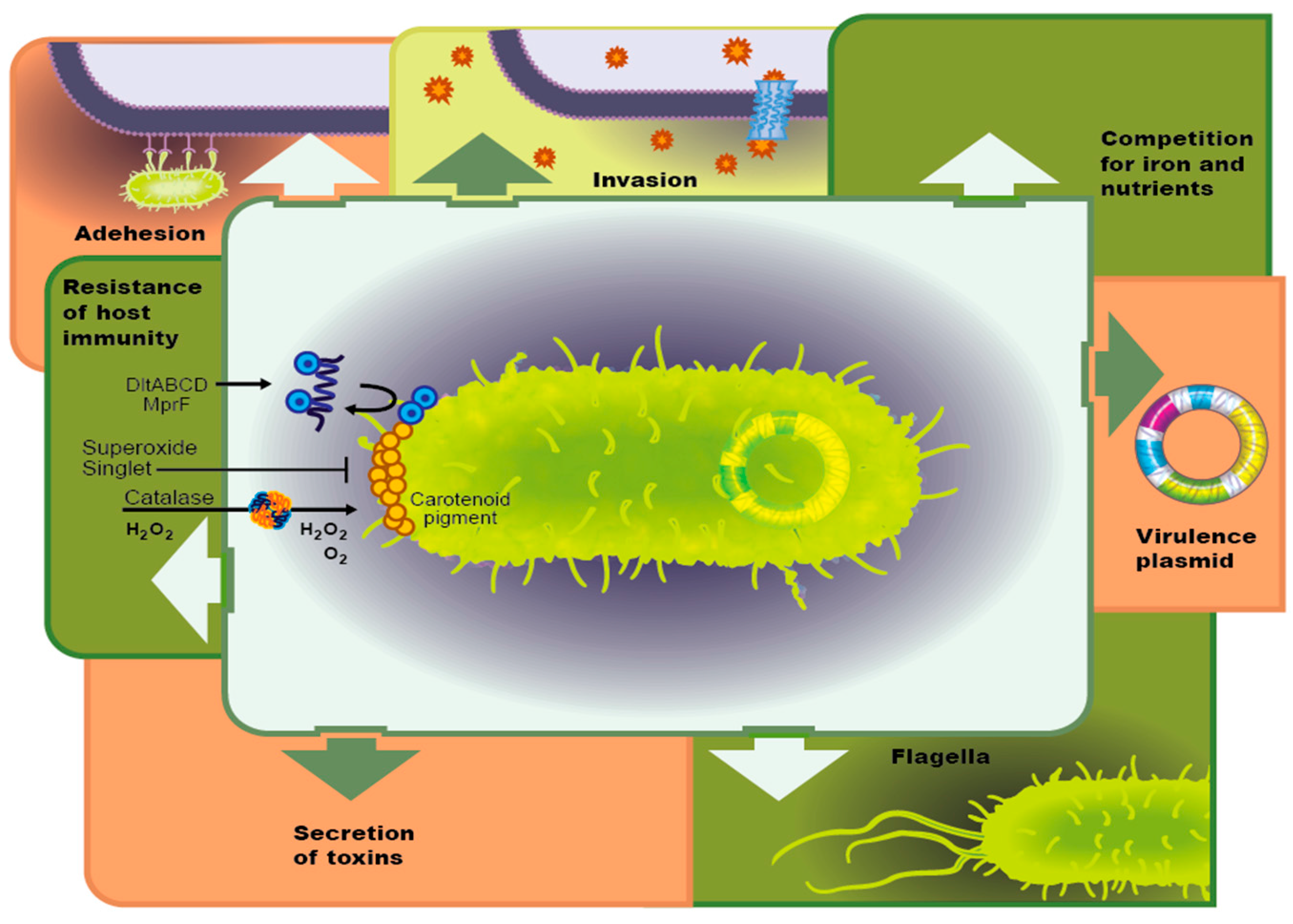
3.1. Virulence Factors
3.1.1. Adhesion Molecules
3.1.2. Host Tissue Invasion
3.1.3. Competition for Resources and Iron
3.1.4. Host Immune Evasion
3.1.5. Bacterial Toxin Secretion
3.1.6. Bacterial Motility
3.2. Virulence Plasmid
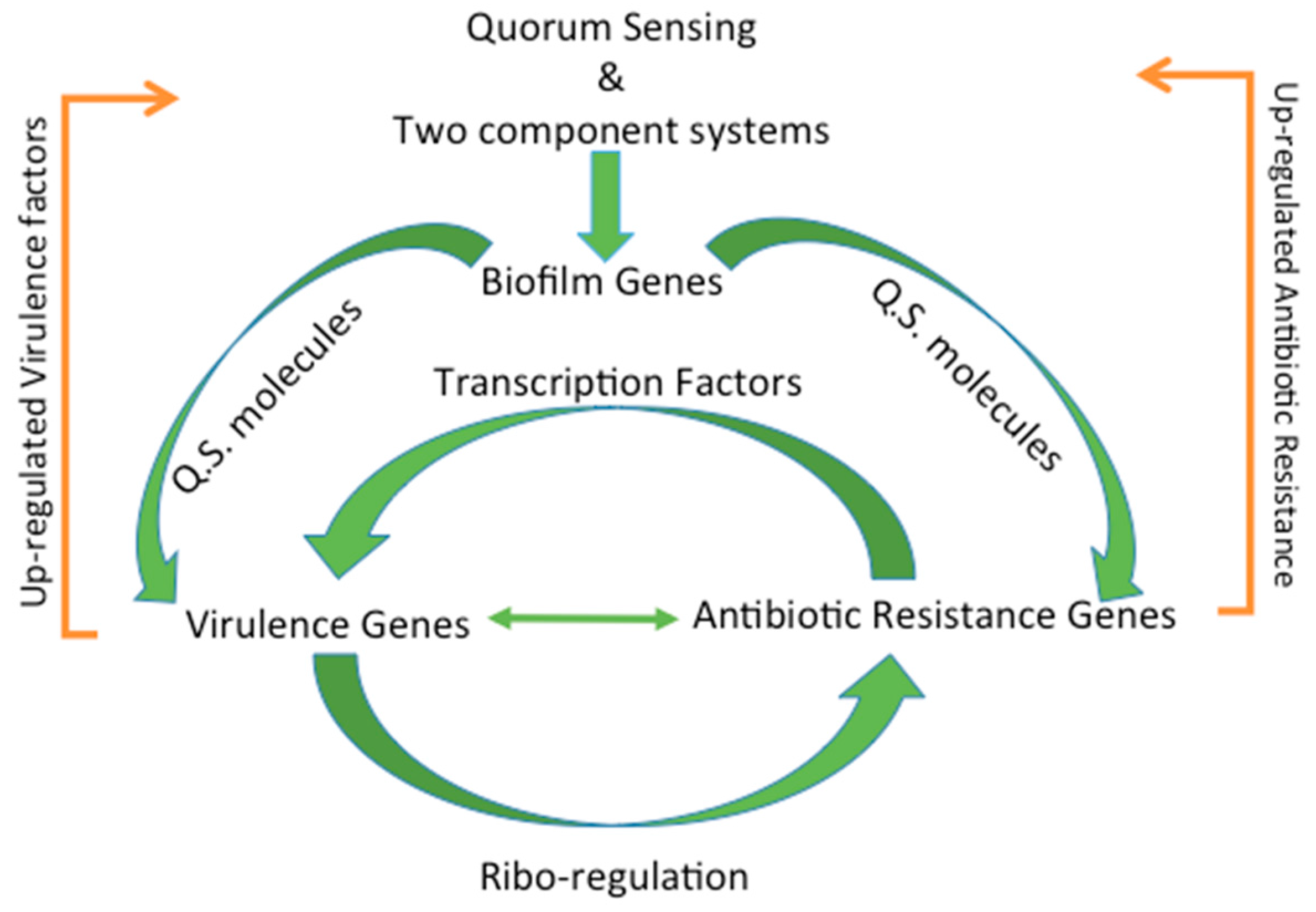
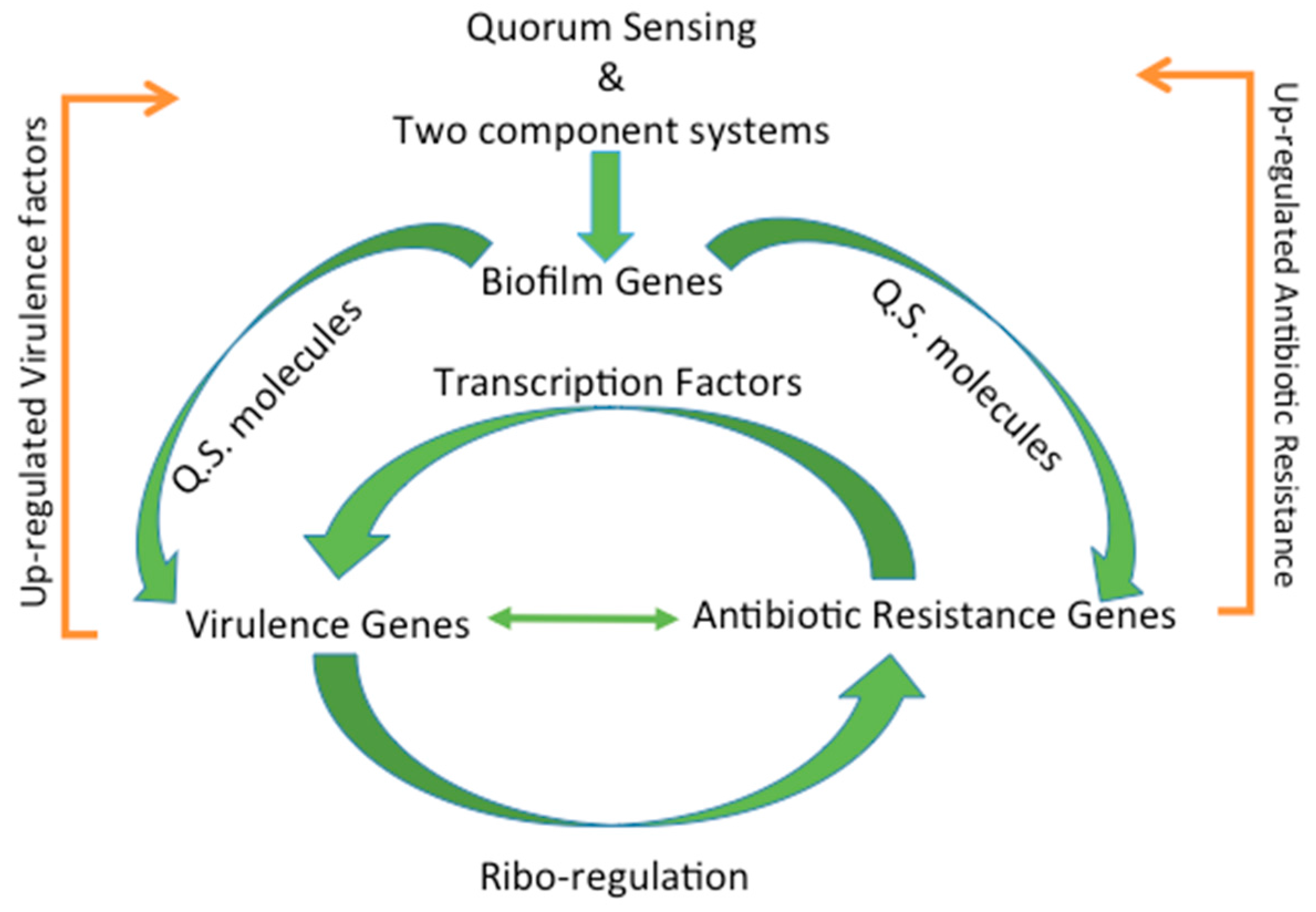
4. Regulation of Virulence and Antibiotic Resistance
4.1. Types of Regulation
4.2. Transcription Factors
4.3. Post-Transcriptional Modifications
4.4. Ribo-Regulation
4.5. Environmental Flux Sensing
4.6. Stress Response
4.7. Complex Multi-Regulation Networks
4.8. Biofilm Formation through Quorum Sensing
5. Biofilms Enhance the Transmission of Virulence and Antibiotic Resistance
5.1. Biofilm Formation
5.2. Bacterial Communication
5.3. Horizontal Transfer of Virulence Genes
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Melnyk, A.H.; Wong, A.; Kassen, R. The fitness costs of antibiotic resistance mutations. Evol. Appl. 2015, 8, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Roux, D.; Danilchanka, O.; Guillard, T.; Cattoir, V.; Aschard, H.; Fu, Y.; Angoulvant, F.; Messika, J.; Ricard, J.-D.; Mekalanos, J.J.; et al. Fitness cost of antibiotic susceptibility during bacterial infection. Sci. Transl. Med. 2015, 7, 297ra114. [Google Scholar] [CrossRef] [PubMed]
- Guillard, T.; Pons, S.; Roux, D.; Pier, G.B.; Skurnik, D. Antibiotic resistance and virulence: Understanding the link and its consequences for prophylaxis and therapy. BioEssays 2016, 38, 682–693. [Google Scholar] [CrossRef] [PubMed]
- Tay, S.B.; Yew, W.S. Development of quorum-based anti-virulence therapeutics targeting Gram-negative bacterial pathogens. Int. J. Mol. Sci. 2013, 14, 16570–16599. [Google Scholar] [CrossRef] [PubMed]
- Von Dohren, H. Antibiotics: Actions, origins, resistance, by C. Walsh. 2003. Washington, DC: ASM Press. 345 pp. $99.95 (hardcover). Protein Sci. Publ. Protein Soc. 2004, 13, 3059–3060. [Google Scholar] [CrossRef]
- Walsh, C. Molecular mechanisms that confer antibacterial drug resistance. Nature 2000, 406, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Neu, H.C. The crisis in antibiotic resistance. Science 1992, 257, 1064–1073. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.H.; Molla, M.N.; Cantor, C.R.; Collins, J.J. Bacterial charity work leads to population-wide resistance. Nature 2010, 467, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Toprak, E.; Veres, A.; Michel, J.-B.; Chait, R.; Hartl, D.L.; Kishony, R. Evolutionary paths to antibiotic resistance under dynamically sustained drug selection. Nat. Genet. 2012, 44, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Yurtsev, E.A.; Chao, H.X.; Datta, M.S.; Artemova, T.; Gore, J. Bacterial cheating drives the population dynamics of cooperative antibiotic resistance plasmids. Mol. Syst. Biol. 2013, 9, 683. [Google Scholar] [CrossRef] [PubMed]
- Fernández, L.; Hancock, R.E.W. Adaptive and mutational resistance: role of porins and efflux pumps in drug resistance. Clin. Microbiol. Rev. 2012, 25, 661–681. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Romero, M.A.; Casadesús, J. Contribution of phenotypic heterogeneity to adaptive antibiotic resistance. Proc. Natl. Acad. Sci. USA 2013. [Google Scholar] [CrossRef] [PubMed]
- Baquero, F.; Alvarez-Ortega, C.; Martinez, J.L. Ecology and evolution of antibiotic resistance. Environ. Microbiol. Rep. 2009, 1, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.I.; Hughes, D. Persistence of antibiotic resistance in bacterial populations. FEMS Microbiol. Rev. 2011, 35, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.; Davies, D. Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. MMBR 2010, 74, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Vranakis, I.; Goniotakis, I.; Psaroulaki, A.; Sandalakis, V.; Tselentis, Y.; Gevaert, K.; Tsiotis, G. Proteome studies of bacterial antibiotic resistance mechanisms. J. Proteomics 2013. [Google Scholar] [CrossRef] [PubMed]
- Berendonk, T.U.; Manaia, C.M.; Merlin, C.; Fatta-Kassinos, D.; Cytryn, E.; Walsh, F.; Bürgmann, H.; Sørum, H.; Norström, M.; Pons, M.-N.; et al. Tackling antibiotic resistance: the environmental framework. Nat. Rev. Microbiol. 2015, 13, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Sabouri Ghannad, M.; Mohammadi, A. Bacteriophage: time to re-evaluate the potential of phage therapy as a promising agent to control multidrug-resistant bacteria. Iran. J. Basic Med. Sci. 2012, 15, 693–701. [Google Scholar] [PubMed]
- Kester, J.C.; Fortune, S.M. Persisters and beyond: Mechanisms of phenotypic drug resistance and drug tolerance in bacteria. Crit. Rev. Biochem. Mol. Biol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Binnewies, T.T.; Motro, Y.; Hallin, P.F.; Lund, O.; Dunn, D.; La, T.; Hampson, D.J.; Bellgard, M.; Wassenaar, T.M.; Ussery, D.W. Ten years of bacterial genome sequencing: comparative-genomics-based discoveries. Funct. Integr. Genomics 2006, 6, 165–185. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Rojas, A.; Rodríguez-Beltrán, J.; Couce, A.; Blázquez, J. Antibiotics and antibiotic resistance: A bitter fight against evolution. Int. J. Med. Microbiol. 2013, 303, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Juhas, M. Horizontal gene transfer in human pathogens. Crit. Rev. Microbiol. 2015, 41, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Von Wintersdorff, C.J.H.; Penders, J.; van Niekerk, J.M.; Mills, N.D.; Majumder, S.; van Alphen, L.B.; Savelkoul, P.H.M.; Wolffs, P.F.G. Dissemination of Antimicrobial Resistance in Microbial Ecosystems through Horizontal Gene Transfer. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Novick, R.P. Phage-mediated intergeneric transfer of toxin genes. Science 2009, 323, 139–141. [Google Scholar] [CrossRef] [PubMed]
- Cirz, R.T.; Chin, J.K.; Andes, D.R.; de Crecy-Lagard, V.; Craig, W.A.; Romesberg, F.E. Inhibition of Mutation and Combating the Evolution of Antibiotic Resistance. PLoS Biol. 2005, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandon, S.; Vale, P.F. The evolution of resistance against good and bad infections. J. Evol. Biol. 2013. [Google Scholar] [CrossRef] [PubMed]
- Norman, A.; Hansen, L.H.; Sørensen, S.J. Conjugative plasmids: Vessels of the communal gene pool. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2009, 364, 2275–2289. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.I.; Hughes, D. Antibiotic resistance and its cost: is it possible to reverse resistance? Nat. Rev. Microbiol. 2010, 8, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Nikaido, H. Multidrug resistance in bacteria. Annu. Rev. Biochem. 2009, 78, 119–146. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-Z.; Nikaido, H. Efflux-mediated drug resistance in bacteria: an update. Drugs 2009, 69, 1555. [Google Scholar] [CrossRef] [PubMed]
- Andersson, D.I.; Hughes, D. Evolution of antibiotic resistance at non-lethal drug concentrations. Drug Resist. Updat. 2012, 15, 162–172. [Google Scholar] [CrossRef] [PubMed]
- Blázquez, J.; Couce, A.; Rodríguez-Beltrán, J.; Rodríguez-Rojas, A. Antimicrobials as promoters of genetic variation. Curr. Opin. Microbiol. 2012, 15, 561–569. [Google Scholar] [CrossRef] [PubMed]
- Silva, P.E.A.D.; Palomino, J.C. Molecular basis and mechanisms of drug resistance in Mycobacterium tuberculosis: classical and new drugs. J. Antimicrob. Chemother. 2011, 66, 1417–1430. [Google Scholar] [CrossRef]
- Gillespie, S.H.; Basu, S.; Dickens, A.L.; O’Sullivan, D.M.; McHugh, T.D. Effect of subinhibitory concentrations of ciprofloxacin on Mycobacterium fortuitum mutation rates. J. Antimicrob. Chemother. 2005, 56, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Cox, H.S.; Niemann, S.; Ismailov, G.; Doshetov, D.; Orozco, J.D.; Blok, L.; Rüsch-Gerdes, S.; Kebede, Y. Risk of acquired drug resistance during short-course directly observed treatment of tuberculosis in an area with high levels of drug resistance. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2007, 44, 1421–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mariam, S.H.; Werngren, J.; Aronsson, J.; Hoffner, S.; Andersson, D.I. Dynamics of antibiotic resistant Mycobacterium tuberculosis during long-term infection and antibiotic treatment. PLoS ONE 2011, 6, e21147. [Google Scholar] [CrossRef] [PubMed]
- de Toro, M.; Rojo-Bezares, B.; Vinué, L.; Undabeitia, E.; Torres, C.; Sáenz, Y. In vivo selection of aac(6’)-Ib-cr and mutations in the gyrA gene in a clinical qnrS1-positive Salmonella enterica serovar Typhimurium DT104B strain recovered after fluoroquinolone treatment. J. Antimicrob. Chemother. 2010, 65, 1945–1949. [Google Scholar] [CrossRef] [PubMed]
- Cattoir, V.; Lesprit, P.; Lascols, C.; Denamur, E.; Legrand, P.; Soussy, C.-J.; Cambau, E. In vivo selection during ofloxacin therapy of Escherichia coli with combined topoisomerase mutations that confer high resistance to ofloxacin but susceptibility to nalidixic acid. J. Antimicrob. Chemother. 2006, 58, 1054–1057. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.S.; Kim, J.A.; Shin, J.H.; Chang, C.L.; Jeong, J.; Cho, J.-H.; Kim, M.-N.; Kim, S.; Kim, Y.R.; Lee, C.H.; et al. Prevalence of plasmid-mediated quinolone resistance and mutations in the gyrase and topoisomerase IV genes in Salmonella isolated from 12 tertiary-care hospitals in Korea. Microb. Drug Resist. Larchmt. N 2011, 17, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Leotard, S.; Poirel, L.; Leblanc, P.E.; Nordmann, P. In vivo selection of oxacillinase-mediated ceftazidime resistance in Pseudomonas aeruginosa. Microb. Drug Resist. Larchmt. N 2001, 7, 273–275. [Google Scholar] [CrossRef] [PubMed]
- Le Thomas, I.; Couetdic, G.; Clermont, O.; Brahimi, N.; Plésiat, P.; Bingen, E. In vivo selection of a target/efflux double mutant of Pseudomonas aeruginosa by ciprofloxacin therapy. J. Antimicrob. Chemother. 2001, 48, 553–555. [Google Scholar] [CrossRef] [PubMed]
- Oliver, A.; Baquero, F.; Blázquez, J. The mismatch repair system (mutS, mutL and uvrD genes) in Pseudomonas aeruginosa: molecular characterization of naturally occurring mutants. Mol. Microbiol. 2002, 43, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Rushdy, A.A.; Mabrouk, M.I.; Abu-Sef, F.A.-H.; Kheiralla, Z.H.; Abdel-All, S.M.; Saleh, N.M. Contribution of different mechanisms to the resistance to fluoroquinolones in clinical isolates of Salmonella enterica. Braz. J. Infect. Dis. Off. Publ. Braz. Soc. Infect. Dis. 2013, 17, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Foster, P.L. Stress-induced mutagenesis in bacteria. Crit. Rev. Biochem. Mol. Biol. 2007, 42, 373–397. [Google Scholar] [CrossRef] [PubMed]
- Kohanski, M.A.; DePristo, M.A.; Collins, J.J. Sublethal antibiotic treatment leads to multidrug resistance via radical-induced mutagenesis. Mol. Cell 2010, 37, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, D.J.; Kohanski, M.A.; Collins, J.J. Role of reactive oxygen species in antibiotic action and resistance. Curr. Opin. Microbiol. 2009, 12, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Boles, B.R.; Singh, P.K. Endogenous oxidative stress produces diversity and adaptability in biofilm communities. Proc. Natl. Acad. Sci. USA 2008, 105, 12503–12508. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.D.; Brooks, A.E. Therapeutic strategies to combat antibiotic resistance. Adv. Drug Deliv. Rev. 2014, 78, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Algburi, A.; Comito, N.; Kashtanov, D.; Dicks, L.M.T.; Chikindas, M.L. Control of Biofilm Formation: Antibiotics and Beyond. Appl. Environ. Microbiol. 2016. AEM.02508-16. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.D.; Brooks, A.E.; Grainger, D.W. Antimicrobial Medical Devices in Preclinical Development and Clinical Use. In Biomaterials Associated Infection; Moriarty, T.F., Zaat, S.A.J., Busscher, H.J., Eds.; Springer: New York, NY, USA, 2013; pp. 307–354. [Google Scholar]
- Ravn, C.; Tafin, U.F.; Bétrisey, B.; Overgaard, S.; Trampuz, A. Reduced ability to detect surface-related biofilm bacteria after antibiotic exposure under in vitro conditions. Acta Orthop. 2016, 87, 644–650. [Google Scholar] [CrossRef] [PubMed]
- Bayramov, D.F.; Neff, J.A. Beyond conventional antibiotics—New directions for combination products to combat biofilm. Adv. Drug Deliv. Rev. 2016. [Google Scholar] [CrossRef] [PubMed]
- Høiby, N.; Bjarnsholt, T.; Givskov, M.; Molin, S.; Ciofu, O. Antibiotic resistance of bacterial biofilms. Int. J. Antimicrob. Agents 2010, 35, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Bjarnsholt, T.; Givskov, M. The role of quorum sensing in the pathogenicity of the cunning aggressor Pseudomonas aeruginosa. Anal. Bioanal. Chem. 2007, 387, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Hirakawa, H.; Tomita, H. Interference of bacterial cell-to-cell communication: a new concept of antimicrobial chemotherapy breaks antibiotic resistance. Front. Microbiol. 2013, 4, 114. [Google Scholar] [CrossRef] [PubMed]
- Camilli, A.; Bassler, B.L. Bacterial Small-Molecule Signaling Pathways. Science 2006, 311, 1113–1116. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.B.; Bassler, B.L. Quorum sensing in bacteria. Annu. Rev. Microbiol. 2001, 55, 165–199. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Grainger, D.W. Drug/device combinations for local drug therapies and infection prophylaxis. Biomaterials 2006, 27, 2450–2467. [Google Scholar] [CrossRef] [PubMed]
- Khardori, N.; Yassien, M. Biofilms in device-related infections. J. Ind. Microbiol. 1995, 15, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Cavallaro, A.; Taheri, S.; Vasilev, K. Responsive and “smart” antibacterial surfaces: Common approaches and new developments. Biointerphases 2014, 9, 29005. [Google Scholar] [CrossRef] [PubMed]
- Vasilev, K.; Cook, J.; Griesser, H.J. Antibacterial surfaces for biomedical devices. Expert Rev. Med. Devices 2009, 6, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Stewart, P.S.; Costerton, J.W. Antibiotic resistance of bacteria in biofilms. The Lancet 2001, 358, 135–138. [Google Scholar] [CrossRef]
- Schmidt, S.; Wallbrecher, R.; van Kuppevelt, T.; Brock, R. Methods to Study the Role of the Glycocalyx in the Uptake of Cell-Penetrating Peptides. In Cell-Penetrating Peptides; Langel, Ü., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2015; pp. 123–131. [Google Scholar]
- Costerton, J.W.; Stewart, P.S.; Greenberg, E.P. Bacterial Biofilms: A Common Cause of Persistent Infections. Science 1999, 284, 1318–1322. [Google Scholar] [CrossRef] [PubMed]
- Arciola, C.R.; Campoccia, D.; Gamberini, S.; Donati, M.E.; Pirini, V.; Visai, L.; Speziale, P.; Montanaro, L. Antibiotic resistance in exopolysaccharide-forming Staphylococcus epidermidis clinical isolates from orthopaedic implant infections. Biomaterials 2005, 26, 6530–6535. [Google Scholar] [CrossRef] [PubMed]
- Campoccia, D.; Montanaro, L.; Speziale, P.; Arciola, C.R. Antibiotic-loaded biomaterials and the risks for the spread of antibiotic resistance following their prophylactic and therapeutic clinical use. Biomaterials 2010, 31, 6363–6377. [Google Scholar] [CrossRef] [PubMed]
- Thallinger, B.; Prasetyo, E.N.; Nyanhongo, G.S.; Guebitz, G.M. Antimicrobial enzymes: An emerging strategy to fight microbes and microbial biofilms. Biotechnol. J. 2013, 8, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Stewart, P.S. Mechanisms of antibiotic resistance in bacterial biofilms. Int. J. Med. Microbiol. 2002, 292, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Driffield, K.; Miller, K.; Bostock, J.M.; O’Neill, A.J.; Chopra, I. Increased mutability of Pseudomonas aeruginosa in biofilms. J. Antimicrob. Chemother. 2008, 61, 1053–1056. [Google Scholar] [CrossRef] [PubMed]
- Conibear, T.C.R.; Collins, S.L.; Webb, J.S. Role of mutation in Pseudomonas aeruginosa biofilm development. PLoS ONE 2009, 4, e6289. [Google Scholar] [CrossRef] [PubMed]
- Goto, T.; Nakame, Y.; Nishida, M.; Ohi, Y. In vitro bactericidal activities of beta-lactamases, amikacin, and fluoroquinolones against Pseudomonas aeruginosa biofilm in artificial urine. Urology 1999, 53, 1058–1062. [Google Scholar] [CrossRef]
- Brooun, A.; Liu, S.; Lewis, K. A dose-response study of antibiotic resistance in Pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother. 2000, 44, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Tenover, F.C. Mechanisms of antimicrobial resistance in bacteria. Am. J. Infect. Control 2006, 34, S3–S10. [Google Scholar] [CrossRef] [PubMed]
- Gradelski, E.; Kolek, B.; Bonner, D.; Fung-Tomc, J. Bactericidal mechanism of gatifloxacin compared with other quinolones. J. Antimicrob. Chemother. 2002, 49, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Finkel, S.E. Long-term survival during stationary phase: evolution and the GASP phenotype. Nat. Rev. Microbiol. 2006, 4, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Conlon, B.P.; Rowe, S.E.; Lewis, K. Persister Cells in Biofilm Associated Infections. In Biofilm-based Healthcare-associated Infections; Donelli, G., Ed.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2015; pp. 1–9. [Google Scholar]
- Jayaraman, R. Bacterial persistence: some new insights into an old phenomenon. J. Biosci. 2008, 33, 795–805. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.D. Bacterial resistance to antibiotics: enzymatic degradation and modification. Adv. Drug Deliv. Rev. 2005, 57, 1451–1470. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, M.S.; Tolmasky, M.E. Aminoglycoside Modifying Enzymes. Drug Resist. Updat. Rev. Comment. Antimicrob. Anticancer Chemother. 2010, 13, 151–171. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.N. Ribosome-targeting antibiotics and mechanisms of bacterial resistance. Nat. Rev. Microbiol. 2013, 12, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Worthington, R.J.; Melander, C. Overcoming resistance to β-lactam antibiotics. J. Org. Chem. 2013, 78, 4207–4213. [Google Scholar] [CrossRef] [PubMed]
- Llano-Sotelo, B.; Azucena, E.F., Jr.; Kotra, L.P.; Mobashery, S.; Chow, C.S. Aminoglycosides modified by resistance enzymes display diminished binding to the bacterial ribosomal aminoacyl-tRNA site. Chem. Biol. 2002, 9, 455–463. [Google Scholar] [CrossRef]
- Llaneza, J.; Villar, C.J.; Salas, J.A.; Suarez, J.E.; Mendoza, M.C.; Hardisson, C. Plasmid-mediated fosfomycin resistance is due to enzymatic modification of the antibiotic. Antimicrob. Agents Chemother. 1985, 28, 163–164. [Google Scholar] [CrossRef] [PubMed]
- Gates, C.A.; Northrop, D.B. Substrate specificities and structure-activity relationships for the nucleotidylation of antibiotics catalyzed by aminoglycoside nucleotidyltransferase 2"-I. Biochemistry (Mosc.) 1988, 27, 3820–3825. [Google Scholar] [CrossRef]
- Krueger, K.M.; Barbieri, J.T. The family of bacterial ADP-ribosylating exotoxins. Clin. Microbiol. Rev. 1995, 8, 34–47. [Google Scholar] [PubMed]
- Cundliffe, E. Glycosylation of macrolide antibiotics in extracts of Streptomyces lividans. Antimicrob. Agents Chemother. 1992, 36, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Disney, M.D. Studying Modification of Aminoglycoside Antibiotics by Resistance-Causing Enzymes via Microarray. Methods Mol. Biol. Clifton NJ 2012, 808. [Google Scholar] [CrossRef] [PubMed]
- Tillotson, G.S.; Theriault, N. New and alternative approaches to tackling antibiotic resistance. F1000prime Rep. 2013, 5, 51. [Google Scholar] [CrossRef] [PubMed]
- Shaw, K.J.; Rather, P.N.; Hare, R.S.; Miller, G.H. Molecular genetics of aminoglycoside resistance genes and familial relationships of the aminoglycoside-modifying enzymes. Microbiol. Rev. 1993, 57, 138. [Google Scholar] [PubMed]
- Yang, W.; Moore, I.F.; Koteva, K.P.; Bareich, D.C.; Hughes, D.W.; Wright, G.D. TetX Is a Flavin-dependent Monooxygenase Conferring Resistance to Tetracycline Antibiotics. J. Biol. Chem. 2004, 279, 52346–52352. [Google Scholar] [CrossRef] [PubMed]
- Mukhtar, T.A.; Koteva, K.P.; Hughes, D.W.; Wright, G.D. Vgb from Staphylococcus aureus Inactivates Streptogramin B Antibiotics by an Elimination Mechanism Not Hydrolysis†. Biochemistry (Mosc.) 2001, 40, 8877–8886. [Google Scholar] [CrossRef]
- Allignet, J.; Loncle, V.; Mazodier, P.; el Solh, N. Nucleotide sequence of a staphylococcal plasmid gene, vgb, encoding a hydrolase inactivating the B components of virginiamycin-like antibiotics. Plasmid 1988, 20, 271–275. [Google Scholar] [CrossRef]
- Worthington, R.J.; Melander, C. Combination approaches to combat multidrug-resistant bacteria. Trends Biotechnol. 2013, 31, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Blair, J.M.A.; Webber, M.A.; Baylay, A.J.; Ogbolu, D.O.; Piddock, L.J.V. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 2015, 13, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Munita, J.; Arias, C. Mechanisms of Antibiotic Resistance. In Virulence Mechanisms of Bacterial Pathogens, Fifth Edition; American Society of Microbiology, 2016; pp. 481–511. [Google Scholar]
- Hassan, K.A.; Skurray, R.A.; Brown, M.H. Active export proteins mediating drug resistance in staphylococci. J. Mol. Microbiol. Biotechnol. 2007, 12, 180–196. [Google Scholar] [CrossRef] [PubMed]
- Soto, S.M. Role of efflux pumps in the antibiotic resistance of bacteria embedded in a biofilm. Virulence 2013, 4, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Collu, F.; Cascella, M. Multidrug resistance and efflux pumps: insights from molecular dynamics simulations. Curr. Top. Med. Chem. 2013, 13, 3165–3183. [Google Scholar] [CrossRef] [PubMed]
- Lubelski, J.; Konings, W.N.; Driessen, A.J.M. Distribution and physiology of ABC-type transporters contributing to multidrug resistance in bacteria. Microbiol. Mol. Biol. Rev. MMBR 2007, 71, 463–476. [Google Scholar] [CrossRef] [PubMed]
- Pao, S.S.; Paulsen, I.T.; Saier, M.H., Jr. Major facilitator superfamily. Microbiol. Mol. Biol. Rev. MMBR 1998, 62, 1–34. [Google Scholar] [PubMed]
- Kuroda, T.; Tsuchiya, T. Multidrug efflux transporters in the MATE family. Biochim. Biophys. Acta 2009, 1794, 763–768. [Google Scholar] [CrossRef] [PubMed]
- Jack, D.L.; Yang, N.M.; Saier, M.H., Jr. The drug/metabolite transporter superfamily. Eur. J. Biochem. FEBS 2001, 268, 3620–3639. [Google Scholar] [CrossRef]
- Tseng, T.T.; Gratwick, K.S.; Kollman, J.; Park, D.; Nies, D.H.; Goffeau, A.; Saier, M.H., Jr. The RND permease superfamily: an ancient, ubiquitous and diverse family that includes human disease and development proteins. J. Mol. Microbiol. Biotechnol. 1999, 1, 107–125. [Google Scholar] [PubMed]
- van Veen, H.W. Structural biology: Last of the multidrug transporters. Nature 2010, 467, 926–927. [Google Scholar] [CrossRef] [PubMed]
- Higgins, C.F. Multiple molecular mechanisms for multidrug resistance transporters. Nature 2007, 446, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Poole, K.; Russell, A.; Lambert, P. Mechanisms of antimicrobial resistance: opportunities for new targeted therapies. Adv. Drug Deliv. Rev. 2005, 57, 1443–1445. [Google Scholar] [CrossRef]
- Alekshun, M.N.; Levy, S.B. Molecular mechanisms of antibacterial multidrug resistance. Cell 2007, 128, 1037–1050. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zolli-Juran, M.; Cechetto, J.D.; Daigle, D.M.; Wright, G.D.; Brown, E.D. Multicopy suppressors for novel antibacterial compounds reveal targets and drug efflux susceptibility. Chem. Biol. 2004, 11, 1423–1430. [Google Scholar] [CrossRef] [PubMed]
- Casadevall, A.; Pirofski, L. Host-Pathogen Interactions: Redefining the Basic Concepts of Virulence and Pathogenicity. Infect. Immun. 1999, 67, 3703–3713. [Google Scholar] [PubMed]
- Hacker, J.; Blum-Oehler, G.; Mühldorfer, I.; Tschäpe, H. Pathogenicity islands of virulent bacteria: structure, function and impact on microbial evolution. Mol. Microbiol. 1997, 23, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Bekal, S.; Brousseau, R.; Masson, L.; Prefontaine, G.; Fairbrother, J.; Harel, J. Rapid identification of Escherichia coli pathotypes by virulence gene detection with DNA microarrays. J. Clin. Microbiol. 2003, 41, 2113–2125. [Google Scholar] [CrossRef] [PubMed]
- Faraji, F.; Mahzounieh, M.; Ebrahimi, A.; Fallah, F.; Teymournejad, O.; Lajevardi, B. Molecular detection of virulence genes in Pseudomonas aeruginosa isolated from children with Cystic Fibrosis and burn wounds in Iran. Microb. Pathog. 2016, 99, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Gomes, F.; Teixeira, P.; Cerca, N.; Ceri, H.; Oliveira, R. Virulence Gene Expression by Staphylococcus epidermidis Biofilm Cells Exposed to Antibiotics. Microb. Drug Resist. 2011, 17, 191–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kao, C.Y.; Lin, W.H.; Tseng, C.C.; Wu, A.B.; Wang, M.C.; Wu, J.J. The complex interplay among bacterial motility and virulence factors in different Escherichia coli infections. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 2157–2162. [Google Scholar] [CrossRef] [PubMed]
- Cossart, P.; Vicente, M.F.; Mengaud, J.; Baquero, F.; Perez-Diaz, J.C.; Berche, P. Listeriolysin O is essential for virulence of Listeria monocytogenes: direct evidence obtained by gene complementation. Infect. Immun. 1989, 57, 3629–3636. [Google Scholar] [PubMed]
- Kim, M.-J.; Han, J.-K.; Park, J.-S.; Lee, J.-S.; Lee, S.-H.; Cho, J.-I.; Kim, K.-S. Various Enterotoxin and Other Virulence Factor Genes Widespread Among Bacillus cereus and Bacillus thuringiensis Strains. J. Microbiol. Biotechnol. 2015, 25, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Burnside, K.; Lembo, A.; de los Reyes, M.; Iliuk, A.; BinhTran, N.-T.; Connelly, J.E.; Lin, W.-J.; Schmidt, B.Z.; Richardson, A.R.; Fang, F.C.; et al. Regulation of Hemolysin Expression and Virulence of Staphylococcus aureus by a Serine/Threonine Kinase and Phosphatase. PLoS ONE 2010, 5, e11071. [Google Scholar] [CrossRef] [PubMed]
- Emaneini, M.; Jabalameli, F.; Mirsalehian, A.; Ghasemi, A.; Beigverdi, R. Characterization of virulence factors, antimicrobial resistance pattern and clonal complexes of group B streptococci isolated from neonates. Microb. Pathog. 2016, 99, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Giaouris, E.; Heir, E.; Desvaux, M.; Hébraud, M.; Møretrø, T.; Langsrud, S.; Doulgeraki, A.; Nychas, G.-J.; Kačániová, M.; Czaczyk, K.; et al. Intra- and inter-species interactions within biofilms of important foodborne bacterial pathogens. Microb. Physiol. Metab. 2015, 841. [Google Scholar] [CrossRef] [PubMed]
- Silverman, M.; Simon, M. Positioning flagellar genes in Escherichia coli by deletion analysis. J. Bacteriol. 1974, 117, 73–79. [Google Scholar] [PubMed]
- Bartlett, D.H.; Frantz, B.B.; Matsumura, P. Flagellar transcriptional activators FlbB and FlaI: gene sequences and 5’ consensus sequences of operons under FlbB and FlaI control. J. Bacteriol. 1988, 170, 1575–1581. [Google Scholar] [CrossRef] [PubMed]
- Thakur, P.; Chawla, R.; Tanwar, A.; Chakotiya, A.S.; Narula, A.; Goel, R.; Arora, R.; Sharma, R.K. Attenuation of adhesion, quorum sensing and biofilm mediated virulence of carbapenem resistant Escherichia coli by selected natural plant products. Microb. Pathog. 2016, 92, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Prüss, B.M.; Liu, X.; Hendrickson, W.; Matsumura, P. FlhD/FlhC-regulated promoters analyzed by gene array and lacZ gene fusions. FEMS Microbiol. Lett. 2001, 197, 91–97. [Google Scholar] [CrossRef]
- Prüss, B.M.; Matsumura, P. A regulator of the flagellar regulon of Escherichia coli, flhD, also affects cell division. J. Bacteriol. 1996, 178, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Ribet, D.; Cossart, P. How bacterial pathogens colonize their hosts and invade deeper tissues. Microbes Infect. 2015, 17, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Holden, V.I.; Bachman, M.A. Diverging roles of bacterial siderophores during infection. Metallomics 2015, 7, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Tu, J.; Lu, F.; Miao, S.; Ni, X.; Jiang, P.; Yu, H.; Xing, L.; Yu, S.; Ding, C.; Hu, Q. The siderophore-interacting protein is involved in iron acquisition and virulence of Riemerella anatipestifer strain CH3. Vet. Microbiol. 2014, 168, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Wang, L.; Li, J.; Xu, G.; He, M.; Li, Y.; Huang, R. Salmonella spv locus suppresses host innate immune responses to bacterial infection. Fish Shellfish Immunol. 2016, 58, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Andrade, W.A.; Firon, A.; Schmidt, T.; Hornung, V.; Fitzgerald, K.A.; Kurt-Jones, E.A.; Trieu-Cuot, P.; Golenbock, D.T.; Kaminski, P.-A. Group B Streptococcus Degrades Cyclic-di-AMP to Modulate STING-Dependent Type I Interferon Production. Cell Host Microbe 2016, 20, 49–59. [Google Scholar] [CrossRef] [PubMed]
- McFarland, A.P.; Woodward, J.J. Flying Under the Radar: Immune Evasion by Group B Streptococcus. Cell Host Microbe 2016, 20, 4–6. [Google Scholar] [CrossRef] [PubMed]
- Avondt, K.V.; van Sorge, N.M.; Meyaard, L. Bacterial Immune Evasion through Manipulation of Host Inhibitory Immune Signaling. PLoS Pathog 2015, 11, e1004644. [Google Scholar]
- Cohen, T.S.; Parker, D.; Prince, A. Pseudomonas aeruginosa Host Immune Evasion. In Pseudomonas; Ramos, J.-L., Goldberg, J.B., Filloux, A., Eds.; Springer Netherlands, 2015; pp. 3–23. [Google Scholar]
- Ali, S.R.; Karin, M.; Nizet, V. Signaling cascades and inflammasome activation in microbial infections. Inflammasome 2016, 2. [Google Scholar] [CrossRef]
- Sperandio, B.; Fischer, N.; Sansonetti, P.J. Mucosal physical and chemical innate barriers: Lessons from microbial evasion strategies. Semin. Immunol. 2015, 27, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Kong, C.; Neoh, H.; Nathan, S. Targeting Staphylococcus aureus Toxins: A Potential form of Anti-Virulence Therapy. Toxins 2016, 8, 72. [Google Scholar] [CrossRef] [PubMed]
- Overhage, J.; Bains, M.; Brazas, M.D.; Hancock, R.E.W. Swarming of Pseudomonas aeruginosa Is a Complex Adaptation Leading to Increased Production of Virulence Factors and Antibiotic Resistance. J. Bacteriol. 2008, 190, 2671–2679. [Google Scholar] [CrossRef] [PubMed]
- Beutin, L.; Montenegro, M.A.; Orskov, I.; Orskov, F.; Prada, J.; Zimmermann, S.; Stephan, R. Close association of verotoxin (Shiga-like toxin) production with enterohemolysin production in strains of Escherichia coli. J. Clin. Microbiol. 1989, 27, 2559–2564. [Google Scholar] [PubMed]
- Schmidt, H.; Henkel, B.; Karch, H. A gene cluster closely related to type II secretion pathway operons of gram-negative bacteria is located on the large plasmid of enterohemorrhagic Escherichia coli O157 strains. FEMS Microbiol. Lett. 1997, 148, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Brunder, W.; Schmidt, H.; Karch, H. KatP, a novel catalase-peroxidase encoded by the large plasmid of enterohaemorrhagic Escherichia coli O157:H7. Microbiology 1996, 142, 3305–3315. [Google Scholar] [CrossRef] [PubMed]
- Tatsuno, I.; Horie, M.; Abe, H.; Miki, T.; Makino, K.; Shinagawa, H.; Taguchi, H.; Kamiya, S.; Hayashi, T.; Sasakawa, C. toxB Gene on pO157 of Enterohemorrhagic Escherichia coli O157:H7 Is Required for Full Epithelial Cell Adherence Phenotype. Infect. Immun. 2001, 69, 6660–6669. [Google Scholar] [CrossRef] [PubMed]
- Lathem, W.W.; Grys, T.E.; Witowski, S.E.; Torres, A.G.; Kaper, J.B.; Tarr, P.I.; Welch, R.A. StcE, a metalloprotease secreted by Escherichia coli O157:H7, specifically cleaves C1 esterase inhibitor. Mol. Microbiol. 2002, 45, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.W.; Lim, J.Y.; Park, Y.H.; Hovde, C.J. Involvement of the Escherichia coli O157:H7(pO157) ecf operon and lipid A myristoyl transferase activity in bacterial survival in the bovine gastrointestinal tract and bacterial persistence in farm water troughs. Infect. Immun. 2005, 73, 2367–2378. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.Y.; Yoon, J.; Hovde, C.J. A brief overview of Escherichia coli O157:H7 and its plasmid O157. J. Microbiol. Biotechnol. 2010, 20, 5–14. [Google Scholar] [PubMed]
- Losada, L.; DebRoy, C.; Radune, D.; Kim, M.; Sanka, R.; Brinkac, L.; Kariyawasam, S.; Shelton, D.; Fratamico, P.M.; Kapur, V.; et al. Whole genome sequencing of diverse Shiga toxin-producing and non-producing Escherichia coli strains reveals a variety of virulence and novel antibiotic resistance plasmids. Plasmid 2016, 83, 8–11. [Google Scholar] [CrossRef] [PubMed]
- Neidig, A.; Yeung, A.T.Y.; Rosay, T.; Tettmann, B.; Strempel, N.; Rueger, M.; Lesouhaitier, O.; Overhage, J. TypA is involved in virulence, antimicrobial resistance and biofilm formation in Pseudomonas aeruginosa. BMC Microbiol. 2013, 13, 77. [Google Scholar] [CrossRef] [PubMed]
- Sauer, K.; Camper, A.K.; Ehrlich, G.D.; Costerton, J.W.; Davies, D.G. Pseudomonas aeruginosa displays multiple phenotypes during development as a biofilm. J. Bacteriol. 2002, 184, 1140–1154. [Google Scholar] [CrossRef] [PubMed]
- Lebreton, A.; Cossart, P. RNA- and protein-mediated control of Listeria monocytogenes virulence gene expression. RNA Biol. 2016, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Cameron, D.R.; Jiang, J.-H.; Kostoulias, X.; Foxwell, D.J.; Peleg, A.Y. Vancomycin susceptibility in methicillin-resistant Staphylococcus aureus is mediated by YycHI activation of the WalRK essential two-component regulatory system. Sci. Rep. 2016, 6, 30823. [Google Scholar] [CrossRef] [PubMed]
- Agladze, K.; Jackson, D.; Romeo, T. Periodicity of Cell Attachment Patterns during Escherichia coli Biofilm Development. J. Bacteriol. 2003, 185, 5632–5638. [Google Scholar] [CrossRef] [PubMed]
- Stewart, P.S.; Franklin, M.J.; Williamson, K.S.; Folsom, J.P.; Boegli, L.; James, G.A. Contribution of stress responses to antibiotic tolerance in Pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother. 2015, 59, 3838–3847. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Schurr, M.J.; Sauer, K. The MerR-like regulator BrlR confers biofilm tolerance by activating multidrug efflux pumps in Pseudomonas aeruginosa biofilms. J. Bacteriol. 2013, 195, 3352–3363. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, B.K.; Kushner, S.R. The majority of Escherichia coli mRNAs undergo post-transcriptional modification in exponentially growing cells. Nucleic Acids Res. 2006, 34, 5695–5704. [Google Scholar] [CrossRef] [PubMed]
- Kerrinnes, T.; Zelas, Z.B.-B.; Streckel, W.; Faber, F.; Tietze, E.; Tschäpe, H.; Yaron, S. CsrA and CsrB are required for the post-transcriptional control of the virulence-associated effector protein AvrA of Salmonella enterica. Int. J. Med. Microbiol. IJMM 2009, 299, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Reales-Calderón, J.A.; Corona, F.; Monteoliva, L.; Gil, C.; Martínez, J.L. Quantitative proteomics unravels that the post-transcriptional regulator Crc modulates the generation of vesicles and secreted virulence determinants of Pseudomonas aeruginosa. J. Proteomics 2015, 127, 352–364. [Google Scholar] [CrossRef] [PubMed]
- Warrier, T.; Kapilashrami, K.; Argyrou, A.; Ioerger, T.R.; Little, D.; Murphy, K.C.; Nandakumar, M.; Park, S.; Gold, B.; Mi, J.; et al. N-methylation of a bactericidal compound as a resistance mechanism in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E4523–E4530. [Google Scholar] [CrossRef] [PubMed]
- Schiano, C.A.; Lathem, W.W. Post-Transcriptional Regulation of Gene Expression in Yersinia Species. Front. Cell. Infect. Microbiol. 2012, 2. [Google Scholar] [CrossRef] [PubMed]
- Toh, S.M. Target Site Modifications in the Ribosome as Antibiotic Resistance Mechanisms, in Pharmacognosy. University of Illinois at Chicago: ProQuest LLC., University of Illisois: Chicago, IL, USA, 2008. [Google Scholar]
- Dar, D.; Shamir, M.; Mellin, J.R.; Koutero, M.; Stern-Ginossar, N.; Cossart, P.; Sorek, R. Term-seq reveals abundant ribo-regulation of antibiotics resistance in bacteria. Science 2016, 352, aad9822. [Google Scholar] [CrossRef] [PubMed]
- Bjarnsholt, T.; Tolker-Nielsen, T.; Høiby, N.; Givskov, M. Interference of Pseudomonas aeruginosa signalling and biofilm formation for infection control. Expert Rev. Mol. Med. 2010, 12, e11. [Google Scholar] [CrossRef] [PubMed]
- Oliva, G.; Sahr, T.; Buchrieser, C. Small RNAs, 5’ UTR elements and RNA-binding proteins in intracellular bacteria: impact on metabolism and virulence. FEMS Microbiol. Rev. 2015, 39, 331–349. [Google Scholar] [CrossRef] [PubMed]
- Fritz, G.; Dintner, S.; Treichel, N.S.; Radeck, J.; Gerland, U.; Mascher, T.; Gebhard, S. A New Way of Sensing: Need-Based Activation of Antibiotic Resistance by a Flux-Sensing Mechanism. mBio 2015, 6, e00975-15. [Google Scholar] [CrossRef] [PubMed]
- Dintner, S.; Staroń, A.; Berchtold, E.; Petri, T.; Mascher, T.; Gebhard, S. Coevolution of ABC Transporters and Two-Component Regulatory Systems as Resistance Modules against Antimicrobial Peptides in Firmicutes Bacteria. J. Bacteriol. 2011, 193, 3851–3862. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, D.; Natarajan, J. Network analysis of S. aureus response to ramoplanin reveals modules for virulence factors and resistance mechanisms and characteristic novel genes. Gene 2015, 574, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Kjos, M.; Miller, E.; Slager, J.; Lake, F.B.; Gericke, O.; Roberts, I.S.; Rozen, D.E.; Veening, J.-W. Expression of Streptococcus pneumoniae Bacteriocins Is Induced by Antibiotics via Regulatory Interplay with the Competence System. PLoS Pathog. 2016, 12, e1005422. [Google Scholar] [CrossRef] [PubMed]
- Grant, A.J.; Farris, M.; Alefounder, P.; Williams, P.H.; Woodward, M.J.; O’Connor, C.D. Co-ordination of pathogenicity island expression by the BipA GTPase in enteropathogenic Escherichia coli (EPEC). Mol. Microbiol. 2003, 48, 507–521. [Google Scholar] [CrossRef] [PubMed]
- Guragain, M.; King, M.M.; Williamson, K.S.; Pérez-Osorio, A.C.; Akiyama, T.; Khanam, S.; Patrauchan, M.A.; Franklin, M.J. The Pseudomonas aeruginosa PAO1 Two-Component Regulator CarSR Regulates Calcium Homeostasis and Calcium-Induced Virulence Factor Production through Its Regulatory Targets CarO and CarP. J. Bacteriol. 2016, 198, 951–963. [Google Scholar] [CrossRef] [PubMed]
- Aedo, S.; Tomasz, A. Role of the Stringent Stress Response in the Antibiotic Resistance Phenotype of Methicillin-Resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2016, 60, 2311–2317. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Bittner, A.N.; Wang, J.D. Diversity in (p)ppGpp metabolism and effectors. Curr. Opin. Microbiol. 2015, 24, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Heroven, A.K.; Böhme, K.; Dersch, P. The Csr/Rsm system of Yersinia and related pathogens. RNA Biol. 2012, 9, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Bartoli, C.; Lamichhane, J.R.; Berge, O.; Varvaro, L.; Morris, C.E. Mutability in Pseudomonas viridiflava as a programmed balance between antibiotic resistance and pathogenicity. Mol. Plant Pathol. 2015, 16, 860–869. [Google Scholar] [CrossRef] [PubMed]
- Gebhardt, M.J.; Gallagher, L.A.; Jacobson, R.K.; Usacheva, E.A.; Peterson, L.R.; Zurawski, D.V.; Shuman, H.A. Joint Transcriptional Control of Virulence and Resistance to Antibiotic and Environmental Stress in Acinetobacter baumannii. mBio 2015, 6, e01660-15. [Google Scholar] [CrossRef] [PubMed]
- Skurnik, D.; Roux, D.; Cattoir, V.; Danilchanka, O.; Lu, X.; Yoder-Himes, D.R.; Han, K.; Guillard, T.; Jiang, D.; Gaultier, C.; et al. Enhanced in vivo fitness of carbapenem-resistant oprD mutants of Pseudomonas aeruginosa revealed through high-throughput sequencing. Proc. Natl. Acad. Sci. 2013, 110, 20747–20752. [Google Scholar] [CrossRef] [PubMed]
- Davey, M.E.; O’toole, G.A. Microbial biofilms: from ecology to molecular genetics. Microbiol. Mol. Biol. Rev. MMBR 2000, 64, 847–867. [Google Scholar] [CrossRef] [PubMed]
- Déziel, E.; Gopalan, S.; Tampakaki, A.P.; Lépine, F.; Padfield, K.E.; Saucier, M.; Xiao, G.; Rahme, L.G. The contribution of MvfR to Pseudomonas aeruginosa pathogenesis and quorum sensing circuitry regulation: multiple quorum sensing-regulated genes are modulated without affecting lasRI, rhlRI or the production of N-acyl-L-homoserine lactones. Mol. Microbiol. 2005, 55, 998–1014. [Google Scholar] [CrossRef] [PubMed]
- Stoodley, P.; Sauer, K.; Davies, D.G.; Costerton, J.W. Biofilms as complex differentiated communities. Annu. Rev. Microbiol. 2002, 56, 187–209. [Google Scholar] [CrossRef] [PubMed]
- Mah, T.F.; O’Toole, G.A. Mechanisms of biofilm resistance to antimicrobial agents. Trends Microbiol. 2001, 9, 34–39. [Google Scholar] [CrossRef]
- Drenkard, E.; Ausubel, F.M. Pseudomonas biofilm formation and antibiotic resistance are linked to phenotypic variation. Nature 2002, 416, 740–743. [Google Scholar] [CrossRef] [PubMed]
- Kapur, N.; Grimwood, K.; Masters, I.B.; Morris, P.S.; Chang, A.B. Lower airway microbiology and cellularity in children with newly diagnosed non-CF bronchiectasis. Pediatr. Pulmonol. 2012, 47, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.W.K.; Periasamy, S.; Mukherjee, M.; Xie, C.; Kjelleberg, S.; Rice, S.A. Biofilm development and enhanced stress resistance of a model, mixed-species community biofilm. ISME J. 2014, 8, 894–907. [Google Scholar] [CrossRef] [PubMed]
- Seth, A.K.; Geringer, M.R.; Hong, S.J.; Leung, K.P.; Galiano, R.D.; Mustoe, T.A. Comparative analysis of single-species and polybacterial wound biofilms using a quantitative, in vivo, rabbit ear model. PLoS ONE 2012, 7, e42897. [Google Scholar] [CrossRef] [PubMed]
- Pastar, I.; Nusbaum, A.G.; Gil, J.; Patel, S.B.; Chen, J.; Valdes, J.; Stojadinovic, O.; Plano, L.R.; Tomic-Canic, M.; Davis, S.C. Interactions of methicillin resistant Staphylococcus aureus USA300 and Pseudomonas aeruginosa in polymicrobial wound infection. PLoS ONE 2013, 8, e56846. [Google Scholar] [CrossRef] [PubMed]
- Brunsing, R.L.; La Clair, C.; Tang, S.; Chiang, C.; Hancock, L.E.; Perego, M.; Hoch, J.A. Characterization of sporulation histidine kinases of Bacillus anthracis. J. Bacteriol. 2005, 187, 6972–6981. [Google Scholar] [CrossRef] [PubMed]
- Tatke, G.; Kumari, H.; Silva-Herzog, E.; Ramirez, L.; Mathee, K. Pseudomonas aeruginosa MifS-MifR Two-Component System Is Specific for α-Ketoglutarate Utilization. PLoS ONE 2015, 10, e0129629. [Google Scholar] [CrossRef] [PubMed]
- Lou, Q.; Ma, Y.; Qu, D. Two-component signal transduction system SaeRS is involved in competence and penicillin susceptibility in Staphylococcus epidermidis. J. Basic Microbiol. 2016, 56, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Davies, D.G.; Parsek, M.R.; Pearson, J.P.; Iglewski, B.H.; Costerton, J.W.; Greenberg, E.P. The Involvement of Cell-to-Cell Signals in the Development of a Bacterial Biofilm. Science 1998, 280, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Fuqua, C.; Greenberg, E.P. Self perception in bacteria: quorum sensing with acylated homoserine lactones. Curr. Opin. Microbiol. 1998, 1, 183–189. [Google Scholar] [CrossRef]
- Bassler, B.L.; Wright, M.; Showalter, R.E.; Silverman, M.R. Intercellular signalling in Vibrio harveyi: sequence and function of genes regulating expression of luminescence. Mol. Microbiol. 1993, 9, 773–786. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.G.; Meighen, E.A. Purification and structural identification of an autoinducer for the luminescence system of Vibrio harveyi. J. Biol. Chem. 1989, 264, 21670–21676. [Google Scholar] [PubMed]
- Chen, X.; Schauder, S.; Potier, N.; Van Dorsselaer, A.; Pelczer, I.; Bassler, B.L.; Hughson, F.M. Structural identification of a bacterial quorum-sensing signal containing boron. Nature 2002, 415, 545–549. [Google Scholar] [CrossRef] [PubMed]
- Neiditch, M.B.; Federle, M.J.; Miller, S.T.; Bassler, B.L.; Hughson, F.M. Regulation of LuxPQ receptor activity by the quorum-sensing signal autoinducer-2. Mol. Cell 2005, 18, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Ji, G.; Beavis, R.C.; Novick, R.P. Cell density control of staphylococcal virulence mediated by an octapeptide pheromone. Proc. Natl. Acad. Sci. USA 1995, 92, 12055–12059. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Guan, W.; Huang, Q.; Yang, Y.; Yan, W.; Sun, B.; Zhao, T. Quorum-sensing contributes to virulence, twitching motility, seed attachment and biofilm formation in the wild type strain Aac-5 of Acidovorax citrulli. Microb. Pathog. 2016, 100, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Delauné, A.; Dubrac, S.; Blanchet, C.; Poupel, O.; Mäder, U.; Hiron, A.; Leduc, A.; Fitting, C.; Nicolas, P.; Cavaillon, J.-M.; et al. The WalKR system controls major staphylococcal virulence genes and is involved in triggering the host inflammatory response. Infect. Immun. 2012, 80, 3438–3453. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Inouye, M. Requirement of both kinase and phosphatase activities of an Escherichia coli receptor (Taz1) for ligand-dependent signal transduction. J. Mol. Biol. 1993, 231, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Hoch, J.A. Two-component and phosphorelay signal transduction. Curr. Opin. Microbiol. 2000, 3, 165–170. [Google Scholar] [CrossRef]
- Ji, Q.; Chen, P.J.; Qin, G.; Deng, X.; Hao, Z.; Wawrzak, Z.; Yeo, W.-S.; Quang, J.W.; Cho, H.; Luo, G.-Z.; et al. Structure and mechanism of the essential two-component signal-transduction system WalKR in Staphylococcus aureus. Nat. Commun. 2016, 7, 11000. [Google Scholar] [CrossRef] [PubMed]
- Hausner, M.; Wuertz, S. High rates of conjugation in bacterial biofilms as determined by quantitative in situ analysis. Appl. Environ. Microbiol. 1999, 65, 3710–3713. [Google Scholar] [PubMed]
- Anderl, J.N.; Franklin, M.J.; Stewart, P.S. Role of antibiotic penetration limitation in Klebsiella pneumoniae biofilm resistance to ampicillin and ciprofloxacin. Antimicrob. Agents Chemother. 2000, 44, 1818–1824. [Google Scholar] [CrossRef] [PubMed]
- Jitwasinkul, T.; Suriyaphol, P.; Tangphatsornruang, S.; Hansen, M.A.; Hansen, L.H.; Sørensen, S.J.; Permpikul, C.; Rongrungruang, Y.; Tribuddharat, C. Plasmid metagenomics reveals multiple antibiotic resistance gene classes among the gut microbiomes of hospitalised patients. J. Glob. Antimicrob. Resist. 2016, 6, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Chen, Y.; Zheng, X.; Su, Y.; Wan, R.; Yang, S. Distribution of tetracycline resistance genes in anaerobic treatment of waste sludge: The role of pH in regulating tetracycline resistant bacteria and horizontal gene transfer. Bioresour. Technol. 2016, 218, 1284–1289. [Google Scholar] [CrossRef] [PubMed]
- Ternent, L.; Dyson, R.J.; Krachler, A.-M.; Jabbari, S. Bacterial fitness shapes the population dynamics of antibiotic-resistant and -susceptible bacteria in a model of combined antibiotic and anti-virulence treatment. J. Theor. Biol. 2015, 372, 1–11. [Google Scholar] [CrossRef] [PubMed]


© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schroeder, M.; Brooks, B.D.; Brooks, A.E. The Complex Relationship between Virulence and Antibiotic Resistance. Genes 2017, 8, 39. https://doi.org/10.3390/genes8010039
Schroeder M, Brooks BD, Brooks AE. The Complex Relationship between Virulence and Antibiotic Resistance. Genes. 2017; 8(1):39. https://doi.org/10.3390/genes8010039
Chicago/Turabian StyleSchroeder, Meredith, Benjamin D. Brooks, and Amanda E. Brooks. 2017. "The Complex Relationship between Virulence and Antibiotic Resistance" Genes 8, no. 1: 39. https://doi.org/10.3390/genes8010039