
Detecting the Common and Individual Effects of Rare Variants on Quantitative Traits by Using Extreme Phenotype Sampling


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
3. Simulation and Results
3.1. Simulation Design
3.2. Evaluation on Type I Error Rates

{kind=link}
{kind=link}
{kind=link}
| Tails | Sample Size | α | JOINT | RS_Fisher | RS_min-p | ES_Fisher | ES_min-p | RS_Burden | ES_Burden |
|---|---|---|---|---|---|---|---|---|---|
| 0.1 | 500 | 0.01 | 0.015 | 0.013 | 0.013 | 0.014 | 0.013 | 0.010 | 0.016 |
| 1000 | 0.01 | 0.011 | 0.010 | 0.018 | 0.008 | 0.003 | 0.015 | 0.008 | |
| 2000 | 0.01 | 0.011 | 0.012 | 0.012 | 0.011 | 0.013 | 0.012 | 0.016 | |
| 500 | 0.05 | 0.047 | 0.051 | 0.045 | 0.043 | 0.043 | 0.048 | 0.047 | |
| 1000 | 0.05 | 0.057 | 0.048 | 0.052 | 0.050 | 0.052 | 0.042 | 0.047 | |
| 2000 | 0.05 | 0.050 | 0.049 | 0.050 | 0.050 | 0.050 | 0.049 | 0.052 | |
| 0.2 | 500 | 0.01 | 0.008 | 0.005 | 0.007 | 0.009 | 0.011 | 0.009 | 0.015 |
| 1000 | 0.01 | 0.012 | 0.012 | 0.012 | 0.011 | 0.010 | 0.010 | 0.013 | |
| 2000 | 0.01 | 0.009 | 0.013 | 0.013 | 0.014 | 0.011 | 0.018 | 0.018 | |
| 500 | 0.05 | 0.053 | 0.049 | 0.054 | 0.041 | 0.044 | 0.054 | 0.037 | |
| 1000 | 0.05 | 0.046 | 0.040 | 0.039 | 0.052 | 0.061 | 0.036 | 0.052 | |
| 2000 | 0.05 | 0.041 | 0.049 | 0.052 | 0.050 | 0.045 | 0.050 | 0.049 |
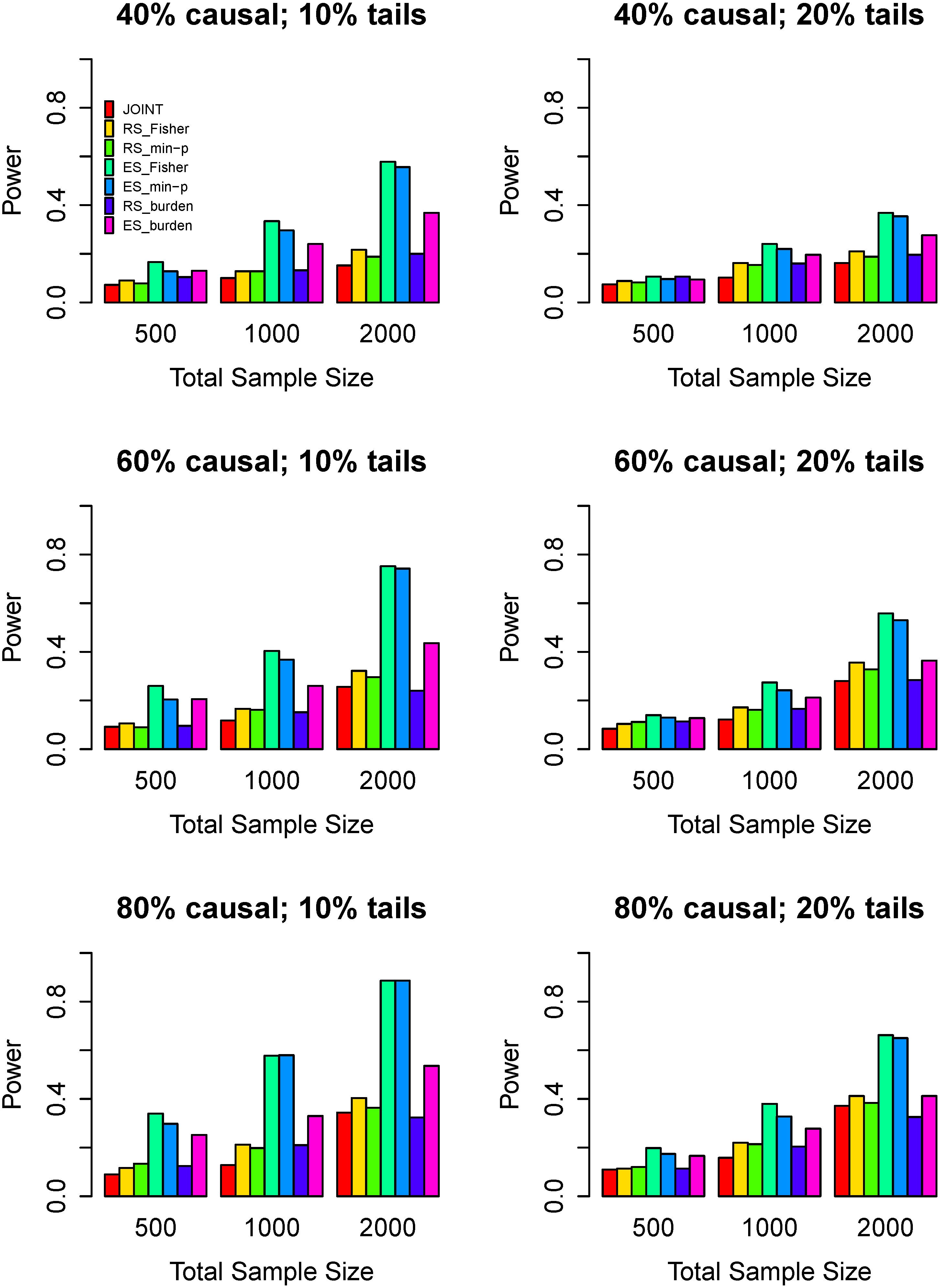
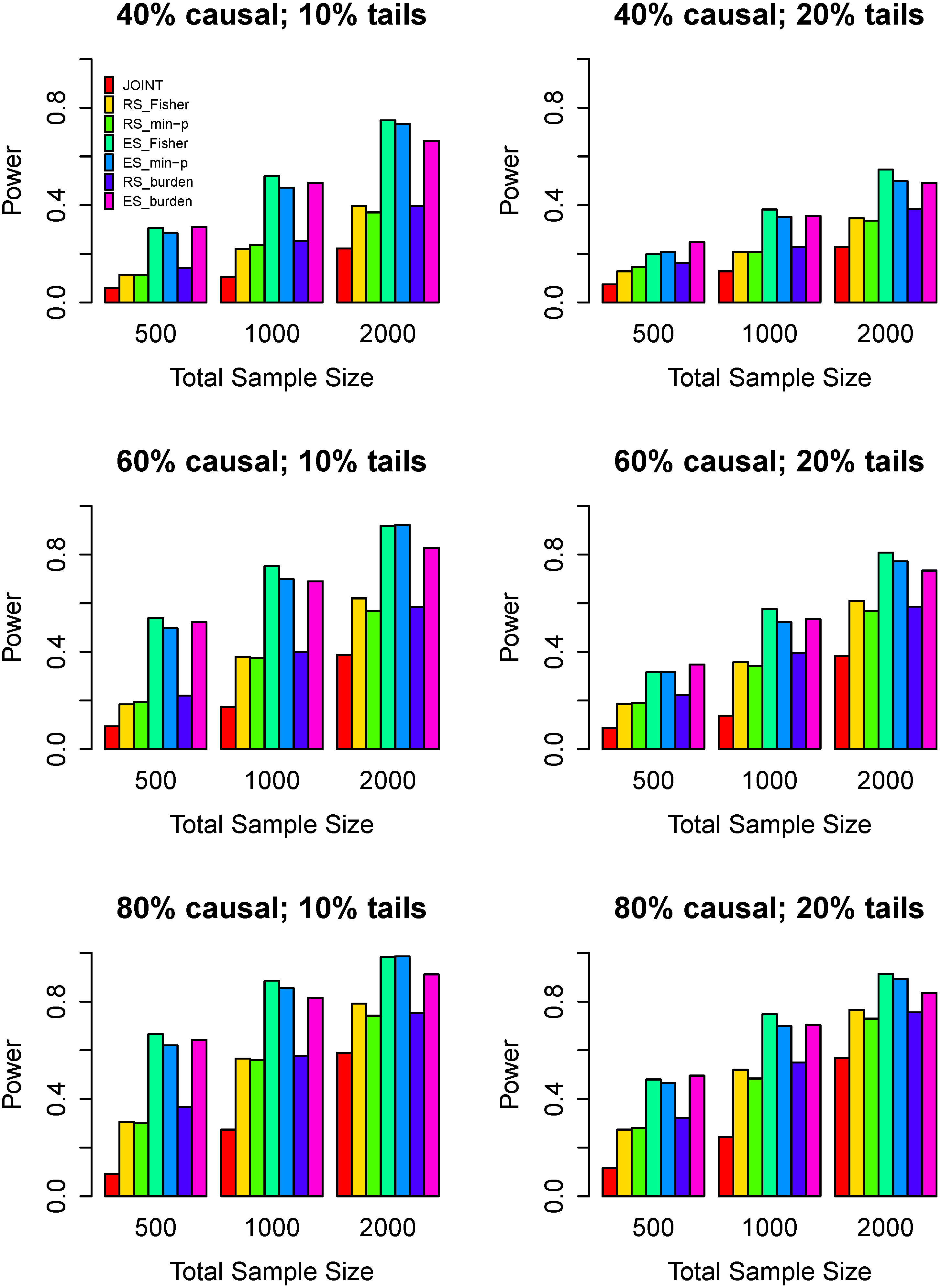
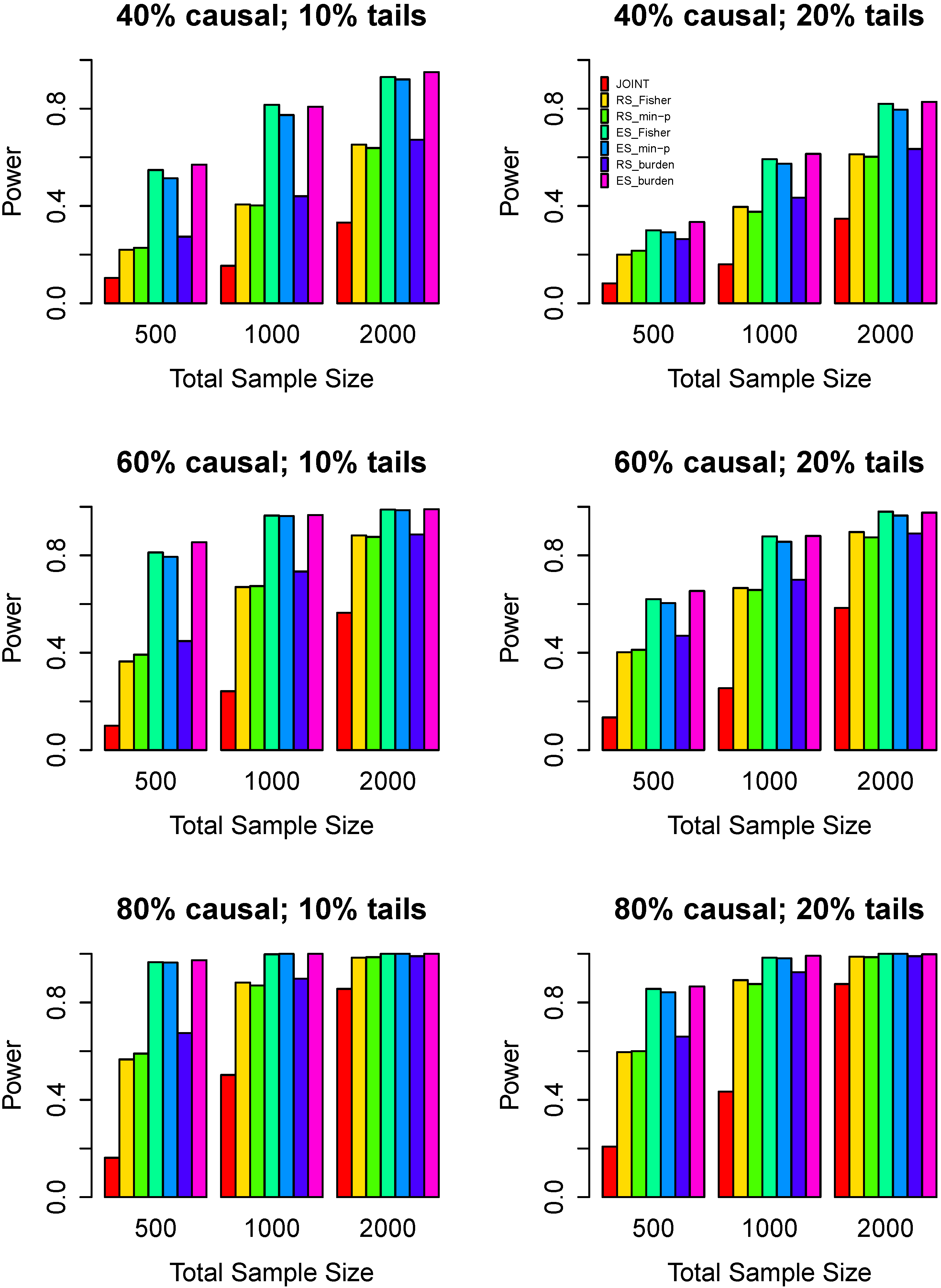
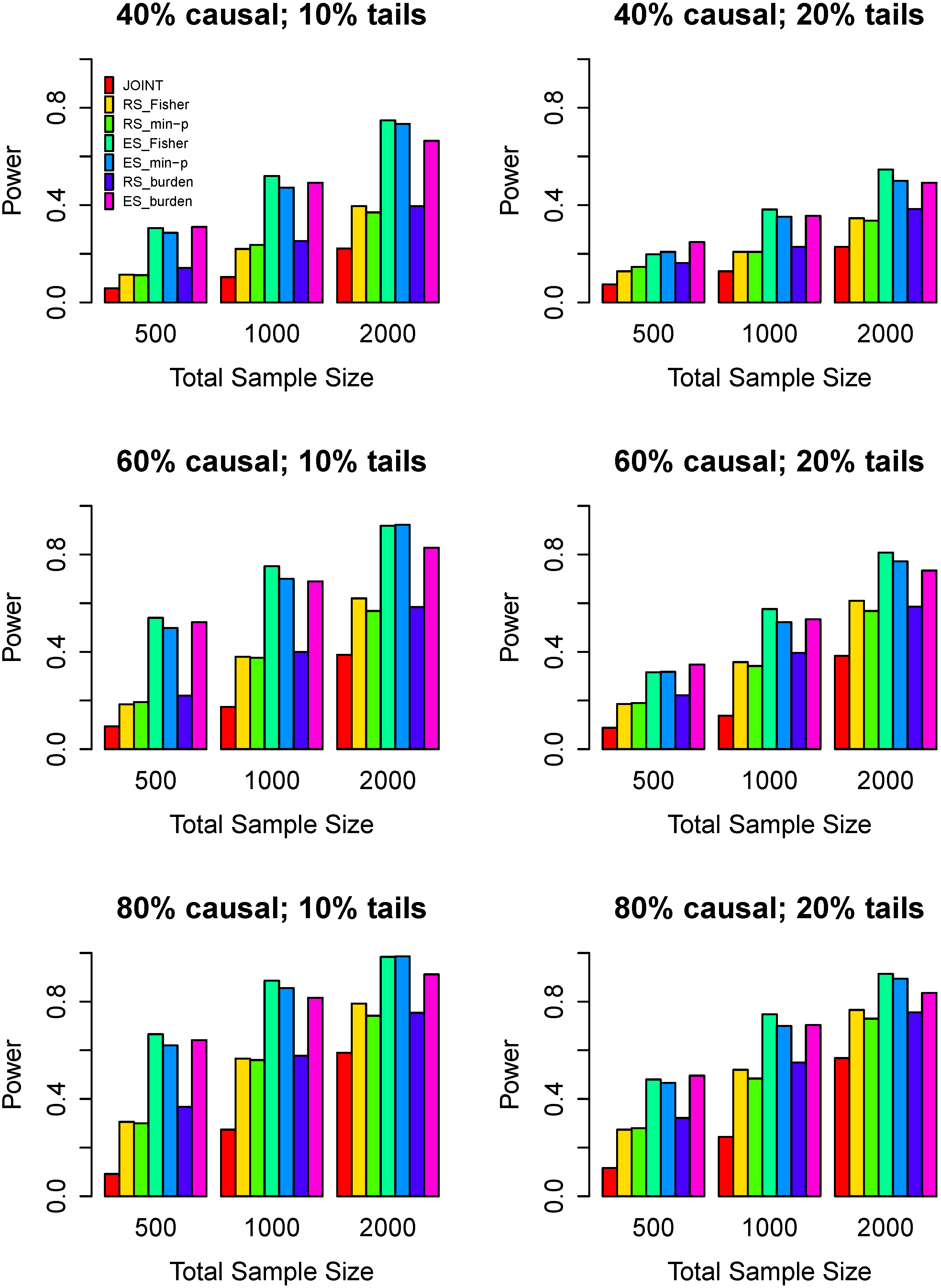
3.3. Power Comparisons
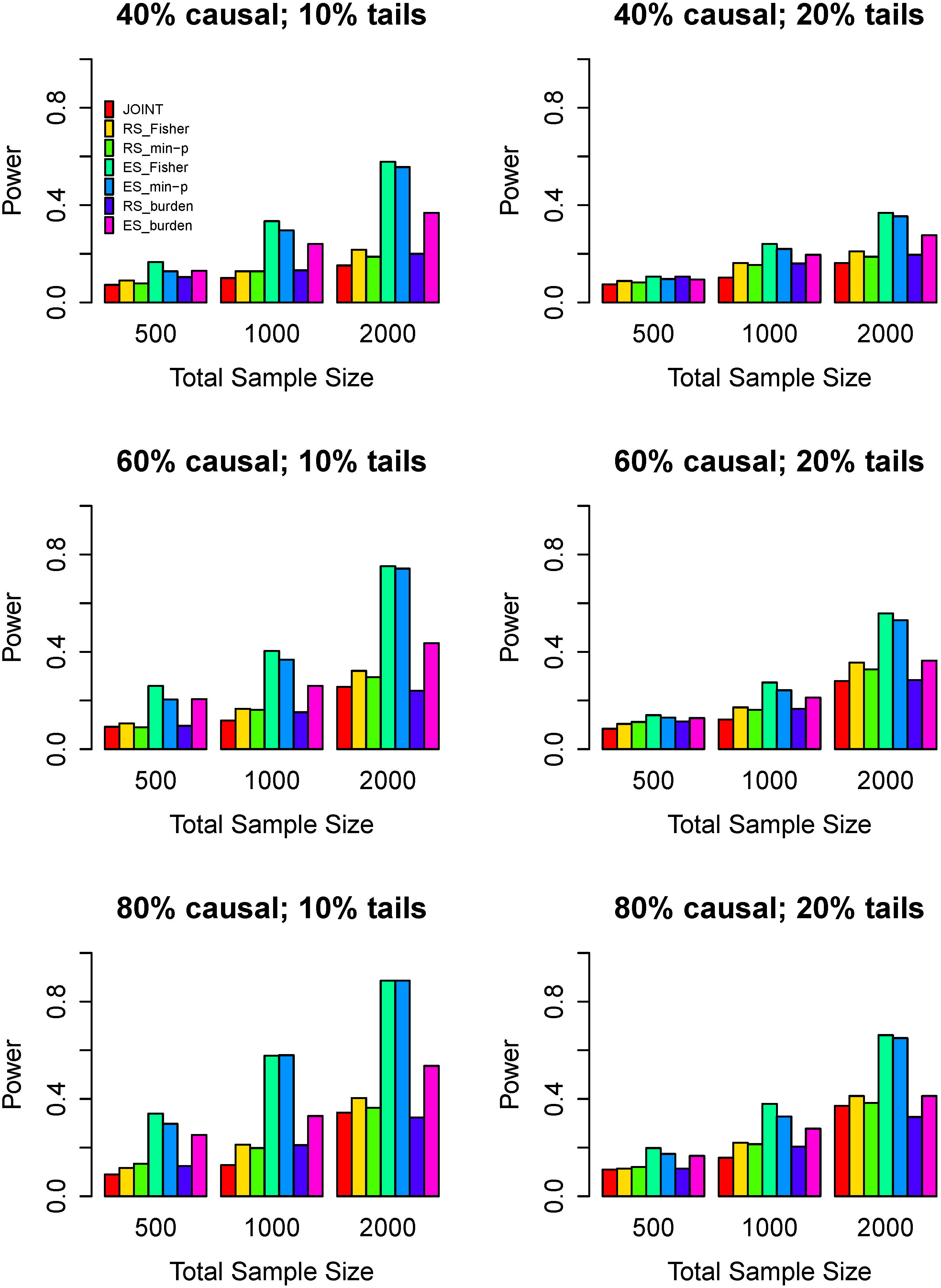

4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Appendix. Score Vector
References
- Bansal, V.; Libiger, O.; Torkamani, A.; Schork, N.J. Statistical analysis strategies for association studies involving rare variants. Nat. Rev. Genet. 2010, 11, 773–785. [Google Scholar] [CrossRef] [PubMed]
- Maher, B. Personal genomes: The case of the missing heritability. Nature 2008, 456, 18–21. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.I.; Abecasis, G.R.; Cardon, L.R.; Goldstein, D.B.; Little, J.; Ioannidis, J.P.; Hirschhorn, J.N. Genome-wide association studies for complex traits: Consensus, uncertainty and challenges. Nat. Rev. Genet. 2008, 9, 356–369. [Google Scholar] [CrossRef] [PubMed]
- Schork, N.J.; Murray, S.S.; Frazer, K.A.; Topol, E.J. Common vs. rare allele hypotheses for complex diseases. Curr. Opin. Genet. Dev. 2009, 19, 212–219. [Google Scholar] [CrossRef] [PubMed]
- Bodmer, W.; Bonilla, C. Common and rare variants in multifactorial susceptibility to common diseases. Nat. Genet. 2008, 40, 695–701. [Google Scholar] [CrossRef] [PubMed]
- Gorlov, I.P.; Gorlova, O.Y.; Sunyaev, S.R.; Spitz, M.R.; Amos, C.I. Shifting paradigm of association studies: Value of rare single-nucleotide polymorphisms. Am. J. Hum. Genet. 2008, 82, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Ji, W.; Foo, J.N.; Oa̧ŕRoak, B.J.; Zhao, H.; Larson, M.G.; Simon, D.B.; Newton-Cheh, C.; State, M.W.; Levy, D.; Lifton, R.P. Rare independent mutations in renal salt handling genes contribute to blood pressure variation. Nat. Genet. 2008, 40, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Manolio, T.A.; Collins, F.S.; Cox, N.J.; Goldstein, D.B.; Hindorff, L.A.; Hunter, D.J.; McCarthy, M.I.; Ramos, E.M.; Cardon, L.R.; Chakravarti, A.; et al. Finding the missing heritability of complex diseases. Nature 2009, 461, 747–753. [Google Scholar] [CrossRef] [PubMed]
- Nejentsev, S.; Walker, N.; Riches, D.; Egholm, M.; Todd, J.A. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science 2009, 324, 387–389. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K. Are rare variants responsible for susceptibility to complex diseases? Am. J. Hum. Genet. 2001, 69, 124–137. [Google Scholar] [CrossRef] [PubMed]
- Morgenthaler, S.; Thilly, W.G. A strategy to discover genes that carry multi-allelic or mono-allelic risk for common diseases: A cohort allelic sums test (CAST). Mutat. Res. 2007, 615, 28–56. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Leal, S.M. Methods for detecting associations with rare variants for common diseases: Application to analysis of sequence data. Am. J. Hum. Genet. 2008, 83, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Madsen, B.E.; Browning, S.R. A groupwise association test for rare mutations using a weighted sum statistic. PLoS Genet. 2009, 5, e1000384. [Google Scholar] [CrossRef] [PubMed]
- Price, A.L.; Kryukov, G.V.; de Bakker, P.I.; Purcell, S.M.; Staples, J.; Wei, L.J.; Sunyaev, S.R. Pooled association tests for rare variants in exon-resequencing studies. Am. J. Hum. Genet. 2010, 86, 832–838. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Pan, W. Comparison of statistical tests for disease association with rare variants. Genet. Epidemiol. 2011, 35, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Sha, Q.; Zhang, S. Two adaptive weighting methods to test for rare variant associations in family-based designs. Genet. Epidemiol. 2012, 36, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Feng, T.; Elston, R.C.; Zhu, X. Detecting rare and common variants for complex traits: Sibpair and odds ratio weighted sum statistics (SPWSS, ORWSS). Genet. Epidemiol. 2011, 35, 398–409. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.Y.; Tang, Z.Z. A general framework for detecting disease associations with rare variants in sequencing studies. Am. J. Hum. Genet. 2011, 89, 354–367. [Google Scholar] [CrossRef] [PubMed]
- Neale, B.M.; Rivas, M.A.; Voight, B.F.; Altshuler, D.; Devlin, B.; Orho-Melander, M.; Kathiresan, S.; Purcell, S.M.; Roeder, K.; Daly, M.J. Testing for an unusual distribution of rare variants. PLoS Genet. 2011, 7, e1001322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.C.; Lee, S.; Cai, T.; Li, Y.; Boehnke, M.; Lin, X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am. J. Hum. Genet. 2011, 89, 82–93. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Emond, M.J.; Bamshad, M.J.; Barnes, K.C.; Rieder, M.J.; Nickerson, D.A.; Christiani, D.C.; Wurfel, M.M.; Lin, X. Optimal unified approach for rare variant association testing with application to small sample case-control whole-exome sequencing studies. Am. J. Hum. Genet. 2012, 91, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Zheng, Y.; Hsu, L. A unified mixed-effects model for rare-variant association in sequencing studies. Genet. Epidemiol. 2013, 37, 334–344. [Google Scholar] [CrossRef] [PubMed]
- Sha, Q.; Wang, X.; Wang, X.; Zhang, S. Detecting association of rare and common variants by testing an optimally weighted combination of variants. Genet. Epidemiol. 2012, 36, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Chen, Y.-H.; Yang, Q. Joint rare variant association test of the average and individual effects for sequencing studies. PLoS ONE 2012, 7, e32485. [Google Scholar] [CrossRef] [PubMed]
- Barnett, I.J.; Lee, S.; Lin, X. Detecting rare variant effects using extreme phenotype sampling in sequencing association studies. Genet. Epidemiol. 2013, 37, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Zhong, Y.; Hao, Y.; Luo, M.; Zhou, Y.; Guo, H.; Liao, W.; Wan, D.; Wei, H.; Gao, Y.; et al. Novel rare alleles of ABCA1 are exclusively associated with extreme high-density lipoprotein-cholesterol levels among the Han Chinese. Clin. Chem. Lab. Med. 2009, 47, 1239–1245. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.E.; Lin, D.Y. Efficient association mapping of quantitative trait loci with selective genotyping. Am. J. Hum. Genet. 2007, 80, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Lewinger, J.P.; Gauderman, W.J.; Murcray, C.E.; Conti, D. Using extreme phenotype sampling to identify the rare causal variants of quantitative traits in association studies. Genet. Epidemiol. 2011, 35, 790–799. [Google Scholar] [CrossRef] [PubMed]
- Wallace, C.; Chapman, J.M.; Clayton, D.G. Improved power offered by a score test for linkage disequilibrium mapping of quantitative-trait loci by selective genotyping. Am. J. Hum. Genet. 2006, 78, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Derkach, A.; Lawless, J.F.; Sun, L. Robust and powerful tests for rare variants using Fishera̧ŕs method to combine evidence of association from two or more complementary tests. Genet. Epidemiol. 2013, 37, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.-Y.; Lou, X.-Y.; Gao, G.; Liu, N. Rare variant association testing by adaptive combination of p-values. PLoS ONE 2014, 9, e85728. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Chan, A.T.; Fuchs, C.S.; Giovannucci, E. Molecular pathological epidemiology of colorectal neoplasia: An emerging transdisciplinary and interdisciplinary field. Gut 2011, 60, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Lochhead, P.; Chan, A.T.; Nishihara, R.; Cho, E.; Wolpin, B.M.; Meyerhardt, J.A.; Meissner, A.; Schernhammer, E.S.; Fuchs, C.S.; et al. Molecular pathological epidemiology of epigenetics: Emerging integrative science to analyze environment, host, and disease. Mod. Pathol. 2013, 26, 465–484. [Google Scholar] [CrossRef]
- Ogino, S.; Campbell, P.T.; Nishihara, R.; Phipps, A.I.; Beck, A.H.; Sherman, M.E.; Chan, A.T.; Troester, M.A.; Bass, A.J.; Fitzgerald, K.C.; et al. Proceedings of the second international molecular pathological epidemiology (MPE) meeting. Cancer Causes Control 2015, 26, 959–972. [Google Scholar] [PubMed]
- Risch, N.; Zhang, H. Extreme discordant sib pairs for mapping quantitative trait loci in humans. Science 1995, 268, 1584–1589. [Google Scholar] [CrossRef] [PubMed]
- Goeman, J.J.; van de Geer, S.A.; van Houwelingen, H.C. Testing against a high dimensional alternative. J. R. Stat. Soc. B 2006, 68, 477–493. [Google Scholar] [CrossRef]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.-J.; Wang, Y.; Chen, L.-L. Detecting the Common and Individual Effects of Rare Variants on Quantitative Traits by Using Extreme Phenotype Sampling. Genes 2016, 7, 2. https://doi.org/10.3390/genes7010002
Zhou Y-J, Wang Y, Chen L-L. Detecting the Common and Individual Effects of Rare Variants on Quantitative Traits by Using Extreme Phenotype Sampling. Genes. 2016; 7(1):2. https://doi.org/10.3390/genes7010002
Chicago/Turabian StyleZhou, Ya-Jing, Yong Wang, and Li-Li Chen. 2016. "Detecting the Common and Individual Effects of Rare Variants on Quantitative Traits by Using Extreme Phenotype Sampling" Genes 7, no. 1: 2. https://doi.org/10.3390/genes7010002