Three-Dimensional Mapping of mRNA Export through the Nuclear Pore Complex

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Single-Molecule Study of mRNA Nuclear Export in Live Cells
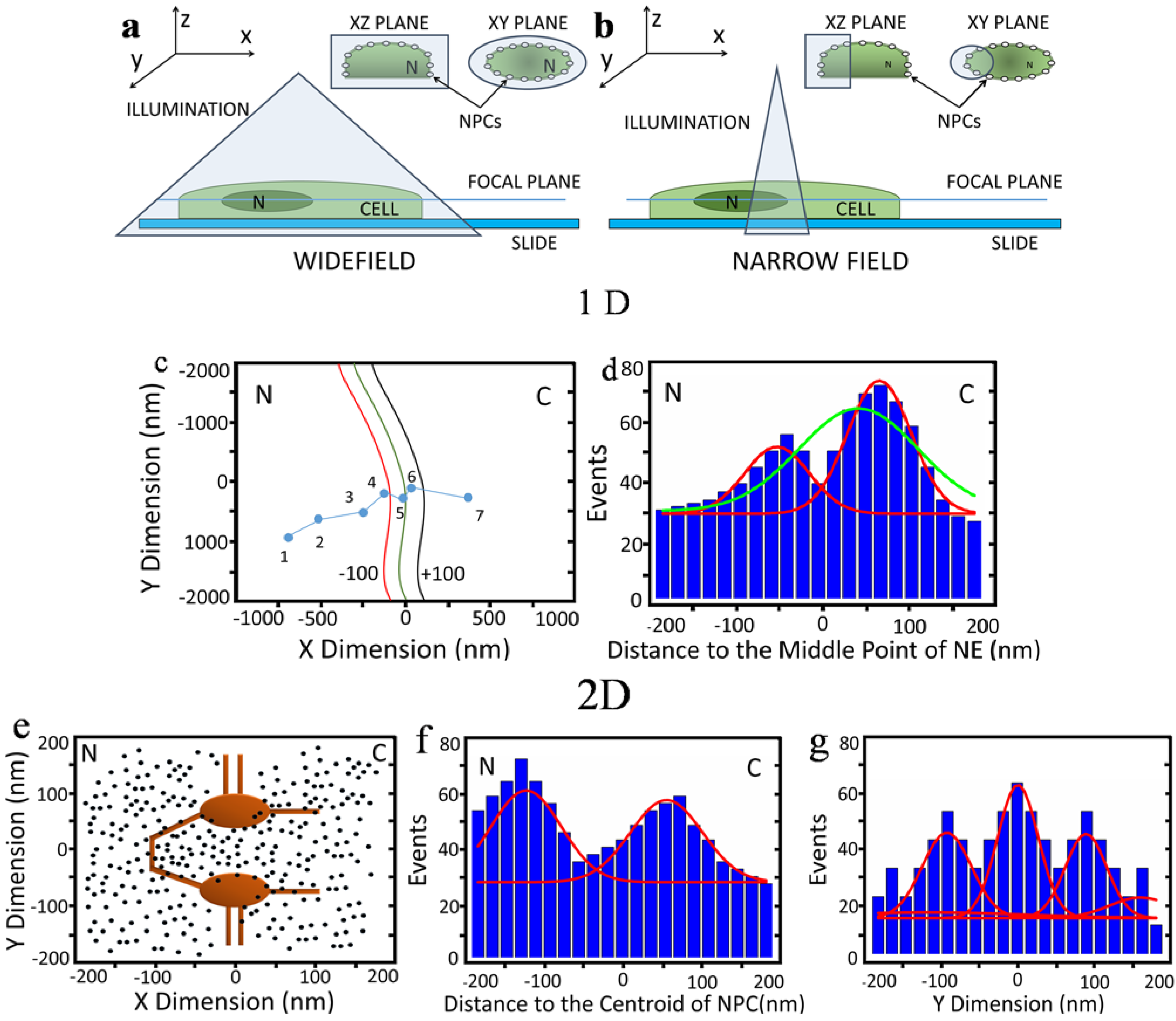
2.1. 1D and 2D Single-Molecule Studies of mRNA Nuclear Export
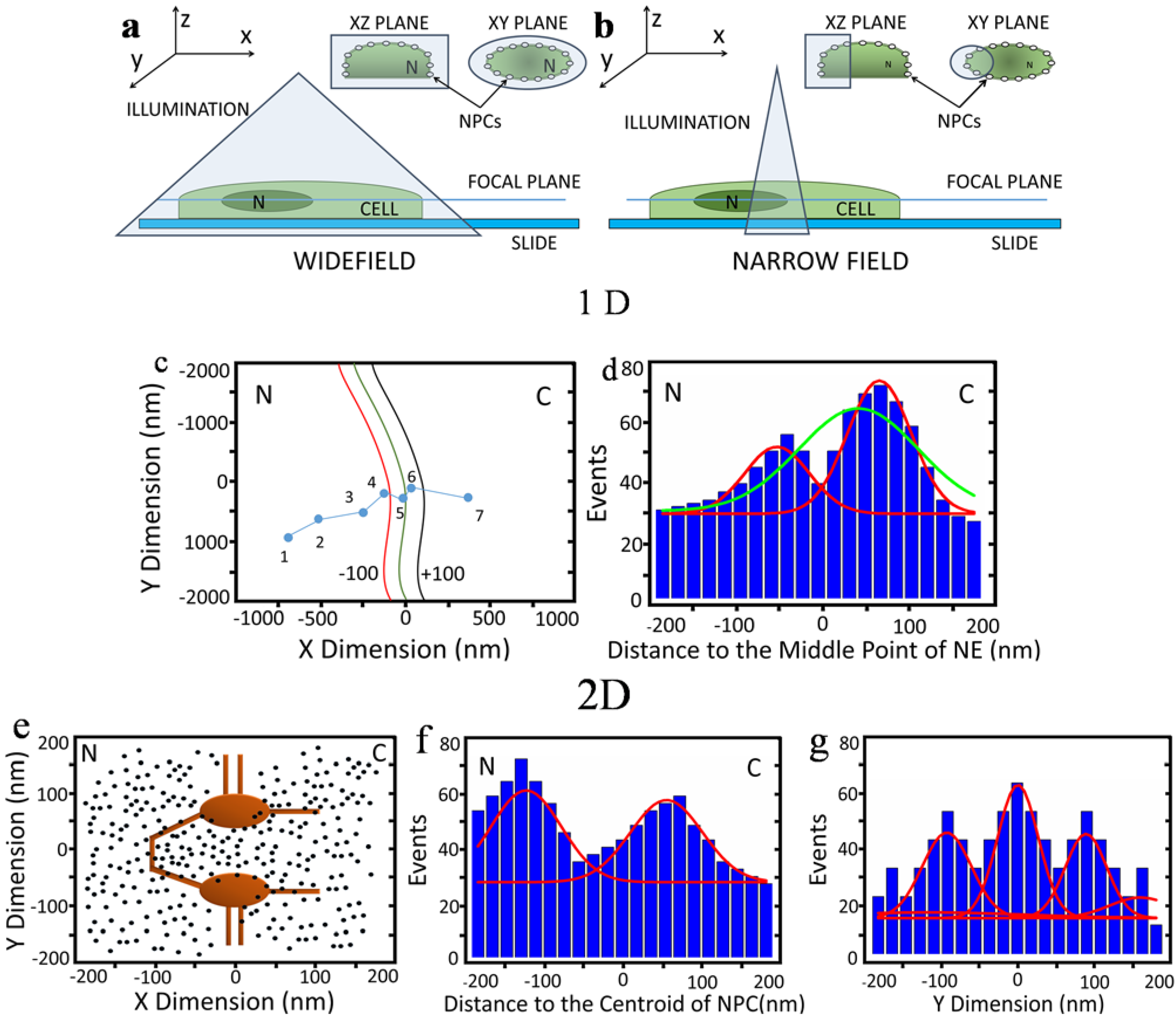
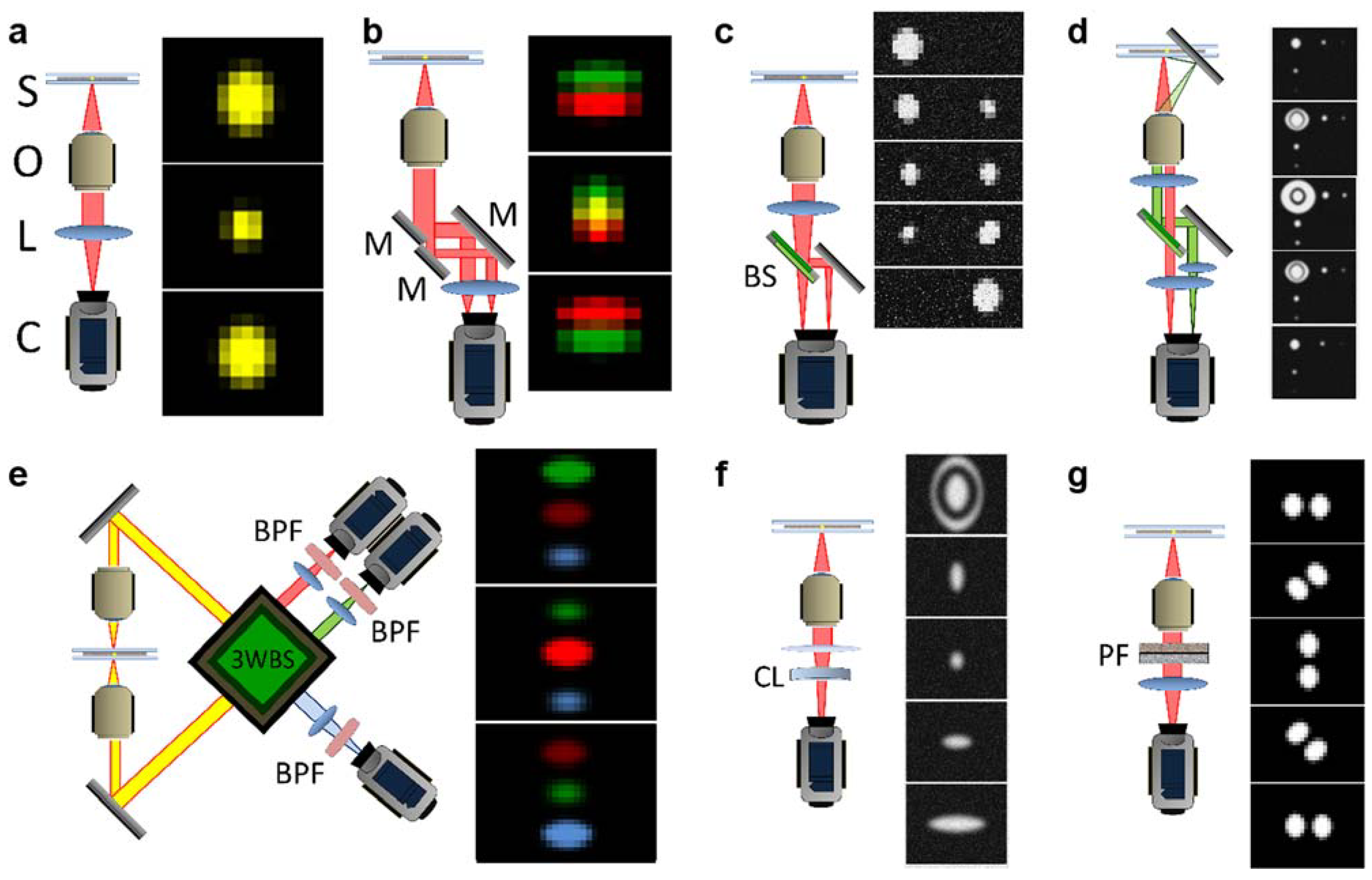
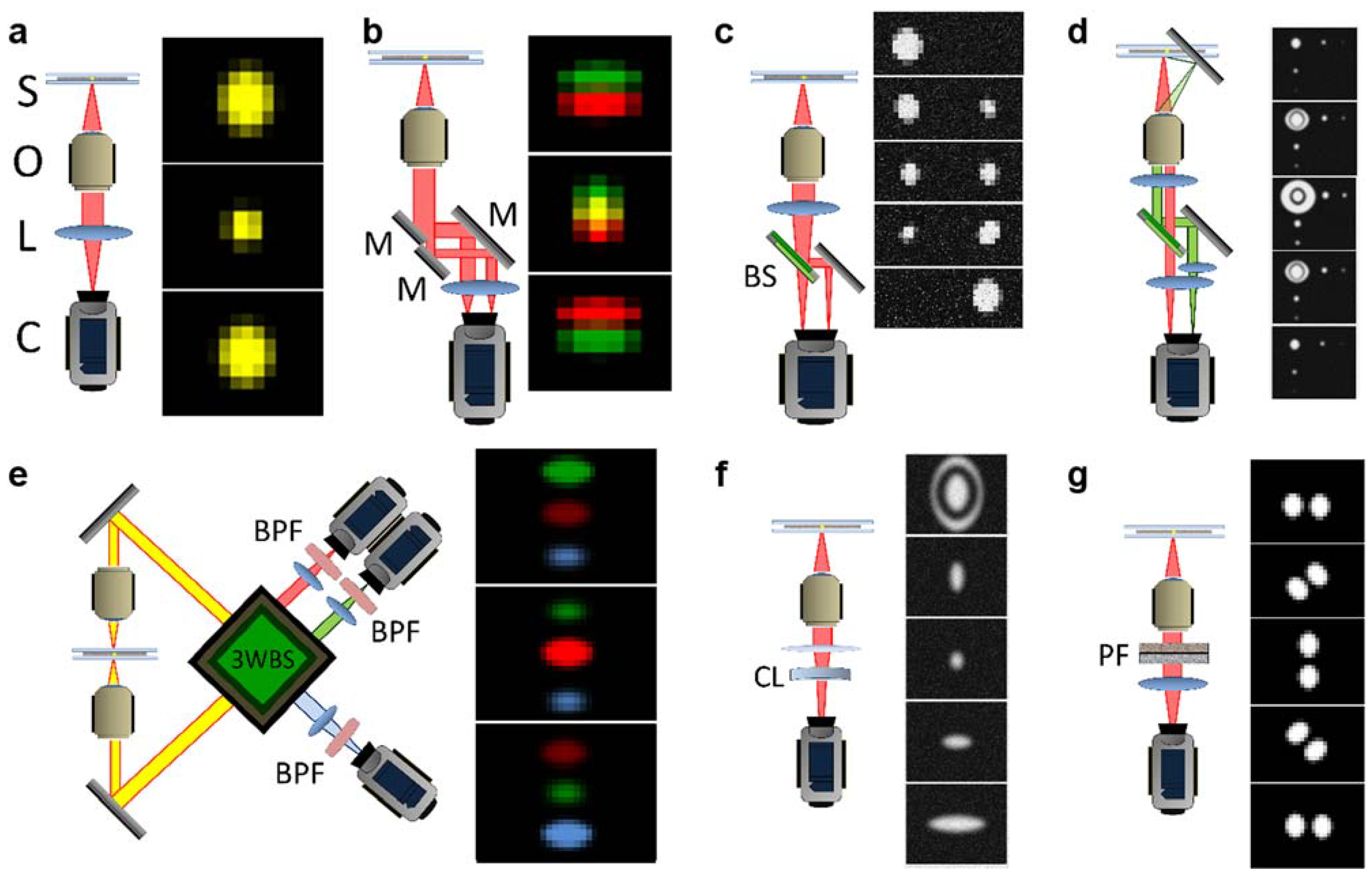
2.2. A Brief Summary of Currently Developed 3D Single-Particle Tracking (SPT) Techniques
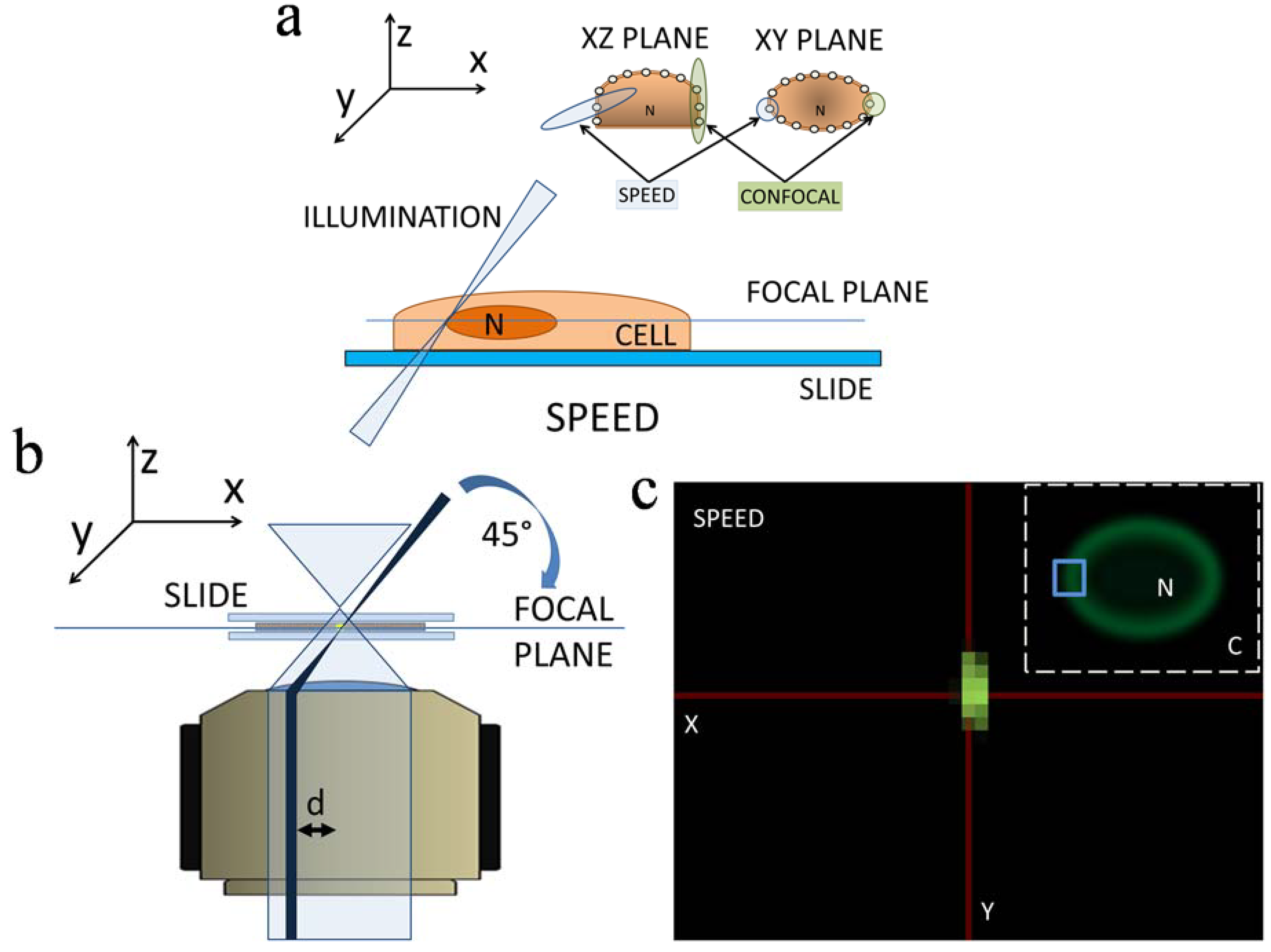
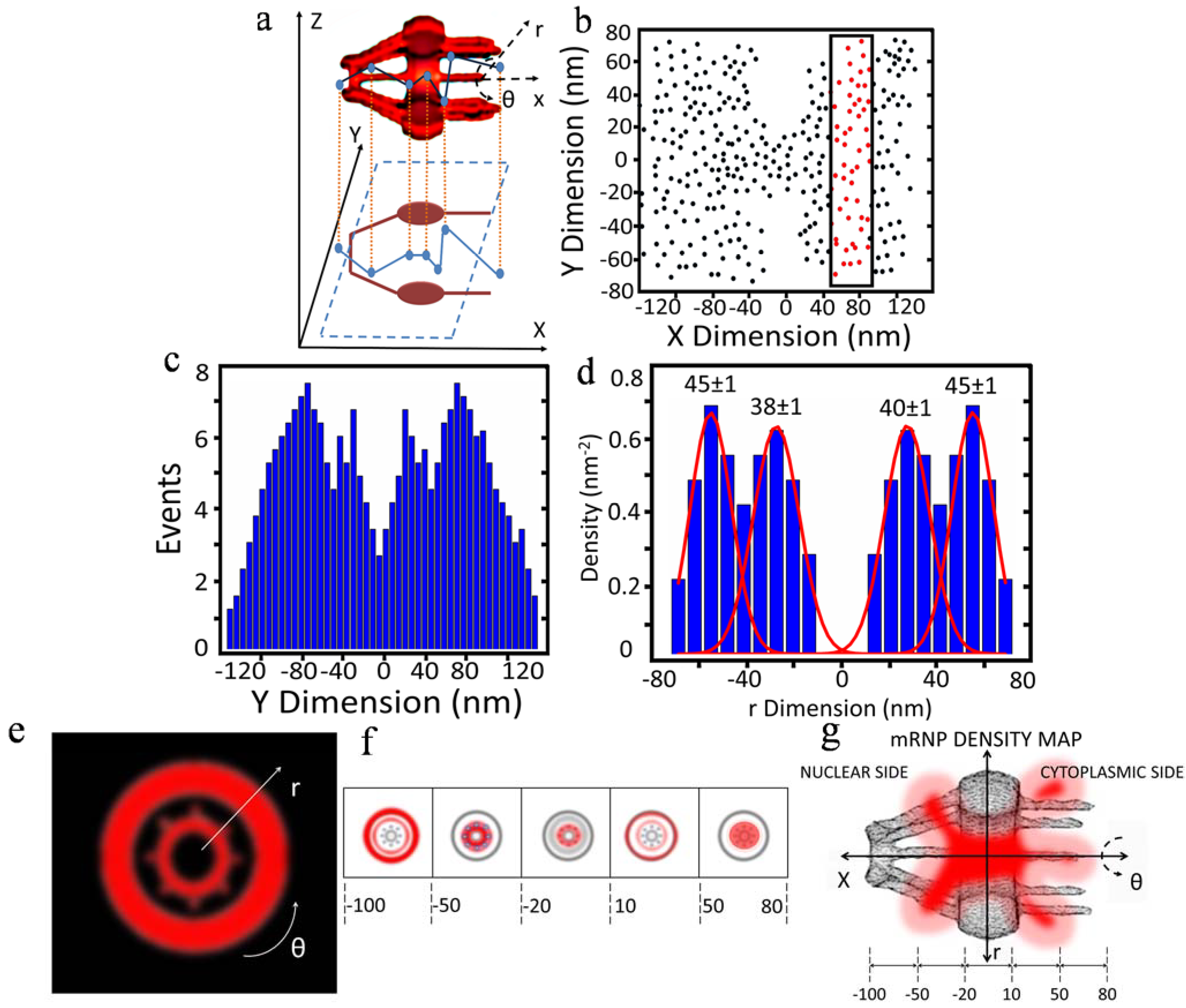
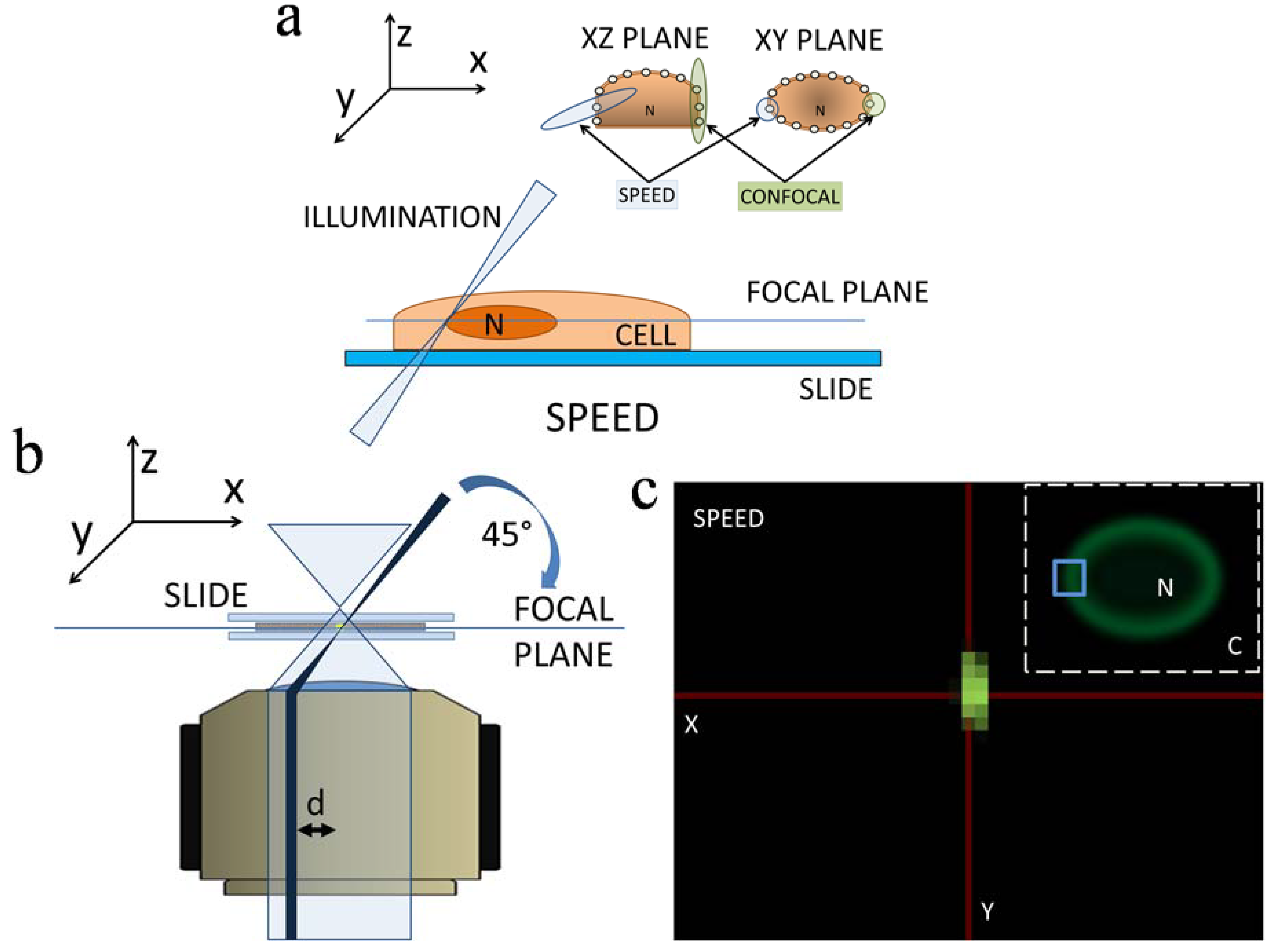
2.3. 3D Mapping of mRNA Nuclear Export with SPEED Microscopy

3. New Features of Nuclear Export of mRNA Obtained with 3D Mapping
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rout, M.P.; Blobel, G. Isolation of the yeast nuclear pore complex. J. Cell. Biol. 1993, 123, 771–783. [Google Scholar] [CrossRef] [PubMed]
- Gorlich, D.; Kutay, U. Transport between the nucleus and cytoplasm. Annu. Rev. Cell Dev. Biol. 1999, 15, 607–660. [Google Scholar] [CrossRef] [PubMed]
- Maul, G.G. Nuclear pore complexes. Elimination and reconstruction during Mitosis. J. Cell Biol. 1977, 74, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Macara, I.G. Transport into and out of the Nucleus. Microbiol. Mol. Biol. Rev. 2001, 65, 570–594. [Google Scholar] [CrossRef] [PubMed]
- Yang, W. Distinct, but not completely separate spatial transport routes in the nuclear pore complex. Nucleus 2013, 4, 166–175. [Google Scholar] [CrossRef] [PubMed]
- Maimon, T.; Nadav, E.; Dahan, I.; Medalia, O. The human nuclear pore complex as revealed by cryo-electron tomography. Structure 2012, 20, 998–1006. [Google Scholar] [CrossRef] [PubMed]
- Maeshima, K.; Iino, H.; Hihara, S.; Funakoshi, T.; Watanabe, A.; Nishimura, M.; Nakatomi, R.; Yahata, K.; Imamoto, F.; Hashikawa, T.; et al. Nuclear pore formation but not nuclear growth is governed by cyclin-dependent kinases (CDKS) during interphase. Nat. Struct. Mol. Biol. 2010, 17, 1065–1072. [Google Scholar] [CrossRef]
- Kohler, A.; Hurt, E. Exporting RNA from the nucleus to the cytoplasm. Nat. Rev. Mol. Cell Biol. 2007, 8, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Goryaynov, A.; Ma, J.; Yang, W. Single-molecule studies of nucleocytoplasmic transport: From one dimension to three dimensions. Integr. Biol. 2012, 4, 10–21. [Google Scholar] [CrossRef]
- Arias, I.M. Nuclear pore complex. In Chichester; John Wiley & Sons: West Sussex, UK; pp. 147–155.
- Bilokapic, S.; Schwartz, T.U. 3D ultrastructure of the nuclear pore complex. Curr. Opin. Cell Biol. 2012, 24, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Hoelz, A.; Debler, E.W.; Blobel, G. The structure of the nuclear pore complex. Annu. Rev. Biochem. 2011, 80, 613–643. [Google Scholar] [CrossRef] [PubMed]
- Wente, S.R.; Rout, M.P. The nuclear pore complex and nuclear transport. Cold Spring Harb. Perspect. Biol. 2010, 2. [Google Scholar] [CrossRef]
- Strambio-De-Castillia, C.; Niepel, M.; Rout, M.P. The nuclear pore complex: Bridging nuclear transport and gene regulation. Nat. Rev. Mol. Cell Biol. 2010, 11, 490–501. [Google Scholar] [CrossRef] [PubMed]
- Fahrenkrog, B.; Aebi, U. The nuclear pore complex: Nucleocytoplasmic transport and beyond. Nat. Mol. Cell Biol. 2003, 4, 757–766. [Google Scholar] [CrossRef]
- Chatel, G.; Fahrenkrog, B. Dynamics and diverse functions of nuclear pore complex proteins. Nucleus 2012, 3, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, M.R.; Cozzolino, L.; Malva, C.; Graziani, F.; Gigliotti, S. nup154 genetically interacts with cup and plays a cell-type-specific function during Drosophila melanogaster egg-chamber development. Genetics 2007, 175, 1751–1759. [Google Scholar] [CrossRef]
- Tang, S.; Presgrabes, D.C. Evolution of the Drosophila nuclear pore complex results in multiple hybrid incompatibilities. Science 2009, 323, 779–782. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, P.; Wang, Y.; Wei, J.H.; van Deursen, J.; Yu, H.; Malureanu, L.; Dasso, M.; Forbes, D.J.; Levy, D.E.; et al. Nucleoporin levels regulate cell cycle progression and phase-specific gene expression. Dev. Cell 2008, 15, 657–667. [Google Scholar] [CrossRef]
- Lim, R.Y.; Fahrenkrog, B. The nuclear pore complex up close. Curr. Opin. Cell Biol. 2006, 18, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Mattaj, I.W.; Englmeier, L. Nuclecytoplasmic transport: The soluble phase. Annu. Rev. Biochem. 1998, 67, 265–306. [Google Scholar] [CrossRef] [PubMed]
- Vasu, S.K.; Forbes, D.J. Nuclear pores and nuclear assembly. Curr. Opin. Cell Biol. 2001, 13, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Strawn, L.A.; Shen, T.; Shulga, N.; Goldfarb, D.S.; Wente, S.R. Minimal nuclear pore complexes define FG repeat domains essential for transport. Nat. Cell Biol. 2004, 6, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Milles, S.; Lemke, E.A. Single molecule study of the intrinsically disordered FG-repeat nucleoporin 153. Biophys. J. 2011, 101, 1710–1719. [Google Scholar] [CrossRef] [PubMed]
- Lim, R.Y.; Huang, N.P.; Koser, J.; Deng, J.; Lau, K.A.; Schwarz-Herion, K.; Fahrenkrog, B.; Aebi, U. Flexible phenylalanine-glycine nucleoporins as entropic barriers to nucleocytoplasmic transport. Proc. Natl. Acad. Sci. USA 2006, 103, 9512–9517. [Google Scholar] [CrossRef] [PubMed]
- Miao, L.; Schulten, K. Transport-related structures and processes of the nuclear pore complex studied through molecular dynamics. Structure 2009, 17, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Terry, L.J.; Shows, E.B.; Wente, S.R. Crossing the nuclear envelope: Hierarchical regulation of nucleocytoplasmic transport. Science 2007, 318, 1412–1416. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.S.; Belmont, B.J.; Sante, J.M.; Rexach, M.F. Natively unfolded nucleoporins gate protein diffusion across the nuclear pore complex. Cell 2007, 129, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Yang, W. Three-dimensional distribution of transient interactions in the nuclear pore complex obtained from single-molecule snapshots. Proc. Natl. Acad. Sci. USA 2010, 107, 7305–7310. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Liu, Z.; Michelotti, N.; Pitchiaya, S.; Veerapaneni, R.; Androsavich, J.R.; Walter, N.G.; Yang, W. High-resolution three-dimensional mapping of mRNA export through the nuclear pore. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef]
- Shahin, V.; Danker, T.; Enss, K.; Ossig, R.; Oberleithner, H. Evidence for Ca2+- and ATP-sensitive peripheral channels in nuclear pore complexes. FASEB J. 2001, 15, 1895–1901. [Google Scholar] [CrossRef] [PubMed]
- Weiss, K. Regulating access to the genome: Nucleocytoplasmic transport throughout the cell cycle. Cell 2003, 112, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Dieppois, G.; Stutz, F. Connecting the transcription site to the nuclear pore: A multi-tether process that regulates gene expression. J. Cell Sci. 2010, 123, 1989–1999. [Google Scholar] [CrossRef] [PubMed]
- Stewart, M. Nuclear export of mRNA. Trends Biochem. Sci. 2010, 35, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Kloc, M.; Foreman, V.; Reddy, S.A. Binary function of mRNA. Biochimie 2011, 93, 1955–1961. [Google Scholar] [CrossRef] [PubMed]
- Campos, S.; Jacobs-Wagner, C. Cellular organization of the transfer of genetic information. Curr. Opin. Microbiol. 2013, 16, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Bjork, P.; Wieslander, L. Mechanisms of mRNA export. Semin. Cell Dev. Biol. 2014, 32, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Natalizio, B.J.; Wente, S.R. Postage for the messenger: designating routes for nuclear mRNA export. Trends Cell Biol. 2013, 23, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Grünwald, D.; Singer, R.H.; Rout, M. Nuclear export dynamics of RNA-protein complexes. Nature 2011, 475, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Mehlin, H.; Daneholt, B.; Skoglund, U. Translocation of a specific premessenger ribonucleoprotein particle through the nuclear-pore studied with electron-microscope tomography. Cell 1992, 69, 605–613. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, N.; Stutz, F. Regulation of mRNP dynamics along the export pathway. FEBS Lett. 2008, 582, 1987–1996. [Google Scholar] [CrossRef] [PubMed]
- Katahira, J. mRNA export and the TREX complex. Biochim. Biophys. Acta 2012, 1819, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Stutz, F.; Izaurralde, E. The interplay of nuclear mRNP assembly, mRNA surveillance and export. Trends Cell Biol. 2003, 13, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.J.; Wente, S.R. Dynamic nuclear pore complexes: Life on the edge. Cell 2006, 125, 1041–1053. [Google Scholar] [CrossRef] [PubMed]
- Braun, I.C.; Herold, A.; Rode, M.; Izaurralde, E. Nuclear export of mRNA by TAP/NXF1 Requires two nucleoporin-binding sites but not p15. Mol. Cell Biol. 2002, 22, 5405–5418. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Dufu, K.; Lee, C.S.; Hsu, J.L.; Dias, A.; Reed, R. Human mRNA export machinery recruited to the 5' end of mRNA. Cell 2006, 127, 1389–1400. [Google Scholar] [CrossRef]
- Strasser, K.; Masuda, S.; Mason, P.; Pfannstiel, J.; Oppizzi, M.; Rodriguez-Navarro, S.; Rondón, A.G.; Aguilera, A.; Struhl, K.; Reed, R.; et al. TREX is a conserved complex coupling transcription with messenger RNA export. Nature 2002, 417, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Masuda, S.; Das, R.; Cheng, H.; Hrut, E.; Dorman, N.; Reed, R. Recruitment of the human TREX complex to mRNA during splicing. Genes Dev. 2005, 19, 1512–1517. [Google Scholar] [CrossRef]
- Wickramasinghe, V.O.; McMurtrie, P.I.; Mills, A.D.; Takei, Y.; Penrhyn-Lowe, S.; Amagase, Y.; Main, S.; Marr, J.; Stewart, M.; Laskey, R.A. mRNA export from mammalian cell nuclei is dependent on GANP. Curr. Biol. 2010, 20, 25–31. [Google Scholar] [CrossRef]
- Jani, D.; Lutz, S.; Hurt, E.; Laskey, R.A.; Stewart, M.; Wickramasinghe, V.O. Functional and structural characterisation of the mammalian TREX-2 complex that links transcription with nuclear messenger RNA export. Nucleic Acids Res. 2012, 40, 4562–4573. [Google Scholar] [CrossRef] [PubMed]
- Wickramasinghe, V.O.; Savill, J.M.; Chavali, S.; Jonsdottir, A.B.; Rajendra, E.; Grüner, T.; Laskey, R.A.; Babu, M.M.; Venkitaraman, A.R. Human inositol polyphosphate multikinase regulates transcript-selective nuclear mRNA export to preserve genome integrity. Mol. Cell 2013, 51, 737–750. [Google Scholar] [CrossRef] [PubMed]
- Saiardia, A.; Caffreyb, J.J.; Snydera, S.H.; Shears, S.B. Inositol polyphosphate multikinase (ArgRIII) determines nuclear mRNA export in Saccharomyces cerevisiae. FEBS Lett. 2000, 468, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Baumann, K. Gene expression: Beyond pores. Nat. Rev. Mol. Cell Biol. 2010, 11. [Google Scholar] [CrossRef]
- Ptak, C.; Aitchison, J.D.; Wozniak, R.W. The multifunctional nuclear pore complex: A platform for controlling gene expression. Curr. Opin. Cell Biol. 2014, 28, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Pascual-Garcia, P.; Capelson, M. Nuclear pores as versatile platforms for gene regulation. Curr. Opin. Genet. Dev. 2014, 25, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.R.; Silver, P.A. Transcriptional regulation at the nuclear pore complex. Curr. Opin. Genet. Dev. 2007, 17, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Arib, G.; Akhtar, A. Multiple facets of nuclear periphery in gene expression control. Curr. Opin. Cell Biol. 2011, 23, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Elliott, D.J.; Stutz, F.; Lescure, A.; Rosbash, M. mRNA nuclear export. Curr. Opin. Genet. Dev. 1994, 4, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Gilboa, E.; Vieweg, J. Cancer immunotherapy with mRNA-transfected dendritic cells. Immunol. Rev. 2004, 199, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Schlake, T.; Thess, A.; Fotin-Mleczek, M.; Kallen, K. Developing mRNA-vaccine technologies. RNA Biol. 2012, 9, 1319–1330. [Google Scholar] [CrossRef] [PubMed]
- Malina, A.; Mills, J.R.; Pelletier, J. Emerging therapeutics targeting mRNA translation. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef]
- McIvor, R.S. Therapeutic Delivery of mRNA: The Medium Is the Message. Mol. Ther. 2011, 19, 822–823. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.; Cautain, B.; de Pedro, N.; Link, W. Targeting nucleocytoplasmic transport in cancer therapy. Oncotarget 2013, 5, 11–28. [Google Scholar]
- Rodriguez, J.A.; Schüchner, S.; Au, W.W.; Fabbro, M.; Henderson, B.R. Nuclear-cytoplasmic shuttling of BARD1 contributes to its proapoptotic activity and is regulated by dimerization with BRCA1. Oncogene 2004, 23, 1809–1820. [Google Scholar] [CrossRef] [PubMed]
- Komiya, Y.; Habas, R. Wnt signal transduction pathways. Organogenesis 2008, 4, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Komiya, Y.; Mandrekar, N.; Sato, A.; Dawid, I.B.; Habas, R. Custos controls β-catenin to regulate head development during vertebrate embryogenesis. Proc. Natl. Acad. Sci. USA 2014, 36, 13099–13104. [Google Scholar] [CrossRef]
- Jamali, T.; Jamali, Y.; Mehrbod, M.; Mofrad, M.R.K. Nuclear pore complex: Biochemistry and biophysics of nucleocytoplasmic transport in health and disease. Int. Rev. Cell Mol. Biol. 2011, 287, 233–286. [Google Scholar] [PubMed]
- Chahine, M.N.; Pierce, G.N. Therapeutic targeting of nuclear protein import in pathological cell conditions. Pharmacol. Rev. 2009, 61, 358–372. [Google Scholar] [CrossRef] [PubMed]
- Snow, C.M.; Senior, A.; Gerace, L. Monoclonal antibodies identify a group of nuclear pore complex glycoproteins. J. Cell Biol. 1987, 104, 1143–1156. [Google Scholar] [CrossRef] [PubMed]
- Gasiorowski, J.Z.; Dean, D.A. Mechanisms of nuclear transport and interventions. Adv. Drug Deliv. Rev. 2003, 55, 703–716. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.G.; Dawson, J.; Cubitt, C.L.; Bazc, R.; Sullivana, D.M. Inhibition of CRM1-dependent nuclear export sensitizes malignant cells to cytotoxic and targeted agents. Semin. Cancer Biol. 2014, 27, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Hell, S.W.; Wichmann, J. Breaking the diffraction resolution limit by stimulated emission: Stimulated-emission-depletion fluorescence microscopy. Opt. Lett. 1994, 19, 780–782. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Bates, M.; Zhuang, X. Super-resolution fluorescence microscopy. Annu. Rev. Biochem. 2009, 78, 993–1016. [Google Scholar] [CrossRef] [PubMed]
- Schermelleh, L.; Heintzmann, R.; Leonhardt, H. A guide to super-resolution fluorescence microscopy. J. Cell Biol. 2010, 190, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Rice, J.H. Far-field Fluorescence Microscopy beyond the Diffraction Limit: Fluorescence Imaging with Ultrahigh Resolution; Cornell University Library: Ithaca, NY, USA, 2007. [Google Scholar]
- Klar, T.A.; Engel, E.; Hell, S.W. Breaking Abbe’s diffraction resolution limit in fluorescence microscopy with stimulated emission depletion beams of various shapes. Phys. Rev. 2001, 64. [Google Scholar] [CrossRef]
- Klar, T.A.; Hell, S.W. Subdiffraction resolution in far-field fluorescence microscopy. Opt. Lett. 1999, 24, 954–956. [Google Scholar] [CrossRef] [PubMed]
- Von Middendorff, C.; Egner, A.; Geisler, C.; Hell, S.W.; Schönle, A. Isotropic 3D nanoscopy based on single emitter switching. Opt. Express 2008, 16, 20774–20788. [Google Scholar] [CrossRef] [PubMed]
- Grunwald, D.; Singer, R. In vivo imaging of labelled endogenous beta-actin mRNA during nucleocytoplasmic transport. Nature 2010, 467, 604–607. [Google Scholar] [CrossRef] [PubMed]
- Kisielowski, C.; Erni, R.R.; Marta, D.; Dahmen, U. Atomic-resolution imaging with a sub-50-pm electron probe. Phys. Rev. Lett. 2009, 102. [Google Scholar] [CrossRef] [PubMed]
- Milazzo, A.-C.; Cheng, A.; Moeller, A.; Lyumkis, D.; Jacovetty, E.; Polukas, J.; Ellisman, M.H.; Xuong, N.H.; Carragher, B.; Potter, C.S.; et al. Initial evaluation of a direct detection device detector for single particle cryo-electron microscopy. J. Struct. Biol. 2011, 176, 404–408. [Google Scholar] [CrossRef] [PubMed]
- Bammes, B.E.; Rochat, R.H.; Jakana, J.; Chen, D.H.; Chiu, W. Direct electron detection yields cryo-EM reconstructions at resolutions beyond ¾ Nyquist frequency. J. Struct. Biol. 2012, 177, 589–601. [Google Scholar] [CrossRef] [PubMed]
- Maimon, T.; Medalia, O. Perspective on the metazoan nuclear pore complex. Nucleus 2010, 1, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.A.; Biteen, J.S.; Lord, S.J.; Conley, N.R.; Moerner, W.E. Molecules and methods for super-resolution imaging. Methods Enzymol. 2010, 475, 27–59. [Google Scholar] [PubMed]
- Mor, A.; Suliman, S.; Ben-Yishay, R.; Yunger, S.; Brody, Y.; Shav-Tal, Y. Dynamics of single mRNP nucleocytoplasmic transport and export through the nuclear pore in living cells. Nat. Cell Biol. 2010, 12, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Siebrasse, J.P.; Kaminski, T.; Kubitscheck, U. Nuclear export of single native mRNA molecules observed by light sheet fluorescence microscopy. Proc. Natl. Acad. Sci. USA 2012, 109, 9426–9431. [Google Scholar] [CrossRef] [PubMed]
- Greger, K.; Swoger, J.; Stelzer, E. Basic building units and properties of a fluorescence single plane illumination microscope. Rev. Sci. Instrum. 2007, 78, 023705–023707. [Google Scholar] [CrossRef] [PubMed]
- Daigle, N.; Beaudouin, J.; Hartnell, L.; Imreh, G.; Hallberg, E.; Lippincott-Schwartz, J.; Ellenberg, J. Nuclear pore complexes form immobile networks and have a very low turnover in live mammalian cells. J. Cell Biol. 2001, 154, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Feldherr, C.-M.; Kallenbach, E.; Schultz, N. Movement of a karyophilic protein through the nuclear pores of oocytes. J. Cell Biol. 1984, 99, 2216–2222. [Google Scholar] [CrossRef] [PubMed]
- Kubitscheck, U.; Wedekind, P.; Zeidler, O.; Grote, M.; Peters, R. Single nuclear pores visualized by confocal microscopy and image processing. Biophys. J. 1996, 70, 2067–2077. [Google Scholar] [CrossRef] [PubMed]
- Deschout, H.; Cella Zanacchi, F.; Mlodzianoski, M.; Diaspro, A.; Bewersdorf, J.; Hess, S.T.; Braeckmans, K. Precisely and accurately localizing single emitters in fluorescence microscopy. Nat. Methods 2014, 11, 253–266. [Google Scholar] [CrossRef] [PubMed]
- Lichtman, J.W.; Conchello, J.A. Fluorescence microscopy. Nat. Methods 2005, 2, 910–919. [Google Scholar] [CrossRef] [PubMed]
- Hibbs, A.R. Confocal Microscopy for Biologists; Kluwer Academic/Plenum Publishers: New York, NY, USA, 2004. [Google Scholar]
- Ploem, J.S.; Tanke, H.J. Introduction to Fluorescence Microscopy; Oxford University Press: Oxford, UK, 1987. [Google Scholar]
- Yuste, R. Fluorescence microscopy today. Nat. Methods 2005, 2, 902–904. [Google Scholar] [CrossRef] [PubMed]
- Ebenstein, Y.; Bentolila, L.A. Placing colloidal spheres in the immediate proximity of fluorescent molecules makes it possible to achieve singlemolecule imaging at high temperatures with a low-cost system. Nat. Nanotechnol. 2010, 5, 99–100. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; McKenna, J.D.; Murray, J.M.; Ostap, E.M.; Goldman, Y.E. Parallax: High accuracy three-dimensional single molecule tracking using split images. Nano Lett. 2009, 9, 2676–2682. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.T.; Bewersdorf, J.; Gould, T.J.; Hess, S.T.; Juette, M.F.; Lessard, M.D.; Mlodzianoski, M.J.; Nagpure, B.S. Three-dimensional sub-100 nm resolution fluorescence microscopy of thick samples. Nat. Methods 2008, 5, 527–529. [Google Scholar] [CrossRef] [PubMed]
- Prabhat, P.; Ram, S.; Ward, E.S.; Ober, R.J. Simultaneous imaging of different focal planes in fluorescence microscopy for the study of cellular dynamics in three dimensions. IEEE Trans. Nanobioscience 2004, 3, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Toprak, E.; Balci, H.; Blehm, B.H.; Selvin, P.R. Three-dimensional particle tracking via bifocal imaging. Nano Lett. 2007, 7, 2043–2045. [Google Scholar] [CrossRef] [PubMed]
- McMahon, M.D.; Berglund, A.J.; Carmichael, P.; McClelland, J.J.; Liddle, J.A. 3D particle trajectories observed by orthogonal tracking microscopy. ACS Nano 2009, 3, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Akerboom, J.; Vaziri, A.; Looger, L.L.; Shank, C.V. Near-isotropic 3D optical nanoscopy with photon-limited chromophores. Proc. Natl. Acad. Sci. USA 2010, 107, 10068–10073. [Google Scholar] [CrossRef] [PubMed]
- Aquino, D.; Schonle, A.; Geisler, C.; von Middendorff, C.; Wurm, C.; Okamura, Y; Lang, T.; Hell, S.W.; Egner, A. Two-color nanoscopy of three-dimensional volumes by 4Pi detection of stochastically switched fluorophores. Nat. Methods 2011, 8, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Shtengel, G.; Davidson, M.W.; Fetter, R.D.; Hess, H.F.; Galbraith, J.A.; Galbraith, C.G.; Lippincott-Schwartz, J.; Gillette, J.M.; Manley, S.; Sougrat, R.; et al. Interferometric fluorescent super-resolution microscopy resolves 3D cellular ultrastructure. Proc. Natl. Acad. Sci. USA 2009, 106, 3125–3130. [Google Scholar] [CrossRef] [PubMed]
- Kao, H.P.; Verkman, A.S. Tracking of single fluorescent particles in three dimensions: Use of cylindrical optics to encode particle position. Biophys. J. 1994, 67, 1291–1300. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Wang, W.; Bates, M.; Zhuang, X. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science 2008, 319, 810–813. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.A.; Lew, M.D.; Badieirostami, M.; Moerner, W.E. Localizing and tracking single nanoscale emitters in three dimensions with high spatiotemporal resolution using a double-helix point spread function. Nano Lett. 2010, 10, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.A.; Casolari, J.M.; Badieirostami, M.; Brown, P.O.; Moerner, W.E. Three-dimensional tracking of single mRNA particles in saccharomyces cerevisiae using a double-helix point spread function. Proc. Natl. Acad. Sci. USA 2010, 107, 17864–17871. [Google Scholar] [CrossRef] [PubMed]
- Pavani, S.R.P.; Thompson, M.A.; Biteen, J.S.; Lord, S.J.; Liu, N.; Twieg, R.J.; Piestun, R.; Moerner, W.E. Three-dimensional, single-molecule fluorescence imaging beyond the diffraction limit by using a double-helix point spread function. Proc. Natl. Acad. Sci. USA 2009, 106, 2995–2999. [Google Scholar] [CrossRef] [PubMed]
- Hiraoka, Y.; Sedat, J.W.; Agard, D.A. Determination of three-dimensional imaging properties of a light microscope system. Partial confocal behavior in epifluorescence microscopy. Biophys. J. 1990, 57, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Wickramasinghe, V.O.; Stewart, M.; Laskey, R.A. GANP enhances the efficiency of mRNA nuclear export in mammalian cells. Nucleus 2010, 1, 393–396. [Google Scholar] [CrossRef] [PubMed]
- Ellsidon, A.M.; Dimitrova, L.; Hurt, E.; Stewart, M. Structural basis for the assembly and nucleic acid binding of the TREX-2 transcription-export complex. Nat. Struct. Mol. Biol. 2012, 19, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Folkmann, A.W.; Noble, K.N.; Cole, C.N.; Wente, S.R. Dbp5, Gle1-IP6 and Nup159: A working model for mRNP export. Nucleus 2011, 2, 540–548. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schnell, S.J.; Ma, J.; Yang, W. Three-Dimensional Mapping of mRNA Export through the Nuclear Pore Complex. Genes 2014, 5, 1032-1049. https://doi.org/10.3390/genes5041032
Schnell SJ, Ma J, Yang W. Three-Dimensional Mapping of mRNA Export through the Nuclear Pore Complex. Genes. 2014; 5(4):1032-1049. https://doi.org/10.3390/genes5041032
Chicago/Turabian StyleSchnell, Steven J., Jiong Ma, and Weidong Yang. 2014. "Three-Dimensional Mapping of mRNA Export through the Nuclear Pore Complex" Genes 5, no. 4: 1032-1049. https://doi.org/10.3390/genes5041032