A Novel Function for the Conserved Glutamate Residue in the Walker B Motif of Replication Factor C

Abstract
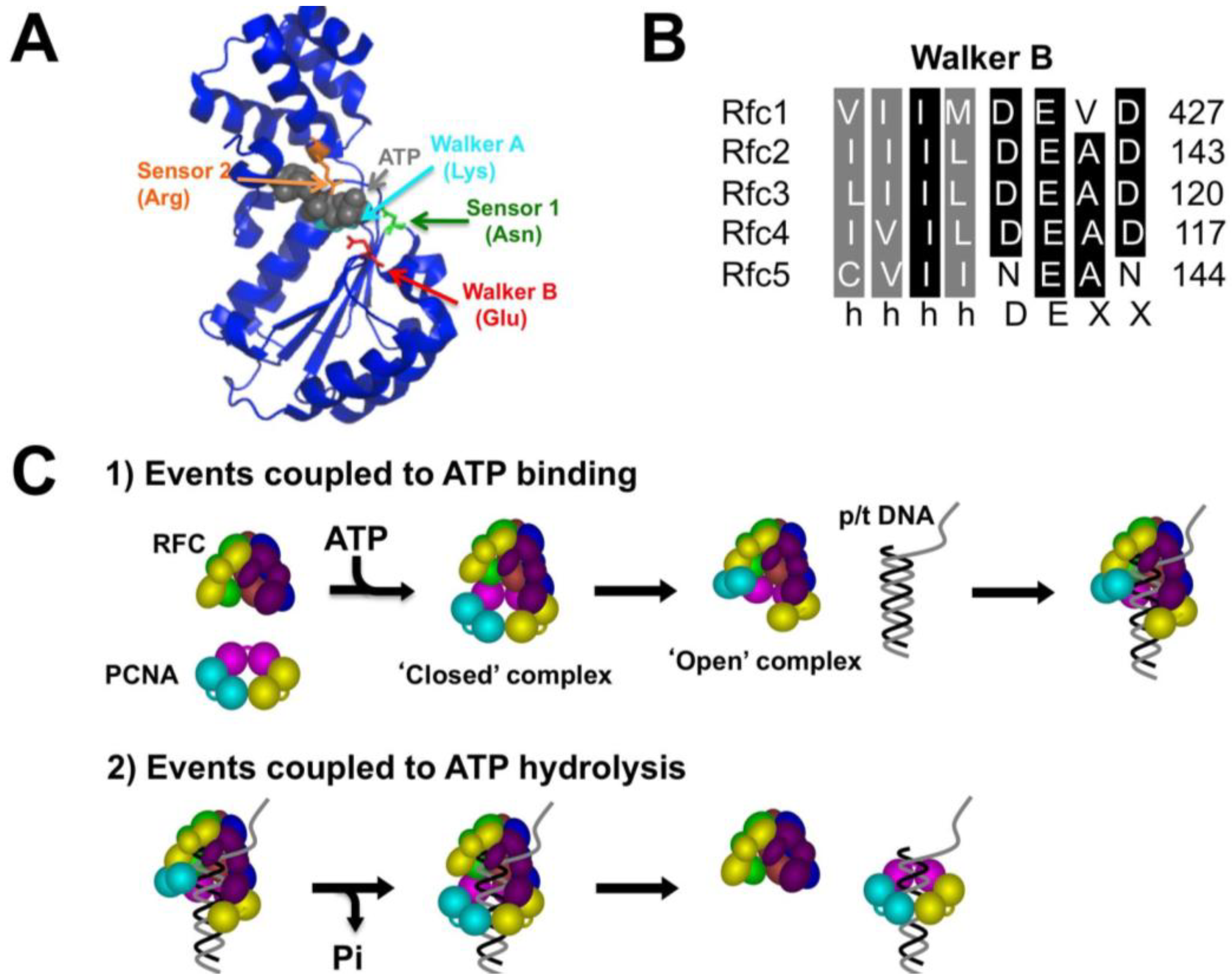
:1. Introduction
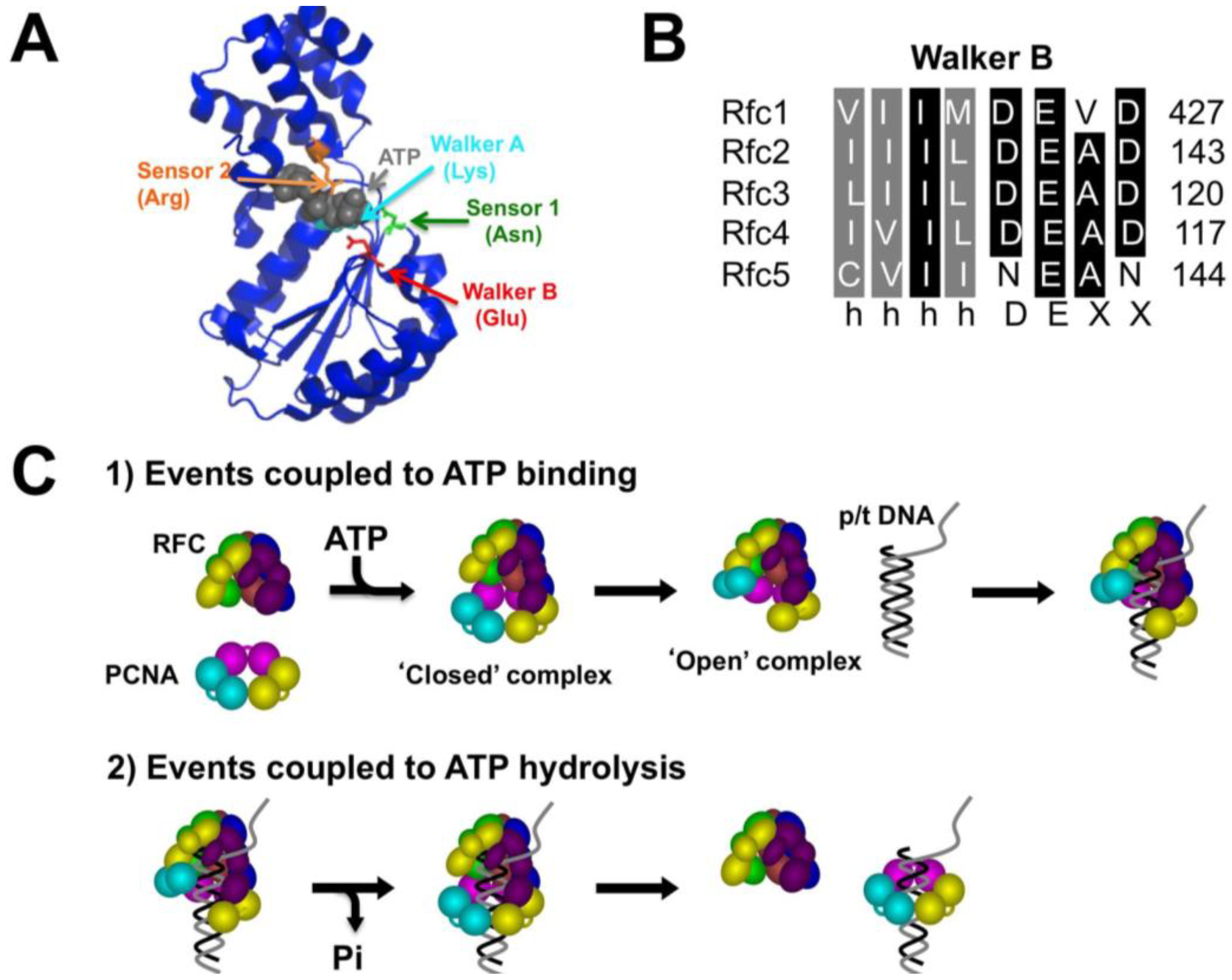
2. Results and Discussion
2.1. Generation of RFC Walker B Mutants
2.2. Measurement of PCNA Binding and Opening
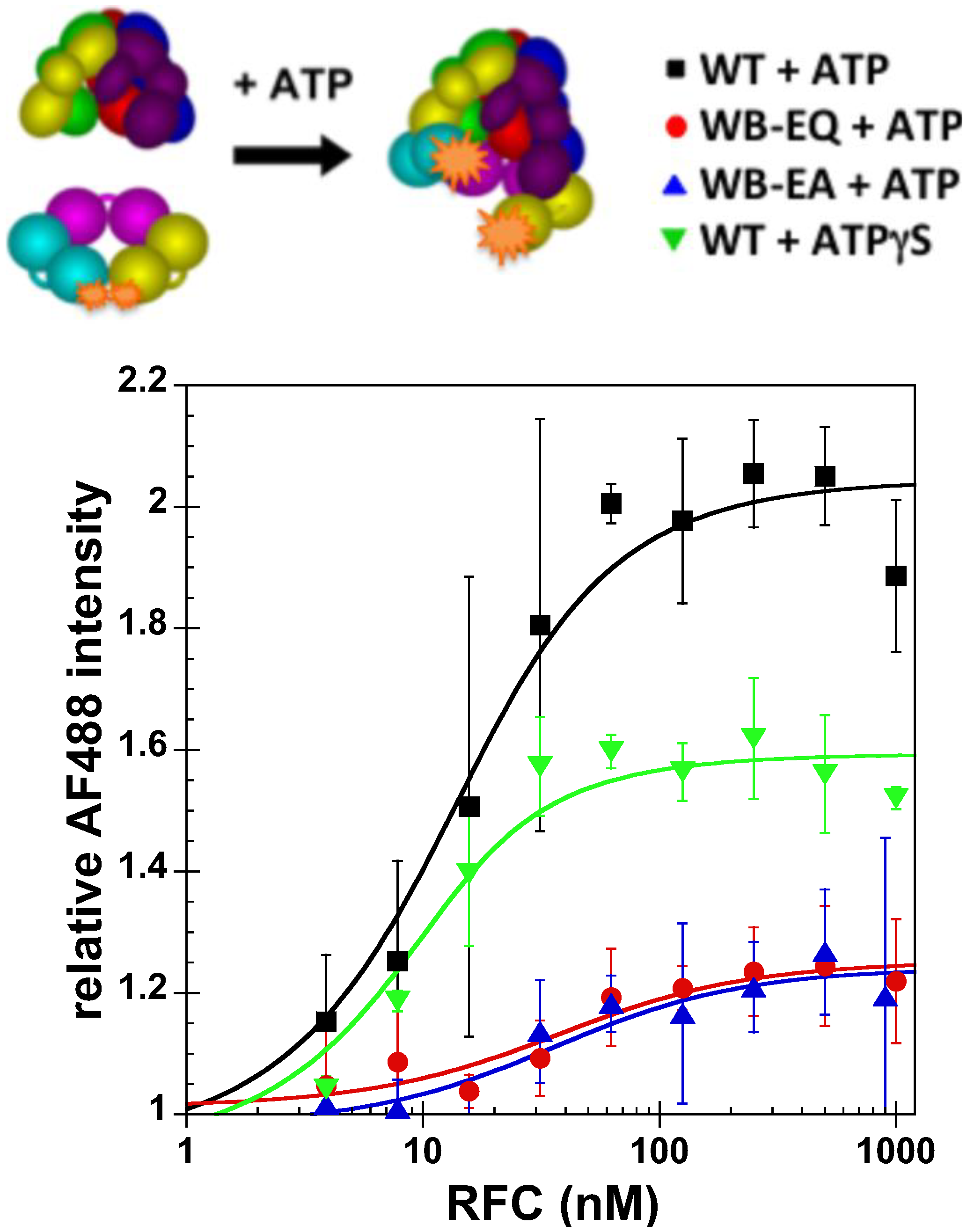
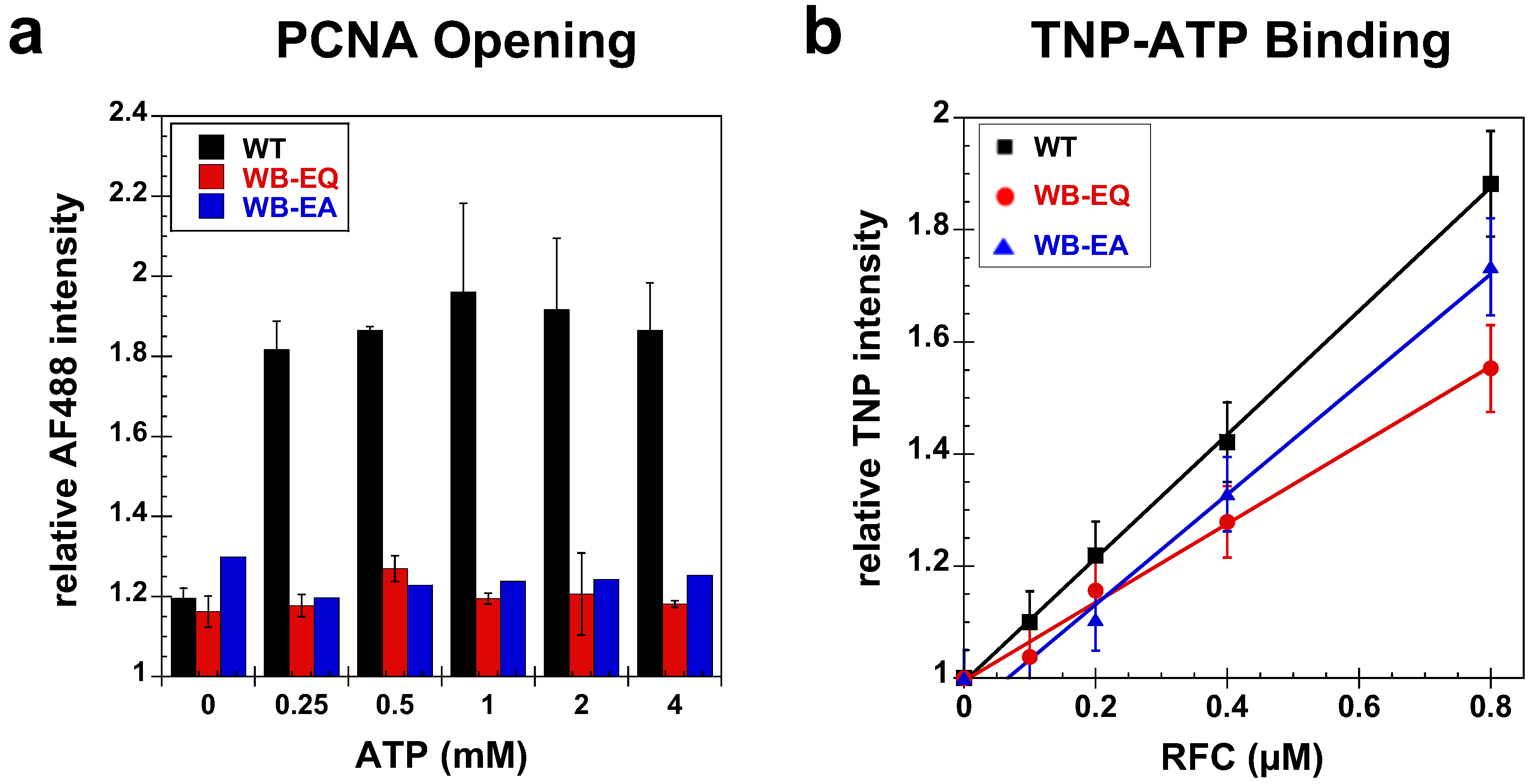
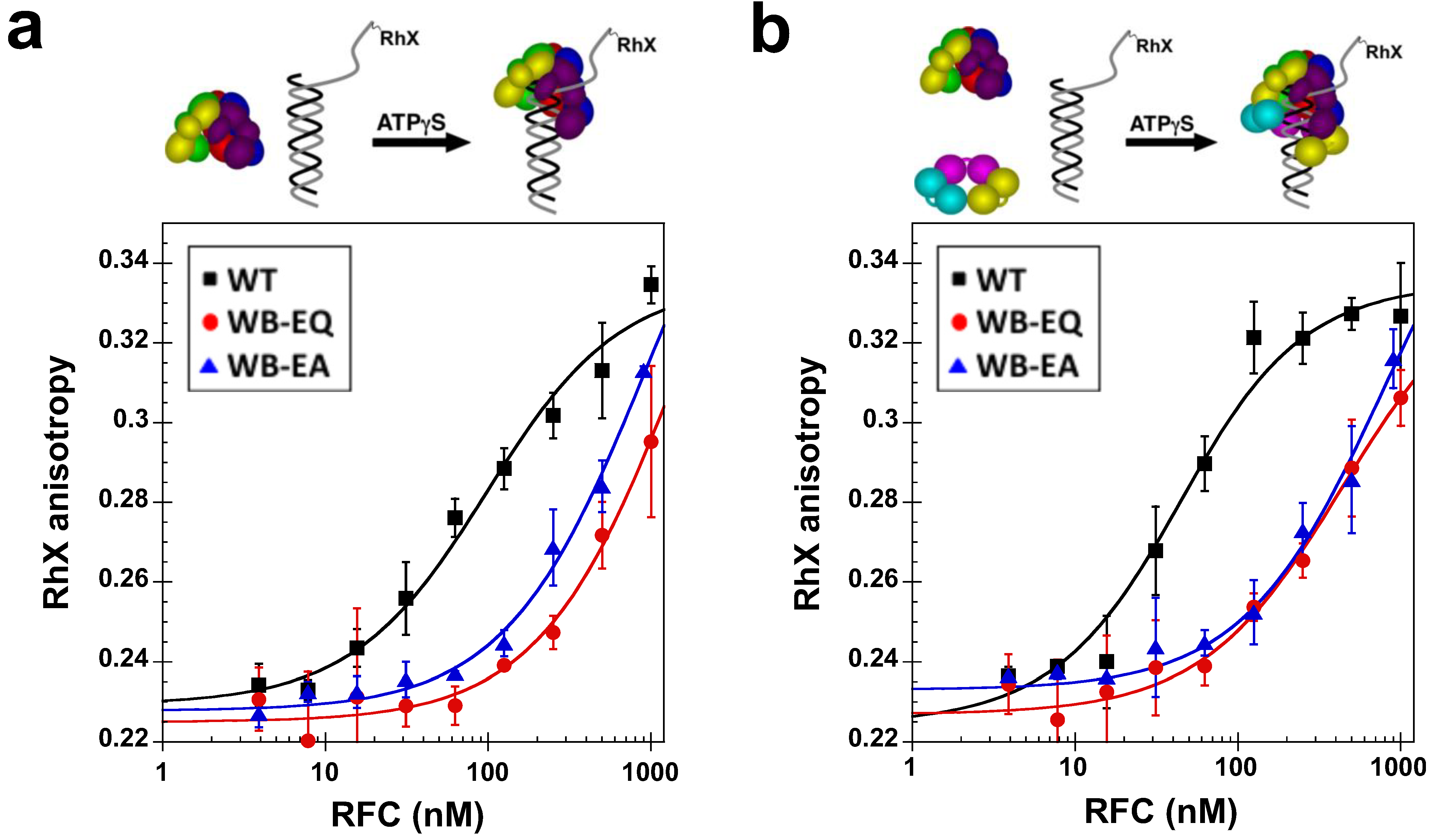
2.2.1. PCNA Binding
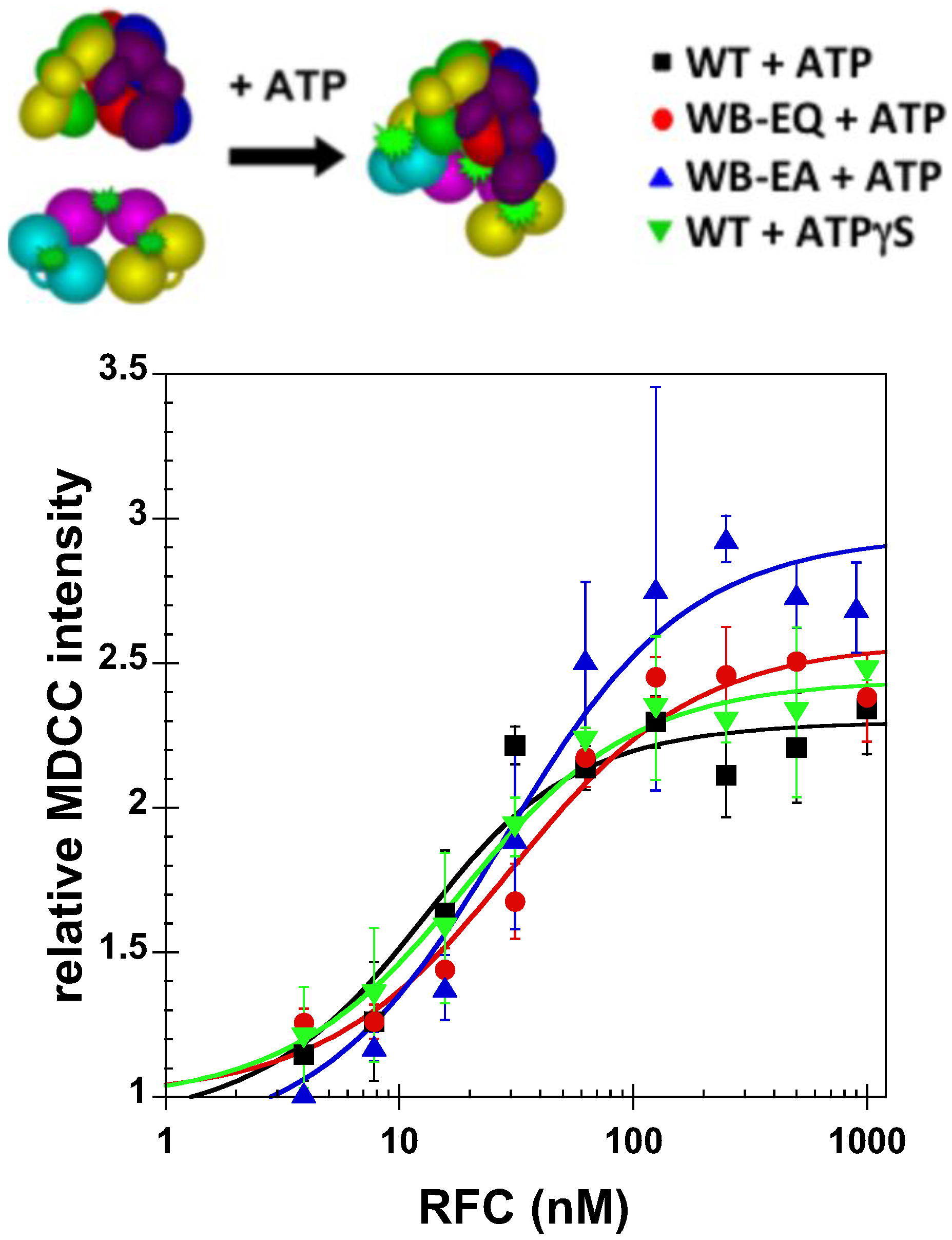
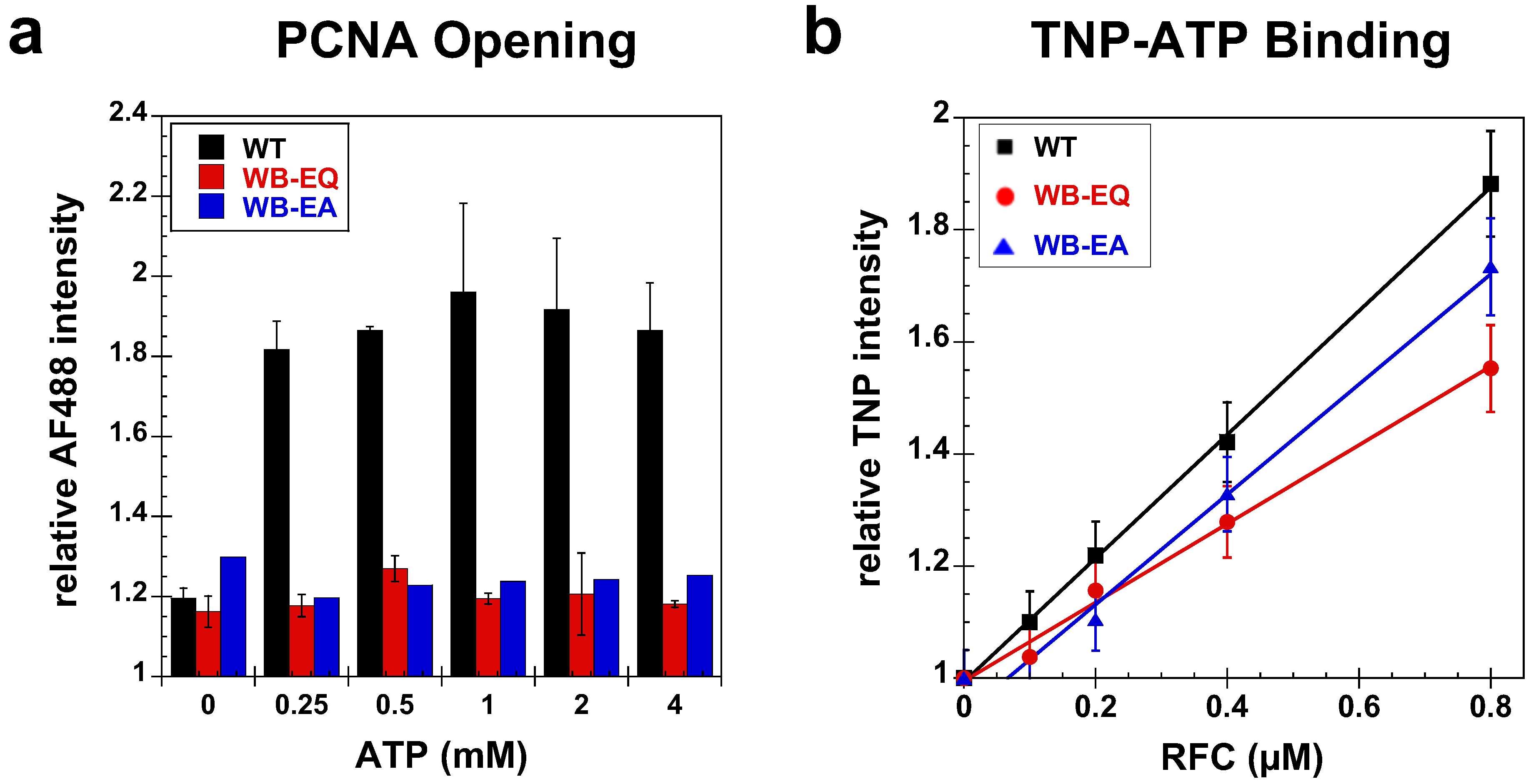
2.2.2. PCNA Opening

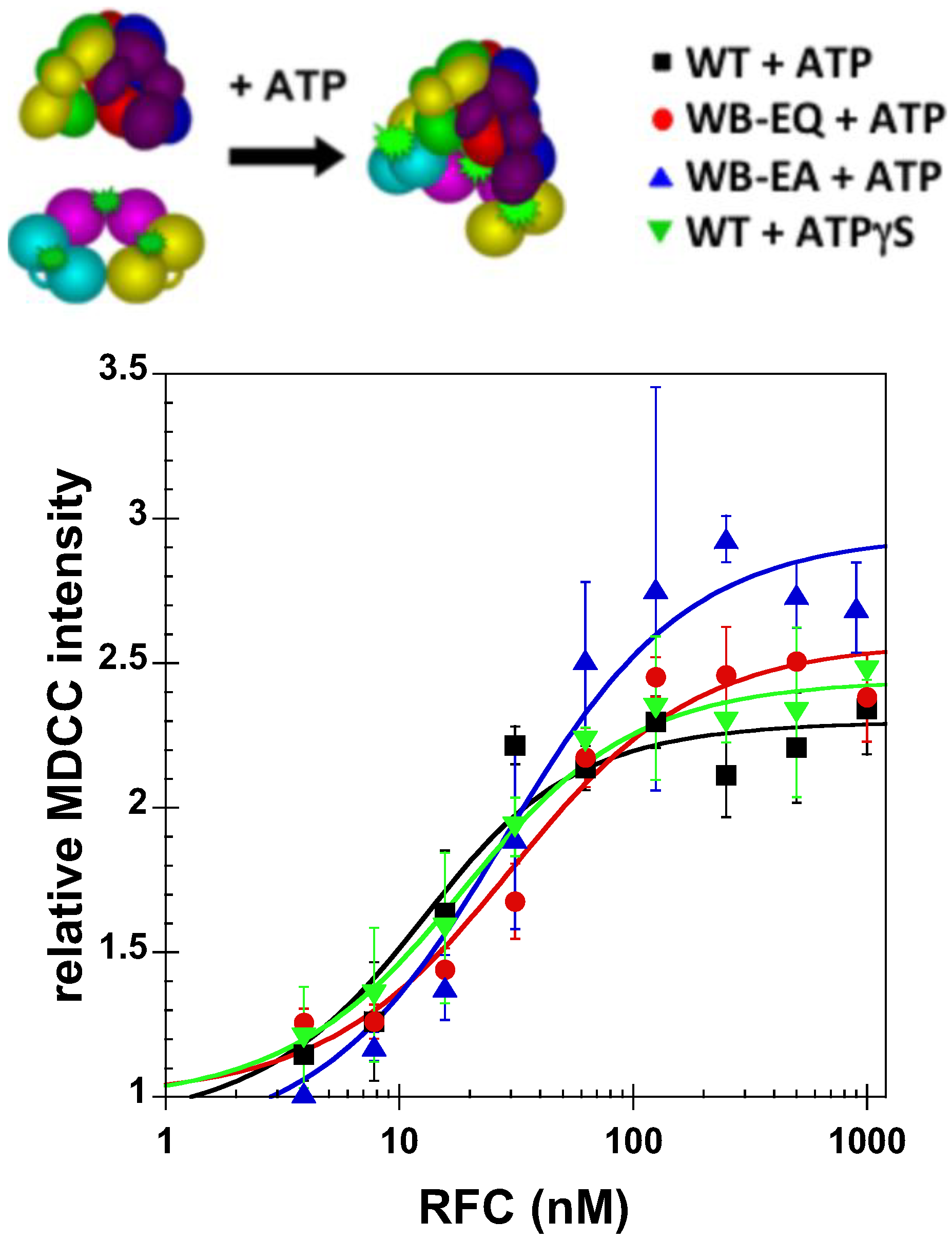
2.3. Measurement of ATP Binding
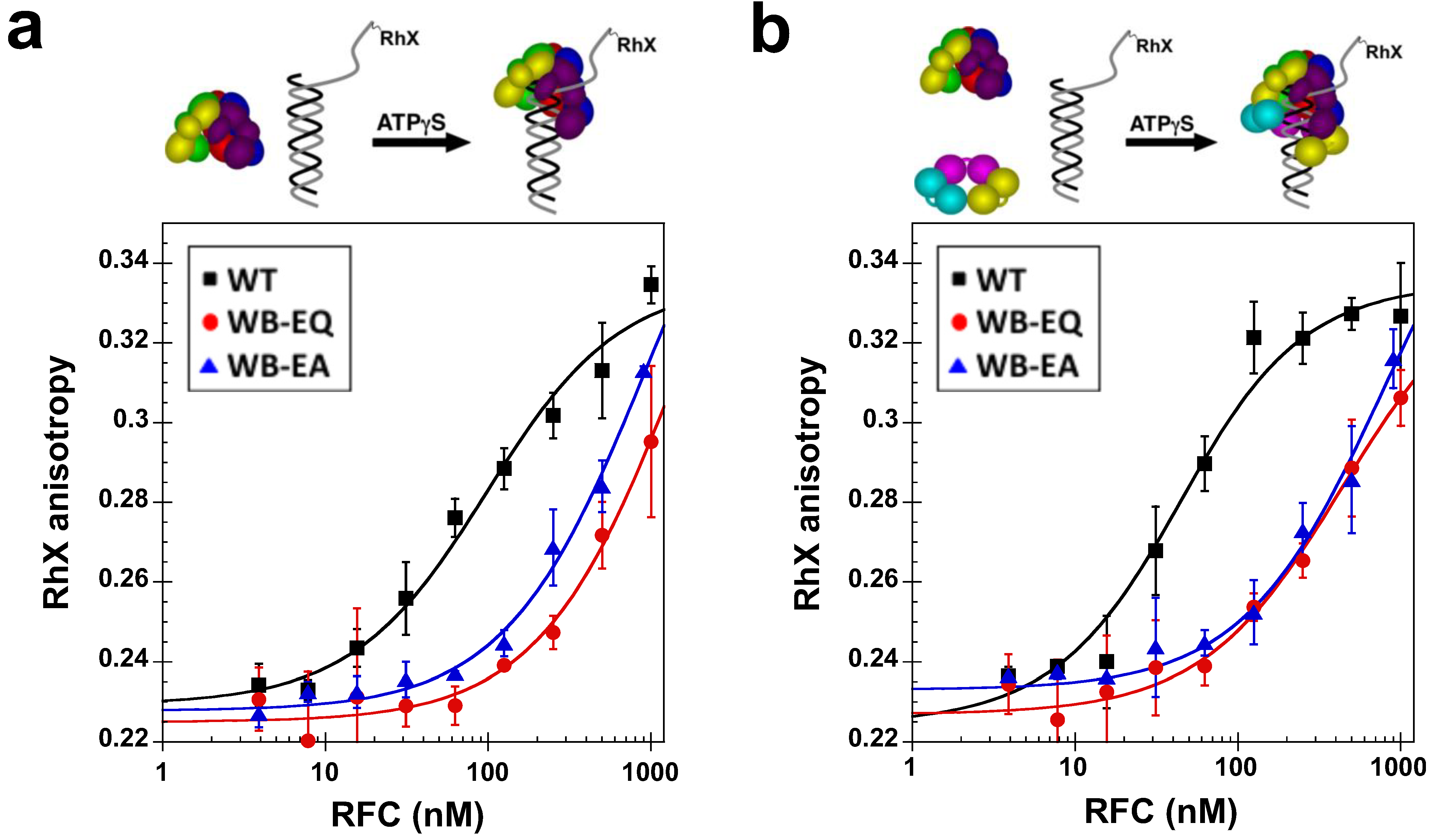
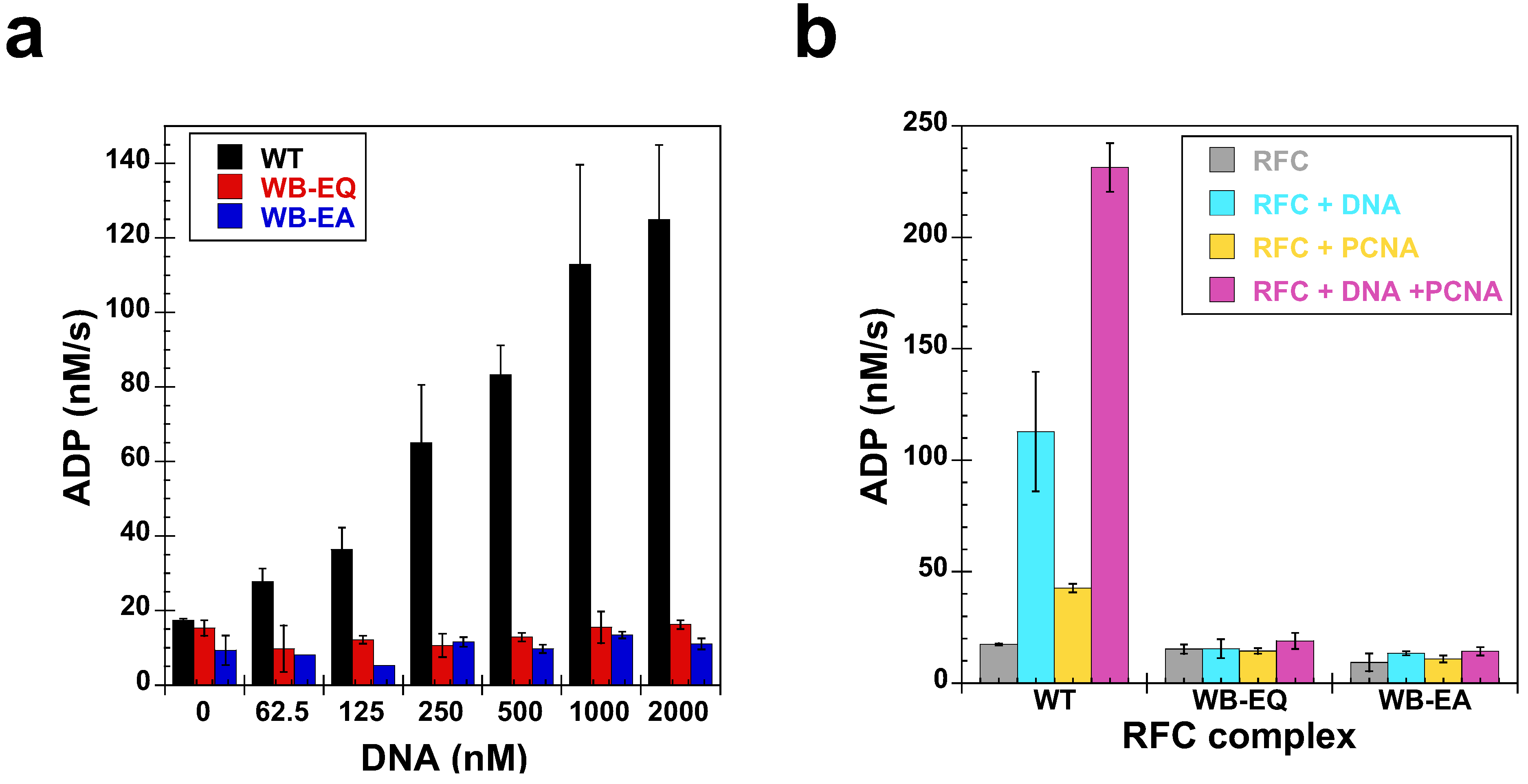
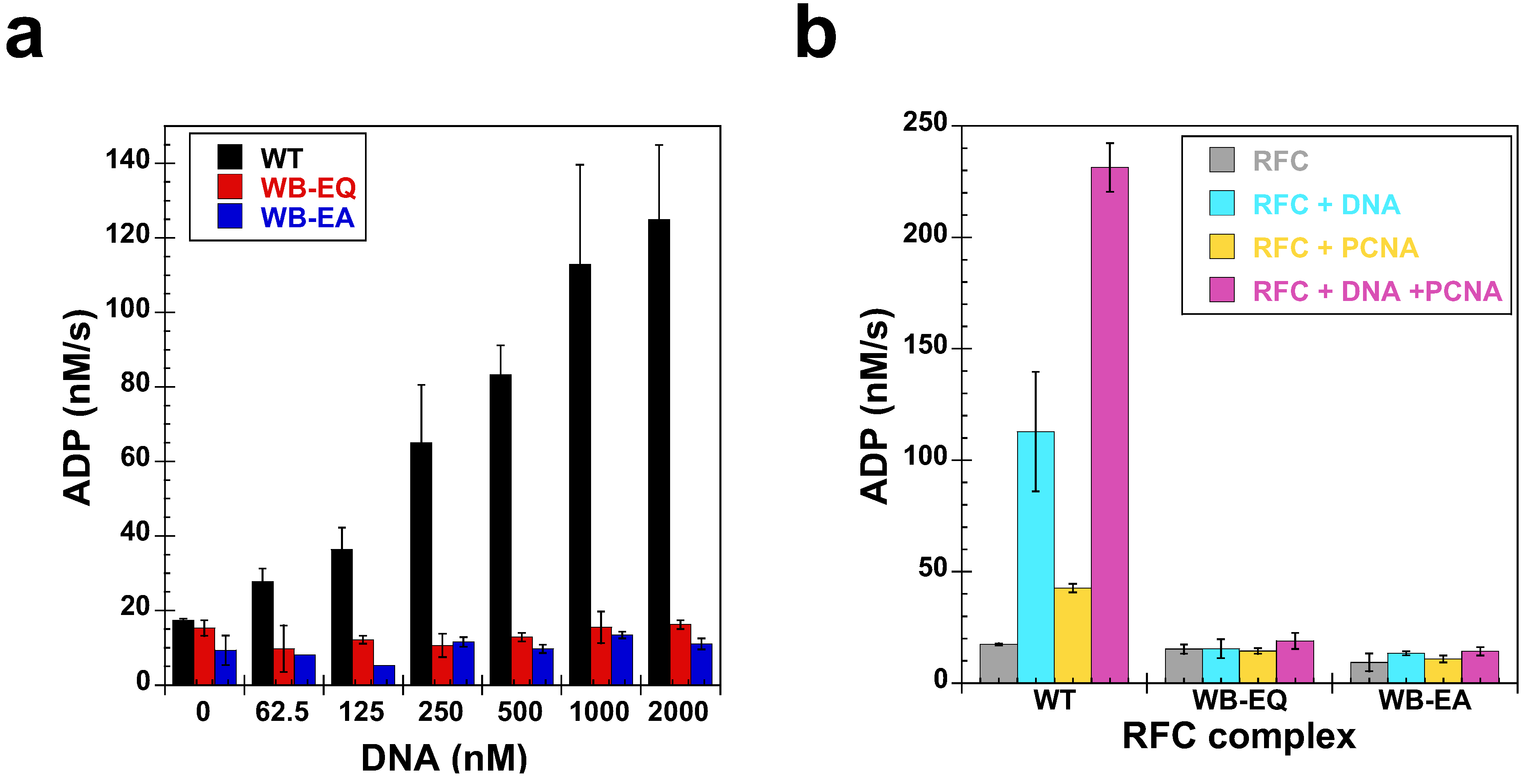
2.4. Measurement of DNA Binding
2.5. Measurement of ATP Hydrolysis
3. Experimental Section
3.1. Buffers
3.2. Construction of RFC Expression Vectors

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RFC Complex | Expression Vector | Coding Sequences |
|---|---|---|
| Wild-type RFC | pLANT/RIL | RFC1 and RFC5 |
| pET-11 | RFC2, RFC3, and RFC4 | |
| RFC WB-EQ | pLANT/RIL | RFC1 (E425Q) and RFC5 |
| pET-11 | RFC3 (E118Q) and RFC4(E115Q) | |
| pCDFDuet | RFC2 (E141Q) | |
| RFC WB-EA | pLANT/RIL | RFC1(E425A) and RFC5 |
| pET-Duet1 | RFC3(E118A) and RFC4(E115A) | |
| pCDFDuet | RFC2(E141A) |
3.3. Proteins
3.4. Oligonucleotides
3.5. Equilibrium PCNA Binding and Opening Measurements
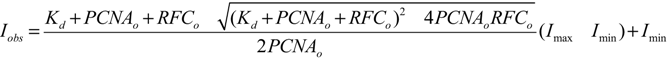
3.6. TNP-ATP Binding Measurements
3.7. DNA Binding Assay
3.8. ATP Hydrolysis Assay
4. Conclusions
Acknowledgments
References and Notes
- Davey, M.J.; Jeruzalmi, D.; Kuriyan, J.; O’Donnell, M. Motors and switches: Aaa+ machines within the replisome. Nat. Rev. Mol. Cell. Biol. 2002, 3, 826–835. [Google Scholar] [CrossRef]
- Duderstadt, K.E.; Berger, J.M. Aaa+ atpases in the initiation of DNA replication. Critical Rev. Biochem. Mol. Biol. 2008, 43, 163–187. [Google Scholar] [CrossRef]
- Kong, X.-P.; Onrust, R.; O’Donnell, M.; Kuriyan, J. Three-dimensional structure of the β subunit of E. Coli DNA polymerase iii holoenzyme: A sliding DNA clamp. Cell 1992, 69, 425–437. [Google Scholar] [CrossRef]
- Stukenberg, P.T.; Studwell-Vaughan, P.S.; O’Donnell, M. Mechanism of the sliding b-clamp of DNA polymerase iii holoenzyme. J. Biol. Chem. 1991, 266, 11328–11334. [Google Scholar]
- Gomes, X.V.; Burgers, P.M. Atp utilization by yeast replication factor c. I. Atp-mediated interaction with DNA and with proliferating cell nuclear antigen. J. Biol. Chem. 2001, 276, 34768–34775. [Google Scholar] [CrossRef]
- Gomes, X.V.; Schmidt, S.L.; Burgers, P.M. Atp utilization by yeast replication factor c. II. Multiple stepwise atp binding events are required to load proliferating cell nuclear antigen onto primed DNA. J. Biol. Chem. 2001, 276, 34776–34783. [Google Scholar]
- Hingorani, M.M.; O’Donnell, M. Atp binding to the Escherichia coli clamp loader powers opening of the ring-shaped clamp of DNA polymerase iii holoenzyme. J. Biol. Chem. 1998, 273, 24550–24563. [Google Scholar] [CrossRef]
- Naktinis, V.; Onrust, R.; Fang, F.; O’Donnell, M. Assembly of a chromosomal replication machine: Two DNA polymerases, a clamp loader, and sliding clamps in one holoenzyme particl. II. Intermediate complex between the clamp loader and its clamp. J. Biol. Chem. 1995, 270, 13358–13365. [Google Scholar] [CrossRef]
- Tsurimoto, T.; Stillman, B. Replication factors required for sv40 DNA replication in vitro. I. DNA structure-specific recognition of a primer-template junction by eukaryotic DNA polymerases and their accessory proteins. J. Biol. Chem. 1991, 266, 1950–1960. [Google Scholar]
- Bertram, J.G.; Bloom, L.B.; Hingorani, M.M.; Beechem, J.M.; O’Donnell, M.; Goodman, M.F. Molecular mechanism and energetics of clamp assembly in Escherichia coli. The role of atp hydrolysis when γ complex loads β on DNA. J. Biol. Chem. 2000, 275, 28413–28420. [Google Scholar]
- Hingorani, M.M.; Bloom, L.B.; Goodman, M.F.; O’Donnell, M. Division of labor-sequential atp hydrolysis drives assembly of a DNA polymerase sliding clamp around DNA. EMBO J. 1999, 18, 5131–5144. [Google Scholar] [CrossRef]
- Erzberger, J.P.; Berger, J.M. Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu. Rev. Biophys. Biomol. Struct. 2006, 35, 93–114. [Google Scholar] [CrossRef]
- Cullmann, G.; Fien, K.; Kobayashi, R.; Stillman, B. Characterization of the five replication factor c genes of saccharomyces cerevisiae. Mol. Cell Biol. 1995, 15, 4661–4671. [Google Scholar]
- Guenther, B.; Onrust, R.; Sali, A.; O’Donnell, M.; Kuriyan, J. Crystal structure of the delta' subunit of the clamp-loader complex of E. Coli DNA polymerase III. Cell 1997, 91, 335–345. [Google Scholar] [CrossRef]
- Jeruzalmi, D.; O’Donnell, M.; Kuriyan, J. Crystal structure of the processivity clamp loader gamma (γ) complex of E. Coli DNA polymerase III. Cell 2001, 106, 429–441. [Google Scholar] [CrossRef]
- Neuwald, A.F.; Aravind, L.; Spouge, J.L.; Koonin, E.V. Aaa+: A class of chaperone-like atpases associated with the assembly, operation, and disassembly of protein complexes. Genome Res. 1999, 9, 27–43. [Google Scholar]
- Walker, J.R.; Hervas, C.; Ross, J.D.; Blinkova, A.; Walbridge, M.J.; Pumarega, E.J.; Park, M.O.; Neely, H.R. Escherichia coli DNA polymerase III tau- and gamma-subunit conserved residues required for activity in vivo and in vitro. J. Bacteriol. 2000, 182, 6106–6113. [Google Scholar] [CrossRef]
- Bowman, G.D.; O’Donnell, M.; Kuriyan, J. Structural analysis of a eukaryotic sliding DNA clamp-clamp loader complex. Nature 2004, 429, 724–730. [Google Scholar] [CrossRef]
- Kelch, B.A.; Makino, D.L.; O’Donnell, M.; Kuriyan, J. How a DNA polymerase clamp loader opens a sliding clamp. Science 2011, 334, 1675–1680. [Google Scholar] [CrossRef]
- Gorbalenya, A.E.; Koonin, E.V.; Donchenko, A.P.; Blinov, V.M. Two related superfamilies of putative helicases involved in replication, recombination, repair and expression of DNA and rna genomes. Nucleic Acids Res. 1989, 17, 4713–4730. [Google Scholar] [CrossRef]
- Saraste, M.; Sibbald, P.R.; Wittinghofer, A. The p-loop—A common motif in atp- and gtp-binding proteins. Trends Biochem. Sci. 1990, 15, 430–434. [Google Scholar] [CrossRef]
- Walker, J.E.; Saraste, M.; Runswick, M.J.; Gay, N.J. Distantly related sequences in the alpha- and beta-subunits of atp synthase, myosin, kinases and other atp-requiring enzymes and a common nucleotide binding fold. EMBO J. 1982, 1, 945–951. [Google Scholar]
- Hodgman, T.C. A new superfamily of replicative proteins. Nature 1988, 333, 22–23. [Google Scholar] [CrossRef]
- Linder, P.; Lasko, P.F.; Ashburner, M.; Leroy, P.; Nielsen, P.J.; Nishi, K.; Schnier, J.; Slonimski, P.P. Birth of the d-e-a-d box. Nature 1989, 337, 121–122. [Google Scholar]
- Subramanya, H.S.; Bird, L.E.; Brannigan, J.A.; Wigley, D.B. Crystal structure of a dexx box DNA helicase. Nature 1996, 384, 379–383. [Google Scholar]
- Abrahams, J.P.; Leslie, A.G.; Lutter, R.; Walker, J.E. Structure at 2.8 a resolution of f1-atpase from bovine heart mitochondria. Nature 1994, 370, 621–628. [Google Scholar]
- Story, R.M.; Steitz, T.A. Structure of the reca protein-adp complex. Nature 1992, 355, 374–376. [Google Scholar] [CrossRef]
- Seybert, A.; Wigley, D.B. Distinct roles for atp binding and hydrolysis at individual subunits of an archaeal clamp loader. EMBO J. 2004, 23, 1360–1371. [Google Scholar] [CrossRef]
- Kim, D.M.; Zheng, H.; Huang, Y.J.; Montelione, G.T.; Hunt, J.F. Atpase active-site electrostatic interactions control the global conformation of the 100 kda seca translocase. J. Am. Chem. Soc. 2012, 135, 2999–3010. [Google Scholar]
- Bowman, G.D.; Goedken, E.R.; Kazmirski, S.L.; O’Donnell, M.; Kuriyan, J. DNA polymerase clamp loaders and DNA recognition. FEBS Lett. 2005, 579, 863–867. [Google Scholar] [CrossRef]
- Simonetta, K.R.; Kazmirski, S.L.; Goedken, E.R.; Cantor, A.J.; Kelch, B.A.; McNally, R.; Seyedin, S.N.; Makino, D.L.; O’Donnell, M.; Kuriyan, J. The mechanism of atp-dependent primer-template recognition by a clamp loader complex. Cell 2009, 137, 659–671. [Google Scholar] [CrossRef]
- Fotedar, R.; Mossi, R.; Fitzgerald, P.; Rousselle, T.; Maga, G.; Brickner, H.; Messier, H.; Kasibhatla, S.; Hubscher, U.; Fotedar, A. A conserved domain of the large subunit of replication factor c binds pcna and acts like a dominant negative inhibitor of DNA replication in mammalian cells. EMBO J. 1996, 15, 4423–4433. [Google Scholar]
- Halligan, B.D.; Teng, M.; Guilliams, T.G.; Nauert, J.B.; Halligan, N.L. Cloning of the murine cdna encoding vdjp, a protein homologous to the large subunit of replication factor c and bacterial DNA ligases. Gene 1995, 161, 217–222. [Google Scholar] [CrossRef]
- Gomes, X.V.; Gary, S.L.; Burgers, P.M. Overproduction in escherichia coli and characterization of yeast replication factor c lacking the ligase homology domain. J. Biol. Chem. 2000, 275, 14541–14549. [Google Scholar] [CrossRef]
- Podust, V.N.; Tiwari, N.; Stephan, S.; Fanning, E. Replication factor c disengages from proliferating cell nuclear antigen (pcna) upon sliding clamp formation, and pcna itself tethers DNA polymerase delta to DNA. J. Biol. Chem. 1998, 273, 31992–31999. [Google Scholar]
- Uhlmann, F.; Cai, J.; Gibbs, E.; O’Donnell, M.; Hurwitz, J. Deletion analysis of the large subunit p140 in human replication factor c reveals regions required for complex formation and replication activities. J. Biol. Chem. 1997, 272, 10058–10064. [Google Scholar]
- Finkelstein, J.; Antony, E.; Hingorani, M.M.; O’Donnell, M. Overproduction and analysis of eukaryotic multiprotein complexes in escherichia coli using a dual-vector strategy. Anal. Biochem. 2003, 319, 78–87. [Google Scholar] [CrossRef]
- Kelman, Z.; Yao, N.; O’Donnell, M. Escherichia coli expression vectors containing a protein kinase recognition motif, his6-tag and hemagglutinin epitope. Gene 1995, 166, 177–178. [Google Scholar] [CrossRef]
- Chen, S.; Levin, M.K.; Sakato, M.; Zhou, Y.; Hingorani, M.M. Mechanism of atp-driven pcna clamp loading by s. Cerevisiae rfc. J. Mol. Biol. 2009, 388, 431–442. [Google Scholar] [CrossRef]
- Yao, N.Y.; Johnson, A.; Bowman, G.D.; Kuriyan, J.; O’Donnell, M. Mechanism of proliferating cell nuclear antigen clamp opening by replication factor c. J. Biol. Chem. 2006, 281, 17528–17539. [Google Scholar]
- Zhuang, Z.; Yoder, B.L.; Burgers, P.M.; Benkovic, S.J. The structure of a ring-opened proliferating cell nuclear antigen-replication factor c complex revealed by fluorescence energy transfer. Proc. Natl. Acad. Sci. USA 2006, 103, 2546–2551. [Google Scholar]
- Marzahn, M.R.; Bloom, L.B. Improved solubility of replication factor c (rfc) walker a mutants. Protein Expr. Purif. 2012, 83, 135–144. [Google Scholar] [CrossRef]
- Thompson, J.A.; Marzahn, M.R.; O’Donnell, M.; Bloom, L.B. Replication factor c is a more effective proliferating cell nuclear antigen (pcna) opener than the checkpoint clamp loader, rad24-rfc. J. Biol. Chem. 2012, 287, 2203–2209. [Google Scholar]
- Hiratsuka, T. Fluorescence properties of 2' (or 3')-o-(2,4,6-trinitrophenyl) adenosine 5'-triphosphate and its use in the study of binding to heavy meromyosin atpase. Biochim. Biophys. Acta 1976, 453, 293–297. [Google Scholar] [CrossRef]
- Hiratsuka, T. New ribose-modified fluorescent analogs of adenine and guanine nucleotides available as substrates for various enzymes. Biochim. Biophys. Acta 1983, 742, 496–508. [Google Scholar] [CrossRef]
- Sakato, M.; O’Donnell, M.; Hingorani, M.M. A central swivel point in the rfc clamp loader controls pcna opening and loading on DNA. J. Mol. Biol. 2012, 416, 163–175. [Google Scholar] [CrossRef]
- Snyder, A.K.; Williams, C.R.; Johnson, A.; O’Donnell, M.; Bloom, L.B. Mechanism of loading the Escherichia coli DNA polymerase III sliding clamp: II. Uncoupling the β and DNA binding activites of the γ complex. J. Biol. Chem. 2004, 279, 4386–4393. [Google Scholar]
- Paschall, C.O.; Thompson, J.A.; Marzahn, M.R.; Chiraniya, A.; Hayner, J.N.; O’Donnell, M.; Robbins, A.H.; McKenna, R.; Bloom, L.B. The Escherichia coli clamp loader can actively pry open the beta-sliding clamp. J. Biol. Chem. 2011, 286, 42704–42714. [Google Scholar] [CrossRef]
- Schmidt, S.L.; Gomes, X.V.; Burgers, P.M. Atp utilization by yeast replication factor c. III. The atp-binding domains of rfc2, rfc3, and rfc4 are essential for DNA recognition and clamp loading. J. Biol. Chem. 2001, 276, 34784–34791. [Google Scholar] [CrossRef]
- Tsurimoto, T.; Stillman, B. Functions of replication factor c and proliferating-cell nuclear antigen: Functional similarity of DNA polymerase accessory proteins from human cells and bacteriophage t4. Proc. Natl. Acad. Sci. USA 1990, 87, 1023–1027. [Google Scholar] [CrossRef]
- Yoder, B.L.; Burgers, P.M. Saccharomyces cerevisiae replication factor c. I. Purification and characterization of its atpase activity. J. Biol. Chem. 1991, 266, 22689–22697. [Google Scholar]
- Yao, N.; Coryell, L.; Zhang, D.; Georgescu, R.E.; Finkelstein, J.; Coman, M.M.; Hingorani, M.M.; O’Donnell, M. Replication factor C clamp loader subunit arrangement within the circular pentamer and its attachment points to proliferating cell nuclear antigen. J. Biol. Chem. 2003, 278, 50744–50753. [Google Scholar] [CrossRef]
- Johnson, A.; Yao, N.Y.; Bowman, G.D.; Kuriyan, J.; O’Donnell, M. The replication factor c clamp loader requires arginine finger sensors to drive DNA binding and proliferating cell nuclear antigen loading. J. Biol. Chem. 2006, 281, 35531–35543. [Google Scholar]
- Perez-Howard, G.M.; Weil, P.A.; Beechem, J.M. Yeast tata binding protein interaction with DNA: Fluorescence determination of oligomeric state, equilibrium binding, on-rate, and dissociation kinetics. Biochemistry 1995, 34, 8005–8017. [Google Scholar] [CrossRef]
- Ason, B.; Bertram, J.G.; Hingorani, M.M.; Beechem, J.M.; O’Donnell, M.; Goodman, M.F.; Bloom, L.B. A model for Escherichia coli DNA polymerase III holoenzyme assembly at primer/template ends: DNA triggers a change in binding specificity of the γ complex clamp loader. J. Biol. Chem. 2000, 275, 3006–3015. [Google Scholar]
- Zhang, X.; Wigley, D.B. The “glutamate switch” provides a link between atpase activity and ligand binding in AAA+ proteins. Nat. Struct. Mol. Biol. 2008, 15, 1223–1227. [Google Scholar] [CrossRef]
- Neuwald, A.F. Bayesian shadows of molecular mechanisms cast in the light of evolution. Trends Biochem. Sci. 2006, 31, 374–382. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chiraniya, A.; Finkelstein, J.; O'Donnell, M.; Bloom, L.B. A Novel Function for the Conserved Glutamate Residue in the Walker B Motif of Replication Factor C. Genes 2013, 4, 134-151. https://doi.org/10.3390/genes4020134
Chiraniya A, Finkelstein J, O'Donnell M, Bloom LB. A Novel Function for the Conserved Glutamate Residue in the Walker B Motif of Replication Factor C. Genes. 2013; 4(2):134-151. https://doi.org/10.3390/genes4020134
Chicago/Turabian StyleChiraniya, Ankita, Jeff Finkelstein, Mike O'Donnell, and Linda B. Bloom. 2013. "A Novel Function for the Conserved Glutamate Residue in the Walker B Motif of Replication Factor C" Genes 4, no. 2: 134-151. https://doi.org/10.3390/genes4020134